 Ryan S. Lane1
Ryan S. Lane1 Amanda W. Lund1,2,3,4*
Amanda W. Lund1,2,3,4*- 1Department of Cell, Developmental and Cancer Biology, Oregon Health and Science University, Portland, OR, United States
- 2Department of Molecular Microbiology and Immunology, Oregon Health and Science University, Portland, OR, United States
- 3Department of Dermatology, Oregon Health and Science University, Portland, OR, United States
- 4Knight Cancer Institute, Oregon Health and Science University, Portland, OR, United States
In response to pathological challenge, the host generates rapid, protective adaptive immune responses while simultaneously maintaining tolerance to self and limiting immune pathology. Peripheral tissues (e.g., skin, gut, lung) are simultaneously the first site of pathogen-encounter and also the location of effector function, and mounting evidence indicates that tissues act as scaffolds to facilitate initiation, maintenance, and resolution of local responses. Just as both effector and memory T cells must adapt to their new interstitial environment upon infiltration, tissues are also remodeled in the context of acute inflammation and disease. In this review, we present the biochemical and biophysical mechanisms by which non-hematopoietic stromal cells and extracellular matrix molecules collaborate to regulate T cell behavior in peripheral tissue. Finally, we discuss how tissue remodeling in the context of tumor microenvironments impairs T cell accumulation and function contributing to immune escape and tumor progression.
Introduction
Immune surveillance and protective immunity is dependent upon sequential, rapid activation, and mobilization of hematopoietic cells that undergo multiple intercellular interactions to mediate immune control. Rather than being stochastic, these interactions are guided by non-hematopoietic cells that generate and maintain tissue scaffolds. The microenvironments through which leukocytes traffic (e.g., blood, lymphoid organs, peripheral tissues) differ significantly with respect to cellular and protein composition and remodel in the context of disease and with age. Thus, in order to perform their protective function, both effector and memory T cells must adapt to continuously changing physical, biochemical, and metabolic tissue environments.
De novo T cell priming is initiated in lymph nodes (LN) that drain peripheral sites of infection, inflammation, and tumors. Within lymphoid organs, non-hematopoietic cells direct cellular interactions and increase the probability of immune activation. Lymph-borne antigen is transported to LNs through afferent lymphatic vessels that connect to the subcapsular sinus allowing delivery of large particulate antigens (>70 kDa) to interfollicular dendritic cells (DC) and subcapsular macrophages (1, 2). Small antigens (<70 kDa) enter fibroblast reticular cell-lined (FRC) conduits and are sampled by resident DCs (3). Both the packing of collagen fibers within FRC-conduits and direct filtration by lymphatic endothelial cells (LEC)-lining the lymphatic sinus floor determine LN size exclusion properties and thus dictate antigen delivery (3, 4). While lymph flow is constitutive at steady state, lymphatic fluid transport is rapidly reduced following cutaneous infection, indicating that peripheral tissue context and lymphatic vessel function dictate antigen delivery (5).
Within LNs, non-hematopoietic stromal cells generate and maintain chemokine gradients to direct leukocyte recruitment and positioning. Afferent lymphatic vessels direct DC homing and express adhesion molecules that permit transendothelial migration, while specialized blood vessels, high endothelial venules, facilitate naïve lymphocyte entry. FRCs provide a physical scaffold within the paracortex, express homeostatic chemokines that bring mature, antigen-loaded DCs in close proximity with naive T cells (6), and modulate their contractile phenotype to permit LN enlargement and lymphocyte expansion (7). Following activation, T cells egress LNs along shingosine-1-phosphate (S1P) gradients actively maintained by efferent LECs and ultimately recirculate into blood (8). These newly T cell receptor (TCR)-stimulated effector T cells are now proficient to recognize inflamed blood endothelium in peripheral, non-lymphoid tissues and are restricted from re-entry into LNs (9). Importantly, while naïve T cells require TCR stimulation in lymphoid organs for activation, pre-existing memory T cells acquire tissue-homing capability independent of TCR-stimulation and are rapidly mobilized to sites of inflammation where they exert their protective function (5, 10, 11). Thus, though effector and memory cells are subject to the same peripheral tissue microenvironments and barriers upon arrival, the signals required to activate mechanisms of homing and tissue adaptation may be distinct (9).
Just as in secondary lymphoid organs, non-hematopoietic cells in peripheral, non-lymphoid tissue provide a functional scaffold that determines T cell infiltration, motility, effector function, and retention. Tissue remodeling in chronic diseases, such as cancer, significantly alters requirements for T cell behavior and function. Here we discuss the current state of knowledge regarding interaction between T cells and non-hematopoietic stromal components in peripheral, non-lymphoid tissue. How effector and memory T cells adapt within and navigate through these non-hematopoietic barriers is poorly understood, and yet the heterogeneity of tissue structure and function within which T cells impart immune control must necessitate an array of adaptive mechanisms. A more detailed understanding of mechanisms used by effector and memory T cells to adapt to their peripheral tissue environment will provide crucial insight into the ways in which solid tumors inhibit T cell function and mediate immune escape.
Getting in: T Cell Extravasation Across the Vascular Endothelium
Inflamed Endothelial Cells Provide Signal Two for Tissue Infiltration
Though activated effector and memory T cells acquire the machinery necessary for homing to inflamed tissue in response to TCR and inflammatory stimuli in circulation and lymphoid organs (9), activated vascular endothelial cells (EC) that line post capillary venules in tissue provide the critical signal 2 necessary for infiltration. Lymphocytes home to sites of inflammation following a cascade of adhesive and signaling events mediated by sequential ligation and activation of selectins, integrins, and chemokines on ECs. EC activation and expression of these necessary adhesive molecules occurs only at sites of inflammation, thus ensuring specific infiltration of inflamed tissue (12) and sparing normal, uninflamed tissues from unnecessary lymphocyte infiltration, such that ECs act as key determinants for the anatomic tissue distribution of stimulated lymphocytes (Figure 1).
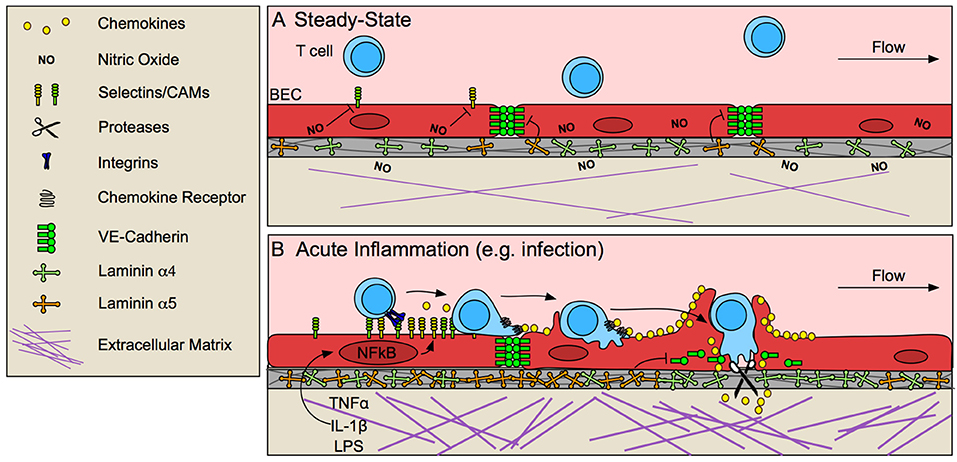
Figure 1. Blood endothelial cells control T cell entry into inflamed tissue. (A) The vascular endothelium limits T cell infiltration at steady-state by low expression of selectins and cell adhesion molecules (CAMs), and stabilized endothelial cell-cell junctions, due in part to tonic nitric oxide (NO) signaling and laminin α5-mediated VE-cadherin stabilization. (B) In response to pathological challenge and inflammatory stimulus (e.g., TNFα, IL-1β, LPS), blood endothelial cells (BECs) become activated and increase expression of selectins, CAMs and chemokines, which promote lymphocyte adhesion and migration to sites permissible for transmigration. In some cases, BECs form a transmigratory cup that provides a perpendicular scaffold to direct T cell transmigration. Inflammatory remodeling of the basement membrane contributes to lymphocyte access through destabilization of VE-cadherin at endothelial junctions and by generating low-density sites permissive to lymphocyte migration.
At steady-state, low levels of lymphocyte adhesion molecule expression (13, 14) is maintained by tonic nitric oxide signaling (15) and lack of inflammatory stimuli. In response to challenge, tissue-resident macrophages, mast cells, and damaged fibroblasts (16) produce tumor necrosis factor α (TNFα) and interleukin-1 (IL-1) (17, 18), which are sufficient to activate local but not systemic ECs (19). Activation of nuclear factor-κB (NF-κB) in ECs by these inflammatory stimuli upregulates P- and E-selectins, intracellular adhesion molecule 1 (ICAM-1), vascular cell adhesion protein 1 (VCAM-1), and chemokines, and EC-specific loss of NF-κB is sufficient to prevent lymphocyte infiltration into tissue (17). Selectins bind to carbohydrate moieties on glycoproteins expressed by effector and memory T cells (9). Selectin binding initiates T cell rolling along the inflamed endothelium (20), allowing for subsequent chemokine detection. Chemokines produced by ECs then direct actin-dependent spreading, polarization, and lateral migration of arrested lymphocytes across the endothelial surface, presumably to identify sites permissive to transmigration, marked by clustered cell adhesion molecule (CAM) expression. High-affinity adhesive interactions between ICAM-1 and VCAM-1 and their respective integrins (LFA-1/αLβ2, and VLA-4/α4β1) ultimately lead to lymphocyte arrest (18).
While the endothelium rapidly responds to inflammatory cues to recruit circulating lymphocytes, it may also inhibit T cell adhesion and migration under certain conditions. T cells have decreased adhesion to inflamed ECs co-cultured with dermal fibroblasts, but not fibroblasts isolated from synovial joints of rheumatoid arthritis patients (21), indicating that fibroblasts help to maintain the endothelial barrier to lymphocyte infiltration in healthy tissue while their dysfunction may promote disease. PEPITEM, a small peptide released from adiponectin-stimulated B cells, binds to cadherin-15 on ECs and triggers production and release of sphingosine 1 phosphate (S1P), which reduces T cell trafficking across endothelium (22), and low expression of adiponectin receptor on B cells is associated with chronic lymphocyte infiltration in diseases such as type 1 diabetes, rheumatoid arthritis, and aging (22).
