 Christophe Chevillard1*†
Christophe Chevillard1*† João Paulo Silva Nunes2,3,4†
João Paulo Silva Nunes2,3,4† Amanda Farage Frade2,4,5
Amanda Farage Frade2,4,5 Rafael Ribeiro Almeida2,3,4
Rafael Ribeiro Almeida2,3,4 Ramendra Pati Pandey2,3,4
Ramendra Pati Pandey2,3,4 Marilda Savóia Nascimento2,3,4
Marilda Savóia Nascimento2,3,4 Jorge Kalil2,3,4
Jorge Kalil2,3,4 Edecio Cunha-Neto2,3,4*
Edecio Cunha-Neto2,3,4*- 1INSERM UMR 1090, TAGC, Aix-Marseille Université, Marseille, France
- 2Laboratorio de Imunologia, Instituto do Coracao, Hospital das Clinicas da Faculdade de Medicina da Universidade de Sao Paulo, São Paulo, Brazil
- 3Disciplina de Imunologia Clínica e Alergia, Faculdade de Medicina da Universidade de Sao Paulo, São Paulo, Brazil
- 4Institute for Investigation in Immunology (iii), INCT, São Paulo, Brazil
- 5Department of Bioengineering, Brazil University, São Paulo, Brazil
Chagas disease is caused by infection with the protozoan Trypanosoma cruzi and affects over 8 million people worldwide. In spite of a powerful innate and adaptive immune response in acute infection, the parasite evades eradication, leading to a chronic persistent infection with low parasitism. Chronically infected subjects display differential patterns of disease progression. While 30% develop chronic Chagas disease cardiomyopathy (CCC)—a severe inflammatory dilated cardiomyopathy—decades after infection, 60% of the patients remain disease-free, in the asymptomatic/indeterminate (ASY) form, and 10% develop gastrointestinal disease. Infection of genetically deficient mice provided a map of genes relevant for resistance to T. cruzi infection, leading to the identification of multiple genes linked to survival to infection. These include pathogen resistance genes (PRG) needed for intracellular parasite destruction, and genes involved in disease tolerance (protection against tissue damage and acute phase death—DTG). All identified DTGs were found to directly or indirectly inhibit IFN-γ production or Th1 differentiation. We hypothesize that the absolute need for DTG to control potentially lethal IFN-γ PRG activity leads to T. cruzi persistence and establishment of chronic infection. IFN-γ production is higher in CCC than ASY patients, and is the most highly expressed cytokine in CCC hearts. Key DTGs that downmodulate IFN-γ, like IL-10, and Ebi3/IL27p28, are higher in ASY patients. Polymorphisms in PRG and DTG are associated with differential disease progression. We thus hypothesize that ASY patients are disease tolerant, while an imbalance of DTG and IFN-γ PRG activity leads to the inflammatory heart damage of CCC.
Introduction
Chagas disease is caused by infection with Trypanosoma cruzi, an obligatory intracellular parasite in the mammalian host. It is endemic in Latin America and affects over 8 million people worldwide, causing thousands of deaths each year. The pathogen naturally infects animals from central Argentina to the southern United States. Acute infection may lead to death in a small proportion of hosts, and survivors live with persistent infection with low parasitism. Chronically infected patients display differential progression. Nearly 30% of infected patients may develop life-threatening chronic heart disease due to an excessive inflammatory response, most others remaining in the asymptomatic/indeterminate (ASY) form with no heart disease, associated with a more immunomodulatory profile. Genes encoding resistance strategies that are shared against intracellular pathogens (1) have evolved for hundreds of millions of years, including the pathogen resistance receptors, TNF-α, and the IFN-γ-dependent immune response, among others (2). Most pathogen resistance genes (PRG) inhibit infection by directly reducing pathogen burden, and are related to immune-driven mechanisms—which, when in excess, can lead to death. Disease tolerance is an alternative strategy to avoid death after infection, whereby the pathogen's damaging effect on the host is mitigated. Disease tolerance is defined as the situation where an organism can bear a pathogen load without tissue damage and in the absence of a disease state. Disease tolerance genes (DTG)—which do not limit infection, but reduce its fitness costs—operate to minimize tissue damaging effects of the pathogen, leading to stress and damage reduction responses; DTG can also operate by counteracting excessive, tissue-damaging PRG activity (1, 3, 4). One such stress response pathways involved in disease tolerance is the oxidative stress response, whose master regulator is nuclear respiratory factor 2 (Nrf2) (3). By not limiting infection, the host remains a pathogen reservoir enabling transmission, in the absence of health consequences for itself, providing an evolutionary advantage for both host and pathogen. Disease tolerance is frequent in pathogens and hosts who have coevolved; while African monkeys which coevolved with the African virus Simian Immunodeficiency Virus (SIV) develop chronic infection with no disease, the same virus causes deadly disease in Asian macaques (5, 6). Evolutionary selection of DTG can be even more effective than that of PRG (4). A possible example of evolutionary selection of DTG against T. cruzi are the South American didelphid marsupials which coevolved with T. cruzi for over 40 million years and maintain high and long-lasting T. cruzi parasitemias in the absence of disease (7). We here hypothesize that the absolute need for DTG to control potentially lethal PRG activity against T. cruzi leads to parasite persistence and establishment of chronic infection. Our second hypothesis is that PRG and DTG also determine the differential progression of chronic Chagas disease toward tissue damage (CCC). According to this hypothesis, ASY patients are disease tolerant, while tissue damage in CCC is a consequence of insufficient DTG and/or excessive PRG activity. Along the review, we will provide evidence supporting both hypotheses.
Pathogen Resistance Genes in T. cruzi Infection
Most known pathogen resistance mechanisms against T. cruzi are immune-driven, directed at the intracellular forms of the parasite, and can be harmful if excessive. T. cruzi evades the powerful immune response and establishes a persistent infection with low parasitism. In order to obtain a list of known PRG and DTG, we surveyed the literature on T. cruzi infection of genetically deficient knockout mice. PRG were defined as genes essential for control of T. cruzi parasitism and needed for survival of infection; operationally, we identified as PRG those genes whose knockout led to increased pathogen load and mortality. DTG were defined as genes whose presence reduced mortality without any effect on T. cruzi control. We identified as DTG those genes whose knockout led to reduced parasitism and increased mortality. Table 1 lists the PRG and DTG identified in our literature review. Most PRG belong to the TLR-MYD88-IL12-IFNG pathway, IL17 pathway, cell migration, inflammasome and other pathways involved in restriction of intracellular pathogen growth. Mice genetically deficient on TLR4, TLR7, and TLR9, MYD88, and UNC93B1 display increased blood parasitism and mortality (8–13). Likewise, mice genetically deficient of IL12A, IL12B, and STAT4, essential for the differentiation of IFN-γ-producing Th1 cells, also display intense tissue and blood parasitism with increased mortality (15, 33). Along with TLR genes, IFNG is one of the main PRG involved in T. cruzi parasite control (43). Mice genetically deficient on IFNG or STAT1 display drastically augmented T. cruzi parasitism and 100% mortality 13 days after infection (20, 21, 24). It was shown that T. cruzi amastigotes themselves dephosphorylate STAT1 serine residues, inhibiting IFN-γ signaling; evasion of IFN-γ signaling is further proof of the importance of the IFN-γ in the control of intracellular parasitism (44). IFN-γ-dependent PRG, like TNFA and NOS2, play a major role in resistance to T. cruzi (45, 46). The key role of TNF-α in T. cruzi control has been shown in TNFA-receptor 1 knockout mice (TNFRSFA1−/−), which display an increased number of blood and tissue parasites and shortened survival time (26). Platelet-activating factor (PAF) KO mice are more susceptible to T. cruzi infection than wildtype mice, and its protective effects depend on TNF-α-dependent NO production. In the context of protection against T. cruzi, IFN-γ, and TNF-α synergistically induce NF-kB activation to control T. cruzi parasitism and mortality in mice, by upregulating the expression of the PRG inducible nitric oxide synthase (NOS2), leading to the production of large amounts of NO and microbicidal reactive nitrogen species (RNS) (45, 46). NOS2-KO mice are susceptible to T. cruzi infection, with increased parasite burden and mortality due to lack of NO production (15, 47). Interestingly, constitutive NOS1-KO mice also showed increased parasitism and mortality (27). Genetic deficiency of macrophage PI3KCG increases susceptibility to T. cruzi infection; PI3KCG expression correlates with IFNG expression in CCC myocardium (38). IFN-γ increases ROS generation through induction of NADP oxidases (NOX2) and mitochondrial ROS via NF-kB activation (47, 48). Mice knockout for NOX2 displayed increased T. cruzi tissue parasitism and mortality due to the lack of type 1 cytotoxic T cells (29). IFN-γ-induced ROS enhances

Table 1. T. cruzi pathogen resistance and disease tolerance genes.
peroxynitrite anion (ONOO−) production, a strong oxidant arising from the reaction of NO with superoxide radical (O) (49). ONOO− induces damage to multiple molecules and is one of the ultimate effectors of parasite killing. Peroxynitrite promotes morphological disruption of internalized parasites, and induces severe alterations of energy metabolism, calcium homeostasis, and trypanothione depletion, severely impairing parasite redox homeostasis (50, 51). In addition, IFN-γ can also exert its protective effects in vivo in a NO-independent manner, through complementary events of protection against T. cruzi, such as production of other macrophage-derived effector molecules, MHC class II induction, CD4+ T cell polarization, IgG isotype switch to IgG2a and TLR induction (52–54). IRGM1, an IFN-γ-dependent GTPase, plays a key role in phagosome maturation and in the killing of intracellular pathogens contained in vacuoles (55).
In addition to inflammatory cells, several other cell types, including cardiomyocytes, fibroblasts and astrocytes, bear IFN-γ receptors and respond to the cytokine (56, 57). IFN-γ activates T. cruzi-infected macrophages and cardiomyocytes to produce TNF-α, NO, microbicidal ROS, and RNS. In vitro treatment with IFN-γ/TNF-α and IL1-β of mouse cardiomyocytes infected with T. cruzi resulted in NO production and elevated trypanocidal activity (58, 59). The primary role of IFN-γ mediating protection against intracellular T. cruzi infection is depicted in Figure 1.
