 Raphaël Mattiuz1
Raphaël Mattiuz1 Christian Wohn1
Christian Wohn1 Sonia Ghilas1
Sonia Ghilas1 Marc Ambrosini1Yannick O. Alexandre1Cindy Sanchez1Anissa Fries1
Marc Ambrosini1Yannick O. Alexandre1Cindy Sanchez1Anissa Fries1 Thien-Phong Vu Manh1
Thien-Phong Vu Manh1 Bernard Malissen1,2
Bernard Malissen1,2 Marc Dalod1*
Marc Dalod1* Karine Crozat1*
Karine Crozat1*- 1Centre d'Immunologie de Marseille-Luminy, Turing Center for Living Systems, CNRS, INSERM, Aix Marseille Univ, Marseille, France
- 2Centre d'Immunophénomique, Aix Marseille Univ, CNRS, INSERM, Marseille, France
Type 1 conventional DCs (cDC1) excel in the cross-priming of CD8+ T cells, which is crucial for orchestrating efficient immune responses against viruses or tumors. However, our understanding of their physiological functions and molecular regulation has been limited by the lack of proper mutant mouse models allowing their conditional genetic targeting. Because the Xcr1 and A530099j19rik (Karma/Gpr141b) genes belong to the core transcriptomic fingerprint of mouse cDC1, we used them to engineer two novel Cre-driver lines, the Xcr1Cre and KarmaCre mice, by knocking in an IRES-Cre expression cassette into their 3′-UTR. We used genetic tracing to characterize the specificity and efficiency of these new models in several lymphoid and non-lymphoid tissues, and compared them to the Clec9aCre mouse model, which targets the immediate precursors of cDCs. Amongst the three Cre-driver mouse models examined, the Xcr1Cre model was the most efficient and specific for the fate mapping of all cDC1, regardless of the tissues examined. The KarmaCre model was rather specific for cDC1 when compared with the Clec9aCre mouse, but less efficient than the Xcr1Cre model. Unexpectedly, the Xcr1Cre model targeted a small fraction of CD4+ T cells, and the KarmaCre model a significant proportion of mast cells in the skin. Importantly, the targeting specificity of these two mouse models was not changed upon inflammation. A high frequency of germline recombination was observed solely in the Xcr1Cre mouse model when both the Cre and the floxed alleles were brought by the same gamete irrespective of its gender. Xcr1, Karma, and Clec9a being differentially expressed within the cDC1 population, the three CRE-driver lines examined showed distinct recombination patterns in cDC1 phenotypic subsets. This advances our understanding of cDC1 subset heterogeneity and the differentiation trajectory of these cells. Therefore, to the best of our knowledge, upon informed use, the Xcr1Cre and KarmaCre mouse models represent the best tools currently reported to specifically and faithfully target cDC1 in vivo, both at steady state and upon inflammation. Future use of these mutant mouse models will undoubtedly boost our understanding of the biology of cDC1.
Introduction
Dendritic cells (DCs) constitute a heterogeneous population of antigen presenting cells (APCs) which are instrumental for the orchestration of innate and adaptive immune responses. In mice and in humans, three distinct types of DCs differing in their phenotype, localization and functions populate all lymphoid and most non-lymphoid tissues at steady state. Plasmacytoid DCs (pDCs) are the major source of type I interferon (IFN) upon many viral infections. Conventional DCs (cDCs) consist of two populations, coined as type 1 and type 2 cDCs and which excel in the cross-priming of CD8+ T cells or in the promotion of CD4+ T cell and humoral immunity, respectively. The functions of cDCs and their molecular regulation have been studied in vivo by using a wealth of mouse models that enable their depletion or genetic manipulation, namely Cd11c (Itgax)hDTR (1) or Cd11c (Itgax)Cre (2, 3) and more recently the Zbtb46hDTR (4) or Zbtb46Cre (5). However, interpretation of the results obtained using those mice can be difficult due to the expression of Cd11c by many other cell types than cDCs and of Zbtb46 by committed erythroid progenitors and endothelial cell populations (6). Moreover, these mutant mouse models are not suited to study the respective functions of each of the two cDC types. This goal requires the use of refined mutant mouse models enabling specific targeting of either cDC1 or cDC2.
Constitutive (Batf3-KO mice) or conditional (ItgaxCre; Irf8fl/fl mice) genetic inactivation of transcription factors required for the differentiation of cDC1 allowed to study their specific functions in vivo (7, 8). However, interpretation of the results obtained with these models can be difficult because they are not targeting solely cDC1 (7, 9–11). Moreover, cDC1 are replenished in Batf3-KO mice under inflammatory conditions, due to expression of other Batf transcription factors that compensate for Batf3 loss (12). Finally, these models do not allow the editing of cDC1 genome, which would be a powerful method to decipher the molecular regulation of their functions. Hence, novel mutant mouse models are needed to reach this goal.
In all tissues with the exception of the intestine, cDC1 can be defined as CD24+ SIRPα/CD172a− cDCs (13–15). In addition, lymphoid-tissue resident cDC1 express CD8α, whereas the cDC1 residing in the parenchyma of non-lymphoid tissues and their counterparts that have migrated in secondary lymphoid organs express CD103. CLEC9A, a C type lectin receptor that allows efficient cross-presentation by cDC1 of dying cell-associated antigens (16) has been identified as a good candidate to generate mice enabling selective targeting of cDC1 in vivo due to its selective expression in these cells and to a lesser extent in pDCs (17–20). However, a thorough analysis of mice expressing a Cre recombinase under the Clec9a promoter showed that Cre-driven recombination occurred not only in cDC1 and to some extent in pDCs, but also in cDC2, leading to the discovery that Clec9a is expressed in a progenitor cell common to both cDC types (21). Hence, the Clec9aCre mouse is not suitable for specific targeting of cDC1.
A major breakthrough in the field of cDC1 was the identification of XCR1 as a universal marker of all cDC1 regardless of their tissues of residency, and present in all the warm-blooded vertebrate species studied to date (22–27). Xcr1 encodes the chemokine receptor XCR1, which ligand XCL1 is strongly upregulated in natural killer (NK) cells, CD8+ T cells and memory T cells upon activation in mice (24, 26, 28–31). Recently, a mouse model based on the expression of the Cre recombinase under the control of the Xcr1 promoter has been generated to specifically manipulate gene expression in cDC1. This mutant mouse model was engineered by replacing the single coding exon of Xcr1 by the Cre gene (32). This strategy assumes that the Xcr1 gene is haplosufficient. However, this hypothesis has to be tested considering that XCR1 promotes the cross-talk between cDC1 and NK cells or CD8+ T lymphocytes, by facilitating their reciprocal recruitment and/or activation (24, 26, 29). Regardless of its potential limitation, this Xcr1tm4(cre)Ksho mouse model has been useful to decipher the role of cDC1 in intraepithelial T cell homeostasis in the intestine (32). However, to the best of our knowledge, it has not been used yet for conditional gene targeting of the cDC1 lineage. Besides Xcr1, the A530099j19rik gene (named Karma hereafter) has also been identified as selectively expressed in cDC1 by bulk transcriptomic analysis on immune cell subsets and organs (10, 33). The Karma gene encodes a protein with 7 transmembrane domains, likely corresponding to a G protein-coupled receptor, leading to its recent denomination as Gpr141b by the Mouse Genome Informatics. Recently, we generated the Karma knock-in reporter/deleter mouse model, which expresses in the Karma locus a construct encoding both the fluorescent tandem dimer Tomato (tdTomato) and the human diphtheria toxin receptor (hDTR), allowing specific tracking and conditional depletion of cDC1 in vivo. Results obtained with this reporter mouse validated the Karma locus as highly reliable to functionally target cDC1 in vivo (33).
To match the unmet need of a mouse model allowing specific and efficient in vivo genetic manipulation of cDC1, we generated two novel Cre-driver lines, the Xcr1Cre and KarmaCre mice, by knocking in an IRES-Cre expression cassette in the 3′-UTR of the Xcr1 or Karma locus, respectively. In this study, we used genetic tracing to characterize the specificity and efficiency of the Cre-mediated recombination in these new models at steady state and upon infection, and compared them with the Clec9aCre model. This study also advanced our understanding of the phenotypic heterogeneity of cDC1 with regard to their differentiation trajectory.