Upon adhesion to inflamed endothelium, lymphocytes next traverse the endothelial barrier. Endothelial cells actively support and guide lymphocytes to sites permissive to transmigration while still maintaining barrier integrity via integrin-dependent mechanisms of actin remodeling (23). At sites of transmigration, ICAM-1/LFA-1 and VCAM-1/VLA-4 clustering forms an immunological synapse-like interaction between ECs and T cells (24), concentrating adhesion molecules into a ring structure (25). ECs often extend microvilli symmetrically around T cells to form a “transmigratory cup” (26) which further strengthens adhesion and provides a perpendicular scaffold to promote transmigration (24). Ultimately, T cells pass through the endothelium in one of two ways, either between ECs at intercellular junctions (paracellular route), or directly through individual ECs (transcellular route). Transcellular migration seems to be initiated by invadosome-like protrusions on lymphocytes (27). Paracellular migration, on the other hand, requires EC-mediated destabilization of vascular endothelial cadherin (VE-cadherin) at endothelial cell-cell junctions (28) and is further mediated by integrins, CAMs, and other adhesion molecules such as PECAM-1, JAM-1, and CD99 (18).
Destabilization of VE-cadherin at EC cell-cell junctions seems to be necessary for lymphocyte transmigration (29). ECs expressing a mutant form of VE-cadherin that is not endocytosed and therefore retained at cell-cell junctions, prevents lymphocyte recruitment to inflamed skin (28). Blockade of VE-cadherin stabilizing integrins, β1 and β3 (29) or dephosphorylation of tyrosine 731 by SHP-2 targets VE-cadherin for endocytosis and subsequently increases neutrophil transmigration in vitro (30). Interestingly, lymphocyte binding to ECs induces SHP-2-mediated VE-cadherin destabilization (30), indicating that lymphocyte adhesion may prime ECs to be permissive of transmigration. VE-cadherin is also cleaved by a disintegrin and metalloproteinase 10 (ADAM10) and tetrospanin 5 and 17, expressed by inflamed ECs, and EC-specific loss of ADAM-10 delays T cell, but not neutrophil or B cell, transmigration in vitro (31). Interestingly, proteolytically active leukocytes, such as neutrophils, may mediate cleavage necessary for lymphocyte transmigration in the absence of EC proteolysis (31). Thus, even if not intrinsically proteolytic, leukocyte protease activity may positively promote lymphocyte transmigration across inflamed endothelium in vivo.
Antigen-Dependence of T Cell Recruitment and Extravasation
Peripheral effector (12) and memory T cells (11) are recruited to inflamed tissue in an antigen-independent manner, indicating that local presentation of cognate antigen is not necessary for tissue infiltration. The antigen-independence of T cell recruitment is exemplified by recent studies that demonstrate abundant bystander, pathogen-specific T cells, in solid tumors (32). Interestingly, however, homing of insulin-specific CD8+ T cells to pancreatic islets, but not other tissues, is reduced with loss of major histocompatibility complex class I (MHC-I) in vivo (33) and antigen-loaded MHC-I presented on luminal surfaces of the blood-brain barrier was functionally required for antigen-specific T cell trafficking to the brain (34). These observations have led to the hypothesis that antigen presentation by ECs may amplify antigen-specific T cell recruitment in certain tissues and disease states. ECs dynamically express MHC-I and MHC-II during inflammatory processes and possess antigen-processing machinery necessary for cross-presentation of exogenous antigens (35). Human ECs scavenge and cross-present the type I diabetes islet autoantigen GAD65 on MHC-II and this enhances the transmigration of antigen-specific T cells in vitro (36). Further in vitro evidence supports both inhibitory (37) and promotional (38, 39) roles for EC antigen presentation in lymphocyte trafficking, indicating that antigen-presented by ECs may provide context-dependent “go” or “stop” signals that tune T cell infiltration.
Interestingly, ECs express a variety of T cell costimulatory and coinhibitory molecules (24), and as such, may represent semi-professional APCs strategically placed to interact with activated effector and memory T cell populations. In addition to tuning transmigration, data from various tissues indicate that ECs may employ their repertoire of immune checkpoints and APC-like function to mediate peripheral tolerance and modify T cell behavior as they transmigrate or arrest at the vascular interface. For example, liver sinusoidal endothelial cells scavenge and cross-present food-borne antigens and induce tolerance through T cell adhesion and sequestration in the liver (40, 41), and tumor-associated LECs cross-present exogenous antigens (42, 43) and maintain peripheral tolerance to self-antigens in LNs (44, 45) dependent on constitutive expression of programmed death-ligand 1 (PD-L1) (45). The relative significance of EC antigen presentation in vivo, however, is likely both tissue and disease specific. Further testing is needed and specifically, EC-specific knockdown strategies, to determine the functional relevance of EC antigen-processing and presentation in vivo.
Overcoming the Basement Membrane
The final and rate-limiting step in lymphocyte extravasation is crossing the basement membrane (46). The basement membrane is a 20–200 nm thick dense proteinaceous substrate composed of laminins, collagen type IV, and sulfated proteoglycans (47), that separates the vascular endothelium from extracellular matrix (ECM) in the tissue parenchyma. Laminins and collagen IV produced by ECs self-assemble into a dense sheet that is crosslinked by perlecans and nidogen and contains 2–5 μm-in-diameter pore-like regions of low protein density (48), presumed sites of lymphocyte passage. Basement membrane composition differs between developmental stage, vessel type, and activation state of the endothelium (47), with particular variability of laminin isoforms. Laminins are composed of alpha, beta, and gamma chains (e.g., laminin α4, β1, γ1 is denoted as laminin 411), and presence in basement membrane is context and location dependent. In the central nervous system (CNS), laminin α4 is ubiquitous (49), while laminin α5 expression is patchy and irregular (50), but both are increased upon inflammation (47, 51). In murine experimental autoimmune encephalomyelitis extravasation occurs predominantly at sites of low laminin α5 density (29, 51) and laminin α5 is sufficient to inhibit T cell transmigration in a dose-dependent manner in vitro (50). Additionally, laminin α4-deficient mice increase expression of laminin α5 in the CNS leading to decreased T cell migration across the blood brain barrier in EAE (50), suggesting that the composition of laminins in the basement membrane may selectively regulate T cell transmigration.
The mechanisms by which different laminin isoforms regulate T cell transmigration are unclear. Laminin α5 binds to integrin β1 and β3 on ECs and stabilizes VE-cadherin at EC junctions (29). Activated lymphocytes also express integrin β1 (18), however, and it is possible that laminin α5 may signal directly to infiltrating lymphocytes and instruct transmigration, although this has not been investigated. Regardless of how T cells get across the EC layer, the basement membrane is a dense, proteinaceous barrier that they must penetrate to complete diapedesis. Neutrophils express elastase to remodel regions of low basement membrane density allowing for their tissue infiltration (52), however, the specific mechanisms of lymphocyte migration through the basement membrane is unclear. The small size and pliability of lymphocytes and their nuclei may permit movement through the 2–5 μm pore-like regions of the basement membrane. However, T cell intrinsic loss of granzyme B (GrzB), which degrades both collagenous and non-collagenous ECMs (53, 54), reduces extravasation in vivo (54), indicating proteolysis may be required for basement membrane penetration. Further studies are needed to evaluate the contribution of the basement membrane to selective lymphocyte extravasation in acute and chronically inflamed tissue.
Moving Around: T Cell Interstitial Migration in Homeostatic and Inflamed Peripheral Tissues
Following extravasation from the vasculature, effector and memory T cells encounter the complex heterogenous interstitial matrix through which they must traverse and locate target cells (Figure 2). The ECM defines the 3D structure of tissues and exhibits heterogeneity across tissue types and disease states. The topography of the interstitial matrix is determined by the combination of structural and non-structural glycoproteins such as fibrillar collagen, elastin, fibronectin, laminin, and tenascin, decorated by associating proteoglycans (e.g., decorin and versican), which contain glycosaminoglycans (GAGs; e.g., heparan sulfate and chondroitin sulfate). The physical spacing and orientation of fibrillar proteins, as well as net charge of decorating GAGs, determines matrix porosity, rigidity, and bioactive molecule presentation. Both the physical and molecular properties of the ECM determine necessary modes of leukocyte migration and thus the efficiency with which leukocytes survey tissue. Compared to innate immune cells (e.g., neutrophil and DC), the mechanisms that govern the interstitial motility and homing behavior of T cells are poorly defined, yet adaptation to and utilization of chemical and physical signals in heterogenous tissues is necessary to rapidly identify rare APCs and mediate their local effector function. Tissue biophysics, matrix rigidity and interstitial fluid flux, may be an important control point for tissue T cell dysfunction. Whether T cell motility is simply a function of the existing microenvironment, or if rather T cells may exert force within interstitial tissues to direct their movement is critical to understanding diseases where T cell infiltration is impaired, such as cancer.
