 Istéfani L. da Silva
Istéfani L. da Silva Lucía Montero-Montero
Lucía Montero-Montero Enio Ferreira
Enio Ferreira Miguel Quintanilla
Miguel Quintanilla- 1Center of Biological Sciences and Health, Federal University of the West of Bahia, Barreiras, Brazil
- 2Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas, Universidad Autónoma de Madrid, Madrid, Spain
- 3Laboratory of Compared Pathology, Department of General Pathology, Biological Science Institute, Federal University of Minas Gerais, Belo Horizonte, Brazil
It is well established that the immune system can identify and destroy neoplastic transformed cells in a process known as immunosurveillance. Most studies have focused on the classical major histocompatibility complex (MHC) class Ia molecules, which are known to play an important role on the presentation of tumor antigens to the immune system in order to activate a response against tumor cells. However, a larger family of non-classical MHC class Ib-related molecules has received less attention. In this mini-review, we discuss the role of class Ib murine Qa-2 and its proposed human HLA-G homolog on immunosurveillance during embryogenesis and cancer. Whereas, both HLA-G and Qa-2 are involved in immune tolerance in pregnancy, the current evidence suggests that they play opposite roles in cancer. HLA-G appears to promote tumor progression while Qa-2 acts as a tumor suppressor awaking the immune system to reject tumor cells, as suggested by studies on different cancer cell models, such as melanoma, lymphoma, lung carcinoma, and our own results in mammary carcinoma.
Introduction
The concept of cancer immunoediting with its three distinct phases: elimination (immunosurveillance), equilibrium (persistence/dormancy) and escape (progression) integrates the capacity of the immune system to both protect the host from cancer and promote cancer development (1). Tumor antigens are presented by the major histocompatibility complex (MHC) class I molecules on antigen-presenting cells (APCs) and recognized by CD8+ T cells, which differentiate into cytotoxic T lymphocytes under co-stimulation of CD4+ T cells. In addition, tumor cells are targets of innate immune cells, such as macrophages and natural killer (NK) cells. Thus, neoplastic cells can be recognized and destroyed by both the innate and adaptive immune systems. But if antitumor immunity is unable to completely eliminate them, tumor variants may survive and enter into the equilibrium phase, where cells and cytokines of adaptive immunity restrain tumor outgrowth. These dormant tumor cells may eventually escape the control of immune cells and progress to clinically detectable malignancies. The immune system contributes to tumor progression by selecting more aggressive tumor variants and allowing cancer cells to survive and growth in immunocompetent hosts. Tumor cells that escape the control of immune cells secrete factors that inhibit effector immune cell function and recruit inflammatory and regulatory immune cells that generate an immunosuppressive microenvironment and promote cancer progression (2).
The Human HLA-G Complex in Physiology and Cancer
The human leukocyte antigen G (HLA-G) belongs to the non-classical MHC complex, or class Ib, that has a potent immunomodulatory activity in pathophysiological situations requiring immune tolerance, such as fetus tolerance during pregnancy, autoimmune and inflammatory diseases, and acceptance of allograft transplantation in patients (3). The diversity of HLA-G products occur by alternative splicing of a primary transcript, which gives rise to four membrane-bound (G1-G4) and three soluble (G5-G7) protein isoforms (4). In addition, soluble isoforms can be generated by proteolytic cleavage of membrane-bound HLA-G forms. While the basic structure of HLA-G is similar to that of classical HLA class Ia molecules, they differ in that HLA-G is less polymorphic (5). HLA-G modulates the innate and adaptive immune systems by interacting with inhibitory receptors on the surface of immune cells, such as the immunoglobulin-like transcript 2 (ILT2) and ILT4 on dendritic cells, ILT2 and the killer immunoglobulin-like receptor 2DL4 (KIR2DL4) on NK cells, and ILT2 on T cells and monocyte/macrophages (4, 6, 7). By binding to these receptors, HLA-G directly inhibits immune cells. However, it has been reported that upon binding to KIR2DL4 HLA-G activates NK cells and promotes cytotoxicity and IFN-γ secretion (8, 9). Furthermore, other studies did not find evidence for a functional interaction between HLA-G and KIR2DL4 in NK cells (10). Therefore, whether HLA-G is a ligand of KIR2DL4 is at present a matter of controversy (11). In addition, several immune cells, including T cells, APCs and a subset of dendritic cells can express or secrete HLA-G. Thus, APCs expressing HLA-G1 induces CD4+ T cell differentiation into regulatory T suppressor cells that block cytotoxic T lymphocyte function (12). These Treg cells are important to sustain immune tolerance and prevent autoimmune diseases. In normal tissues, the most abundant expression of HLA-G is on the surface of trophoblasts in the placenta where can effectively suppress the local immune response in the uterus and promote maternal tolerance to the fetus (13). Apart from this specific function, HLA-G shares with classical HLA class Ia molecules the ability to present antigens (14, 15), suggesting a role for HLA-G on the immune defense against infection and anti-cancer response.