Materials and Methods
Generation of cDC1 Targeting Cre Constructs and Mice
Xcr1Cre (B6-Xcr1tm1Ciphe) and KarmaCre (B6-Gpr141btm2Ciphe) mice were made according to a standard gene targeting approach in C57BL/6N-derived ES cells. They were constructed by inserting, through ET homologous recombination, a cassette containing the internal ribosome entry site (IRES) followed by a gene encoding the codon-improved version of Cre recombinase (34), into the 3′-UTR of the Xcr1 or A530099j19rik/Gpr141b genes, 34 and 98 bp after the stop codon, respectively. These mice were outcrossed for three generations with wild type (Wt) C57BL/6J mice purchased from Charles River Laboratories. All experiments were performed with sex-matched littermate mice at 6–12 weeks of age. Clec9aCre (Clec9atm2.1(icre)Crs) (21) knock-in mice [kindly provided by Caetano Reis e Sousa (The Francis Crick Institute, UK)], Karma-tdTomato-hDTR (Gp141btm1Ciphe) (33) were maintained on the C57BL/6J background. Rosa26lox−stop−lox−tdRFP (Gt(ROSA)26Sortm1Hjf) mice in which expression of the tandem dimer Red Fluorescent Protein (tdRFP) is driven through the deletion of a “lox–stop–lox” sequence (35) were purchased from the Jackson Laboratory and maintained on the C57BL/6J background. Rosa26lox−stop−lox−DTA (Gt(ROSA)26Sortm1(DTA)Lky) mice in which expression of active domain of the diphtheria toxin (DTA) is driven through the deletion of a “lox–stop–lox” sequence (36) were obtained from Prs. David Voehringer and Richard M. Locksley, and maintained on the C57BL/6J background. Mice were bred and maintained in our specific pathogen—free animal facility. This study was carried out in accordance with institutional guidelines and with protocols approved by the Comité National de Réflexion Ethique sur l'Expérimentation Animale #14.
Preparation of Cell Suspension From Blood and Tissues, and Analysis by Flow Cytometry
Splenocytes were prepared by infusing spleens with an enzymatic cocktail made of Collagenase D (1 mg/ml) and DNase I (70 μg/ml, both Roche) in plain RPMI 1640, and further incubation for 25 min at 37°C. Ice cold EDTA (2 mM) was added for additional 5 min. Cells were filtered through a 70-μm nylon sieve, and exposed to 0.155 M NH4Cl, 10 mM KHCO3, 0.127 M EDTA to lyse red blood cells. Liver and lungs were minced in an enzymatic cocktail (1 mg/ml of Collagenase D and 70 μg/ml of DNase I), incubated for 25 min at 37°C. Ice-cold EDTA (2 mM) was added for additional 5 min, then digested tissues were filtered through a 70 μm nylon sieve (BD Falcon). Low-density cells were further enriched by centrifugation over a 1.069 g/ml density gradient (OptiPrep, Axis-Shield), washed and resuspended in PBS, EDTA 2 mM, 2% BSA, and red blood cells were lysed as detailed above. Cutaneous LNs (inguinal and axillar LNs) were cut into small pieces and digested for 25 min at 37°C with a mixture of type II collagenase (Worthington Biochemical) and DNase I (Sigma-Aldrich) in plain RPMI 1640. The resulting cell suspension was treated with 5 mM EDTA and filtered through a 70 μm nylon sieve (BD Falcon). For the skin, ears were split into a ventral and dorsal parts and incubated for 105 min at 37°C in RPMI containing 0.25 mg/ml Liberase TL (Roche Diagnostic Corp.) and 0.5 mg/ml DNase I (Sigma Aldrich). Digested tissue was homogenized using Medicons and Medimachine (Becton Dickinson) to obtain homogenous cell suspensions. For skin mast cells, we used a protocol recently described (37). To test the germline recombination in blood cells, peripheral blood mononuclear cells (PBMCs) were enriched by centrifugation over a 1.077 g/ml density gradient (Ficoll-Paque Plus, GE Healthcare), washed and resuspended in PBS, EDTA 2 mM, 2% BSA before staining. Staining of cells for flow cytometry started with a pre-incubation with 2.4G2 mAb to block unspecific binding to Fc-receptors. Staining with mAb (Supplementay Table S1) was then performed in PBS, 2% BSA, 2 mM EDTA for 25 min on ice. For exclusion of dead cells 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific) was added 5 min before acquisition. Data were acquired on a LSRFortessa X-20 flow cytometer (BD Biosciences), and analyzed using FlowJo (Tree Star, Inc.).
Bone Marrow-Derived DC Differentiation
FLT3-L-BMDCs were generated as described (38) with some modifications. BM cell suspensions were prepared and red blood cells were lysed as detailed in the previous section. After washing in complete RPMI 1640 medium, cells were cultured at 3 × 106 cells/ml in 24 well plates, with 10% FBS, RPMI 1640 medium containing murine FLT3-L (in house supernatant from B16-Flt3l cells, used at 1/20 final) at 37°C in 5% CO2. Four days after, half of the culture medium was replaced by fresh FLT3-L. Cells were harvested at indicated times for flow cytometry analysis after staining for CD11c, SiglecH, CD24, SIRPα, XCR1, and CD11b (Supplementay Table S1).
Microarray Data Generation and Analysis
DCs were generated in vitro from mouse BM FLT3-L cultures and sorting by flow cytometry to over 98% purity, as live, singlet, CD11c+ cells that were SiglecH+ for eq-pDCs, SIRPα−CD24high for eq-cDC1 and SIRPα+CD24−/low for eq-cDC2. Total RNA (50 ng) was used as starting material for each sample to synthesize biotinylated probes, using the NuGen protocol as described previously (39). Affymetrix Mouse Gene 1.0 ST raw. CEL files were analyzed in the R statistical environment (version 3.4.1). Data were RMA normalized using the oligo package and processed as described previously (40). Heatmaps of Log2-normalized expression values of selected genes were performed using the Morpheus website from the Broad Institute (https://software.broadinstitute.org/morpheus/). Hierarchical clusterings were performed using the One-Pearson correlation as a metric and the average linkage as a clustering method for samples and genes, except for Figure 5B where the complete linkage method was used for the genes. The microarray data have been deposited in the GEO database under the series accession number GSE121859.
Mouse Cytomegalovirus Infection
Animals were infected intraperitoneally with 2 × 105 PFU of salivary gland-extracted MCMV Smith strain (3rd in vivo passage). Forty-Eight hours later, spleen and liver were harvested and prepared for flow cytometry analysis as described above.
Analysis of Germline Recombination
Four females and 2 males of each Xcr1Cre/wt; Rosa26tdRFP/wt and KarmaCre/wt; Rosa26tdRFP/wt genotype were backcrossed to C57BL/6J mice. Their progeny was genotyped and bled to analyse tdRFP expression in circulating T and B cells as a sign of germline recombination.
Results
Generation of New cDC1-Targeting Cre-Driver Lines
By comparative gene expression profiling, we and other have previously identified Xcr1 and a530099j19rik (Gpr141b/Karma) genes as specifically expressed by mouse cDC1 in different tissues throughout the body (10, 22–26, 31, 33). We used such unique gene expression profile to genetically target cDC1 in vivo by generating Xcr1Cre (Figure 1A) and KarmaCre (Figure 1B) knock-in mouse models. The insertion of an IRES-Cre cassette after the STOP codon of Xcr1 and Karma genes allows the translation of two separate proteins resulting in the expression of the Cre recombinase. Expression of endogenous XCR1 was not significantly altered in the Xcr1Cre mouse model (Supplementary Figure S1A, top).
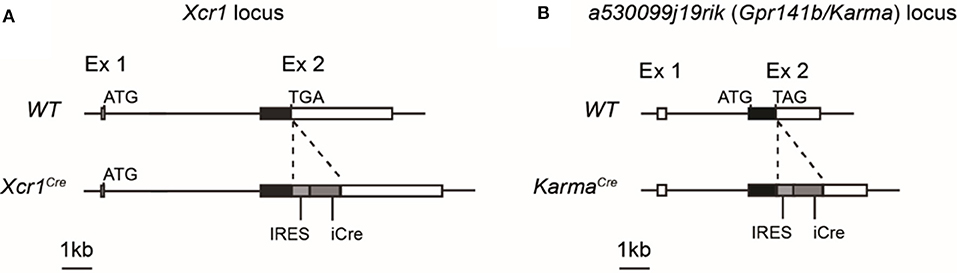
Figure 1. Schematic representations of Xcr1Cre and KarmaCre genetic constructions. A cassette containing an IRES sequence upstream of a gene encoding a codon-improved CRE recombinase was inserted by homologous ET recombination downstream of the stop codon of Xcr1 exon 2 (A) and of a530099j19rik/Karma/Gpr141b exon 2 (B) genes, to produce Xcr1Cre and KarmaCre mouse mutants, respectively, on a C57BL/6J background.