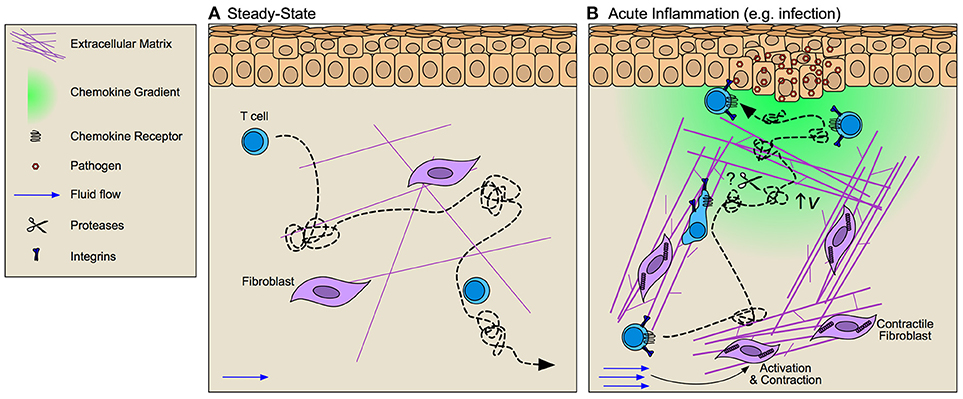
Figure 2. Interstitial matrices control T cell migration through inflamed tissue. (A) At steady-state T cells exhibit integrin-independent amoeboid-like Lévy walk behavior (dotted line) that facilitates their surveillance of peripheral tissue. (B) During inflammation fibroblast activation alters tissue tension through increased deposition, bundling, and cross-linking of extracellular matrix (ECM) components thereby altering the scaffold across which T cells must migrate. Increased interstitial fluid flows that result from vascular permeability further activate fibroblasts and promote directional fiber alignment. These inflammation-induced ECM changes activate integrin-dependent lymphocyte migration along collagen bundles. Whether T cell migration is dependent upon ECM organization or if rather lymphocytes may activate proteolytic activity to promote tissue invasion remains largely unclear. Facilitating T cell position at the site of challenge are chemokine gradients (e.g., CXCL9/10) that increase lymphocyte velocity (v) thereby improving their Lévy walk search efficiency and permitting accumulation at and around target cells.
Distinct Mechanisms of T Cell Interstitial Migration in Naïve and Inflamed Tissue
Molecular mechanisms of T cell interstitial migration (55, 56) have largely been determined in the context of 2D and 3D in vitro experimental systems that allow for control of physical and chemical cues to determine specific effects on T cell motility. These studies indicate that in naïve matrices lymphocytes primarily utilize amoeboid modes of movement independent of focalized adhesions and pericellular proteolysis (57, 58). Nuclear squeezing and deformation permits lymphocyte movement along preformed structures at velocities 10–40 fold higher than adhesion-dependent, mesenchymal migration (59). These studies have importantly established guiding principles for understanding T cell behavior in 3D, however, fail to capture the full complexity of inflamed interstitial matrices, which exhibit altered collagen fiber density, orientation, and composition relative to naïve in a tissue-specific and challenge-specific manner. Highlighting the discrepancies found between naïve matrices in vitro and inflamed tissue in vivo, intravital imaging in inflamed skin indicate that Th1 T cell motility is integrin-dependent, where specifically αv integrins (paired either with β1 or β3) facilitate T cell motility along matrix fibers (60). Matrix remodeling, therefore, necessitates T cell adaptation and activation of adhesion-dependent modes of migration. Importantly, effector and memory T cells do express an array of matrix-binding integrins that increase in expression upon activation and provide ligand specificity for various matrix components (61). Furthermore, T cells extracted from gut are more adhesive to ECM in vitro than circulating lymphocytes (62) and CD4+ effector T cells display distinct integrin repertoires when extracted from skin or lung, consistent with the differences of ECM composition in each tissue (60). Whether T cells tune their integrin repertoire in the context of their resident ECM, are imprinted at priming to prefer certain matrices, or a combination of both, remains an open question.
Interestingly, intravital imaging largely demonstrates that T cells preferentially follow pre-formed networks of fibrillar structures (55, 60, 63). 3D confinement studies in vitro indicate that limiting the space through which T cells can migrate significantly alters migratory speeds (64) and thus within a heterogenous interstitial matrix, it has been proposed that T cells may simply follow the path of least resistance (65). While in healthy tissue, pre-formed ECM tracks may facilitate the directional migration of T cells toward their target, under pathological conditions such as fibrosis and cancer, increased ECM density and rigidity (66) may act as a barrier to T cell infiltration and motility (discussed in more detail later). Consistent with this hypothesis, dynamic imaging reveals that matrix fiber density and orientation at tumor borders directs peripheral but not intratumoral T cell motility (63), and liver fibrosis inhibits T cell-mediated killing of infected hepatocytes (67), suggesting that T cells are incapable of proteolytically invading dense matrix structures.
Peripheral T cells, however, do express a limited array of proteases upon antigen stimulation and cytokine exposure. Matrix metalloproteinase 2 (MMP-2) is upregulated following VCAM-dependent adhesion to ECs (68), likely facilitating T lymphocyte invasion across the basement membrane. Inflammatory mediators [e.g., CCL5, prostaglandin E2, leukotriene B4 (LTB4), TNFα, transforming growth factor β (TGFβ), and IL-2] induce MMP-9 expression while type I and II interferons (IFN) are suppressive (68). Granzyme B, secreted by activated T cells, has protease activity (53, 54) and urokinase degrades laminins and fibronectin and activates some latent form of MMPs (69). Importantly, evidence for protease-dependent tissue invasion of T cells was identified in rheumatoid arthritis patients. T cells isolated from peripheral blood of rheumatoid arthritis patients display increased invasive capacity in vitro and in vivo dependent upon elevated expression Tks5, a scaffold protein required for the formation of matrix-degrading invadopodia (70). Thus, at least in some diseased states, T cells may activate intrinsic proteolysis that supports their pathologic activity.
While the ECM provides an instructional scaffold upon which leukocytes migrate through the interstitium, diffuse matrix-bound chemokine gradients are proposed to determine positioning and directionality. Intravital imaging, however, fails to identify persistent and directional T cell homing in vivo and T cells rather exhibit a Lévy walk motility pattern characterized by straight runs at fixed velocity interspersed with pauses (71). Interestingly, blockade of the CXCR3 ligand, CXCL10, in a model of Toxoplasma infection of CNS resulted in reduced CD8+ T cell velocity but did not alter Lévy walk patterns, indicating that CXCL10 may function to improve protective immunity by increasing T cell speed rather than through directional migration (71). Thus, reduced velocity impairs searching efficiency and reduces lymphocyte recruitment to and positioning at infectious foci. Consistently, CXCR3 expression on T cells was dispensable for vascular extravasation but required for localization to infected foci in cutaneous vaccinia infection (72). Continued understanding of the ways in which chemokines affect T lymphocyte behavior in tissues requires further intravital imaging studies to directly assay the dynamic behavior of T cells in the presence or absence of specific chemoattractant and chemorepellant molecules.
Organization of the Interstitial Matrix by Fluid Flows
Constitutive, interstitial fluid flows are kept in constant motion by hydrostatic and osmotic pressure differences between blood vessels, the interstitium, and lymphatic vessels (73). Interstitial fluid flow is slow, ranging from 0.1–2 μm/s at steady state and increased during inflammation and in tumors (73). Lymphatic vessels dynamically regulate fluid transport and consequently influence levels of interstitial fluid flow, which may in turn affect interstitial immunity. Cutaneous lymphatic vessels rapidly shut down fluid transport in a type I IFN-dependent manner preventing passive viral dissemination to LNs (5). Furthermore, mosquito saliva is sufficient to induce local edema and an inflammatory influx of neutrophils that when delivered in combination with virus, promotes viral retention at the bite site and enhances infection (74). Thus, modulation of fluid transport phenomena (either through enhanced vascular leakiness or altered lymphatic transport) may be a critical feature of tissue infection, which remains to be explored more carefully.
Dynamic regulation of fluid flux through tissue impacts ECM density, stiffness, and alignment and thus the scaffold within which cell motility is directed. Increased interstitial fluid pressure activates fibroblasts through integrin signaling and TGF-β upregulation of α-smooth muscle actin (αSMA) thereby increasing fibroblast contractility and subsequent alignment and bundling of collagen fibers (75). Flow additionally influences the expression and distribution of soluble and matrix bound factors along this scaffold to inform interstitial cell motility. Pericellular gradients can be established via multiple mechanisms including proteolytic release from ECM, degradation, metabolism, or removal by decoy receptors of a local source of attractants (76). Additionally, interstitial fluid flow introduces directional bias in chemokine distribution when expressed by migrating cells setting up functional gradients in the direction of flow. Termed autologous chemotaxis, this mechanism may support directional migration of tumor cells (77) and DCs to draining peripheral lymphatic vessels. How interstitial fluid flows influence T cell motility either directly, or indirectly remains to be experimentally tested.
Getting Out: T Cell Egress via Lymphatic Vessels
Lymphocytes Exit Peripheral, Non-Lymphoid Tissues
Following entry into and surveillance of tissue at least a subset of T cells continue on and egress out through lymphatic vessels (Figure 3). Parabiosis experiments demonstrate that most endogenous memory T cells in peripheral tissue reach equilibrium with migratory blood-borne donor T cells indicating rapid turnover in peripheral tissue (78) [with the notable exception of resident memory lymphocytes 79). In sheep, where lymph can be readily sampled, afferent lymph contains 106 cells/ml (80). Furthermore, the number of leukocytes in lymph are increased by sometimes as much as 100 fold during acute and chronic inflammatory signals (80), indicating tissue egress is influenced by context. Whether the cellular component of afferent lymph is simply a reflection of the tissue it drains (e.g., passive, random transport) or rather represents a subset of tissue lymphocytes (e.g., active, selective transport) remains largely unknown.