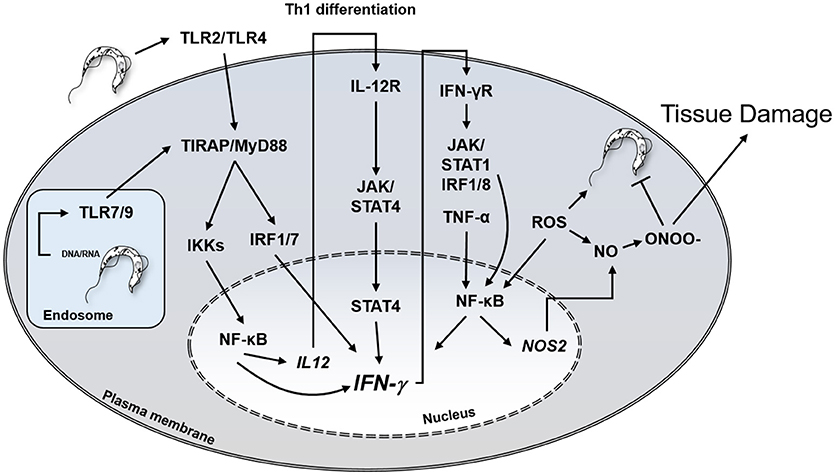
Figure 1. Central role of IFN-γ in protection against T. cruzi and potential host cell damage by peroxynitrite. IFN-γ, interferon-gamma; TLR, Toll-like receptor, IL12R, IL-12 receptor; IFNγR, IFN-γ receptor; JAK, Janus Associated Kinase; NO, nitric oxide; ONOO-, peroxynitrite; IRF, Interferon regulatory factor; STAT, Signal Transducer And Activator Of Transcription; ROS, reactive oxygen species.
IL17A is also a key PRG, and Th17 T cells may confer stronger protection against T. cruzi-related mortality than Th1 cells (60). IL-17A signaling is mainly dependent on TNF-α receptor associated factor 6 (TRAF6), but it can strongly promote TNF-α-induced NF-kB signaling by stabilizing pro-inflammatory mRNAs (61). Indeed, IL17A KO mice displayed an increase in blood and tissue parasitism with increased mortality. This was related to an impairment of leukocyte migration and activation of immune cells in the sites of parasite infection, as well as by reduced production of NO (18, 19). IL-17A also increases the persistence time of T. cruzi in the parasitophorous vacuole, enhancing exposure time of T. cruzi to the antimicrobial environment of endolysosomes, which can be further enhanced by IFN-γ-induced mechanisms (18). Genetic depletion of key inflammasome-related genes CASP1 and PYCARD/Asc induce enhanced mortality and increased parasitism (28). Mice knockout for genes involved in migration pathways, like chemokines/receptors CCL2, CCR5, and adhesion molecule ICAM1 also develop increased parasitism and decreased survival, in line with the impaired recruitment of leukocytes to sites of parasite replication (30–33). CD28 KO mice display T cell activation defects, and NCF1 (p47phox/NOX2) display a reduced type 1 CD8+ T cell response, leading to impaired control of parasitism and increased susceptibility to infection (29, 34). Although IL27RA KO mice develop increased parasitism and mortality and was a priori classified as a PRG, these mice develop grossly increased IFN-γ and inflammatory cytokine production, and death was related to the IFN-γ dependent tissue inflammation, a phenotype similar to that of IL10 KO mice (62). This is in line with the known effect of IL27RA signaling in the control of IFN-γ and inflammatory cytokines. Phospholipase A2 γ knockout mice show increased susceptibility to T. cruzi infection, and decreased arachidonic acid and prostaglandin E2 production; the mechanism of protection is still obscure (37). Galectin-1 KO mice displayed increased parasitism and mortality upon infection with the Tulahuen strain, classifying it as a PRG (36). However, subsequent studies with infection of Galectin-1 with the RA strain showed reduced parasitism and mortality, which is not a PRG profile (63). This suggests the phenotypes may change according to the T. cruzi strain being tested and site of infection.
Disease Tolerance Genes in T. cruzi Infection
Table 1 shows the T. cruzi DTG. Remarkably, all T. cruzi DTG (IL10, Ebi-IL27p28, IL17RA, IL23, IL6) shared as a common feature the ability to reduce IFN-γ production or Th1 differentiation. In addition to decreased parasitism and increased mortality, inflammation was upregulated in all DTG-deficient mice. Infection of IL-10 deficient mice is accompanied by increased release of IFN-γ, TNF-α, IL-12, and RNS (40). Mechanistically, IL-10 is a potent inhibitor of monocyte-macrophage activation and NK cell activity and can inhibit the synthesis of TNF-α and IL-12 and IFN-γ (64, 65). Mice genetically deficient of IL-17RA or IL-23 showed increased mortality due to a shift to a Th1 profile after infection and augmented IFN-γ and TNF-α levels in the heart (39, 42, 66). IL17RA signaling downregulated T-bet expression, and reduced Th1 T cell differentiation, and further downregulated IFN-γ production in acute infection by recruiting IL-10-producing suppressive neutrophils (42). Paradoxically, IL-17A is a PRG (18). It is possible that a engagement of the IL-17 receptor by a different IL-17 family member can be responsible for the DTG effect of IL17RA. IL-23 negatively regulates IL-12-induced IFN-γ production in CD8+ T cells by reducing STAT4 phosphorylation, independently of IL-17A, IL-17F, or IL-22 (18, 25, 67, 68). In addition, IL-23 is a key stimulatory cytokine for Th17 and innate “type 17” cells that can respond immediately to pathogenic insults; IL-23 may thus also suppress IFN-γ by promoting IL17RA signaling (69). Infection of Ebi3/IL-27p28 deficient mice is accompanied by increased IFN-γ production, with augmented Th1 immune response (70). Mechanistically, Ebi3 signaling modulates overproduction of IFN-γ, by inducing a population of IL-10 producing Tr1 T cells (39). On the other hand, IL-6 can be both a PRG and a DTG, depending on the model of T. cruzi infection. IL-6's DTG activity may be secondary to inhibition of Th1 differentiation through enhancing IL-4 production in CD4+ T cells (71), while its PRG activity may be explained by its ability to upregulate endothelial adhesion molecules, facilitating lymphocyte migration into non-lymphoid tissues (14, 71). We will discuss evidence that IFNG, a major PRG that operates as a key player in pathogen protection and is the culprit of tissue damage in Chagas disease, is the main target of modulation by DTGs with relevance in both the acute infection and differential progression of chronic disease.
Immune Dynamics in Acute T. cruzi Infection
T. cruzi subverts a highly conserved cellular pathway for the repair of plasma membrane lesions and explores endogenous cellular machinery for invasion, escape from the parasitophorous vacuole, which allows intracytoplasmic survival, and replication (72). The intracellular life cycle of T. cruzi is a major target of the antiparasite response (73). Extracellular T. cruzi components, such as trypomastigote-derived glycosylphosphatidylinositol (tGPI) and glycoinositolphospholipid (eGIPL) engage membrane Toll-like receptors (TLR) 2 and 4 (74). Trypanosoma cruzi is internalized by several different mechanisms, but end up in the phagolysosomal compartment, where T. cruzi DNA and RNA engage TLR7 and TLR9. TLR engagement promotes the Myd88-mediated activation of NF-kB. Lysosome acidification promotes escape of T. cruzi to the cytoplasm, where it differentiates into the replicative amastigote forms. Amastigote replication in the cytoplasm leads to activation of inflammasomes that can induce inflammatory cytokines and NF-kB activation. This induces pro-inflammatory cytokines including IL-12, a PRG which elicits differentiation of IFN-γ-producing Th1 cells soon after infection, promoting Th1 cell differentiation. IFN-γ induces expression of multiple other PRGs, such as TNFA and NOS2 (45, 46). Recent studies have reported that IL17A is a key PRG, plays a major protective role during the acute phase of infection (60). A strong antibody response is also triggered, but apparently has a lower effect on parasitism than innate immunity and acquired IFN-γ-dependent CD4+ and CD8+ T cell responses (53).
Although powerful, the immune response that occurs during acute infection leads to partial parasite control. T. cruzi evades complete eradication, leading to the establishment of a chronic persistent infection with low parasitism. T. cruzi-infected individuals maintain increased production of inflammatory/Th1 cytokines like IFN-γ and TNF-α as compared to healthy individuals, as a result of persistent stimulus of innate and specific immunity (75).
Immune Dynamics and Differential Disease Progression to CCC
Chronically infected individuals display differential disease progression. While 60% of infected individuals remain disease-free for life, 30% develop life-threatening CCC—a severe inflammatory dilated cardiomyopathy, years after infection. Ten percent develop gastrointestinal disease. CCC has a worse prognosis than cardiomyopathies of non-inflammatory etiologies, such as ischemic or idiopathic cardiomyopathy (76). CCC patients show an increased number of IFN-γ-producing Th1 T cells and plasma TNF-α levels as compared with ASY. Conversely, numbers of IL-10-producing CD4+CD25+ regulatory T cells (Tregs) CD4+CD25+ FoxP3+ Tregs and Th17 cells, as well as Ebi3/IL-27p28 levels are lower as compared with ASY (28, 31, 39, 44, 60, 77) (Figure 2). The exacerbated Th1 response observed in the peripheral blood of CCC patients is reflected on the Th1-rich inflammatory infiltrate predominantly secreting IFN-γ and TNF-α. A lower, but significant, production t of IL-4, IL-6, IL-7, IL-15, IL-18 was found in their heart tissue (57, 78–83). Indeed, IFN-γ is the most upregulated cytokine in CCC heart tissue; concordantly, we observed significant expression of T-bet, the hallmark Th1 transcription factor, in the CCC myocardium (84). We found a positive correlation between Tbet expression and left ventricular dilation, corroborating the pathogenic role of Tbet positive/IFN-γ producing T cells toward CCC. Conversely, mRNA expression of GATA3, RORγT, and FoxP3, hallmark transcription factors of Th1-antagonizing Th2, Th17, and Treg populations was low or undetectable. In agreement, mRNA expression of their signature cytokines IL4, IL13, IL17, IL10, and Treg molecular markers FOXP3 and CTLA4 was also low or undetectable (84). IFN-γ-producing CCR5+CXCR3+ Th1 T cells are more abundant in CCC than ASY (85), and the same cells were identified in CCC heart tissue, along with their chemokine ligands (CCL3, CCL4, CCL5, CXCL9, and CXCL10, respectively). CCL5 and CXCL9 were the most highly expressed chemokine mRNAs, and the intensity of the myocardial inflammation was positively correlated with CXCL9 mRNA expression (86, 87). Together, this suggests that locally produced Th1 T cell-attracting chemokines play a role in the selective accumulation of Th1 T cells in CCC hearts. Moreover, it indicates that the Th1 infiltrate in CCC myocardium is essentially unopposed by regulatory cells or cytokines, suffering little regulation. This lack of regulation could explain the destructiveness of the inflammatory infiltrate, most likely due to excessive collateral damage by IFN-γ-producing T cells as described in acutely T. cruzi-infected mice. IFN-γ is thus considered the culprit of CCC. We believe the unopposed IFN-γ action is linked to the fact that DTGs that reduce IFN-γ production and/or Th1 T cell differentiation IL10, Ebi3/IL27p28, and genes involved in IL-17 signaling are downregulated in CCC patients.