The XCR1Cre Mouse Model Allows Selective and Efficient Recombination of loxP Sequences in Migratory and Resident cDC1 in All Tissues Examined
To determine the specificity of the Cre-induced recombination in Xcr1Cre (Figure 1A) and KarmaCre (Figure 1B) mice, we bred them with the Cre-reporter line Rosa26lox−stop−lox−tdRFP (named hereafter Rosa26tdRFP) (35), and analyzed the tdRFP expression pattern in immune cells of lymphoid organs [spleen and cutaneous lymph-nodes (CLNs)] (Supplementary Figures S1A,B) and non-lymphoid tissues (lungs, liver, and skin) (Supplementary Figures S1C–E). To define DC cell populations, we applied gating strategies adapted from (41). As a control Cre-driver line, we used the Clec9aCre mice (21) bred to Rosa26tdRFP. Regardless of the tissues examined, we found that all three mouse models achieved effective targeting of the cDC1-lineage, with Xcr1Cre, and Clec9aCre being the most efficient (Figure 2). In contrary to Clec9aCre, the Xcr1Cre and KarmaCre mouse models did not show any significant Cre activity, neither in pDCs nor in macrophages (Figure 2). However, Cre recombination (tdRFP signal) was detected in a fraction of other cell types. In Xcr1Cre/wt; Rosa26tdRFP/wt mice, a minute proportion (< 1%) of CD4+ T cells expressed tdRFP in the spleen, CLNs, lung and liver, which increased to a much higher fraction of CD4+ T cells in the skin (8.4 ± 6.4%). The CD4+ T cells harboring Xcr1-driven Cre recombination lacked detectable level of XCR1 (Supplementary Figure S1E, bottom). Hence, they likely derived from progenitors cells that transiently expressed Xcr1. In the skin, lungs and CLNs of KarmaCre/wt; Rosa26tdRFP/wt mice, a fraction of cDC2 had undergone recombination, although to a lesser extent than in Clec9aCre/wt; Rosa26tdRFP/wt mice where cDC2 were targeted in all tissues (Figure 2). Surprisingly, in the skin of KarmaCre/wt; Rosa26tdRFP/wt mice, a large proportion of mast cells (61.1 ± 12.8%) also expressed the tdRFP (Figure 2).
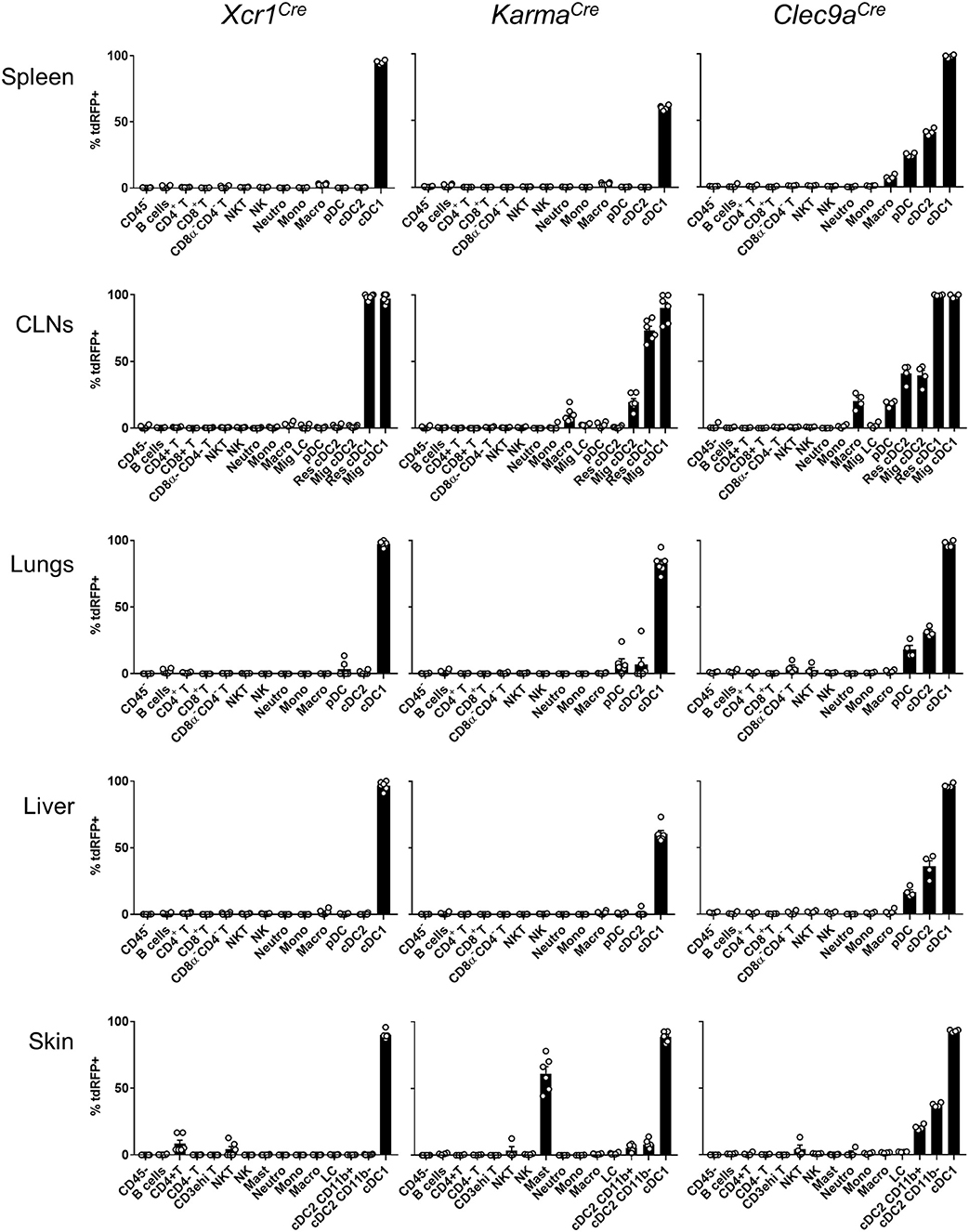
Figure 2. Xcr1Cre and KarmaCre mouse models target with high specificity cDC1 in lymphoid and non-lymphoid tissues. Flow cytometry analysis of the tdRFP expression by different immune cell populations (as defined in Supplementary Figure S1), from spleen, CLNs, lungs, liver, and skin of Xcr1Cre/wt; Rosa26tdRFP/wt, KarmaCre/wt; Rosa26tdRFP/wt and Clec9aCre/wt; Rosa26tdRFP/wt mice. Gating strategies are detailed in Supplementary Figure S1. Data show one dot per individual value, mean +/– SEM per group, and are from two pooled experiments with at least two mice per group.
To further assess cDC1-targeting specificity in the three Cre-driver mouse strains, we analyzed the proportion of different immune cell types within the tdRFP+ cells (Figure 3, Supplementary Figure S2). As expected on the basis of previous report (21), other cells than cDC1, in particular cDC2 and to some extent pDCs, represented the major fraction of the cells targeted in Clec9aCre mice (Figure 3). In contrast, in all examined organs, except for the CLNs, cDC1 represented the major fraction of the cells targeted in Xcr1Cre mice, CD4+ T cells constituting the second most frequent cell types expressing tdRFP in these organs, and the most frequent in the CLNs. This reflects the higher numbers of CD4+ T cells as compared to cDC1s in all these organs. Finally, cDC1 represented the major fraction of targeted cells in KarmaCre mice in all examined organs, except for the skin where 64.4 ± 5.8% of the tdRFP+ cells were mast cells (Figure 3). We assessed the expression pattern of the Karma gene in mast cells and compared it to a variety of other immune cell types, using the database from the Immgen consortium. These results revealed that mast cells express similar levels of the Karma gene as cDC1 in all organs examined, trachea, tongue, esophagus, skin, and peritoneal cavity (Supplementary Figure S3), consistent with the efficient genetic tracing of mast cells in the skin of KarmaCre mice. Altogether, these results show that our novel Xcr1Cre and KarmaCre mouse models constitute the most reliable Cre-driver lines reported to date for selective and efficient in vivo targeting of cDC1, with the Xcr1Cre model performing the best. However, it should be noted that a small fraction of CD4+ T cells is targeted in Xcr1Cre mice and that mast cells are largely targeted in KarmaCre animals.