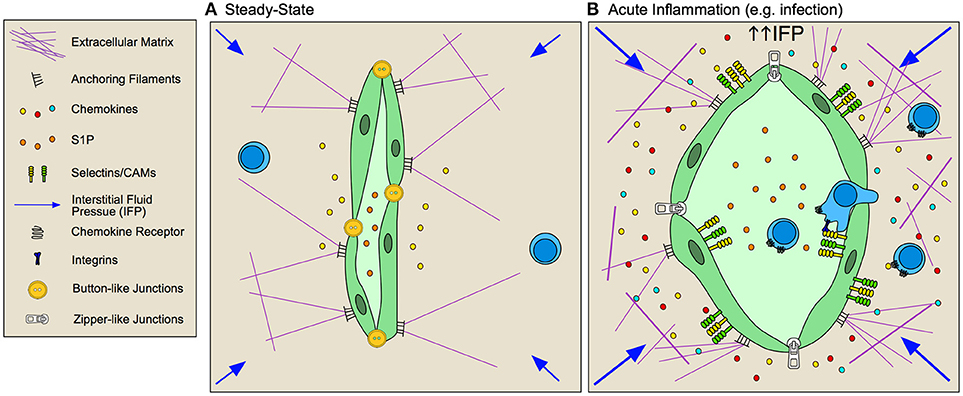
Figure 3. Inflamed lymphatic vessels promote lymphocyte exit from tissue. (A) Steady state lymphatic endothelial cells (LEC) form loose button-like junctions in lymphatic capillaries, and constitutively transport interstitial fluid from peripheral non-lymphoid tissue to draining lymph nodes. Production of homoestatic chemokines, such as CCL21, supports immune surveillance by directing leukocyte homing toward lymphatic vessels and tissue egress. (B) During inflammation, lymphatic vessels respond to both biochemical and biophysical stimuli to adapt their function within the tissue. Lymphatic vessels are activated by inflammatory cytokines and elevated interstitial fluid flow resulting in increased expression of selectins, cell adhesion molecules (CAMs) and context-dependent chemokine secretion that together promotes lymphocyte egress from inflamed tissues. LECs remodel their junctions, going from loose, button-like junctions to tight, zipper-like junctions, which is associated with decreased fluid transport from inflamed tissues and subsequent increased interstitial fluid pressures.
At least two candidate molecules have been proposed as necessary signals for lymphatic directed egress, CCL21/CCR7 and S1P/S1PR1. In models of acute lung and skin infection, T cells egress from inflamed peripheral tissue in a CCR7-dependent manner (81, 82), such that CD4+ T cells accumulate in epithelial tissue of CCR7−/− mice in an age-dependent manner (83). Additionally, forced overexpression of the spingosine-1-phosphate receptor (S1PR1) prevents the establishment of tissue resident memory CD8+ T cells, suggesting that the inability to respond to S1P gradients maintained by lymphatic vessels is necessary for local retention (84). Treatment with FTY720 (S1PR1 agonist) only partially inhibits egress (85), but does improve CD69-deficient CD8+ T cell persistence in skin after HSV infection (86). Thus, this together with low levels of CCR7 and expression of the E-cadherin binding CD103 and β1 integrin may promote retention (12, 87, 88). Conversely, egress from chronically inflamed tissue is pertussis toxin sensitive but CCR7-independent (85), indicating a role for alternative G-protein coupled receptors. CXCR4 might represent an alternative mechanism of lymphatic vessel directed egress. DC trafficking is reduced but not completely eliminated in CCR7−/− mice, and inhibition of CXCR4 further reduced DC trafficking to draining LNs in a model of contact hypersensitivity (89), but evidence for CXCR4-dependent T cell egress is lacking (90). Studies using quantitative models that track endogenous populations of tissue-resident lymphocytes, either through intravital imaging or photoconvertible mice, remain necessary to provide clarity regarding the molecular mechanisms that determine the context-dependence and specificity of leukocyte egress from inflamed tissues.
Lymphatic Vessels Regulate Tissue Exit
To facilitate tissue exit, lymphatic vessels express an array of chemokines in a context-dependent manner. LECs constitutively express CCL21 (91, 92) and further increase expression during chronic lung inflammation (90) and acute inflammation in skin, but not treatment with complete Freund's adjuvent (CFA) (93). TNFα stimulation of LECs causes release of CCL21 stores (94) and de novo production of CCL21 (94) as well as a host of other chemokines including CCL20, CXCL5, CCL5, CXCL2, CX3CL1, and CCL2 (95). Additionally, in vitro analysis indicates that lipoteichoic acid, a component of gram-positive bacterial cell walls, induces TLR2-dependent expression of CXCL1, CXCL3, CXCL6, and CXCL8 (96). In vivo analysis of mRNA from LECs in inflamed skin confirms these in vitro results, and also identified several other chemokines expressed by LECs, including the CD8+ T cell-homing chemokines CXCL9 and CXCL10 (93), all together indicating that the chemokine repertoire produced by LECs in peripheral tissue is context dependent. Consequently, how this diverse repertoire of chemokines produced by inflamed LECs functionally regulates lymphocyte egress from tissue remains a largely open question.
LECs, like BECs, increase expression of the T cell adhesion molecules in response to local inflammation and interstitial fluid flows. ICAM, VCAM, and E-selectin are expressed on the LEC surface rapidly following peripheral challenge in vivo (5, 93, 95) and following stimulation in vitro (97). LFA-1 is necessary for naïve T cell egress from inflamed skin (98) and inhibition of vascular endothelial receptor-1 (CLEVER-1) and macrophage mannose receptor prevented T cell migration through afferent lymphatic vessels to draining LNs (99, 100). The requirement for integrins in LEC transendothelial migration in inflamed tissue may mirror the differential integrin requirement for DCs. While DCs in skin squeeze through overlapping, button-like junctions in naïve lymphatic capillaries (101), transmigration across inflamed vessels requires integrin-mediated adhesion (97). Interestingly, cutaneous viral infection (5) and tracheal bacterial infection (102) induces lymphatic capillary remodeling of naïve button-like junctions to tight, zipper-junctions, typically found in deeper collecting vessels. These reversible changes may generate a less permeable endothelium and thus determine the integrin dependence of cellular transport. The functional relevance of lymphocyte egress at both steady-state and during inflammation remains to be determined, and in particular whether lymphocytes exit tissue to mediate immune resolution or rather enter LNs for re-stimulation by professional APCs remains an open and interesting question.
Adaptation in Tumor Microenvironments
Though tumors were previously thought to be poorly immunogenic and not capable of activating an immune response, we now know that somatic mutations (generated by DNA instability and environmental challenge) generate neo-antigens that are sufficiently distinct from self, such that T cells are capable of expanding and directing tumor-specific killing. Thus, the accumulation of neo-antigens in tumors is likely a prerequisite to anti-tumor immunity across tumor types, and consistently, those tumors that exhibit highest somatic mutational burden, e.g., melanoma, exhibit good overall response to immune checkpoint blockade (103). Even in the presence of potent neo-antigens, however, some tumors still fail to respond to therapy and somatic mutational burden is not sufficient to predict T cell infiltration within and across tumor types (104). Thus, multiple overlapping mechanisms of immune suppression create a more complex immune landscape, such that, as discussed above, processes of T cell recruitment, retention, survival, and exit may underscore intratumoral T cell presence and thus influence response to therapy. Current efforts to define biomarkers that are predictive of response to immune checkpoint blockade reveal an array of factors from myeloid cells to the microbiome, that affect patient response. Here we will focus on the localization of T cells within and around the tumor parenchyma as one indicator of responsiveness and discuss how tumor-induced stromal remodeling may contribute to T cell distribution (Figure 4).
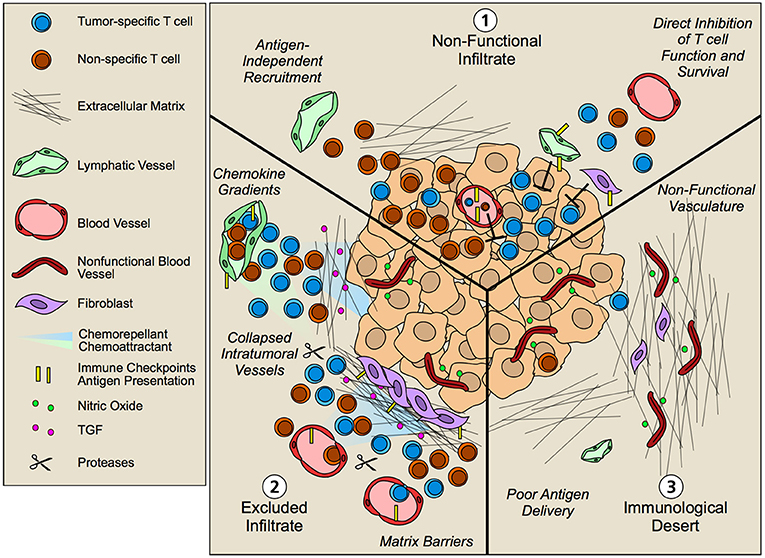
Figure 4. Non-hematopoietic cell contribution to tumor immune landscapes. The geographic distribution of T cells within intratumoral and peritumoral regions is both predictive of overall survival and response to immunotherapy. Patients that fail to respond to immunotherapy often exhibit three patterns of T cell infiltrate that are governed by an array of mechanisms including contributions from tumor cells and infiltrating hematopoietic cells. Non-hematopoetic cells, however, additionally contribute to the infiltration, retention, and function of T lymphocytes in tumor microenvironments. (1) Non-functional infiltrate: possessing an intratumoral but seemingly ineffective infiltrate. Antigen-independent recruitment of both effector and memory T cells subsets by vascular endothelium generates a diverse repertoire of T cells both relevant and irrelevant for tumor killing. Upon tissue entry, non-hematopoietic cells further exert multiple mechanisms of immune suppression, including expression of immune checkpoints such as PD-L1 and FasL that limit local T cell function. (2) Excluded infiltrate: possessing a T cell infiltrate that is restricted to the tumor periphery. Establishment of matrix barriers, collapsed intratumoral vessels, poor expression of adhesion molecules, and collaborating chemoattractant and chemorepellant gradients likely all contribute to the exclusion of T cells at the periphery of tumor nests such that inhibition of these features may improve infiltration. (3) Immunological desert: Completely lacking a T cell infiltrate in both tumor nests and stroma. Impaired lymphatic transport may result in poor antigen delivery to lymph nodes and thus poor priming. However, even in the presence of an activated systemic T cell pool, non-functional vessels driven by the angiogenic and desmoplastic tumor microenvironment may prevent local infiltration leading to lesion-specific differences in immune infiltrates.