Figure 2. Immune profile of CCC patients revolves around the IFN-γ axis. CCC patients display increased production of PRGs IFN-γ and TNF-α, and decreased levels of IFN-γ/Th1 suppressive factors as compared with ASY patients. IL-10 and Ebi3/IL-27p28 are DTGs and IL-17, also reduced in CCC, is included here as the DTG IL17RA signaling showed to suppress the IFN-γ/Th1 axis. CCC, Chagas disease cardiomyopathy; PRG, pathogen resistance gene; ASY, asymptomatic; DTG, disease tolerance gene; IFN-γ, interferon-gamma; TNF-α, tumor necrosis factor alpha; IL, interleukin; Ebi3, Epstein-Barr virus induced gene 3; IL17RA, interleukin 17 receptor A.
The immunomodulatory profile of ASY patients, with increased levels of DTG and lower levels of the PRG IFN-γ, indicates that ASY patients are in a state of disease tolerance. It is interesting to notice that the majority of chronic Chagas disease patients −60%- are disease-tolerant ASY patients. This is in line with evolutionary studies indicating that selection of DTG is dominant over PRG, leading to higher frequencies of individuals expressing DTG than PRG.
Deleterious Effects of IFN-γ
While IFN-γ can control parasites, excessive levels can cause tissue damage and death in the acute and chronic phases. IFN-γ effectively regulates the expression of over 1,000 genes through activation of Janus tyrosine kinase (JAK) and phosphorylation of transducer and activator of transcription 1 (STAT-1) pathway, along with other mechanisms (88, 89). Among key IFN-γ-inducible inflammatory genes are TNF-α, and several other inflammatory cytokines and chemokines, interferon-inducible factor 1 (IRF1) and other PRG, including inducible nitric oxide synthase (NOS2) (90). Multiple findings suggest that IFN-γ plays a central pathogenic role in the myocarditis and heart failure in CCC patients. Systemic T. cruzi persistence drives continued production of IFN-γ by T cells, important for parasite control through NOS production, as well as activating ROS through induction of NADP oxidases and mitochondrial ROS through NF-kB (47, 48). However, T. cruzi is highly resistant to ROS; IFN-γ and the accompanying NOS and ROS can also induce severe disturbances of heart function. IFN-γ can modulate gene expression in immune cells and cardiomyocytes. A significant proportion of genes modulated in the CCC myocardium and hearts from acutely infected mice are inducible by IFN-γ (86, 91). Significantly, transgenic mice overexpressing IFN-γ develop a TNF-α-dependent myocarditis and cardiomyopathy (92, 93). In cardiomyocytes, IFN-γ treatment reduced contractility, induced NO/peroxynitrite-dependent cardiomyocyte apoptosis, reduced cardiomyocyte area, and also induced atrial natriuretic factor and production of chemokines CCL3, CCL5, and CXCL1 (94–96). IFN-γ regulates cardiac fibrosis by increasing fibroblast proliferation, production of hyaluronan and metalloproteinases 2 and 9 (95–98).
IFN-γ And Mitochondria: Dangerous Liaisons
Evidence of mitochondrial dysfunction has been found in hearts of animal models of Chagas disease, as well as the myocardium of CCC patients. This is especially relevant for CCC pathogenesis, since mitochondrial dysfunction is a paramount feature of heart failure of diverse etiologies (99). Nisha Garg and her group pioneered and studied in detail mitochondrial dysfunction in hearts of murine models of acute and chronic Chagas disease and in vitro infection models (100). Regarding mitochondrial damage in human CCC, our group described altered expression of mitochondrial genes and 16S mitochondrial rRNA in CCC heart lesions (57). We also found a selective reduction of protein expression of ATP synthase and creatine kinase activity—key mitochondrial energy metabolism enzymes- in CCC heart lesions (101). In agreement, mitochondrial DNA content was found to be reduced in CCC heart tissue (102), further indicating that mitochondrial function is compromised in CCC. Evidence indicates that many damaging effects of IFN-γ are secondary to promoting peroxynitrite-dependent and independent mitochondrial dysfunction and oxidative stress. IFN-γ effects on mitochondria include inhibition of the oxidative metabolism (103) an increased rate of ATP depletion (104) and inhibition of creatine kinase expression (105, 106). Moreover, IFN-γ+TLR ligand (LPS) treatment of cardiomyocytes– but not T. cruzi infection per se—downregulates expression of multiple mitochondrial genes. These genes were involved in the electron transport chain, mitochondrial fission, mitophagy, mitochondrial, and nuclear gene transcription (51). NF-kB activation is one of the main mechanisms of mitochondrial damage induced by IFN-γ. Although IFN-γ does not directly activate NF-kB, it enhances TNF-α-induced NF-kB nuclear translocation. IFN-γ/TNF-α-driven NF-kB activation is known to cause dissipation of the proton gradient and impairment of the mitochondrial membrane potential (MMP) and ATP synthesis, leading to apoptosis (106, 107). Persistent activation of NF-kB by IFN-γ/TNF-α has been described to potentiate ROS release, which can in turn increase tissue damage and mitochondrial energy imbalance (106). Indeed, inhibition of NF-kB has been shown to improve MMP with substantial decrease of NOS2/NO induction and ROS release. Taken together, these reports strongly suggest that IFN-γ may play a significant role in mitochondrial dysfunction and heart failure. Excessive mitochondrial ROS production by cardiomyocytes is considered as a central cause of heart failure (108, 109).
A significant crosstalk occurs between NF-kB and mitochondrion-protecting proteins. NF-kB signaling down-regulates sirtuin-1 (SIRT1) activity through the expression of IFN-γ, ROS, and NO (110). SIRT1, an antioxidant and anti-inflammatory protein, regulates the oxidative respiration and cellular survival and is highly expressed in the heart, acting as an inhibitor of NF-kB inflammatory signals (111). While NF-kB stimulates glycolytic energy flux in acute inflammation, SIRT1 inhibits NF-kB and enhances mitochondrial oxidative metabolism through 5′ AMP-activated protein kinase (AMPK) resulting in the resolution of inflammation (110). Treatment of T. cruzi-infected mice with SIRT1 and/or AMPK agonists SRT1720, resveratrol and metformin reduced myocardial NF-kB transcriptional activity, inflammation and oxidative stress, resulting in beneficial results for restoration of cardiac function (100, 112). Preserving Nrf2 activity was shown to arrest the mitochondrial and cardiac oxidative stress, cardiac fibrosis, and heart failure in murine T. cruzi infection (113). Nrf2 is the master regulator of the antioxidant response, a transcription factor controlling expression of hundreds of genes (114), and promotes mitochondrial biogenesis (115). HMOX1 (Heme oxygenase 1), a key effector of the Nrf2 antioxidative response, is upregulated in the hearts of acutely infected mice (91), but not in the myocardium of CCC patients. Furthermore, its paralog HMOX2 is downregulated in CCC myocardium (ECN and CC, submitted for publication), indicating that Nrf2-dependent antioxidative defenses are even lower in CCC myocardium than in acute T. cruzi myocarditis (91). It has been recently observed that mitochondrial damage due to any stimulus leads to inflammation. Figure 3 shows the interplay between IFN-γ, DTG, NF-kB, and its antagonists, mitochondrial protective factors SIRT1, Nrf2, and AMPK vs. the outcome of mitochondrial function and inflammation.
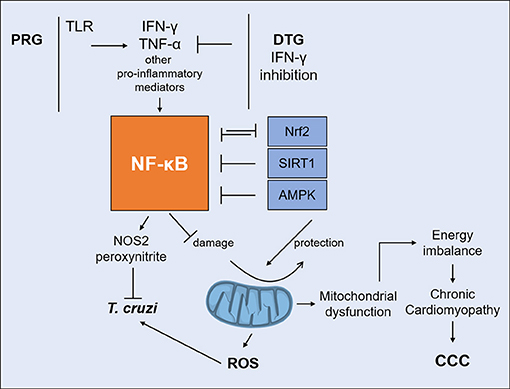
Figure 3. Interplay between IFN-γ and DTG determines the fate of mitochondria. IFN-γ/TNF-α strongly activate NF-kB signaling. Unchecked NF-kB activation leads to mitochondrial dysfunction, including decreased MMP, increased ROS production and reduced ATP production. IFN-γ-lowering DTG can reduce IFN-γ/TNF-α activation of NF-kB and shift the balance favoring SIRT1, Nrf2, and AMPK. Agonists can protect mitochondria and myocardial damage by reverting NF-kB activity in experimental CCC. DTG, DISEASE tolerance gene; IFN-γ, interferon-gamma; TNF-α, tumor necrosis factor alpha; NF-kB, nuclear factor kappa B; SIRT1, Sirtuin-1; Nrf2, nuclear respiratory factor 2; AMPK, 5′ adenosine monophosphate-activated protein kinase; MMP, mitochondrial membrane potential; ROS, reactive oxygen species; ATP, adenosine triphosphate.
ROS activate NF-kB, and dysfunctional or damaged mitochondria release mitochondrial DAMPs (damage-associated molecular patterns) including mitochondrial DNA (mtDNA) leading to the activation of the TLR9/Myd88/NF-kB, cGas/Type I IFN, and NRLP3 inflammasome pathways (116). In summary, the release of mitochondrial DAMPs in response to initial inflammatory stimuli and infection can establish a sterile inflammation process, a self-perpetuating positive feedback loop of mitochondrial damage and inflammation. This process has been found to occur in heart failure of diverse etiologies, and has been called sterile cardiac inflammation (117). Such sterile inflammation could be a contributing factor for the maintenance of inflammation in the absence of T. cruzi and may potentiate inflammatory damage in CCC (Figure 4). Together, these results indicate that mitochondrial dysfunction and mitochondrial ROS production are major pathogenic factors and therapeutic targets in CCC.