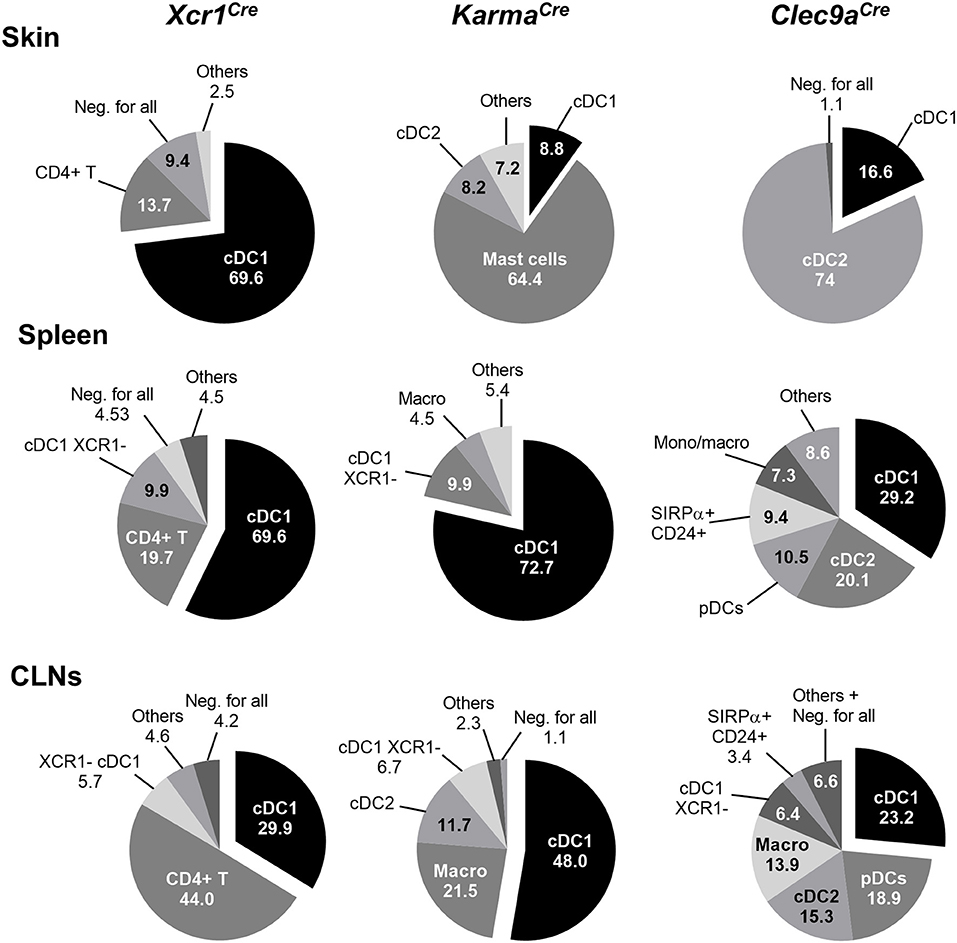
Figure 3. Proportion of cell types within tdRFP+ cells in the skin, spleen, and CLNs of each genotype. The back gating strategy used to define the relative proportions of cell types among tdRFP+ cells was performed by using the antibody staining detailed in the upper side of Supplementary Figure S1. Exceptions were made as follow for Xcr1Cre/wt; Rosa26tdRFP/wt mice: to define further the tdRFP+ cells that fell within the Lin+ (CD19/CD3ε/NKp46/Ly6G) gate, the antibody staining detailed in the bottom side of Supplementary Figure S1 was used. The proportion of CD4+ T lymphocytes within tdRFP+ cells when using the latter (“lymphoid”) antibody panel was matching the proportion of Lin+ cells within tdRFP+ cells when using the former antibody (“myeloid”) panel. For KarmaCre/wt; Rosa26tdRFP/wt mice, mast cells were defined as Lin− CD11b− CD11c− XCR1− MHC-II− CD64− F4/80− FcεRIα+ CD117+. Others: sum of all the other cell subsets not detailed in the pie charts. Neg. for all, negative for all: cells that did not stain positive for the markers used in the upper antibody panel. Data are shown for one experiment representative of three with three mice per group.
The Use of Different cDC1-Specific Promoters for Driving Cre Expression Reveals Heterogeneity in the cDC1 Population Defined as CD24+ SIRPα− cDCs
To try understanding the lower efficiency of the KarmaCre model for cDC1 targeting, as compared to the Xcr1Cre or Clec9aCre mice, we examined the expression pattern of the tdRFP reporter within the splenic cDC1 defined as CD24+SIRPα− cDCs (gated in the CD45+Lin−SiglecH−MerTK−CD64−CD11c+MHC-II+CD26+ cells) (13). CD24+SIRPα− cDC1 can be split into 4 subsets according to their heterogeneous expression of CD8α and XCR1 (Figure 4). XCR1 expression was reported to correlate with a better crosspresentation by cDC1 (23, 42). The CD8α+XCR1+ cDC1 subset was reported to be phenotypically and functionally homogenous (23, 42), and likely corresponds to full-fledged differentiated cDC1 endowed with a high crosspresentation activity (43). However, the three other cDC1 subsets, CD8α+XCR1−, CD8α−XCR1+, and CD8α−XCR1−, have not been extensively characterized. The CD8α−XCR1− cells may encompass pre-cDC1 (13, 44). The CD8α−XCR1+ cells likely correspond to pre-terminally differentiated cDC1. The CD8α+XCR1− cells could correspond to the small fraction of homeostatically matured splenic cDC1 that have downregulated XCR1 expression, similarly to what occurs at steady state in the skin, the intestine or the thymus (25, 32, 39). It could also be possible that XCR1− cells encompass other cell types contaminating the SIRPα−CD24+ cDC1 gate. However, the exclusion of CD3ε+ and SiglecH+ cells in our gating strategy ensured that the CD8α+XCR1− cDC1 subset was not contaminated by CD8+ T cells nor CD8α+ pDCs (45–47). Consistent with the early expression of Clec9a starting at the pre-DC stage (21), Clec9aCre model targeted efficiently all 4 subsets, regardless of XCR1 and CD8α acquisition, with more than 85% of the cells in each subset expressing tdRFP (Figure 4). Although the Xcr1Cre model was more efficient in targeting XCR1+ cDC1 as initially expected, a significant Cre activity was also detected both in XCR1−CD8α− and XCR1−CD8α+ cDC1 subsets, with 37% and 53% of tdRFP expression, respectively (Figure 4). This indicated that a significant proportion of these cells from these two subsets derived from XCR1-expressing precursors, consistent with the hypothesis that they respectively encompass pre-cDC1 and terminally matured cDC1. The KarmaCre model was effective in targeting the XCR1+CD8α+ cDC1 subset, contrasting with no recombination detected in the XCR1−CD8α− subset and with only a weak Cre activity in the XCR1+CD8α− subset (Figure 4). The fraction of XCR1−CD8α+ cDC1 targeted in KarmaCre mice (22.8 ± 6.1%) was lower than that of XCR1+CD8α+ cDC1 (69.0 ± 2.1%). This suggest that most of XCR1−CD8α+ cDC1 do not derive from XCR1+CD8α+ cDC1 contrary to our expectation that the majority of the former cells correspond to an advanced maturation state of the latter ones. Altogether, the combined use of our fate mapping mouse models suggests consecutive expression of the corresponding genes along the differentiation of the cDC1 lineage in the spleen, with Clec9a expressed from the common cDC progenitor stage, Xcr1 likely starting at the pre-cDC1 stage and Karma turned on only at a later stage similarly to CD8α.
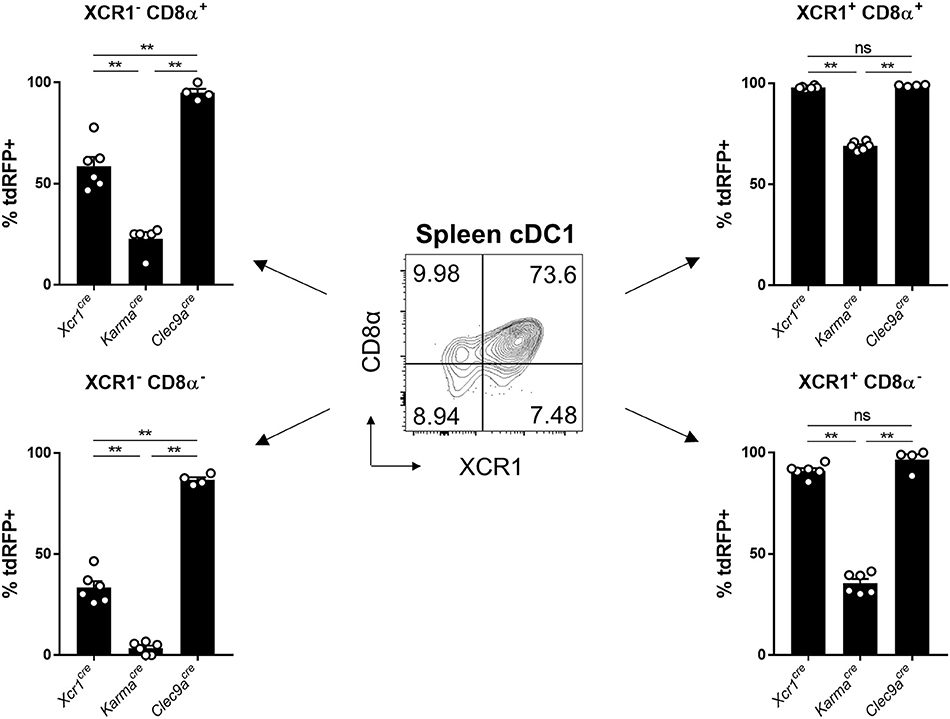
Figure 4. Tracing of Xcr1 and Karma expression in the cDC1 population in spleen. Flow cytometry analysis of the tdRFP expression in the 4 CD24+ cDC1 subsets defined as CD8α+XCR1−, CD8α+XCR1+, CD8α−XCR1+, and CD8α−XCR1−, in the spleen of Xcr1Cre/wt; Rosa26tdRFP/wt, KarmaCre/wt; Rosa26tdRFP/wt, and Clec9aCre/wt; Rosa26tdRFP/wt mice. Data show one dot per individual value, mean +/- SEM per group, and are from two pooled experiments with at least two mice per group. Statistical analyses were performed using nonparametric Mann-Whitney tests in all experiments (**, p < 0.01; n.s, non-significant).