Multiple studies across tumor types now indicate that the presence of T cells within tumor nests is predictive of response to therapy (105). As a consequence, non-responding tumors typically exhibit T cell infiltrates that are described by three main patterns: (1) non-functional immune responses, possessing an intratumoral but seemingly ineffective infiltrate; (2) tissue excluded T cell infiltrates, possessing a T cell infiltrate that is restricted to the tumor periphery; and (3) immunological deserts, completely lacking T cell infiltrate both in the tumor nests and in adjacent stromal (106). The underlying biology that regulates these patterns of T cell infiltration is clearly multifactorial—some of the contributing factors from the perspective of the non-hematopoietic tumor stroma are discussed below.
Non-functional T Cell Infiltrates
Non-functional immune infiltrates (106), refers to tumors containing intratumoral lymphocytes in both pre- and post-therapy biopsies that do not contribute to significant clinical response. Importantly, methods to evaluate intratumoral T cell populations largely quantify changes in bulk T cell populations (CD4 or CD8), and even when enriched for markers of previous antigen exposure (CD45RO) or effector function (GrzB) likely still quantify a heterogenous pool of effector, effector memory, and central memory T cells that represent a range of antigen specificities both relevant and irrelevant to the tumor. Rapid recruitment of effector and memory T cells is antigen-independent (11, 12), and bystander, viral-specific T cells (e.g., HCMV or EBV-specific) are abundant in human tumor tissue (32). Thus, efforts to specifically quantify tumor-reactive T cell clones may be more predictive than bulk T cell populations. Consistent with this hypothesis, CD39 was recently identified as a marker to distinguish tumor antigen-specific CD8+ T cells from bystander T cells across multiple tumor types (32, 107) and stratification of patients based on frequency of CD39+CD103+ double positive CD8+ T cells associated with increased overall survival in head and neck cancer patients (107). Thus, because of the promiscuity of T cell infiltration across the vascular endothelium, the presence of bulk T cells in tumor microenvironments may be insufficient to indicate response. Even when tumors are well infiltrated with antigen-specific T cells, however, multiple additional mechanisms suppress their local effector function mediated by tumor, hematopoietic (108), and non-hematopoietic stromal cells.
Aberrant tumor angiogenesis and disrupted fluid flows in tumor microenvironments generate hypoxia and increased interstitial fluid pressures in solid tumors (73, 109) that influence T cell function. Hypoxia induces Warburg effect by cancer cells, leading to increased acidification and lactate production, both of which inhibit cytotoxic activity of lymphocytes in vivo (110, 111). Furthermore, increased interstitial fluid flow in the tumor microenvironment activates fibroblasts leading to TGFβ production (73) and ECM contraction. ECM contraction together with shear stress activates stromal stores of latent TGFβ (112), which attenuates CD8+ T cell cytotoxicity (113) making them non-responsive to TCR signaling (114). Thus, the disrupted fluid mechanics within tumor tissue may itself participate in the regulation of local T cell function.
Furthermore, non-hematopoietic cells likely exert direct effects on T cells within tumor microenvironments. In the LN, LECs, and FRCs display specific immunological properties that function to maintain peripheral tolerance at steady state, and while we have drawn parallels between the structural role of LN stromal cells and non-hematopoietic cells in peripheral, non-lymphoid tissues, it remains less clear whether peripheral non-hematopoietic cells also acquire immunomodulatory properties characteristic of LN stroma. LN LECs express peripheral tissue antigens in an Aire-independent manner (44, 115); can scavenge and cross present exogenous antigens leading to dysfunctional CD8+ T cell activation (42, 43); and can receive peptide-MHC-II expressing exosomes from DCs and induce CD4+ T cell hyporesponsiveness (116). In tumors LECs are also capable of scavenging tumor-associated antigens and cross-presenting them on MHC-I (42), however, whether LEC antigen presentation functionally contributes to peripheral T cell responses remains unknown. LECs further are capable of inhibiting DC maturation and function (117) and T cell proliferation through the production of nitric oxide (118), demonstrating that LN-resident LECs inhibit T cell activation and proliferation both directly and indirectly. LN LECs also constitutively express PD-L1 and delete naïve, self-reactive CD8+ T cells (45) and peripheral BECs and LECs express PD-L1 in tumors (119–121) and infected tissue (121, 122). Loss of non-hematopoietic PD-L1 and inhibition of IFNγ signaling on peripheral LECs, thus preventing PD-L1 upregulation, improved the persistence of anti-tumor CD8+ T cell-mediated tumor killing and overall survival in melanoma-bearing mice (121). Importantly, loss of IFNγ signaling in LECs also promoted the accumulation of anti-viral T cells in infected skin and exacerbated tissue pathology (121). Thus, tumors may coopt normal non-hematopoietic-based mechanisms of tissue protection for immune escape.
Similarly, FRCs exhibit immunomodulatory function in LNs. FRCs express and present peripheral tissue antigens to T cells in LNs (115), receive peptide-MHC-II loaded exosomes from DCs and induce CD4+ T cell hyporesponsiveness (116), and dampen T cell proliferation through the production of nitric oxide (118, 123, 124). Interestingly, some cancer-associated fibroblasts acquire markers consistent with FRCs, namely expression of podoplanin and ER-TR7 (125). Whether these fibroblasts represent a unique subset that exhibits immune suppressive function remains unclear, though a recent study demonstrates that tumor-associated fibroblasts in melanoma models cross-present tumor antigens and inhibit T cells in a FasL and PD-L2 dependent manner (126). In LNs, migrating DCs inhibit FRC contraction through CLEC-2/PDPN interactions leading to scaffold relaxation and more space for accumulating T cells (7). Whether leukocytes directly modulate fibroblast contraction in peripheral tissue remains to be tested but might have important implications for T cell invasion into dense, desmoplastic stroma. Further work is necessary to determine the functional relevance of stromal relative to hematopoietic or tumor-mediated immune suppression.
Excluded Infiltrates
T cell exclusion, in which T cells are absent from tumor nests and rather retained in adjacent, surrounding stroma (106, 127) is a significant barrier to response to therapy. One leading hypothesis is that tissue desmoplasia, the aberrant synthesis, alignment, and crosslinking of ECM proteins by fibroblasts in tumor microenvironments (128, 129), creates a physical barrier that prevents T cell invasion. Pancreatic ductal adenocarcinoma (PDAC) is particularly fibrotic and breast carcinomas exhibit stiff collagen fibers in parallel alignment tangential to tumor borders that correlate with poor prognosis (66, 130). Furthermore, dynamic intravital imaging reveals T cell migration along collagen fibers and vessels in tumors (63, 131), consistent with their preferred amoeboid-like mode of migration described in non-malignant matrices. Thus it has been proposed that the orientation and density of matrix fibers prevents T cell infiltration into tumor parenchyma (63).
However, strategies to reduce fibrosis in mouse PDAC models have had mixed results. Though cancer-associated fibroblasts are attributed an array of tumor-promoting activities, including immune suppression, their bulk depletion in the context of PDAC did not improve tumor control but instead drove more aggressive tumor invasion and metastasis (132, 133). In contrast, strategies that rather target the composition of the ECM using enzymes that degrade specific components (e.g., hyaluronan) thus reducing interstitial fluid pressures (134), or reduce ECM deposition and alignment (135) improved immune infiltration and therapeutic response. Interestingly, CAR T cells have had limited utility in solid tumors at least in part due to physical tissue barriers that prevent their infiltration. Consistent with this hypothesis, CAR T cells expressing heparanase exhibit enhanced tumor infiltration and antitumor function (136). Finally, a TGF-β signature was specifically identified in a subset of PD-L1 inhibitor resistant patients exhibiting an excluded infiltrate (not dysfunctional or desert) where T cells were restricted to the fibroblast- and collagen-rich peritumoral region characteristic of metastatic urothelial carcinoma (137). Therapeutic co-administration of antibodies targeting TGF-β and PD-L1 facilitated T cell penetration into the tumor center and provoked a vigorous adaptive immune response leading to tumor regression in a large Phase II clinical trial testing atezolizumab in patients with metastatic urothelial carcinoma (137). Thus, while the ECM facilitates T cell migration in naïve or acutely inflamed tissue, tumor-associated matrix remodeling may suppress T cell motility or place new requirements for interstitial proteolysis to mediate intratumoral penetration.
Contradicting the hypothesis that a fibrotic ECM is a sufficient physical barrier to prevent T cell infiltration are desmoplastic melanomas. Desmoplastic melanomas are densely fibrotic but also exceptional responders to immunotherapy, with 70% objective response rates and 32% complete responses (138). While these tumors demonstrate a significantly higher mutational burden than PDAC, and pre-existing adaptive immune responses that correlate with PD-L1 expression, this data clearly indicates that T cells are capable of infiltrating a dense, desmoplastic fibrous stroma and that its presence is not sufficient for T cell exclusion. Furthermore, even without engineered protease expression, adoptively transferred T cells can surmount physical barriers to treat experimental PDAC models (139). While ECM composition and rigidity differs between tissue and tumor types, the specific rate-limiting factors for T cell infiltration into fibrotic tissue remain to be determined.
In addition to the effects of desmoplasia on T cell exclusion, angiogenic growth factors such as vascular endothelial growth factor A (VEGF-A), angiopoietin, basic fibroblast growth factor (bFGF), and endothelin-1 (140, 141) attenuate inflammatory-mediated endothelial activation and thus intratumoral vessels exhibit reduced expression of adhesion molecules that would mediate lymphocyte extravasation, such as ICAM, VCAM, and E-selectin (142, 143). For example, bFGF inhibits TNFα/IL-1α-mediated expression of ICAM, VCAM, and E-selectin in vitro (144), and VEGF-A disrupts their clustering, therefore decreasing T cell adhesion to ECs (13). Endothelin signaling on ECs increases NO production and subsequent downregulation of adhesion molecules, thus blockade of the receptor increases T cell adhesion and infiltration into tumors (142). Angiogenic signaling from tumor cells also induces FasL expression on tumor associated ECs that limits tumor infiltrating CD8+ T cells, presumably through direct killing as demonstrated in vitro (145, 146). Consequently, factors that drive the angiogenic switch in tumors simultaneously establishes immunological barriers to limit immune surveillance and facilitates immune escape.