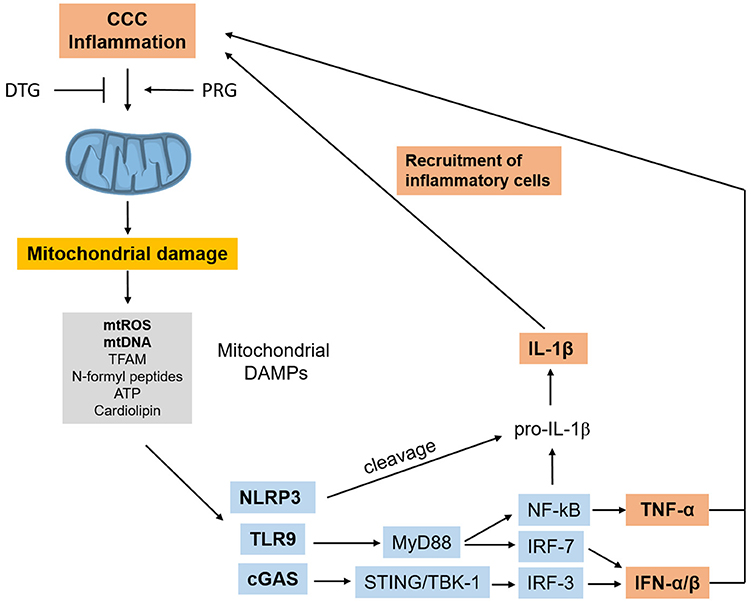
Figure 4. Inflammation-induced mitochondrial dysfunction starts a positive feedback loop. Inflammation in CCC can trigger mitochondrial dysfunction and damage, leading to release of mtROS and other mitochondrial DAMPs which can trigger inflammation cascades themselves, using several pathways. Such mitochondrial DAMP-induced inflammation can perpetuate inflammation and promote further mitochondrial dysfunction in a positive feedback loop. “Sterile” cardiac inflammation mediated by mitochondrial damage could thus perpetuate and/or potentiate inflammation in the hearts or CCC patients. CCC, Chagas disease cardiomyopathy; DAMPs, damage-associated molecular patterns; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; TFAM, transcription factor A, mitochondrial; cGAS, Cyclic GMP-AMP Synthase; NRLP3, NLR family pyrin domain containing 3; TLR9, toll-like receptor 9; MyD88, myeloid differentiation primary response 88; STING, stimulator of interferon genes protein; TBK1, TANK binding kinase 1; IRF, interferon regulatory factor; NF-kB, nuclear factor kappa B; IFN, interferon; TNF-α, tumor necrosis factor alpha.
Host Genetic Factors Associated to Differential Disease Progression
The finding that 30% of Chagas disease patients develop CCC suggested the participation of genetics in differential disease progression. This was reinforced by the finding of familial aggregation of cases of CCC in endemic area settings (118). CCC patients display a more intense inflammatory response than the ASY patients, who seem to have a more regulated immune response. Given the importance of inflammatory mechanisms for CCC pathogenesis, genetic susceptibility to CCC may result from functionally relevant genetic polymorphisms that lead to variations in the intensity of the innate or acquired immune response and in inflammatory cytokines and chemokines involved in the pathogenesis of the disease. Common genetic association studies compare frequencies of a genetic polymorphism (single nucleotide polymorphism, SNP) in a candidate gene (picked by the investigator) in two populations (typically disease and control). If a gene variant is more frequent in the disease group, it is said to be associated with the disease and assumed to confer risk toward developing disease. These studies show genetic contributions that are typically small, explaining < 10% of the phenotype of complex, polygenic diseases like CCC. A SNP found to be associated with a disease may be directly connected to a phenotype (e.g., a polymorphism in a transcription factor-binding sequence in the promoter region, affecting gene expression) or merely a marker of a linked biologically relevant polymorphism. In the case of Chagas disease, we performed a thorough literature search at the PUBMED database and retrieved 145 association studies addressing polymorphisms in 76 genes, which disclosed 62 SNPs from 44 genes to be associated with CCC. From those, 9 SNPs/genes were associated with CCC severity—SNPs were more frequent among severe CCC patients, with significant left ventricular dysfunction (ejection fraction < 40%), as compared to the remaining CCC patients.
In our literature search, we found that 7 genes with polymorphisms associated with CCC or severe CCC were also defined as PRG or DTG in mouse knockout infection studies (Table 2). The 5 PRG belonged to the TLR, Th1, or chemokine pathways/processes. Since TNF-α signaling is key in both the acute infection and CCC, we added TNFA and TNF-receptor alpha (TNFRSF1A) to this table, even though TNFA was only assessed in association studies (120–123, 132, 133, ), and TNFRSF1A only in knockout infection experiments (26). The two DTG Ebi3/IL27p28 and IL-10 both modulate IFN-γ via downregulation of IFN-γ production and Th1 differentiation. Such genes are central in the pathogenesis of CCC, as IFN-γ and TNF-α-producing cells migrate to CCC heart tissue in response to chemokines CCL2 and CCR5. While the−308 variant of TNFA has been associated with increased production, this has not been validated every time. Significantly, polymorphisms in several other genes belonging to the same pathways (TLR-IL12-IL18-IFNG-TNFA-NF-kB, chemokines/receptors, inflammasome including both IL1B, and IL18) have been associated with CCC.
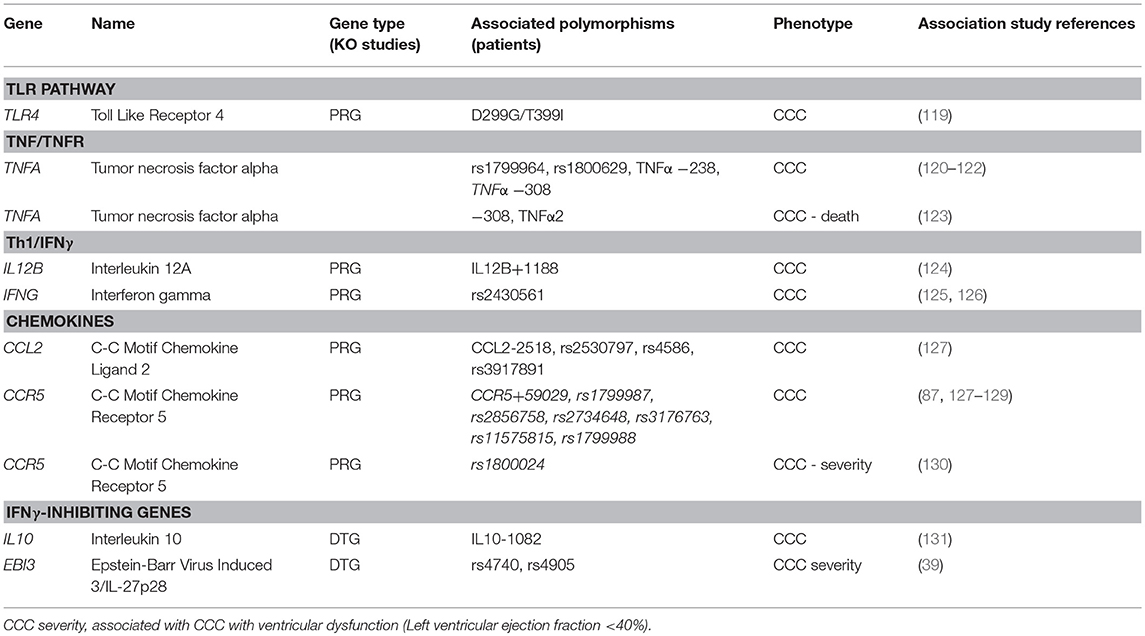
Table 2. PRG and DTG gene polymorphisms associated with CCC, severe CCC, or death.
The heterozygous TLR4-D299G/T399I genotype occurred more frequently in ASY infected subjects than CCC patients (OR = 5.4, 95% CI = 1.03–52.6, P = 0.02) (119). In humans, D299G/T399I reduces TLR4 responsiveness to lipopolysaccharide and associates with increased susceptibility and/or severity of numerous infectious and inflammatory diseases. The TIRAP variant S180L was found to be more prevalent in the ASY group than in the CCC group (OR = 0.31, 95%CI = 0.16–0.60, P = 0.0084) (134). Mutation leads to a decrease in signal transduction upon ligation of TLR2 or TLR4 to their respective ligands. Weitzel et al. have shown a similar trend of association for TLR4 (119). A similar study on a large Brazilian population found that TIRAP rs8177376A/A is associated with an increased susceptibility to CCC (OR = 1.36; 95%CI = 1.19–1.80, P = 0.037) (127). This same TIRAP variant confers protection against malaria, tuberculosis, pneumococcal disease, and bacteraemia (135). Polymorphisms in the TNFA-LTA region have been extensively studied. TNFA –LTA polymorphisms (−308, −244, and −238) were studied on a Peruvian cohort with negative results (132), while it was found to be associated in a Mexican population (OR = 3.03, 95% CI = 1.29–7.12, P = 0.008) (120). Pissetti et al. have shown that the LTA (+252) polymorphism was significantly associated with Chagas disease as compared with healthy seronegative individuals in a Brazilian population (133). Pissetti et al. have shown that this allele is associated with higher TNF-α production in Brazilian Chagas disease patients as compared to healthy seronegative individuals (OR = 1.846, 95%CI = 1.057–3.223, P = 0.03) (121). Similar results were detected on a Colombian cohort (122). Severe CCC patients with significant ventricular dysfunction carrying the susceptibility genotypes TNFA−308 or TNFA A2 microsatellite display a significantly shorter survival time than those carrying other alleles (OR = 0.51, 95%CI = 0.27–0.97, P = 0.04) (123). CCL2 is an important chemokine attracting monocytes/macrophages to inflamed tissue. The plasma concentrations CCL2 are associated with myocardial dysfunction in patients with severe CCC, dilated cardiomyopathy and acute myocardial infarction (55, 136, 137). Ramasawmy et al. have shown on their Brazilian population that the variant at position CCL2−2518 A allele conferred susceptibility to CCC in a Brazilian population (OR = 4.1; 95%CI = 1.7–11; P = 0.001) (138), a result that was subsequently confirmed (rs2530797: OR = 1.46; 95%CI = 1.11–1.92, P = 0.007) (127). Multiple studies have identified associations between different polymorphisms in the CCR5 gene (87, 127–129, 139–142). A polymorphism in IFNG rs2430561 was found to be associated with CCC (126). The IL12B +1188 C allele was found to be associated with CCC in a similar population (OR = 3.39, 95%CI = 1.3–9.15, P = 0.015) (124).
Finally, genes tagged as DTG were also associated in genetic studies. The functional IL10 gene polymorphism −1082G/A is associated with the development of CCC in a Brazilian population (OR = 0.84, 95%CI = 1.44–4.95, p < 0.01) (131). Similar data were obtained in Colombia (143). Ebi3/IL-27p28 regulates IFN-γ -mediated myocarditis by promoting an anti-inflammatory environment through IL-10. Medina et al. has shown that Ebi3 polymorphism (SNP rs4740) is less frequent in patients with severe cardiopathy (6.6%) compared to patients with the indeterminate form (OR 3.157, 95% CI = 1.15–8.64, p = 0.025) (39).
Does T. Cruzi Genetic Variability Play a Role in Determining the Outcome of Human Chagas Disease?
In addition to host genetic susceptibility factors, genetic variation in T. cruzi may also play a role in the outcome of Chagas disease. In animal models of infection, different T. cruzi strains show very distinct patterns of acute phase death, induction/activation of cytokines, PRGs and DTGs, tissue tropism, and development of chronic infection (144, 145).