However, while downregulation of classical HLA class Ia molecules is common to most cancers, overexpression of membrane-bound and soluble HLA-G proteins have been found in tumors, including lymphomas, leukemias, melanoma, and breast, kidney, ovarian, lung, esophageal, gastric, pancreatic and colorectal carcinomas. The majority of studies find HLA-G expression in solid tumors to be associated with malignancy and poor prognosis (3, 4, 16–18). In fact, HLA-G is considered as a target for cancer gene therapy (19). However, there are some reports showing the opposite. Thus, for example, upregulation of HLA-G levels was found a favorable prognosis factor in triple-negative breast carcinomas (20). In contrast to solid tumors, HLA-G levels did not show any clear correlation with patient outcome in hematological malignancies (4). HLA-G can modify the action of innate immune cells by inducing tolerance in APCs and inhibiting NK-cell mediated killing (see Figure 1). Thus, APCs expressing HLA-G in the tumor microenvironment have suppressive properties and are partly responsible for tumor immune escape. On the other hand, soluble HLA-G secreted by both tumor and immune cells may directly inhibit CD4+ and CD8+ T cell proliferation (21). In fact, plasma levels of soluble HLA-G are higher in patients with pancreatic cancer compared to healthy donors, and soluble HLA-G levels inversely correlate with the number of peripheral activated T cells (22). It is worth to mention that soluble HLA-G levels are significantly higher in benign lesions than in malignant tumors and can be used as a diagnosis tool to distinguish premalignant from malignant stages (6, 23). HLA-G molecules can be transferred from tumor cells to activated NK cells and other immune cells by a process called trogocytosis, which involves the delivery of plasma membrane fragments from one cell to another leading to downregulation of the immune response (7, 24). This constitutes an effective mechanism for tumor cells to evade surveillance of surrounding immune cells. On the other hand, HLA-G has been found to be secreted associated to extracellular vesicles; i.e., exosomes originated by the endo-lysosomal pathway, derived from melanoma cells (25) and placental trophoblasts (26), which provide another mechanism for tumors to modulate the host immune response and for trophoblasts to modify the maternal immunological environment.
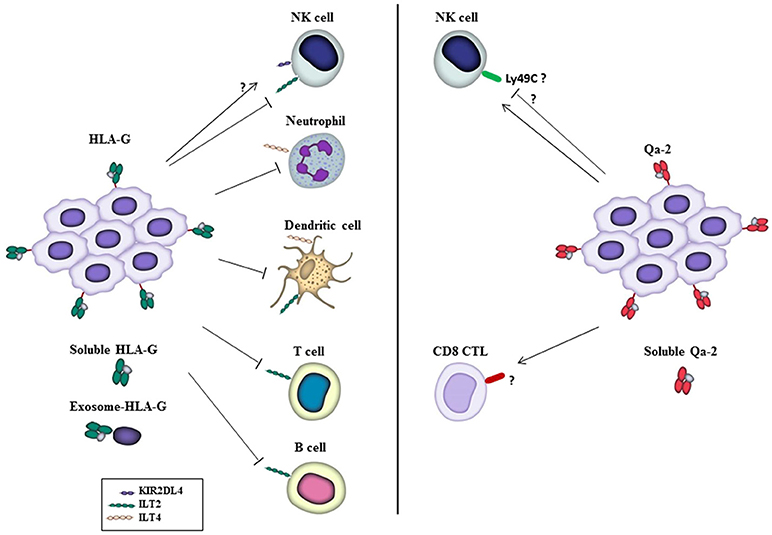
Figure 1. Functions of HLA-G and Qa-2 on immune cells in the tumor microenvironment. HLA-G, either on the surface of tumor cells or in its soluble form or associated with exosomes, inhibits different traits of immune cells through interaction with the inhibitory receptors ILT2 and ILT4, allowing tumor cells to escape from immune surveillance. Also, HLA-G has been reported to interact with KIR2DL4 receptor to activate NK cells, although this is a matter of controversy (see the text). On the other hand, Qa-2 has been reported to activate NK cells and CD8+ CTLs that inhibit tumor cell growth and mediate tumor rejection. Inhibition of NK cells by Qa-2 was also reported in vitro, and Ly49C postulated as a Qa-2 inhibitory receptor, see the text. Receptors on immune cells involved in the response to Qa-2 are presently unknown. Likewise, whether Qa-2 is released to the extracellular milieu associated to exosomes remains to be investigated.