While anti-angiogenic therapy focused on destruction of tumor-associated vessels largely failed in most solid tumor types, adaptation of these strategies utilizing lower, normalizing doses to restore perfusion and adhesion molecule expression has proved more productive (147). Dual angiopoietin and VEGF-A blockade leads to increased T cell accumulation and function in several tumor models and synergizes with anti-PD-1 therapy (120). Furthermore, combination of anti-VEGFR2 and anti-PD-L1 antibodies induced lymphotoxin-dependent emergence of high endothelial venule-like vessels (148), which were necessary for response and are associated with better overall outcome in patients (149, 150). Interestingly, in mouse models, responders to immune checkpoint blockade exhibited rapid reperfusion of intratumoral vessels indicating that intratumoral vascular function may be required for T cell effector function and additionally that checkpoint blockade may directly affect endothelial cells (151). Thus, normalizing the angiogenic tumor vasculature may improve local T cell recruitment generating microenvironments primed to be responsive to immunotherapy. Interestingly, poorly adhesive, angiogenic vessels appear to be largely restricted to intratumoral regions, where they exhibit reduced adhesive properties and elevated expression of immune checkpoints (142). While this geographic vessel heterogeneity may limit infiltration directly into the tumor proper, it still allows infiltration into adjacent stroma perhaps contributing to the dense rings of CD8+ T cells observed around tumor nests.
Additionally, antitumor effector and memory T cells restricted to peritumoral stroma may be unable to locate target tumor cells due disrupted chemokine signals. High expression of the T cell attracting chemokines CXCL9, CXCL10, CXCL12, and CCL5 correlates positively with CD8+ T cell infiltration across several tumor types (152, 153, 154), indicating that if the tumors express the proper chemokines, T cells can get there. Chemokines, however, can be post-translationally modified by proteolytic cleavage, glycosylation, nitration, or deamination which results in dramatically altered activity (127). When CCL2 is nitrated, by reactive nitrogen species in the TME, for example, T cell infiltration into tumors is hindered and rather remain excluded from the tumor mass (155). In addition to the absence of chemoattractants, secretion of chemokines that serve as chemorepellants may protect tumor nests from T cell infiltration. In a mouse model of PDAC, fibroblast activating protein-expressing CAFs produce CXCL12 that coats tumor cells and prevents CXCR4+ CD8+ T cells from infiltrating tumor nests and controlling the tumor (156). Administration of AMD3100 (CXCR4 inhibitor) increased T cell infiltration into tumor nests, and synergized with anti-PD-L1 therapy to reduce tumor growth (156). Thus, competing chemokine gradients, initiated and maintained by multiple cell types within the tumor microenvironment, determine lymphocyte positioning and subsequent function.
Immunological Deserts
Finally, immunological deserts are defined as those tumor microenvironments completely lacking T cell infiltrates within tumor nests and in adjacent stroma. Low somatic mutational burden and tumor immunogenicity is likely a significant driver of failed T cell responses in these tumors. However, even in the presence of immunogenic epitopes lymphatic transport and poor DC migration may limit anti-tumor T cell priming in lymphoid organs and thus prevent systemic T cell expansion. In fact, tumors induced or implanted in mice lacking dermal lymphatic vessels fail to activate and accumulate anti-tumor T cell responses (157, 158) and lymphatic vessel density correlates with T cell infiltration in colorectal cancer and melanoma patients (159, 160). Conversely, overexpression of lymphangiogenic growth factors enhances intratumoral inflammation and response to various immunotherapies (42, 161), indicating that lymphatic transport plays an important role in both adaptive immune priming and setting up an inflammatory tumor microenvironment. Thus, the non-hematopoietic stroma may dictate the systemic expansion of anti-tumor immunity and thereby restrict the pool of T cells available for tumor recruitment. Still, downstream of T cell priming, analysis of T cells in synchronous metastases reveals heterogeneous distribution of the existing systemic repertoire (162) indicating additional mechanisms of control. Furthermore, even in the absence of de novo, tumor-specific T cell priming, recruitment of pre-existing memory populations should lead to intratumoral accumulation of T cells. Thus, additional factors must limit extravasation and tumor residence of bulk T cell populations. Tissue-specific vascular heterogeneity or dysfunction (stromal and intratumoral) may limit T cell infiltration in a lesion-specific manner and thus contribute to immunological deserts in some and not all metastatic lesions.
Future Directions
Non-hematopoietic cells provide context to in situ peripheral tissue immune responses and thus may be critical local signals that determine the switch between protective immunity and immune suppression. Dynamic imaging in animal models continues to reveal the spatiotemporal control tissues exert over infiltrating T cell responses in naïve and inflamed tissues (55). Extension of mechanisms elucidated during normal tissue responses to tumors will provide critical insight into the heterogeneity of T cell recruitment and retention in synchronous metastases in patients (162); may provide novel strategies to improve the efficacy of CAR T cell therapy in solid tumors (163); and provide insight into the multiple immune barriers across solid tumor types. Importantly, careful analysis of tissue-specific differences in immune infiltrate (164, 165), if coupled with tissue-specific vasculature and matrix components through multiplexed imaging technologies, may reveal important environmental context to inform dynamics of intratumoral inflammation and thus response to therapy. Similarly, single cell sequencing, while a powerful tool for extracting novel transcriptional states in tumor, hematopoietic, and stromal cells (143, 166, 167) loses structural information that informs interpretation and thus should be coupled with validation and further discovery in matching tissue sections. The added context may not only improve the prognostic value of extracted biomarker signatures, but will also generate hypotheses for rigorous mechanistic testing in experimental models leading to new strategies for immune modulation and tumor control. Importantly, non-hematopoietic stromal interactions provide inherently local mechanisms of immune control that if targeted, may serve to unleash effector T cell responses and thus revive tumor control.
Author Contributions
RSL and AWL researched the literature, created content, and wrote the manuscript.
Funding
This work was supported by the OHSU Knight Cancer Center Support grant NIH P30-CA069533, the Department of Defense Peer Reviewed Cancer Research Program (W81XWH-15-1-0348), V Foundation for Cancer Research (V2015-024), Cancer Research Institute, and Melanoma Research Alliance (403181). RL received support from the NIH/NCI Ruth L. Kirchstein National Research Service Award (Molecular Basis of Skin/Mucosa Pathobiology Training Grant; T32-CA106195).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the AWL Lab for helpful discussions.
References
1. Scandella E, Fink K, Junt T, Senn BM, Lattmann E, Forster R, et al. Dendritic cell-independent B cell activation during acute virus infection: a role for early CCR7-driven B-T helper cell collaboration. J Immunol. (2007) 178:1468–76. doi: 10.4049/jimmunol.178.3.1468
2. Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. (2008) 9:155–65. doi: 10.1038/ni1557
3. Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity (2005) 22:19–29. doi: 10.1016/j.immuni.2004.11.013
4. Rantakari P, Auvinen K, Jappinen N, Kapraali M, Valtonen J, Karikoski M, et al. The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat Immunol. (2015) 16:386–96. doi: 10.1038/ni.3101
5. Loo CP, Nelson NA, Lane RS, Booth JL, Loprinzi Hardin SC, Thomas A, et al. Lymphatic vessels balance viral dissemination and immune activation following cutaneous viral infection. Cell Rep. (2017) 20:3176–87. doi: 10.1016/j.celrep.2017.09.006
6. Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity (2006) 25:989–1001. doi: 10.1016/j.immuni.2006.10.011
7. Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, et al. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol. (2015) 16:75–84. doi: 10.1038/ni.3035
8. Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. (2010) 207:17–27. doi: 10.1084/jem.20091619
9. Nolz JC. Molecular mechanisms of CD8(+) T cell trafficking and localization. Cell Mol Life Sci. (2015) 72:2461–73. doi: 10.1007/s00018-015-1835-0
10. Nolz JC, Harty JT. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. J Clin Invest. (2014) 124:1013–26. doi: 10.1172/JCI72039
11. Osborn JF, Mooster JL, Hobbs SJ, Munks MW, Barry C, Harty JT, et al. Enzymatic synthesis of core 2 O-glycans governs the tissue-trafficking potential of memory CD8+ T cells. Sci Immunol. (2017) 2:eaan6049. doi: 10.1126/sciimmunol.aan6049
12. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med. (2016) 213:951–66. doi: 10.1084/jem.20151855
13. Bouzin C, Brouet A, De Vriese J, DeWever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol. (2007) 178:1505–11. doi: 10.4049/jimmunol.178.3.1505
14. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
15. Tousoulis D, Kampoli AM, Papageorgiou CTN, Stefanadis C. The role of nitric oxide in endothelial function. Curr Vasc Pharmacol. (2012) 10:4–18. doi: 10.2174/157016112798829760
16. Walzog B, Gaehtgens P. Adhesion molecules: the path to a new understanding of acute inflammation. News Physiol Sci. (2000) 15:107–13. doi: 10.1152/physiologyonline.2000.15.3.107
17. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. (2008) 205:1303–15. doi: 10.1084/jem.20071393
18. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. (2015) 15:692–704. doi: 10.1038/nri3908
19. Munro JM, Pober JS, Cotran RS. tumor necrosis factor and interferon-ey induce distinct patterns of endothelial activation and associated leukocyte accumulation in skin of papio anubis. Am J Pathol. (1989) 135:121–33.
20. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res. (2015) 107:331–9. doi: 10.1093/cvr/cvv154
21. McGettrick HM, Smith E, Filer A, Kissane S, Salmon M, Buckley CD, et al. Fibroblasts from different sites may promote or inhibit recruitment of flowing lymphocytes by endothelial cells. Eur J Immunol. (2009) 39:113–25. doi: 10.1002/eji.200838232
22. Chimen M, McGettrick HM, Apta B, Kuravi SJ, Yates CM, Kennedy A, et al. Homeostatic regulation of T cell trafficking by a B cell-derived peptide is impaired in autoimmune and chronic inflammatory disease. Nat Med. (2015) 21:467–75. doi: 10.1038/nm.3842
23. Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol. (2013) 201:449–65. doi: 10.1083/jcb.201209077
24. Carman CV, Martinelli R. T lymphocyte-endothelial interactions: emerging understanding of trafficking and antigen-specific immunity. Front Immunol. (2015) 6:603. doi: 10.3389/fimmu.2015.00603
25. Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. (2004) 200:1571–80. doi: 10.1084/jem.20040965
26. Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. (2004) 167:377–88. doi: 10.1083/jcb.200404129
27. Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity (2007) 26:784–97. doi: 10.1016/j.immuni.2007.04.015
28. Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J (2011) 30:4157–70. doi: 10.1038/emboj.2011.304
29. Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, et al. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. (2017) 18:1256–69. doi: 10.1016/j.celrep.2016.12.092
30. Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. (2014) 15:223–30. doi: 10.1038/ni.2824
31. Reyat JS, Chimen M, Noy PJ, Szyroka J, Rainger GE, Tomlinson MG. ADAM10-Interacting Tetraspanins Tspan5 and Tspan17 regulate VE-cadherin expression and promote T lymphocyte transmigration. J Immunol. (2017) 199:666–76. doi: 10.4049/jimmunol.1600713
32. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
33. Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+T cells. J Exp Med. (2003) 197:643–56. doi: 10.1084/jem.20021378
34. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. (2007) 204:2023–30. doi: 10.1084/jem.20070064
35. Bagai R, Valujskikh A, Canaday DH, Bailey E, Lalli PN, Harding CV, et al. Mouse endothelial cells cross-present lymphocyte-derived antigen on class I MHC via a TAP1- and proteasome-dependent pathway. J Immunol. (2005) 174:7711–5. doi: 10.4049/jimmunol.174.12.7711
36. Greening JE, Tree TIM, Kotowicz KT, van Halteren AG, Roep BO, Klein NJ, et al. Processing and presentation of the islet autoantigen GAD by vascular endothelial cells promotes transmigration of autoreactive T-cells. Diabetes (2003) 52:717–25. doi: 10.2337/diabetes.52.3.717
37. Sage PT, Varghese LM, Martinelli R, Sciuto TE, Kamei M, Dvorak AM, et al. Antigen recognition is facilitated by invadosome-like protrusions formed by memory/effector T cells. J Immunol. (2012) 188:3686–99. doi: 10.4049/jimmunol.1102594
38. Marelli-Berg FM, James MJ, Dangerfield J, Dyson J, Millrain M, Scott D, et al. Cognate recognition of the endothelium induces HY-specific CD8+ T-lymphocyte transendothelial migration (diapedesis) in vivo. Blood (2004) 103:3111–6. doi: 10.1182/blood-2003-08-2717
39. Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. J Immunol. (2008) 180:8386–92. doi: 10.4049/jimmunol.180.12.8386
40. Limmer A, Ohl J, Kurts C, Ljunggren H, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liber endothelial cells to CD8+ T cells results in antigen-speciic T-cell tolerance. Nat Med (2000) 6:1348–54. doi: 10.1038/82161
41. Schurich A, Berg M, Stabenow D, Bottcher J, Kern M, Schild HJ, et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol. (2010) 184:4107–14. doi: 10.4049/jimmunol.0902580
42. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. (2012) 1:191–9. doi: 10.1016/j.celrep.2012.01.005
43. Hirosue S, Vokali E, Raghavan VR, Rincon-Restrepo M, Lund AW, Corthesy-Henrioud P, et al. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: a new role for lymphatic endothelial cells. J Immunol. (2014) 192:5002–11. doi: 10.4049/jimmunol.1302492
44. Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med. (2010) 207:681–8. doi: 10.1084/jem.20092465
45. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qaio H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood (2012) 120:4772–82. doi: 10.1182/blood-2012-04
46. Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature (2009) 462:94–8. doi: 10.1038/nature08478
47. Korpos E, Wu C, Song J, Hallmann R, Sorokin L. Role of the extracellular matrix in lymphocyte migration. Cell Tissue Res. (2010) 339:47–57. doi: 10.1007/s00441-009-0853-3
48. Voisin MB, Probstl D, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am J Pathol. (2010) 176:482–95. doi: 10.2353/ajpath.2010.090510
49. Petajaniemi N, Korhonen M, Kortesmaa J, Tryggvason K, Sekiguchi K, Fujiwara H, et al. Localization of laminin a4-chain in developint and adult human tissues. J Histochem Cytochem (2002) 50:1113–30. doi: 10.1177/002215540205000813
50. Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. (2009) 15:519–27. doi: 10.1038/nm.1957
51. Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood–brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. (2001) 153:933–46. doi: 10.1083/jcb.153.5.933
52. Voisin MB, Woodfin A, Nourshargh S. Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arterioscler Thromb Vasc Biol. (2009) 29:1193–9. doi: 10.1161/ATVBAHA.109.187450
53. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. (2005) 280:23549–58. doi: 10.1074/jbc.M412001200
54. Prakash MD, Munoz MA, Jain R, Tong PL, Koskinen A, Regner M, et al. Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity (2014) 41:960–72. doi: 10.1016/j.immuni.2014.11.012
55. Gaylo A, Schrock DC, Fernandes NRJ, Fowell DJ. T Cell Interstitial Migration: Motility Cues from the Inflamed Tissue for Micro- and Macro-Positioning. Front Immunol. (2016) 7:428. doi: 10.3389/fimmu.2016.00428
56. Moreau HD, Piel M, Voituriez R, Lennon-Dumenil AM. Integrating physical and molecular insights on immune cell migration. Trends Immunol. (2018) 39:632–43. doi: 10.1016/j.it.2018.04.007
57. Friedl P, Entschladen F, Conrad C, Niggemann B, Zanker KS. CD4+ T lymphocytes migrating in three-dimensional collagen lattices lack focal adhesions and utilize b1 integrin-independent strategies for polarization, interaction with collagen fibers and locomotion. Eur J Immunol. (1998) 28:2331–43.
58. Wolf K, Muller R, Borgmann S, Brocker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood (2003) 102:3262–9. doi: 10.1182/blood-2002-12-3791
59. Friedl P, Wolf K. Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. (2009) 28:129–35. doi: 10.1007/s10555-008-9174-3
60. Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, Lambert K, et al. Inflammation-induced interstitial migration of effector CD4(+) T cells is dependent on integrin alphaV. Nat Immunol. (2013) 14:949–58. doi: 10.1038/ni.2682
61. Tufail S, Badrealam KF, Sherwani A, Gupta UD, Owais M. Tissue specific heterogeneity in effector immune cell response. Front Immunol. (2013) 4:254. doi: 10.3389/fimmu.2013.00254
62. Krivacic KA, Levine AD. Extracellular Matrix Conditions T Cells for Adhesion to Tissue Interstitium. J Immunol. (2003) 170:5034–44. doi: 10.4049/jimmunol.170.10.5034
63. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/JCI45817
64. Jacobelli J, Friedman RS, Conti MA, Lennon-Dumenil AM, Piel M, Sorensen CM, et al. Confinement-optimized three-dimensional T cell amoeboid motility is modulated via myosin IIA-regulated adhesions. Nat Immunol. (2010) 11:953–61. doi: 10.1038/ni.1936
65. Friedl P, Weigelin B. Interstitial leukocyte migration and immune function. Nat Immunol. (2008) 9:960–9. doi: 10.1038/ni.f.212
66. Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. (2006) 4:38. doi: 10.1186/1741-7015-4-38
67. Guidotti LG, Inverso D, Sironi L, Di Lucia P, Fioravanti J, Ganzer L, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell (2015) 161:486–500. doi: 10.1016/j.cell.2015.03.005
68. Edsparr K, Basse PH, Goldfarb RH, Albertsson P. Matrix metalloproteinases in cytotoxic lymphocytes impact on tumour infiltration and immunomodulation. Cancer Microenviron. (2011) 4:351–60. doi: 10.1007/s12307-010-0057-0
69. Froelich CJ, Zhang X, Turbov J, Hudig D, Winkler U, Hanna WL. Human granzyme B degrades aggrecan proteoglycan in matrix synthesized by chondrocytes. J Immunol. (1993) 151:7161–71.
70. Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol. (2017) 18:1025–34. doi: 10.1038/ni.3808
71. Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, et al. Generalized Levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature (2012) 486:545–8. doi: 10.1038/nature11098
72. Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, et al. CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity (2015) 42:524–37. doi: 10.1016/j.immuni.2015.02.009
73. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer (2012) 12:210–9. doi: 10.1038/nrc3186
74. Pingen M, Bryden SR, Pondeville E, Schnettler E, Kohl A, Merits A, et al. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity (2016) 44:1455–69. doi: 10.1016/j.immuni.2016.06.002
75. Ng C. P., Hinz B., Swartz M. A. (2005). Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci 118(Pt 20), 4731–4739. doi: 10.1242/jcs.02605
76. Schumann K, Lammermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity (2010) 32:703–13. doi: 10.1016/j.immuni.2010.04.017
77. Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell (2007) 11:526–38. doi: 10.1016/j.ccr.2007.04.020
78. Klonowski K. D., Williams K. J., Marzo A. L., Blair D. A., Lingenheld E. G., Lefrancois L. (2004). Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity 20:551–62. doi: 10.1016/S1074-7613(04)00103-7
79. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483:227–31. doi: 10.1038/nature10851
80. Seabrook T, Au B, Dickstein J, Zhang X, Ristevski B, Hay JB. The traffic of resting lymphocytes through delayed hypersensitivity and chronic inflammatory lesions: a dynamic equilibrium. Semin Immunol. (1999) 11:115–23. doi: 10.1006/smim.1999.0167
81. Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. (2005) 6:895–901. doi: 10.1038/ni1240
82. Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, et al. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. (2005) 6:889–94. doi: 10.1038/ni1238
83. Hopken UE, Wengner AM, Loddenkemper C, Stein H, Heimesaat MM, Rehm A, et al. CCR7 deficiency causes ectopic lymphoid neogenesis and disturbed mucosal tissue integrity. Blood (2007) 109:886–95. doi: 10.1182/blood-2006-03-013532
84. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
85. Brown MN, Fintushel SR, Lee MH, Jennrich S, Geherin SA, Hay JB, et al. Chemoattractant receptors and lymphocyte egress from extralymphoid tissue: changing requirements during the course of inflammation. J Immunol. (2010) 185:4873–82. doi: 10.4049/jimmunol.1000676
86. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. (2015) 194:2059–63. doi: 10.4049/jimmunol.1402256
87. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
88. Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, and Donda A. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. (2016) 7:573. doi: 10.3389/fimmu.2016.00573
89. Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. (2007) 171:1249–57. doi: 10.2353/ajpath.2007.070225
90. Geherin SA, Wilson RP, Jennrich S, Debes GF. CXCR4 is dispensable for T cell egress from chronically inflamed skin via the afferent lymph. PLoS ONE (2014) 9:e95626. doi: 10.1371/journal.pone.0095626
91. Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science (2013) 339:328–32. doi: 10.1126/science.1228456
92. Russo E, Teijeira A, Vaahtomeri K, Willrodt AH, Bloch JS, Nitschke M, et al. Intralymphatic CCL21 promotes tissue egress of dendritic cells through afferent lymphatic vessels. Cell Rep. (2016) 14:1723–34. doi: 10.1016/j.celrep.2016.01.048
93. Vigl B, Aebischer D, Nitschké M, Iolyeva M, Röthlin T, Antsiferova O, et al. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood (2011) 118:205–15. doi: 10.1182/blood-2010-12-326447
94. Johnson LA, Jackson DG. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int Immunol. (2010) 22:839–49. doi: 10.1093/intimm/dxq435
95. Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. (2006) 203:2763–77. doi: 10.1084/jem.20051759
96. Sawa Y, Tsuruga E, Iwasawa K, Ishikawa H, Yoshida S. Leukocyte adhesion molecule and chemokine production through lipoteichoic acid recognition by toll-like receptor 2 in cultured human lymphatic endothelium. Cell Tissue Res. (2008) 333:237–52. doi: 10.1007/s00441-008-0625-5
97. Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ Res. (2010) 106:920–31. doi: 10.1161/CIRCRESAHA.109.207274
98. Teijeira A, Hunter MC, Russo E, Proulx ST, Frei T, Debes GF, et al. T cell migration from inflamed skin to draining lymph nodes requires intralymphatic crawling supported by ICAM-1/LFA-1 interactions. Cell Rep. (2017) 18:857–65. doi: 10.1016/j.celrep.2016.12.078
99. Karikoski M, Irjala H, Maksimow M, Miiluniemi M, Granfors K, Hernesniemi S, et al. Clever-1/Stabilin-1 regulates lymphocyte migration within lymphatics and leukocyte entrance to sites of inflammation. Eur J Immunol. (2009) 39:3477–87. doi: 10.1002/eji.200939896
100. Salmi M, Karikoski M, Elima K, Rantakari P, Jalkanen S. CD44 binds to macrophage mannose receptor on lymphatic endothelium and supports lymphocyte migration via afferent lymphatics. Circ Res. (2013) 112:1577–82. doi: 10.1161/CIRCRESAHA.111.300476
101. Pflicke H, Sixt M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J Exp Med. (2009) 206:2925–35. doi: 10.1084/jem.20091739
102. Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol. (2012) 180:2561–75. doi: 10.1016/j.ajpath.2012.02.019
103. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
104. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA. (2016) 113:E7759–68. doi: 10.1073/pnas.1609376113
105. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
106. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
107. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. (2018) 9:2724. doi: 10.1038/s41467-018-05072-0
108. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348:74–80. doi: 10.1126/science.aaa6204
109. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
110. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi: 10.1182/blood-2006-07-
111. Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett. (2015) 167:72–86. doi: 10.1016/j.imlet.2015.07.003
112. Ahamed J, Burg N, Yoshinaga K, Janczak CA, Rifkin DB, Coller BS. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood (2008) 112:3650–60. doi: 10.1182/blood-2008-04-151753
113. Ahmadzadeh M, Rosenberg SA. TGF- 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. (2005) 174:5215–23. doi: 10.4049/jimmunol.174.9.5215
114. Broderick L, Bankert RB. Membrane-associated TGF- 1 inhibits human memory T cell signaling in malignant and nonmalignant inflammatory microenvironments. J Immunol. (2006) 177:3082–8. doi: 10.4049/jimmunol.177.5.3082
115. Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med. (2010) 207:689–97. doi: 10.1084/jem.20092642
116. Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, Suter T, et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4(+) T cell tolerance. J Exp Med. (2014) 211:1153–66. doi: 10.1084/jem.20132000
117. Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol. (2009) 183:1767–79. doi: 10.4049/jimmunol.0802167
118. Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. (2011) 12:1096–104. doi: 10.1038/ni.2112
119. Dieterich CD, Ikenberg K, Cetintas T, Kapaklikaya K, Hutmacher C, Detmar M. Tumor-associated lymphatic vessels upregulate PDL1 to inhibit T-Cell activation. Front Immunol. (2017) 8:66. doi: 10.3389/fimmu.2017.00066
120. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Rmili CW, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. (2017) 9:eaak9670. doi: 10.1126/scitranslmed.aak9670
121. Lane RS, Femel J, Breazeale AP, Loo CP, Thibault G, Kaempf A, et al. IFNg-activated dermal lymphatic vessels inhibit cytotoxic T cell in melanoma and inflamed skin. J Exp Med. (2018) 20:3176–87. doi: 10.1084/jem.20180654
122. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
123. Khan O, Headley MB, Gerard A, Wei W, Liu L, Krummel MF. Regulation of T cell priming by lymphoid stroma. PLoS ONE (2011) 6:e26138. doi: 10.1371/10.1371/journal.pone.0026138.g001
124. Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS ONE (2011) 6:e27618. doi: 10.1371/journal.pone.0027618
125. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328:749–52. doi: 10.1126/science.1185837
126. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. (2018) 9:948. doi: 10.1038/s41467-018-03347-0
127. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity (2013) 39:61–73. doi: 10.1016/j.immuni.2013.07.005
128. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6:392–401. doi: 10.1038/nrc1877
129. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. (2017) 66:1037–48. doi: 10.1007/s00262-017-2003-1
130. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. (2015) 7:1120–34. doi: 10.1039/C5IB00040H
131. Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. (2007) 204:345–56. doi: 10.1084/jem.20061890
132. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
133. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
134. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
135. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. (2013) 4:2516. doi: 10.1038/ncomms3516
136. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
137. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
138. Eroglu Z, Zaretsky JM, Hu-Lieskovan S, Kim DW, Algazi A, Johnson DB, et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature (2018) 553:347–50. doi: 10.1038/nature25187
139. Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. (2015) 28:638–52. doi: 10.1016/j.ccell.2015.09.022
140. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
141. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. (2018) 9:115. doi: 10.1038/s41419-017-0061-0
142. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. (2008) 14:28–36. doi: 10.1038/nm1699
143. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
144. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 88:667–73.
145. Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J Neurooncol. (2003) 64:55–61. doi: 10.1007/bf02700020
146. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
147. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. (2001) 7:987. doi: 10.1038/nm0901-987
148. Allen E, Jabouille A, Rivera LB, Lodewijckx O, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. (2017) 9:eaak9679. doi: 10.1126/scitranslmed.aak9679
149. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. (2011) 71:5678–87. doi: 10.1158/0008-5472.CAN-11-0431
150. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
151. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest. (2018) 128:2104–15. doi: 10.1172/JCI96582
152. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
153. Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut (2012) 61:427–38. doi: 10.1136/gutjnl-2011-300509
154. Muthuswamy R, Corman JM, Dahl K, Chatta GS, Kalinski P. Functional reprogramming of human prostate cancer to promote local attraction of effector CD8(+) T cells. Prostate (2016) 76:1095–105. doi: 10.1002/pros.23194
155. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
156. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
157. Alitalo AK, Proulx ST, Karaman S, Aebischer D, Martino S, Jost M, et al. VEGF-C and VEGF-D blockade inhibits inflammatory skin carcinogenesis. Cancer Res. (2013) 73:4212–21. doi: 10.1158/0008-5472.CAN-12-4539
158. Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest. (2016) 126:3389–402. doi: 10.1172/JCI79434
159. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. (2016) 8:327ra326. doi: 10.1126/scitranslmed.aad6352
160. Bordry N, Broggi MAS, de Jonge K, Schaeuble K, Gannon PO, Foukas PG, et al. Lymphatic vessel density is associated with CD8+ T cell infiltration and immunosuppressive factors in human melanoma. OncoImmunology (2018) 7:e1462878. doi: 10.1080/2162402x.2018.1462878
161. Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund AW, et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. (2017) 9:eaal4712. doi: 10.1126/scitranslmed.aal4712
162. Reuben A, Spencer CN, Prieto PA, Gopalakrishnan V, Reddy SM, Miller JP, et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom Med. (2017) 2:10. doi: 10.1038/s41525-017-0013-8
163. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:282. doi: 10.1038/s41419-018-0278-6
164. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
165. Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. (2017) 19:203–17. doi: 10.1016/j.celrep.2017.03.037
166. Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (2016) 352:189–96. doi: 10.1126/science.aad0501
Keywords: non-hematopoietic cells, T cell, immunotherapy, extravasation, interstitial migration, fluid flow, trafficking
Citation: Lane RS and Lund AW (2018) Non-hematopoietic Control of Peripheral Tissue T Cell Responses: Implications for Solid Tumors. Front. Immunol. 9:2662. doi: 10.3389/fimmu.2018.02662
Received: 01 September 2018; Accepted: 29 October 2018;
Published: 15 November 2018.
Edited by:
Carmen Gerlach, Karolinska Institutet (KI), SwedenReviewed by:
Karl Kai McKinstry, University of Central Florida, United StatesTara Marlene Strutt, University of Central Florida, United States
Copyright © 2018 Lane and Lund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda W. Lund, bHVuZGFAb2hzdS5lZHU=