Major subgroups of T. cruzi genetic variability have been classified into 6 discrete type units (DTU)- TcI to TcVI, each with a distinct geographical distribution. It has been reported that TcI (Colombian) and TcII (Y) strains differentially activate human monocytes (146). However, there was no difference in the DTUs found in the CCC and the ASY groups in a same geographical region; heart disease is caused by the DTUs that are prevalent in a given region (147–150) Although DTUs were not clearly associated with Chagas disease outcomes, T. cruzi genetic complexity surpasses the simple DTU classification. It is likely that yet undisclosed genetic variants of T. cruzi may influence the development of CCC, as shown in mice (145), which will certainly interact with the host genome to lead to different outcomes. Infection of BALB/c or C57BL/6 mice leads to differential activation of PRGs (151).
Concluding Remarks
The finding that all identified T. cruzi DTGs converge on the inhibition of IFN-γ production and/or Th1 T cell differentiation indicates the importance of this homeostatic control to avoid IFN-γ-induced death in the acute phase. Downmodulation of the IFN-γ response by DTGs favors T. cruzi evasion and the establishment of chronic infection. Among chronically infected patients, disease progression is associated with distinct immune profiles. In chronically infected ASY patients, IFN-γ production levels are adequately modulated by DTG IL10, Ebi3/IL27p28, Th17/IL17RA signaling, along with Treg, promoting a state of disease tolerance. Conversely, in CCC, an imbalance between DTG and IFNG causes increased IFN-γ production associated with inflammatory cardiac damage. Excessive myocardial IFN-γ signaling can lead to mitochondrial dysfunction. In turn this leads to energy imbalance, impairment of contractile function. Mitochondrial dysfunction induces further inflammation, in a positive feedback loop that can help perpetuate myocardial inflammation in CCC.
Author Contributions
EC-N and CC participated in conceptual design, manuscript drafting, and revision. JN and RA participated in manuscript drafting, and revision. AF, RP, and MN participated in manuscript drafting. JK participated in manuscript revision.
Funding
This work was funded by the following research support agencies: Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Brazil) 2014/50890-5; 2016/15209-0, 2013/50302-3. Co-funded by Agence Nationale de Recherche (ANR France Br-Fr-Chagas), Institut National de la Santé et de la Recherche Médicale (INSERM), Aix-Marseille Université, ARCUS/Institut de Recherche pour le Developpement, USP-COFECUB and from ParaFrap-ANR-11-LABX-0024, France; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001; and NIAID/NIH grant 2U19AI098461-06. EC-N and JK are recipients of CNPq productivity awards.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. He X, Berland R, Mekasha S, Christensen TG, Alroy J, Kramnik I, et al. The sst1 resistance locus regulates evasion of type I interferon signaling by Chlamydia pneumoniae as a disease tolerance mechanism. PLoS Pathog. (2013) 9:e1003569. doi: 10.1371/journal.ppat.1003569
2. Savan R, Ravichandran S, Collins JR, Sakai M, Young HA. Structural conservation of interferon gamma among vertebrates. Cytokine Growth Factor Rev. (2009) 20:115–24. doi: 10.1016/j.cytogfr.2009.02.006
3. Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol. (2014) 35:483–94. doi: 10.1016/j.it.2014.08.001
4. Roy BA, Kirchner JW. Evolutionary dynamics of pathogen resistance and tolerance. Evolution (2000) 54:51–63. doi: 10.1554/0014-3820(2000)054[0051:EDOPRA]2.0.CO;2
5. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol. (2010) 84:7886–91. doi: 10.1128/JVI.02612-09
6. Harris LD, Klatt NR, Vinton C, Briant JA, Tabb B, Ladell K, et al. Mechanisms underlying γδ T-cell subset perturbations in SIV-infected Asian rhesus macaques. Blood (2010) 116:4148–57. doi: 10.1182/blood-2010-05-283549
7. Jansen AM, Xavier SC, Roque AL. The multiple and complex and changeable scenarios of the Trypanosoma cruzi transmission cycle in the sylvatic environment. Acta Trop. (2015) 151:1–15. doi: 10.1016/j.actatropica.2015.07.018
8. Oliveira AC, de Alencar BC, Tzelepis F, Klezewsky W, da Silva RN, Neves FS, et al. Impaired innate immunity in Tlr4(-/-) mice but preserved CD8+ T cell responses against Trypanosoma cruzi in Tlr4-, Tlr2-, Tlr9- or Myd88-deficient mice. PLoS Pathog. (2010) 6:e1000870. doi: 10.1371/journal.ppat.1000870
9. Caetano BC, Carmo BB, Melo MB, Cerny A, dos Santos SL, Bartholomeu DC, et al. Requirement of UNC93B1 reveals a critical role for TLR7 in host resistance to primary infection with Trypanosoma cruzi. J Immunol. (2011) 187:1903–11. doi: 10.4049/jimmunol.1003911
10. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge, TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. (2006) 177:3515-9
11. Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, et al. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. (2004) 172:1711–8. doi: 10.4049/jimmunol.172.3.1711
12. Koga R, Hamano S, Kuwata H, Atarashi K, Ogawa M, Hisaeda H, et al. TLR-dependent induction of IFN-beta mediates host defense against Trypanosoma cruzi. J Immunol. (2006) 177:7059–66. doi: 10.4049/jimmunol.177.10.7059
13. Gonçalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, et al. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PLoS Negl Trop Dis. (2013) 7:e2469. doi: 10.1371/journal.pntd.0002469
14. Gao W, Pereira MA. Interleukin-6 is required for parasite specific response and host resistance to Trypanosoma cruzi. Int J Parasitol. (2002) 32:167–70. doi: 10.1016/S0020-7519(01)00322-8
15. Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and interferon-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol. (2001) 159:1723–33. doi: 10.1016/S0002-9440(10)63019-2
16. Müller U, Köhler G, Mossmann H, Schaub GA, Alber G, Di Santo JP, et al. IL-12-independent IFN-gamma production by T cells in experimental Chagas' disease is mediated by IL-18. J Immunol. (2001) 167:3346–53. doi: 10.4049/jimmunol.167.6.3346
17. Bombeiro AL, Gonçalves LA, Penha-Gonçalves C, Marinho CR, D'Império Lima MR, Chadi G, et al. IL-12p40 deficiency leads to uncontrolled Trypanosoma cruzi dissemination in the spinal cord resulting in neuronal death and motor dysfunction. PLoS ONE (2012) 7:e49022. doi: 10.1371/journal.pone.0049022
18. Erdmann H, Rossnagel C, Bohme J, Iwakura Y, Jacobs T, Schaible UE, et al. IL-17A promotes macrophage effector mechanisms against Trypanosoma cruzi by trapping parasites in the endolysosomal compartment. Immunobiology (2013) 218:910–23. doi: 10.1016/j.imbio.2012.10.005
19. Miyazaki Y, Hamano S, Wang S, Shimanoe Y, Iwakura Y, Yoshida H. IL-17 is necessary for host protection against acute-phase Trypanosoma cruzi infection. J Immunol. (2010) 185:1150–7. doi: 10.4049/jimmunol.0900047
20. Martins GA, Vieira LQ, Cunha FQ, Silva JS. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infection: a possible role in immune response control. Infect Immun. (1999) 67:3864-71
21. Cummings KL, Tarleton RL. Inducible nitric oxide synthase is not essential for control of Trypanosoma cruzi infection in mice. Infect Immun. (2004) 72:4081–9. doi: 10.1128/iai.72.7.4081-4089.2004
22. Arantes RM, Marche HH, Bahia MT, Cunha FQ, Rossi MA, Silva JS. Interferon-gamma-induced nitric oxide causes intrinsic intestinal denervation in Trypanosoma cruzi-infected mice. Am J Pathol. (2004) 164:1361–8. doi: 10.1016/S0002-9440(10)63222-1
23. Santiago HC, Feng CG, Bafica A, Roffe E, Arantes RM, Cheever A, et al. Mice deficient in LRG-47 display enhanced susceptibility to Trypanosoma cruzi infection associated with defective hemopoiesis and intracellular control of parasite growth. J Immunol. (2005) 175:8165–72. doi: 10.4049/jimmunol.175.12.8165
24. Kulkarni MM, Varikuti S, Terrazas C, Kimble JL, Satoskar AR, McGwire BS. Signal transducer and activator of transcription 1 (STAT-1) plays a critical role in control of Trypanosoma cruzi infection. Immunology (2015) 145:225–31. doi: 10.1111/imm.12438
25. Tarleton RL, Grusby MJ, Zhang L. Increased susceptibility of Stat4-deficient and enhanced resistance in Stat6-deficient mice to infection with Trypanosoma cruzi. J Immunol. (2000) 165:1520–5. doi: 10.4049/jimmunol.165.3.1520
26. Pérez AR, Roggero E, Nicora A, Palazzi J, Besedovsky HO, Del Rey A, et al. Thymus atrophy during Trypanosoma cruzi infection is caused by an immuno-endocrine imbalance. Brain Behav Immun. (2007) 21:890–900. doi: 10.1016/j.bbi.2007.02.004
27. Durand JL, Mukherjee S, Commodari F, De Souza AP, Zhao D, Machado FS, et al. Role of NO synthase in the development of Trypanosoma cruzi-induced cardiomyopathy in mice. Am J Trop Med Hyg. (2009) 80:782–7. doi: 10.4269/ajtmh.2009.80.782
28. Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FR, et al. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1β response and host resistance to Trypanosoma cruzi infection. J Immunol. (2013) 191:3373–83. doi: 10.4049/jimmunol.1203293
29. Dhiman M, Garg NJ P47phox−/− mice are compromised in expansion and activation of CD8+ T cells and susceptible to Trypanosoma cruzi infection. PLoS Pathog. (2014) 10:e1004516. doi: 10.1371/journal.ppat.1004516
30. Paiva CN, Figueiredo RT, Kroll-Palhares K, Silva AA, Silvério JC, Gibaldi D, et al. CCL2/MCP-1 controls parasite burden, cell infiltration, and mononuclear activation during acute Trypanosoma cruzi infection. J Leukoc Biol. (2009) 86:1239–46. doi: 10.1189/jlb.0309187
31. Machado FS, Koyama NS, Carregaro V, Ferreira BR, Milanezi CM, Teixeira MM, et al. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. J Infect Dis. (2005) 191:627–36. doi: 10.1086/427515
32. Hardison JL, Wrightsman RA, Carpenter PM, Kuziel WA, Lane TE, Manning JE. The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi. Infect Immun. (2006) 74:135–43. doi: 10.1128/IAI.74.1.135-143.2006
33. Michailowsky V, Celes MR, Marino AP, Silva AA, Vieira LQ, Rossi MA, et al. Intercellular adhesion molecule 1 deficiency leads to impaired recruitment of T lymphocytes and enhanced host susceptibility to infection with Trypanosoma cruzi. J Immunol. (2004) 173:463–70. doi: 10.4049/jimmunol.173.1.463
34. Martins GA, Campanelli AP, Silva RB, Tadokoro CE, Russo M, Cunha FQ, et al. CD28 is required for T cell activation and IFN-gamma production by CD4+ and CD8+ T cells in response to Trypanosoma cruzi infection. Microbes Infect. (2004) 6:1133–44. doi: 10.1016/j.micinf.2004.07.002
35. Talvani A, Santana G, Barcelos LS, Ishii S, Shimizu T, Romanha AJ, et al. Experimental Trypanosoma cruzi infection in platelet-activating factor receptor-deficient mice. Microbes Infect. (2003) 5:789–96. doi: 10.1016/S1286-4579(03)00146-1
36. Benatar AF, García GA, Bua J, Cerliani JP, Postan M, Tasso LM, et al. Galectin-1 prevents infection and damage induced by Trypanosoma cruzi on cardiac cells. PLoS Negl Trop Dis. (2015) 9:e0004148. doi: 10.1371/journal.pntd.0004148
37. Sharma J, Eickhoff CS, Hoft DF, Ford DA, Gross RW, McHowat J. The absence of myocardial calcium-independent phospholipase A2gamma results in impaired prostaglandin E2 production and decreased survival in mice with acute Trypanosoma cruzi infection. Infect Immun. (2013) 81:2278–87. doi: 10.1128/iai.00497-12
38. Silva MC, Davoli-Ferreira M, Medina TS, Sesti-Costa R, Silva GK, Lopes CD, et al. Canonical PI3Kγ signaling in myeloid cells restricts Trypanosoma cruzi infection and dampens chagasic myocarditis. Nat Commun. (2018) 9:1513. doi: 10.1038/s41467-018-03986-3
39. Medina TS, Oliveira GG, Silva MC, David BA, Silva GK, Fonseca DM, et al. Ebi3 prevents Trypanosoma cruzi-induced myocarditis by dampening IFN-γ-driven inflammation. Front Immunol. (2017) 8:1213. doi: 10.3389/fimmu.2017.01213
40. Hölscher C, Mohrs M, Dai WJ, Köhler G, Ryffel B, Schaub GA, et al. Tumor necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. (2000) 68:4075–83. doi: 10.1128/IAI.68.7.4075-4083.2000
41. Hunter CA, Ellis-Neyes LA, Slifer T, Kanaly S, Grünig G, Fort M, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. (1997) 158:3311–6.