The Murine Non-Classical MHC Complex Qa-2. Its Role in Embryogenesis and Autoimmunity
Qa-2 has been reported to be the HLA-G homolog in mice (27). Both HLA-G and Qa-2 molecules have membrane-bound and soluble forms that originate by alternative splicing, display immunoregulatory roles, and are involved in embryonic development (7). There are four major Qa-2 loci: Q6, Q7, Q8, and Q9, which localize at the Qa region of chromosome 17 and are present in different combinations in each mouse haplotype (28). Since Q6 and Q8 loci are almost perfect duplicate of each other, and Q7 and Q9 only differ by a single nucleotide leading to a change of Gln in Q7 to Glu in Q9, they are referred as Q6/Q8 and Q7/Q9 pairs. Therefore, Qa-2 gene clusters seem to have evolved by sequential duplication of a primordial gene pair consisting of one “odd” locus that originated Q7 and Q9 and one “even” locus that gave rise to Q6 and Q8 (27). The pair Q6/Q8 encodes a transmembrane protein while the pair Q7/Q9 encodes a glycosylphosphatidylinositol (GPI)-anchored membrane protein. The GPI linkage facilitates clustering of Qa-2 protein on lipid rafts and suggests a potential role for Qa-2 in cell signaling (29). Q7/Q9 may generate a soluble Qa-2 isoform by alternative splicing, in which loss of exon 5 results in the inability of the product to attach the GPI anchor (30, 31). In addition, alike to HLA-G, soluble Qa-2 isoforms can also be generated by shedding of membrane-bound forms (32).
Qa-2 expression on the cell surface requires the assembly within the endoplasmic reticulum of a trimer. Qa-2 associates with β2 microglobulin and is loaded with a small peptide, which is primarily produced in the cytosol by the ubiquitin–proteasome pathway and then transported into the lumen of the endoplasmic reticulum by the transporter associated with antigen processing (TAP) protein, a heterodimer consisting of TAP1 and TAP2 subunits (33). Qa-2 binds a wide array of TAP-dependent self and non-self-nonameric peptides, which suggests Qa-2 has an immunosurveillance function alerting for the presence of intracellular pathogen infections or neoplastic transformation (34). Thus, Qa-2 restricted CD8+ T cells contribute to protect the host against infection with Mycobacterium tuberculosis (35) and polyoma virus (36), the latter requiring CD4+ T cells for its maintenance phase (37). Likewise, CD8+ T cells are involved in the anti-tumor immune response elicited by Qa-2 (see below).
Qa-2 genes were identified as Ped (preimplantation embryo development) candidate genes that modulate the rate of embryo cleavage division and subsequent fetal survival (38, 39). Embryos from some mouse strains develop faster than those of other strains, depending on the presence of Qa-2 protein (i.e., the products of Q7/Q9 pair) on the surface of preimplantation embryo. This has been named the Ped fast phenotype (39). It was proposed that Qa-2 protein protects the developing fetus from the attack of maternal NK cells and/or macrophages, which could account for the higher rate of fetal loss observed in embryos lacking Qa-2 protein (40). In addition, besides an overall reproductive advantage, the presence of Qa-2 protein in the embryo grants a healthier life to the adult mouse (41).