Comparison of the Three Fate Mapping Mouse Strains Advances Our Understanding of the Differentiation Trajectory of cDC1
We further investigated to which extent our fate mapping mutant mouse models could help refining the differentiation trajectory of cDC1, using as a simple model bone marrow (BM) cells cultured with Fms-like tyrosine kinase 3 ligand (FLT3-L) (38). This model allows in vitro generation of three subsets of DCs, which are phenotypically and functionally equivalent to in vivo cDC1 (eq-cDC1), cDC2 (eq-cDC2) and pDCs (eq-pDCs) (38, 48–50). To refine the differentiation trajectory of cDC1, we followed the acquisition of the tdRFP signal over time in FLT3-L-differentiated DCs generated from BM of our fate mapping mutant mice (Supplementary Figure S4, Figure 5A). Xcr1Cre and Clec9aCre models allowed efficient recombination in eq-cDC1 (56 vs. 90%) (Figure 5A). Interestingly, KarmaCre did not present any recombinase activity in any of the DC populations (Figure 5A). Consistently, whereas gene expression profiling of the eq-DC subsets generated in standard BM FLT3-L cultures confirmed their close homology to their in vivo counterparts isolated from the spleen (Figure 5B), it also showed that eq-cDC1 lacked expression of the Karma, Cd8a and Ly75 (Cd205) genes (Figure 5B, black arrows). This was confirmed using our previously published Karma reporter mouse model knocked-in for tdTomato in the 3′UTR of the Karma gene (33) (Supplementary Figure S5A). However, these eq-cDC1 acquired Karma and CD8α expression upon in vivo transfer (Supplementary Figures S5B,C). Karma was also expressed in eq-cDC1 differentiated from BM cells cultured with FLT3-L on feeder cells expressing the Notch ligand Delta-like 1 (Figure 5C, black arrows), similarly to what has been recently reported for CD8α and CD205 expression (43). Altogether, these results demonstrate a sequential expression of Clec9a, Xcr1 and Karma during cDC1 ontogeny, with Clec9a being induced early starting at the common cDC progenitor stage (21), then followed by Xcr1 which induction might be initiated already at the pre-cDC1 stage. Likewise to CD8α, Karma is acquired at a more advanced differentiation stage that is not reached under classical conditions of DC differentiation from BM in FLT3-L in vitro cultures but can be promoted by Notch signaling. This work thus significantly extends two recent studies showing that cDC1 derived in vitro from mouse or human hematopoietic precursors with a combination of cytokines and growth factors need additional signals to reach a terminal state of differentiation including acquisition of CD8α expression for mouse cDC1 (43, 51).
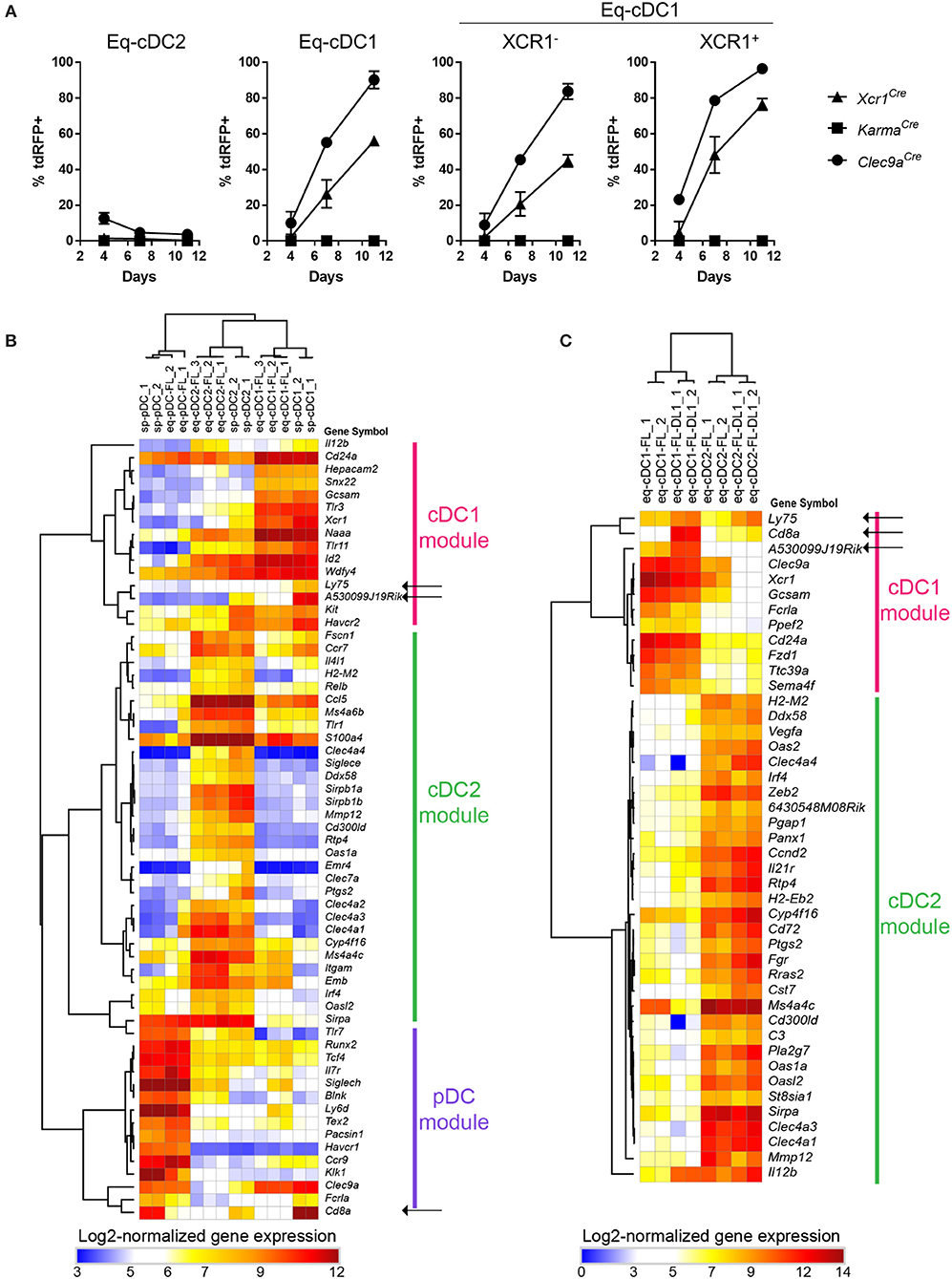
Figure 5. Sequential regulation of Clec9a, Xcr1 and Karma expression during cDC1 differentiation. (A) Kinetic analysis of the tdRFP expression in DCs differentiated in vitro from Xcr1Cre/wt; Rosa26tdRFP/wt, KarmaCre/wt; Rosa26tdRFP/wt and Clec9aCre/wt; Rosa26tdRFP/wt BM cells cultured with FLT3-L. Eq-cDC2 were gated as CD11c+MHC-II+CD24−SIRPα+ cells, and eq-cDC1 as CD11c+ MHC-II+CD24+SIRPα− cells (Supplementay Figure S4). Data are shown for one experiment representative of two, with three mice per group. (B,C) Heatmaps display the expression profiles of archetypical genes previously shown to be selectively expressed in cDC1, cDC2, or pDC. (B) Gene expression across DC types either isolated from murine spleens (sp-pDC, sp-cDC1, and sp-cDC2), or derived in vitro in standard FLT3-L BM cultures (eq-pDC-FL, eq-cDC1-FL, and eq-cDC2-FL), as assessed with microarrays. (C) Gene expression patterns across cDC types derived in vitro from FLT3-L BM cultures under standard conditions (-FL) or on DL1-expression OP9 feeder cells (-FL-DL1), as assessed from public RNA-seq data (GEO accession number GSE110577).
The Cre Expression Under Xcr1 or Karma Promoters Remains cDC1-Specific Upon Infection-Induced Inflammation
The expression of many membrane proteins or transcription factors changes upon inflammation (12, 52). An important incentive for generating Xcr1Cre and KarmaCre models, was that the expression of the Xcr1 and Karma genes was specific for cDC1 both at steady state and under inflammatory conditions (53). To confirm this observation based on transcriptomic studies, we examined cDC1-targeting specificity of the KarmaCre and Xcr1Cre mouse models in an inflammatory context, namely systemic mouse cytomegalovirus (MCMV) infection, using the Rosa26tdRFP reporter as a read out. We adapted a gating strategy adapted from (52) to identify inflammatory DCs (InflDCs) in spleen and liver (Supplementary Figure S6). Although we could observe the appearance of InflDCs upon MCMV infection, tdRFP expression remained unchanged in infected animals as compared to control mice, being still essentially confined to the cDC1 population, both in Xcr1Cre/wt; Rosa26tdRFP/wt and KarmaCre/wt; Rosa26tdRFP/wt mice (Supplementary Figure 6). This demonstrated that both cDC1-targeting models are stable and allow excision of a floxed genomic sequence efficiently and largely selectively in cDC1 at steady state and upon inflammation.