42. Tosello Boari J, Amezcua Vesely MC, Bermejo DA, Ramello MC, Montes CL, Cejas H, et al. IL-17RA signaling reduces inflammation and mortality during Trypanosoma cruzi infection by recruiting suppressive IL-10-producing neutrophils. PLoS Pathog. (2012) 8:e1002658. doi: 10.1371/journal.ppat.1002658
43. Ferreira LR, Frade AF, Baron MA, Navarro IC, Kalil J, Chevillard C, et al. Interferon-γ, and other inflammatory mediators in cardiomyocyte signaling during Chagas disease cardiomyopathy. World J Cardiol. (2014) 6:782–90. doi: 10.4330/wjc.v6.i8.782
44. Stahl P, Ruppert V, Schwarz RT, Meyer T. Trypanosoma cruzi evades the protective role of interferon-gamma-signaling in parasite-infected cells. PLoS ONE (2014) 9:e110512. doi: 10.1371/journal.pone.0110512
45. Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FQ. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun. (1995) 63:4862–7.
46. Vila-del Sol V, Punzón C, Fresno M. IFN-gamma-induced TNF-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol. (2008) 181:4461–70.
47. Hölscher C, Köhler G, Müller U, Mossmann H, Schaub GA, Brombacher F. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect Immu. (1998) 66:1208–15.
48. Wu Y, Antony S, Juhasz A, Lu J, Ge Y, Jiang G, et al. Up-regulation and sustained activation of Stat1 are essential for interferon-gamma (IFN-gamma)-induced dual oxidase 2 (Duox2) and dual oxidase A2 (DuoxA2) expression in human pancreatic cancer cell lines. J Biol Chem, (2011) 286:12245–56. doi: 10.1074/jbc.M110.191031
49. Rakshit S, Chandrasekar BS, Saha B, Victor ES, Majumdar S, Nandi D. Interferon-gamma induced cell death: regulation and contributions of nitric oxide, cJun N-terminal kinase, reactive oxygen species and peroxynitrite. Biochim Biophys Acta. (2014) 1843:2645–61. doi: 10.1016/j.bbamcr.2014.06.014
50. Alvarez MN, Peluffo G, Piacenza L, Radi R. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J Biol Chem. (2011) 286:6627–40. doi: 10.1074/jbc.M110.167247
51. Koo SJ, Chowdhury IH, Szczesny B, Wan X, Garg NJ. Macrophages promote oxidative metabolism to drive nitric oxide generation in response to Trypanosoma cruzi. Infect Immun. (2016) 84:3527–41. doi: 10.1128/IAI.00809-16
52. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. (2004) 75:163–89. doi: 10.1189/jlb.0603252
53. Marinho CR, Nunez-Apaza LN, Martins-Santos R, Bastos KR, Bombeiro AL, Bucci DZ, et al. IFN-gamma, but not nitric oxide or specific IgG, is essential for the in vivo control of low-virulence Sylvio X10/4 Trypanosoma cruzi parasites. Scand J Immunol. (2007) 66:297–308. doi: 10.1111/j.1365-3083.2007.01958.x
54. Schroder K, Lichtinger M, Irvine KM, Brion K, Trieu A, Ross IL, et al. PU.1 and ICSBP control constitutive and IFN-gamma-regulated Tlr9 gene expression in mouse macrophages. J Leukoc Biol. (2007) 81:1577–90. doi: 10.1189/jlb.0107036
55. Talvani A, Rocha MO, Barcelos LS, Gomes YM, Ribeiro AL, Teixeira MM. Elevated concentrations of CCL2 and tumor necrosis factor-alpha in chagasic cardiomyopathy. Clin Infect Dis. (2004) 38:943–50. doi: 10.1086/381892
56. Silva RR, Mariante RM, Silva AA, dos Santos AL, Roffê E, Santiago H, et al. Interferon-gamma promotes infection of astrocytes by Trypanosoma cruzi. PLoS ONE (2015) 10:e0118600. doi: 10.1371/journal.pone.0118600
57. Cunha-Neto E, Dzau VJ, Allen PD, Stamatiou D, Benvenutti L, Higuchi ML, et al. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol. (2005) 167:305–13. doi: 10.1016/S0002-9440(10)62976-8
58. Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation (2000) 102:3003–8. doi: 10.1161/01.cir.102.24.3003
59. Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect Immun. (1994) 62:5177–82.
60. Cai CW, Blase JR, Zhang X, Eickhoff CS, Hoft DF. Th17 cells are more protective than Th1 cells against the intracellular parasite Trypanosoma cruzi. PLoS Pathog. (2016) 12:e1005902. doi: 10.1371/journal.ppat.1005902
61. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64:477–85. doi: 10.1016/j.cyto.2013.07.022
62. Hamano S, Himeno K, Miyazaki Y, Ishii K, Yamanaka A, Takeda A, et al. WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity (2003) 19:657–67.
63. Poncini CV, Ilarregui JM, Batalla EI, Engels S, Cerliani JP, Cucher MA, et al. Trypanosoma cruzi infection imparts a regulatory program in dendritic cells and T cells via galectin-1-dependent mechanisms. J Immunol. (2015) 195:3311–24. doi: 10.4049/jimmunol.1403019
64. Abrahamsohn IA, Coffman RL. Trypanosoma cruzi, IL-10, TNF, IFN-gamma, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol. (1996) 84:231–44. doi: 10.1006/expr.1996.0109
65. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
66. da Matta Guedes PM, Gutierrez FR, Maia FL, Milanezi CM, Silva GK, Pavanelli WR, et al. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLoS Negl Trop Dis. (2010) 4:e604. doi: 10.1371/journal.pntd.0000604
67. Sieve AN, Meeks KD, Lee S, Berg RE. A novel immunoregulatory function for IL-23: inhibition of IL-12-dependent IFN-γ production. Eur J Immunol. (2010) 40:2236–47. doi: 10.1002/eji.200939759
68. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
69. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
70. Böhme J, Roßnagel C, Jacobs T, Behrends J, Hölscher C, Erdmann H. Epstein-Barr virus-induced gene 3 suppresses T helper type 1, type 17 and type 2 immune responses after Trypanosoma cruzi infection and inhibits parasite replication by interfering with alternative macrophage activation. Immunology (2016) 147:338–48. doi: 10.1111/imm.12565
71. Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. (2002) 39:531–6. doi: 10.1016/S0161-5890(02)00210-9
72. Fernandes MC, Andrews NW. Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol Rev. (2012) 36:734–47. doi: 10.1111/j.1574-6976.2012.00333.x
73. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol. (2015) 6:659. doi: 10.3389/fimmu.2015.00659
74. Rodrigues MM, Oliveira AC, Bellio M. The immune response to Trypanosoma cruzi: role of Toll-like receptors and perspectives for vaccine development. J Parasitol Res. (2012) 2012:507874. doi: 10.1155/2012/507874
75. De Alba-Alvarado M, Salazar-Schettino PM, Jiménez-Álvarez L, Cabrera-Bravo M, García-Sancho C, Zenteno E, et al. Th-17 cytokines are associated with severity of Trypanosoma cruzi chronic infection in pediatric patients from endemic areas of Mexico. Acta Trop. (2018) 178:134–41. doi: 10.1016/j.actatropica.2017.11.009
76. Bestetti RB, Muccillo G. Clinical course of Chagas' heart disease: a comparison with dilated cardiomyopathy. Int J Cardiol. (1997) 60:187–93.
77. Sousa GR, Gomes JA, Damasio MP, Nunes MC, Costa HS, Medeiros NI, et al. The role of interleukin 17-mediated immune response in Chagas disease: high level is correlated with better left ventricular function. PLoS ONE (2017) 12:e0172833. doi: 10.1371/journal.pone.0172833
78. Higuchi Mde L, Gutierrez PS, Aiello VD, Palomino S, Bocchi E, Kalil J, et al. Immunohistochemical characterization of infiltrating cells in human chronic chagasic myocarditis: comparison with myocardial rejection process. Virchows Arch A Pathol Anat Histopathol. (1993) 423:157–60.
79. Reis DD, Jones EM, Tostes S Jr, Lopes ER, Gazzinelli G, Colley DG, et al. Characterization of inflammatory infiltrates in chronic chagasic myocardial lesions: presence of tumor necrosis factor-alpha+ cells and dominance of granzyme A+, CD8+ lymphocytes. Am J Trop Med Hyg. (1993) 48:637–44.
80. Reis MM, Higuchi Mde L, Benvenuti LA, Aiello VD, Gutierrez PS, Bellotti G, et al. An in situ quantitative immunohistochemical study of cytokines and IL-2R+ in chronic human chagasic myocarditis: correlation with the presence of myocardial Trypanosoma cruzi antigens. Clin Immunol Immunopathol. (1997) 83:165–72.
81. Fonseca SG, Reis MM, Coelho V, Nogueira LG, Monteiro SM, Mairena EC, et al. Locally produced survival cytokines IL-15 and IL-7 may be associated to the predominance of CD8+ T cells at heart lesions of human chronic Chagas disease cardiomyopathy. Scand J Immunol. (2007) 66:362–71. doi: 10.1111/j.1365-3083.2007.01987.x
82. Abel LC, Rizzo LV, Ianni B, Albuquerque F, Bacal F, Carrara D, et al. Chronic Chagas' disease cardiomyopathy patients display an increased IFN-gamma response to Trypanosoma cruzi infection. J Autoimmun. (2001) 17:99–107. doi: 10.1006/jaut.2001.0523
83. Rocha Rodrigues DB, dos Reis MA, Romano A, Pereira SA, Teixeira Vde P, Tostes S Jr, et al. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas' disease patients with heart failure. Clin Dev Immunol. (2012) 2012:361730. doi: 10.1155/2012/361730
84. Nogueira LG, Santos RH, Fiorelli AI, Mairena EC, Benvenuti LA, Bocchi EA, et al. Myocardial gene expression of T-bet, GATA-3, Ror-γt, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: an essentially unopposed TH1-type response. Mediators Inflamm. (2014) 2014:914326. doi: 10.1155/2014/914326
85. Gomes JA, Bahia-Oliveira LM, Rocha MO, Busek SC, Teixeira MM, Silva JS, et al. Type 1 chemokine receptor expression in Chagas' disease correlates with morbidity in cardiac patients. Infect Immun. (2005) 73:7960–6. doi: 10.1128/IAI.73.12.7960-7966.2005
86. Cunha-Neto E, Dzao VJ, Allen PD, Stamatiou D, Benvenutti L, Higuchi ML, et al. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol. (2005) 167:305–13. doi: 10.1016/s0002-9440(10)62976-8
87. Nogueira LG, Santos RHB, Ianni BM, Fiorelli AI, Mairena EC, Benvenuti LA, et al. Myocardial chemokine expression and intensity of myocarditis in chagas cardiomyopathy are controlled by polymorphisms in CXCL9 and CXCL10. Plos Negl Trop Dis. (2012) 6:e186. doi: 10.1371/journal.pntd.0001867
88. Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, et al. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med (2001) 194:1123–40. doi: 10.1084/jem.194.8.1123
89. Levick SP, Goldspink PH. Could interferon-gamma be a therapeutic target for treating heart failure? Heart Fail Rev. (2014) 19:227–36. doi: 10.1007/s10741-013-9393-8
90. Krause CD, He W, Kotenko S, Pestka S. Modulation of the activation of Stat1 by the interferon-gamma receptor complex. Cell Res. (2006) 16:113–23. doi: 10.1038/sj.cr.7310015
91. Ferreira LRP, Ferreira FM, Laugier L, Cabantous S, Navarro IC, da Silva Cândido D, et al. Integration of miRNA and gene expression profiles suggest a role for miRNAs in the pathobiological processes of acute Trypanosoma cruzi infection. Sci Rep. (2017) 7:17990. doi: 10.1038/s41598-017-18080-9
92. Reifenberg K, Lehr HA, Torzewski M, Steige G, Wiese E, Küpper I, et al. Interferon-gamma induces chronic active myocarditis and cardiomyopathy in transgenic mice. Am J Pathol. (2007) 171:463–72. doi: 10.2353/ajpath.2007.060906
93. Torzewski M, Wenzel P, Kleinert H, Becker C, El-Masri J, Wiese E, et al. Chronic inflammatory cardiomyopathy of interferon γ-overexpressing transgenic mice is mediated by tumor necrosis factor-α. Am J Pathol. (2012) 180:73–81. doi: 10.1016/j.ajpath.2011.09.006
94. Patten M, Krämer E, Bünemann J, Wenck C, Thoenes M, Wieland T, et al. Endotoxin and cytokines alter contractile protein expression in cardiac myocytes in vivo. Pflugers Arch. (2001) 442:920–7. doi: 10.1007/s004240100612
95. Wang Z, Jiang B, Brecher P. Selective inhibition of STAT3 phosphorylation by sodium salicylate in cardiac fibroblasts. Biochem Pharmacol. (2002) 63:1197–207. doi: 10.1016/S0006-2952(02)00853-5
96. Chae HJ, Ha KC, Kim DS, Cheung GS, Kwak YG, Kim HM, et al. Catalase protects cardiomyocytes via its inhibition of nitric oxide synthesis. Nitric Oxide (2006) 14:189–99. doi: 10.1016/j.niox.2005.11.008
97. Dai B, Cui M, Zhu M, Su WL, Qiu MC, Zhang H. STAT1/3 and ERK1/2 synergistically regulate cardiac fibrosis induced by high glucose. Cell Physiol Biochem. (2013) 32:960–71. doi: 10.1159/000354499
98. Wu AJ, Lafrenie RM, Park C, Apinhasmit W, Chen ZJ, Birkedal-Hansen H, et al. Modulation of MMP-2 (gelatinase A) and MMP-9 (gelatinase B) by interferon-gamma in a human salivary gland cell line. J Cell Physiol. (1997) 171:117–24. doi: 10.1002/(SICI)1097-4652(199705)171:2<117::AID-JCP1>3.0.CO;2-R
99. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–250. doi: 10.1038/nrcardio.2016.203
100. Wan X, Wen JJ, Koo SJ, Liang LY, Garg NJ. SIRT1-PGC1α-NFκB pathway of oxidative and inflammatory stress during Trypanosoma cruzi infection: benefits of SIRT1-targeted therapy in improving heart function in Chagas disease. PLoS Pathog. (2016) 12:e1005954. doi: 10.1371/journal.ppat.1005954
101. Teixeira PC, Santos RH, Fiorelli AI, Bilate AM, Benvenuti LA, Stolf NA, et al. Selective decrease of components of the creatine kinase system and ATP synthase complex in chronic Chagas disease cardiomyopathy. PLoS Negl Trop Dis. (2011) 5:e1205. doi: 10.1371/journal.pntd.0001205
102. Wan X, Gupta S, Zago MP, Davidson MM, Dousset P, Amoroso A, et al. Defects of mtDNA replication impaired mitochondrial biogenesis during Trypanosoma cruzi infection in human cardiomyocytes and chagasic patients: the role of Nrf1/2 and antioxidant response. J Am Heart Assoc. (2012) 1:e003855. doi: 10.1161/JAHA.112.003855
103. Luss H, Watkins SC, Freeswick PD, Imro AK, Nussler AK, Billiar TR, et al. Characterization of inducible nitric oxide synthase expression in endotoxemic rat cardiac myocytes in vivo and following cytokine exposure in vitro. J Mol Cell Cardiol. (1995) 27:2015–29.
104. Wang D, McMillin JB, Bick R, Buja LM. Response of the neonatal rat cardiomyocyte in culture to energy depletion: effects of cytokines, nitric oxide, and heat shock proteins. Lab Invest. (1996) 75:809–18.
105. Kalovidouris AE, Plotkin Z, Graesser D. Interferon-gamma inhibits proliferation, differentiation, and creatine kinase activity of cultured human muscle cells. II. A possible role in myositis. J Rheumatol. (1993) 20:1718–23.
106. Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY, Park YS, et al. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J Mol Biol. (2007) 369:967–84. doi: 10.1016/j.jmb.2007.03.072
107. Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, et al. Mitochondrial membrane potential. Anal Biochem. (2017) 552: 50–9. doi: 10.1016/j.ab.2017.07.009
108. Pall ML. The NO/ONOO-cycle as the central cause of heart failure. Int J Mol Sci. (2013) 14:22274–330. doi: 10.3390/ijms141122274
109. Paiva CN, Medei E, Bozza MT. ROS and Trypanosoma cruzi: fuel to infection, poison to the heart. PLoS Pathog. (2018) 14:e1006928. doi: 10.1371/journal.ppat.1006928
110. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. (2013) 25:1939–48. doi: 10.1016/j.cellsig.2013.06.007
111. Caruso R, Marafini I, Franzè E, Stolfi C, Zorzi F, Monteleone I, et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol. (2014) 7:1467–79. doi: 10.1038/mi.2014.35
112. Vilar-Pereira G, Carneiro VC, Mata-Santos H, Vicentino AR, Ramos IP, Giarola NL, et al. Resveratrol reverses functional chagas heart disease in mice. PLoS Pathog. (2016) 12:e1005947. doi: 10.1371/journal.ppat.1005947
113. Wen JJ, Porter C, Garg NJ. Inhibition of NFE2L2-antioxidant response element pathway by mitochondrial reactive oxygen species contributes to development of cardiomyopathy and left ventricular dysfunction in Chagas disease. Antioxid Redox Signal. (2017) 27:550–66. doi: 10.1089/ars.2016.6831
114. Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. (2015) 1850:794–801. doi: 10.1016/j.bbagen.2014.11.021
115. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. (2015) 88(Pt B):179–88. doi: 10.1016/j.freeradbiomed.2015.04.036
116. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. (2011) 11:389–402. doi: 10.1038/nri2975
117. Van Linthout S, Tschöpe C. Inflammation - cause or consequence of heart failure or both? Curr Heart Fail Rep. (2017) 14:251–65. doi: 10.1007/s11897-017-0337-9
118. Zicker F, Smith PG, Netto JC, Oliveira RM, Zicker EM. Physical activity, opportunity for reinfection, and sibling history of heart disease as risk factors for Chagas' cardiopathy. Am J Trop Med Hyg. (1990) 43:498–505.