Germline Recombination of loxP Sequences Is Frequent in the Offspring of Xcr1Cre but Not KarmaCre Mice
Recombination of loxP-flanked genomic sequences in germ cells have been described in many Cre mouse models (54–56). To test whether germline recombination occurs in our cDC1-targeting Cre-driver lines, we backcrossed Xcr1Cre/wt; Rosa26tdRFP/wt and KarmaCre/wt; Rosa26tdRFP/wt mice to C57BL/6J mice, and analyzed their offspring for ubiquitous tdRFP expression, using blood T and B cells as a readout (Figure 7A). Total or partial germline recombination of the Rosa26tdRFP locus occurred in 95% of the offspring who had inherited one loxP-flanked allele from Xcr1Cre/wt; Rosa26tdRFP/wt male mice (Figure 7B). Germline recombination occurred with the same frequency irrespective of the segregation of the paternal Cre and loxP-flanked alleles in the offspring, demonstrating that this process occurred during meiosis rather than in the embryo. No germline recombination was observed when both alleles were from maternal germ cells (Figure 7B). In ongoing crosses using the Xcr1Cre mouse model with different loxP-flanked mouse strains, we could also observe germline recombination in the progeny even when the floxed alleles were brought together with the Xcr1Cre allele by the maternal gamete (data not shown). This indicates that, contrary to what the results of the Rosa26tdRFP/wt backcross appears to suggest, off-target activity of the Cre recombinase in germline is not a gender effect. Additionally, the incidence of germline recombination depended on the loxP-flanked allele (data not shown). No occurrence of germline recombination was detected so far for the KarmaCre model (Figure 7B). Therefore, the KarmaCre model might be more appropriate than the Xcr1Cre model to obtain rapidly mice in which cDC1 are inactivated for candidate genes, through conventional breeding strategies. Germline recombination in the offspring should however always be assessed for any novel loxP-flanked allele.
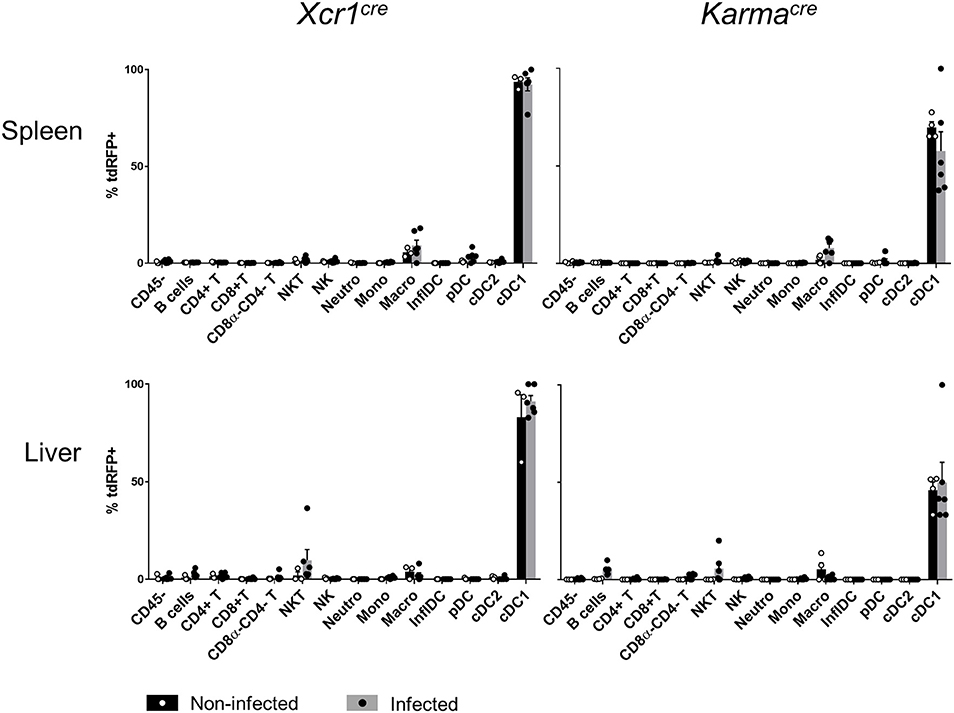
Figure 6. Xcr1 and Karma expressions remain confined to cDC1 upon virus-induced inflammatory responses. Flow cytometry analysis of the tdRFP expression by different immune cell populations as in Figure 2A, from spleen, CLNs, and liver of Xcr1Cre/wt; Rosa26tdRFP/wt, and KarmaCre/wt; Rosa26tdRFP/wt mice 2 days after MCMV infection. Cell population gating strategy detailed in Supplementay Figure S3. Data show one dot per individual value, mean +/- SEM per group, and are from two pooled experiments with at least three individuals per group of infected mice. Statistical analyses were performed using nonparametric Mann-Whitney tests when possible, and the difference between non infected and infected was non-significant in each cell population. NI, non-infected; InflDCs, inflammatory DCs.
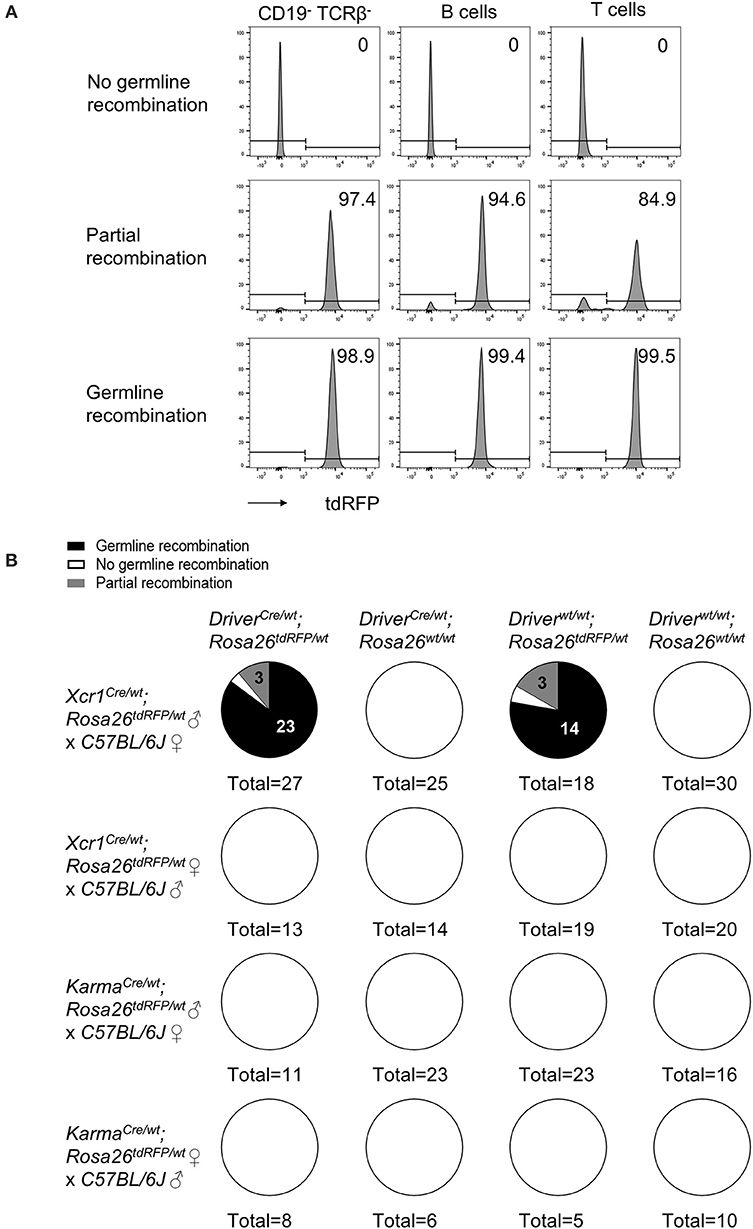
Figure 7. The Cre expression under Xcr1 but not Karma promoter efficiently recombines Lox-sequences in germ cells. (A) Representative flow cytometry analysis of tdRFP expression in CD19− TCRβ−, B and T cells in blood of mice with no germline recombination, partial recombination, and germline recombination. One sample representative of each is shown. (B) Analysis of germline recombination in offspring from backcrosses of Xcr1Cre/wt; Rosa26tdRFP/wt and KarmaCre/wt; Rosa26tdRFP/wt mice of both sexes with wild-type (WT; C57BL/6J) mice. Germline recombination shown here occurred when both Cre and floxed alleles were of paternal origin. However, with other type of flox constructs, we regularly observed occurrences of germline recombination when both alleles were brought together, either by the father or by the mother.
Discussion
Xcr1 and A530099j19rik (Karma/Gpr141b) genes code for the chemokine receptor XCR1 and for the putative G protein-coupled receptor Gpr141b, respectively, and are among the core gene signature specifically identifying mouse cDC1 throughout the organism (10, 22–25, 33). We have inserted an IRES-Cre cassette into the 3′ UTR of the Xcr1 and Karma coding exon to generate Xcr1Cre and KarmaCre mouse models, respectively. In this study, we have characterized the efficiency and specificity of Cre-mediated recombination in these novel mouse models at steady state and upon viral infection, comparing them to the Clec9aCre model. To the best of our knowledge, this study demonstrated that our novel Xcr1Cre and KarmaCre mouse models are the most trustful and robust for the genetic tracking and manipulation of cDC1 in vivo.