119. Weitzel T, Zulantay I, Danquah I, Hamann L, Schumann RR, Apt W, et al. Mannose-binding lectin and Toll-like receptor polymorphisms and Chagas disease in Chile. Am J Trop Med Hyg. (2012) 86:229–32. doi: 10.4269/ajtmh.2012.11-0539
120. Rodríguez-Pérez JM, Cruz-Robles D, Hernández-Pacheco G, Pérez-Hernández N, Murguía LE, Granados J, et al. Tumor necrosis factor-alpha promoter polymorphism in Mexican patients with Chagas' disease. Immunol Lett. (2005) 98:97–102. doi: 10.1016/j.imlet.2004.10.017
121. Pissetti CW, Correia D, de Oliveira RF, Llaguno MM, Balarin MA, Silva-Grecco RL, et al. Genetic and functional role of TNF-alpha in the development Trypanosoma cruzi infection. PLoS Negl Trop Dis. (2011) 5:e976. doi: 10.1371/journal.pntd.0000976
122. Criado L, Flórez O, Martín J, González CI. Genetic polymorphisms in TNFA/TNFR2 genes and Chagas disease in a Colombian endemic population. Cytokine (2012) 57:398–401. doi: 10.1016/j.cyto.2011.12.007
123. Drigo SA, Cunha-Neto E, Ianni B, Cardoso MR, Braga PE, Faé KC, et al. TNF gene polymorphisms are associated with reduced survival in severe Chagas' disease cardiomyopathy patients. Microbes Infect. (2006) 8:598–603. doi: 10.1016/j.micinf.2005.08.009
124. Zafra G, Morillo C, Martín J, González A, González CI. Polymorphism in the 3' UTR of the IL12B gene is associated with Chagas' disease cardiomyopathy. Microbes Infect. (2007) 9:1049–52. doi: 10.1016/j.micinf.2007.04.010
125. Torres OA, Calzada JE, Beraún Y, Morillo CA, González A, González CI, et al. Role of the IFNG +874T/A polymorphism in Chagas disease in a Colombian population. Infect Genet Evol. (2010) 10:682–5. doi: 10.1016/j.meegid.2010.03.009
126. MacMurray J, Comings DE, Napolioni V. The gene-immune-behavioral pathway: Gamma-interferon (IFN-γ) simultaneously coordinates susceptibility to infectious disease and harm avoidance behaviors. Brain Behav Immun. (2014) 35:169–75. doi: 10.1016/j.bbi.2013.09.012
127. Frade AF, Pissetti CW, Ianni BM, Saba B, Lin-Wang HT, Nogueira LG, et al. Genetic susceptibility to Chagas disease cardiomyopathy: involvement of several genes of the innate immunity and chemokine-dependent migration pathways. BMC Infect Dis. (2013) 13:587. doi: 10.1186/1471-2334-13-587
128. Flórez O, Martín J, González CI. Genetic variants in the chemokines and chemokine receptors in Chagas disease. Hum Immunol. (2012) 73:852–8. doi: 10.1016/j.humimm.2012.04.005
129. de Oliveira AP, Bernardo CR, Camargo AV, Ronchi LS, Borim AA, de Mattos CC, et al. Genetic susceptibility to cardiac and digestive clinical forms of chronic Chagas disease, involvement of the CCR5 59029 A/G polymorphism. PLoS ONE (2015) 10:e0141847. doi: 10.1371/journal.pone.0141847
130. Machuca MA, Suárez EU, Echeverría LE, Martín J, González CI. SNP/haplotype associations of CCR2 and CCR5 genes with severity of chagasic cardiomyopathy. Hum Immunol. (2014) 75:1210–5. doi: 10.1016/j.humimm.2014.09.023
131. Costa GC, da Costa Rocha MO, Moreira PR, Menezes CA, Silva MR, Gollob KJ, et al. Functional IL-10 gene polymorphism is associated with Chagas disease cardiomyopathy. J Infect Dis. (2009) 199:451–4. doi: 10.1086/596061
132. Beraun Y, Nieto A, Collado MD, Gonzalez A, Martin J. Polymorphisms at tumor necrosis factor (TNF) loci are not associated with Chagas' disease. Tissue Antigens. (1998) 52:81–3.
133. Pissetti CW, de Oliveira RF, Correia D, Nascentes GA, Llaguno MM, Rodrigues V. Association between the lymphotoxin-alpha gene polymorphism and chagasic cardiopathy. J Interferon Cytokine Res. (2013) 33:130–5. doi: 10.1089/jir.2012.0024
134. Ramasawmy R, Cunha-Neto E, Fae KC, Borba SC, Teixeira PC, Ferreira SC, et al. Heterozygosity for the S180L variant of MAL/TIRAP, a gene expressing an adaptor protein in the Toll-like receptor pathway, is associated with lower risk of developing chronic Chagas cardiomyopathy. J Infect Dis. (2009) 199:1838–45. doi: 10.1086/599212
135. Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. (2007) 39:523–8. doi: 10.1038/ng1976
136. Parissis JT, Adamopoulos S, Venetsanou KF, Mentzikof DG, Karas SM, Kremastinos DT. Serum profiles of C-C chemokines in acute myocardial infarction: possible implication in postinfarction left ventricular remodeling. J Interferon Cytokine Res. (2002) 22:223–9. doi: 10.1089/107999002753536194
137. Aukrust P, Ueland T, Müller F, Andreassen AK, Nordøy I, Aas H, et al. Elevated circulating levels of C-C chemokines in patients with congestive heart failure. Circulation (1998) 97:1136–43.
138. Ramasawmy R, Cunha-Neto E, Fae KC, Martello FG, Müller NG, Cavalcanti VL, et al. The monocyte chemoattractant protein-1 gene polymorphism is associated with cardiomyopathy in human chagas disease. Clin Infect Dis. (2006) 43:305–11. doi: 10.1086/505395
139. Calzada JE, Nieto A, Beraún Y, Martín J. Chemokine receptor CCR5 polymorphisms and Chagas' disease cardiomyopathy. Tissue Antigens. (2001) 58:154–8. doi: 10.1034/j.1399-0039.2001.580302.x
140. Fernández-Mestre MT, Montagnani S, Layrisse Z. Is the CCR5-59029-G/G genotype a protective factor for cardiomyopathy in Chagas disease? Hum Immunol. (2004) 65:725–8. doi: 10.1016/j.humimm.2004.05.002
141. de Oliveira AP, Ayo CM, Bestetti RB, Brandão de Mattos CC, Cavasini CE, de Mattos LC. The role of CCR5 in Chagas disease - a systematic review. Infect Genet Evol. (2016) 45:132–7. doi: 10.1016/j.meegid.2016.08.012
142. Stein RW, Whelan J. Insulin gene enhancer activity is inhibited by adenovirus 5 E1a gene products. Mol Cell Biol. (1989) 9:4531–4.
143. Flórez O, Martín J, González CI. Interleukin 4, interleukin 4 receptor-α and interleukin 10 gene polymorphisms in Chagas disease. Parasite Immunol. (2011) 33:506–11. doi: 10.1111/j.1365-3024.2011.01314.x
144. Sales-Campos H, Kappel HB, Andrade CP, Lima TP, de Castilho A, Giraldo LE, et al. Trypanosoma cruzi DTU TcII presents higher blood parasitism than DTU TcI in an experimental model of mixed infection. Acta Parasitol. (2015) 60:435–41. doi: 10.1515/ap-2015-0060
145. Meza SK, Kaneshima EN, Silva Sde O, Gabriel M, de Araújo SM, Gomes ML, et al. Comparative pathogenicity in Swiss mice of Trypanosoma cruzi IV from northern Brazil and Trypanosoma cruzi II from southern Brazil. Exp Parasitol. (2014) 146:34–42. doi: 10.1016/j.exppara.2014.08.014
146. Magalhães LM, Viana A, Chiari E, Galvão LM, Gollob KJ, Dutra WO. Differential Activation of human monocytes and lymphocytes by distinct strains of Trypanosoma cruzi. PLoS Negl Trop Dis. (2015) 9:e0003816. doi: 10.1371/journal.pntd.0003816
147. Teixeira MM, da Silva FM, Marcili A, Umezawa ES, Shikanai-Yasuda MA, Cunha-Neto E, et al. Short communication: Trypanosoma cruzi lineage I in endomyocardial biopsy from a north-eastern Brazilian patient at end-stage chronic Chagasic cardiomyopathy. Trop Med Int Health. (2006) 11:294–8. doi: 10.1111/j.1365-3156.2006.01575.x
148. Cura CI, Lucero RH, Bisio M, Oshiro E, Formichelli LB, Burgos JM, et al. Trypanosoma cruzi discrete typing units in Chagas disease patients from endemic and non-endemic regions of Argentina. Parasitology (2012) 139:516–21. doi: 10.1017/S0031182011002186
149. Risso MG, Sartor PA, Burgos JM, Briceño L, Rodríguez EM, Guhl F, et al. Immunological identification of Trypanosoma cruzi lineages in human infection along the endemic area. Am J Trop Med Hyg. (2011) 84:78-84. doi: 10.4269/ajtmh.2011.10-0177
150. Muñoz-San Martín C, Zulantay I, Saavedra M, Fuentealba C, Muñoz G, Apt W. Discrete typing units of Trypanosoma cruzi detected by real-time PCR in Chilean patients with chronic Chagas cardiomyopathy. Acta Trop. (2018) 185:280–4. doi: 10.1016/j.actatropica.2018.05.004
151. Carrera-Silva EA, Eugenio Antonio CS, Cano RC, Carolina CR, Guiñazú N, Natalia G, et al. TLR2, TLR4 and TLR9 are differentially modulated in liver lethally injured from BALB/c and C57BL/6 mice during Trypanosoma cruzi acute infection. Mol Immunol. (2008) 45:3580–8. doi: 10.1016/j.molimm.2008.05.004
Keywords: disease tolerance gene, pathogen resistance genes, Tripanosoma cruzi, chagas cardiomyopathy, interferon gamma
Citation: Chevillard C, Nunes JPS, Frade AF, Almeida RR, Pandey RP, Nascimento MS, Kalil J and Cunha-Neto E (2018) Disease Tolerance and Pathogen Resistance Genes May Underlie Trypanosoma cruzi Persistence and Differential Progression to Chagas Disease Cardiomyopathy. Front. Immunol. 9:2791. doi: 10.3389/fimmu.2018.02791
Received: 01 July 2018; Accepted: 13 November 2018;
Published: 03 December 2018.
Edited by:
Alexandre Morrot, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Luciana Oliveira Andrade, Universidade Federal de Minas Gerais, BrazilKarina Andrea Gomez, CONICET Instituto de Investigaciones en Ingeniería Genética y Biología Molecular Dr. Héctor N. Torres (INGEBI), Argentina
Copyright © 2018 Chevillard, Nunes, Frade, Almeida, Pandey, Nascimento, Kalil and Cunha-Neto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edecio Cunha-Neto, ZWRlY3VuaGFAZ21haWwuY29t
Christophe Chevillard, Y2hyaXN0b3BoZS5jaGV2aWxsYXJkQHVuaXYtYW11LmZy
†These authors have contributed equally to this work