Amongst the three Cre-driver mouse models examined, Xcr1Cre model is the most efficient and specific for fate mapping all cDC1 regardless of the tissues examined. The KarmaCre model is rather specific for cDC1 when compared with Clec9aCre mouse, but much less efficient than the Xcr1Cre model. Unexpectedly, a fraction of CD4+ T cells is labeled with tdRFP in the Xcr1Cre;Rosa26tdRFP/wt mouse (Figure 2) without expressing any detectable XCR1 at their cell surface (Supplementary Figure S1E). Further analysis need to be conducted to determine whether XCR1 was transiently turned on in the distant progenitors of these cells or on the contrary during their terminal differentiation. Interestingly, the proportion of Xcr1cre fate-mapped CD4+ T cells was much higher in the skin than in the other organs examined, suggesting that these cells may be polarized toward specific functions and/or develop under instructive signals encountered preferentially in barrier organs. Further studies will be needed to test these hypotheses. In the skin of the KarmaCre; Rosa26tdRFP/wt mouse model, the vast majority of the tdRFP+ cells were of mast cell origin (Figure 3B). Microarray data released recently by the Immgen consortium (https://www.immgen.org) show that mast cells from the skin, peritoneal cavity, trachea and esophagus express high level of the Karma gene (Supplementary Figure S3), confirming our observation. To the best of our knowledge, this is the first report of a gene that is selectively shared by both cDC1 and mast cells. The KarmaCre mice will therefore be of special interest to researchers aiming at genetically manipulating mast cells in tissues. In all tissues and in all mouse models examined, no tdRFP expression was detected in the CD45-negative cells present in cell suspensions (Figure 2). Although, we did not examine tdRFP expression in other non-hematopoietic cells, it is unlikely that some of these cell types would be targeted in Xcr1Cre or KarmaCre mice considering that the Xcr1 and Gp141b genes were not expressed outside of the hematopoietic system in all of the transcriptomic databases we queried.
To inactivate specifically a candidate gene in cDC1, both alleles of this candidate gene should be excised. This requires a breeding strategy in which one Cre allele and one floxed allele of the gene to be inactivated are brought by the same germ cells, where unexpected recombination could occur. We have tested the frequency of germline recombination for both the Xcr1Cre and KarmaCre models. Only the Xcr1Cre model showed adventitious Cre activity in germ cells resulting in progeny with recombined Rosa26tdRFP locus in all their cells. Interestingly, this was paternal inherited in this specific experimental setting. However, this may depend on the loxP-flanked construct, as we had events of germline recombination transmitted by females for other floxed genes than the Rosa26tdRFP reporter. Therefore, to reach specific recombination in cDC1 using the Xcr1Cre model, the Cre allele and the loxP-flanked allele should be inherited from different parents. We recommend breeding one parent homozygous for both the Cre allele and a null allele of the target gene, to another parent homozygous for the floxed allele of the target gene. Each investigator using the Xcr1Cre model should always test their progeny for unexpected off-target recombination. A recent publication strongly suggested to include, in each experimental procedure of publication using Cre mouse models, detailed procedures about the breeding strategies used and the method the investigators applied to detect any unexpected and unspecific recombination (56).
Our genetic tracing of cDC1 in vivo in the spleen (Figure 4), or in vitro in FLT3-L-differentiated BM-DC cultures (Figure 5A) revealed heterogeneity in the cDC1 population. The differential expression of Xcr1 and Karma genes within the cDC1 population qualifies Xcr1Cre and KarmaCre mouse models as powerful tools to describe further these cDC1 subsets in vivo. In vitro, BM-DCs derived from KarmaCre mice did not show any sign of Cre activity (Figure 5A), confirming that the Karma gene is not transcriptionally active in these cells as directly assessed through their gene expression profiling (Figure 5B), akin to Cd8a or Ly75 (Dec205) (43). We show here that expression of the Karma gene on cDC1 requires accessory signals, which can be provided upon in vivo transfer, or in vitro by Notch signaling likewise to what has been recently reported for Cd8a and Ly75 (43). Of note, in FLT3-L in vitro BM-DC culture, detection in eq-cDC1 of the activity of the Cre recombinase as readout by tdRFP expression seemed to be delayed over time as compared to cell surface acquisition of XCR1 (Figure 5), although the Cre and Xcr1 genes were expressed under the same promoter from one bi-cistronic mRNA. This might be explained by a delayed translation of the Cre gene as compared to Xcr1, or because efficient recombination of DNA by the Cre requires time. This latter case might especially apply to the Rosa26tdRFP reporter mouse line used in this study, because it was engineered as requiring two consecutive rounds of Cre-excision to generate detectable tdRFP signal, in order to limit any leaky transcription of the fluorescent reporter gene across the stopper at steady state (35). This contrasts to most reporter lines which require only one sequence of recombination to emit signal (57, 58) and must therefore require lower and/or less sustained Cre activity to allow recombination. Breeding the Xcr1Cre model with a Cre-reporter mouse which is easily recombined (57) might allow a better synchronization of XCR1 surface expression with Cre activity.
Our results advanced our understanding of the differentiation trajectory of cDC1, and validated the Xcr1Cre mouse model as a robust tool to inactivate genes selectively in cDC1 either in vivo or in vitro using BM-derived DC cultures. Future use of these mutant mouse models will undoubtedly boost the advancing of our understanding of the biology of cDC1.
Author Contributions
MD and KC: Conceptualization. RM, CW, BM, MD, and KC: Methodology, Validation, Formal Analysis. RM, CW, SG, MA, YA, CS, and AF: Investigation. T-PV, MD: bioinformatics analyses. BM: Mouse model construction supervision. RM, KC: Visualization. RM, KC, and MD: Writing. All authors: Editing. BM, MD, and KC: Project Supervision and Administration.
Funding
This work was in part carried out in the frame of the Innate Immunocytes in Health and Disease (I2HD) collaborative project between CIML, AVIESAN, and SANOFI. It was supported by grants from the European Research Council under the European Community's Seventh Framework Program [FP7/2007–2013 grant agreement number 281225 to M. Dalod; FP7/2007-2013 grant no. 322465 (Integrate) to BM], from the Agence Nationale de la Recherche (ANR; XCR1-DirectingCells to KC), from the Fondation pour la Recherche Médicale (label Equipe FRM 2011, project number DEQ20110421284 to MD), which supported the KarmaCre mouse generation, from the Program Hubert Curien (PHC) Maïmonide-Israel 2017 project number 38155SJ (to MD) supported by the French Ministries of Foreign Affairs (MAEDI) and of Research and Higher Education (MENESR) and from the Fondation ARC pour la recherche sur le cancer (to KC). CIPHE is supported by the French Ministry of Research via PHENOMIN (French National Infrastructure for mouse Phenogenomics; ANR10-INBS-07 to BM). This work also benefited from institutional funding from CNRS and INSERM. RM was supported by doctoral fellowships from the Biotrail PhD program (Fondation A*MIDEX). RM, CW, SG, and YA were supported by fellowships from Fondation ARC pour la recherche sur le cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Frédéric Fiore and members of the Centre d'Immunophénomique (CIPHE) (Aix Marseille Univ, INSERM US012, CNRS UMS3367, Marseille, France) for generating the Xcr1Cre and KarmaCre mice, and for taking care of mouse breeding; Marilyn Boyron, the cytometry core, and animal house facilities (CIML, UMR7280, France) for critical technical assistance. The authors also thank Rebecca Gentek (Aix Marseille Univ, CNRS, INSERM, CIML, France) for helpful discussion and protocol sharing. This work benefited from data assembled by the ImmGen consortium.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02805/full#supplementary-material
References
1. Jung S, Unutmaz D, Wong P, Sano G-I, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17:211–20. doi: 10.1016/S1074-7613(02)00365-5
2. Caton ML, Smith-Raska MR, Reizis B. Notch–RBP-J signaling controls the homeostasis of CD8 − dendritic cells in the spleen. J Exp Med. (2007) 204:1653–64. doi: 10.1084/jem.20062648
3. Stranges PB, Watson J, Cooper CJ, Choisy-Rossi C-M, Stonebraker AC, Beighton RA, et al. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity (2007) 26:629–41. doi: 10.1016/j.immuni.2007.03.016
4. Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. (2012) 209:1153–65. doi: 10.1084/jem.20112675
5. Loschko J, Schreiber HA, Rieke GJ, Esterházy D, Meredith MM, Pedicord VA, et al. Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J Exp Med. (2016) 213:517–34. doi: 10.1084/jem.20160062
6. Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. (2012) 209:1135–152. doi: 10.1084/jem.20120030
7. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi: 10.1126/science.1164206
8. Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, et al. IRF8 transcription-factor-dependent classical dendritic cells are essential for intestinal T cell homeostasis. Immunity (2016) 44:860–74. doi: 10.1016/j.immuni.2016.02.008
9. Lee W, Kim HS, Hwang SS, Lee GR. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med. (2017) 49:e393. doi: 10.1038/emm.2017.157
10. Robbins SH, Walzer T, Dembélé D, Thibault C, Defays A, Bessou G, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol. (2008) 9:R17. doi: 10.1186/gb-2008-9-1-r17
11. Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity (2016) 45:626–40. doi: 10.1016/j.immuni.2016.08.013
12. Tussiwand R, Lee W-L, Murphy TL, Mashayekhi M, Kc W, Albring JC, et al. Compensatory dendritic cell development mediated by BATF–IRF interactions. Nature (2012) 490:502–7. doi: 10.1038/nature11531
13. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8α(+) conventional DC clonogenic progenitor. Nat Immunol. (2015) 16:708–17. doi: 10.1038/ni.3197
14. Lahoud MH, Proietto AI, Gartlan KH, Kitsoulis S, Curtis J, Wettenhall J, et al. Signal regulatory protein molecules are differentially expressed by CD8– dendritic cells. J Immunol. (2006) 177:372–82. doi: 10.4049/jimmunol.177.1.372
15. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
16. Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature (2009) 458:899–903. doi: 10.1038/nature07750
17. Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin Teh J, Lo JCY, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood (2008) 112:3264–73. doi: 10.1182/blood-2008-05-155176
18. Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem. (2008) 283:16693–701. doi: 10.1074/jbc.M709923200
19. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood (2012) 119:6052–62. doi: 10.1182/blood-2012-01-406967
20. Sancho D, Mourão-Sá D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest. (2008) 2098–110. doi: 10.1172/JCI34584
21. Schraml BU, van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell (2013) 154:843–58. doi: 10.1016/j.cell.2013.07.014
22. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c + CD141 + cells as homologues of mouse CD8 + dendritic cells. J Exp Med. (2010) 207:1273–81. doi: 10.1084/jem.20100348
23. Bachem A, Hartung E, Güttler S, Mora A, Zhou X, Hegemann A, et al. Expression of XCR1 characterizes the Batf3-dependent lineage of dendritic cells capable of antigen cross-presentation. Front Immunol. (2012) 3:214. doi: 10.3389/fimmu.2012.00214
24. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre C-A, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8α + dendritic cells. J Exp Med. (2010) 207:1283–92. doi: 10.1084/jem.20100223
25. Crozat K, Tamoutounour S, Vu Manh T-P, Fossum E, Luche H, Ardouin L, et al. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8 + Type. J Immunol. (2011) 187:4411–5. doi: 10.4049/jimmunol.1101717
26. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Güttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T Cells. Immunity (2009) 31:823–33. doi: 10.1016/j.immuni.2009.08.027
27. Vu Manh T-P, Elhmouzi-Younes J, Urien C, Ruscanu S, Jouneau L, Bourge M, et al. Defining mononuclear phagocyte subset homology across several distant warm-blooded vertebrates through comparative transcriptomics. Front Immunol. (2015) 6:299. doi: 10.3389/fimmu.2015.00299
28. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172:1022–37 e14. doi: 10.1016/j.cell.2018.01.004
29. Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
30. Dorner BG, Smith HRC, French AR, Kim S, Poursine-Laurent J, Beckman DL, et al. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol. (2004) 172:3119–31. doi: 10.4049/jimmunol.172.5.3119
31. Yamazaki C, Miyamoto R, Hoshino K, Fukuda Y, Sasaki I, Saito M, et al. Conservation of a chemokine system, XCR1 and its ligand, XCL1, between human and mice. Biochem Biophys Res Commun. (2010) 397:756–61. doi: 10.1016/j.bbrc.2010.06.029
32. Ohta T, Sugiyama M, Hemmi H, Yamazaki C, Okura S, Sasaki I, et al. Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Sci Rep. (2016) 6:23505. doi: 10.1038/srep23505
33. Alexandre YO, Ghilas S, Sanchez C, Le Bon A, Crozat K, Dalod M. XCR1 + dendritic cells promote memory CD8 + T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. J Exp Med. (2016) 213:75–92. doi: 10.1084/jem.20142350
34. Shimshek DR, Kim J, Hübner MR, Spergel DJ, Buchholz F, Casanova E, et al. Codon-improved Cre recombinase (iCre) expression in the mouse. Genes (2002) 32:19–26. doi: 10.1002/gene.10023
35. Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol. (2007) 37:43–53. doi: 10.1002/eji.200636745
36. Voehringer D, Liang H-E, Locksley RM. Homeostasis and effector function of lymphopenia-induced “Memory-Like” T cells in constitutively T cell-depleted mice. J Immunol. (2008) 180:4742–53. doi: 10.4049/jimmunol.180.7.4742
37. Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity (2018) 48:1160–171.e5. doi: 10.1016/j.immuni.2018.04.025
38. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. (2005) 174:6592–7. doi: 10.4049/jimmunol.174.11.6592
39. Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity (2016) 45:305–18. doi: 10.1016/j.immuni.2016.07.019
40. Baranek T, Manh T-PV, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12:571–84. doi: 10.1016/j.chom.2012.09.002
41. Guilliams M, Dutertre C-A, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity (2016) 45:669–84. doi: 10.1016/j.immuni.2016.08.015
42. Kroczek RA, Henn V. The Role of XCR1 and its ligand XCL1 in antigen cross-presentation by murine and human dendritic cells. Front Immunol. (2012) 3:14. doi: 10.3389/fimmu.2012.00014
43. Kirkling ME, Cytlak U, Lau CM, Lewis KL, Resteu A, Khodadadi-Jamayran A, et al. Notch signaling facilitates in vitro generation of cross-presenting classical dendritic cells. Cell Rep. (2018) 23:3658–72.e6. doi: 10.1016/j.celrep.2018.05.068
44. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HRB, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
45. Bar-On L, Birnberg T, Lewis KL, Edelson BT, Bruder D, Hildner K, et al. CX3CR1+ CD8alpha+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc Natl Acad Sci USA. (2010) 107:14745–50. doi: 10.1073/pnas.1001562107
46. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Brière F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. (2002) 195:517–28. doi: 10.1084/jem.20011672
47. O'Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, et al. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med. (2002) 196:1307–19. doi: 10.1084/jem.20021031
48. Brasel K, De Smedt TD, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand–supplemented bone marrow cultures. Blood (2000) 96:3029–39.
49. Brawand P, Fitzpatrick DR, Greenfield BW, Brasel K, Maliszewski CR, De Smedt T. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol. (2002) 169:6711–6719. doi: 10.4049/jimmunol.169.12.6711
50. Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu X-L, Trinchieri G, et al. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med. (2002) 195:953–8. doi: 10.1084/jem.20020045
51. Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, et al. Large-scale human dendritic cell differentiation revealing notch-dependent lineage bifurcation and heterogeneity. Cell Rep. (2018) 24:1902–15.e6. doi: 10.1016/j.celrep.2018.07.033
52. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity (2012) 36:1031–1046. doi: 10.1016/j.immuni.2012.03.027
53. Manh T-PV, Alexandre Y, Baranek T, Crozat K, Dalod M. Plasmacytoid, conventional, and monocyte-derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur J Immunol. (2013) 43:1706–15. doi: 10.1002/eji.201243106
54. Becher B, Waisman A, Lu L-F. Conditional gene-targeting in mice: problems and solutions. Immunity (2018) 48:835–6. doi: 10.1016/j.immuni.2018.05.002
55. Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. (2007) 8:665–68. doi: 10.1038/ni0707-665
56. Song AJ, Palmiter RD. Detecting and avoiding problems when using the Cre–lox system. Trends Genet. (2018) 34:333–40. doi: 10.1016/j.tig.2017.12.008
57. Liu J, Willet SG, Bankaitis ED, Xu Y, Wright CVE, Gu G. Non-parallel recombination limits cre-loxP-based reporters as precise indicators of conditional genetic manipulation: Cre-Recombinations are Non-Parallel Events. Genesis (2013) 51:436–42. doi: 10.1002/dvg.22384
Keywords: dendritic cells, cDC1, XCR1, Gp141b, Karma, Clec9a, Cre, fate mapping
Citation: Mattiuz R, Wohn C, Ghilas S, Ambrosini M, Alexandre YO, Sanchez C, Fries A, Vu Manh T-P, Malissen B, Dalod M and Crozat K (2018) Novel Cre-Expressing Mouse Strains Permitting to Selectively Track and Edit Type 1 Conventional Dendritic Cells Facilitate Disentangling Their Complexity in vivo. Front. Immunol. 9:2805. doi: 10.3389/fimmu.2018.02805
Received: 29 July 2018; Accepted: 14 November 2018;
Published: 04 December 2018.
Edited by:
Diana Dudziak, Universitätsklinikum Erlangen, GermanyReviewed by:
Natalio Garbi, Universität Bonn, GermanyChristophe Jean Desmet, University of Liege, Belgium
Copyright © 2018 Mattiuz, Wohn, Ghilas, Ambrosini, Alexandre, Sanchez, Fries, Vu Manh, Malissen, Dalod and Crozat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Dalod, ZGFsb2RAY2ltbC51bml2LW1ycy5mcg==
Karine Crozat, Y3JvemF0QGNpbWwudW5pdi1tcnMuZnI=