In adult tissues, Qa-2 is widely distributed but generally at low levels. It is present in APCs, immature and mature thymocytes, thymic epithelial cells, intestinal epithelial cells and immunologically privileged sites/cells, such as the anterior chamber of the eye, hair follicles and sperm in testis (31, 32, 42). Qa-2 has been involved in T lymphocyte proliferation, particularly in CD4+ T cells (43). However, the α3 domain of Qa-2 is unable to interact with CD8 in order to effectively activate cytotoxic T cells (44). Qa-2 has been reported to mediate signal transduction pathways, as antibody-induced crosslinking of Qa-2 promoted proliferation of resting T cells depending on Fyn (a member of the Src family of kinases), PI-3 kinase and Akt (45). Also, Qa-2 is required for the selection of intraepithelial unconventional CD8αα/TCRαβ T cells, which appear to be regulatory cells involved in maintaining intestinal integrity (46).
HLA-G allelic variants inducing a significantly lower expression level of HLA-G products are genetic risk factors for Behcet's disease, a chronic multi-systemic disorder involving gastrointestinal, mucocutaneous, ocular, vascular, central nervous and articular systems (47). A similar role for Qa-2 was also shown in a Behcet's disease-like mouse model induced by the herpex virus simplex, in which silencing of Qa-2 by intravenous injection of specific siRNA worsened the disease symptoms (48).
Acute graft rejection is the main complication in organ transplantation. It involves allograft recognition by the immune system as non-self and graft destruction by reactive immune cells (49). It has been reported that patients with upregulated HLA-G expression in grafts and plasma have better allograft acceptance that HLA-G-negative patients (50, 51). Lu and coworkers have studied the relationship of Qa-2 expression in grafts and peripheral blood lymphocytes (PBLs) with graft rejection in a well-established murine skin transplantation model in the presence or absence of immunosuppressive agents. They found negative or weakly positive Qa-2 expression in mice without immunosuppressive treatment similar to that of control mice. In contrast, treatment with immunosuppressive drugs upregulated Qa-2 expression in both grafts and PBLs and prolonged the survival of skin allografts. Qa-2 expression subsequently decreased during allograft rejection (52). These results clearly point to an inverse relationship between Qa-2 levels and allograft rejection.
Whereas, HLA-G is a well-established ligand of the ILT family of immune inhibitory receptors (4, 6, 7), little is known about the receptors for Qa-2 in immune cells. A potential candidate is paired immunoglobulin-like inhibitory receptor (PIR-B), the murine ortholog of human ILT, which has been shown to interact with MHC class I molecules (53) and HLA-G (54, 55). However, to our knowledge, evidence for a functional interaction between PIR-B and Qa-2 is still lacking. In a recent review, Goodall and co-workers claim that Qa-2 binds Ly49C on NK cells (56). Ly49C is a member of the lectin-like Ly49 family receptors, the functional counterparts in mice of human KIR receptors. However, the experimental demonstration for this interaction has not been published yet.
The Role of Qa-2 In Cancer
Similarly to classical HLA class Ia molecules, Qa-2 expression was found to be downregulated in tumors and derived cell lines, including melanoma, hepatoma, lymphoma, leukemia, and mastocytoma, which suggested to the authors a suppressive role for Qa-2 in cancer (42). In accordance with this hypothesis, Chiang and Stroynowski demonstrated that restoration of Q9 expression in melanoma, lung carcinoma and T-cell lymphoma cell lines that have downregulated Qa-2 expression resulted in a CD8+ cytotoxic T lymphocyte (CTL)-mediated immune response that inhibited in vivo tumor growth in syngeneic hosts (57–59).
The lack of Qa-2 expression in B16 and B16-derived melanoma cell lines with low and high metastatic capacities suggest that downregulation of Qa-2 levels is an early event during melanoma progression. Experiments with genetically manipulated melanoma cells showed that TAP-2-dependent restoration of Q9 expression led to reduced tumorigenic potential in syngeneic mice, and in vivo depletion studies indicated that both NK cells and CD8+ CTLs collaborated in the rejection response (57) (Figure 1). It should be mentioned, however, that Q9 was previously shown to partially protect melanoma cells from lysis by activated NK cells in vitro, suggesting that Q9 may interact with an inhibitory NK receptor (60) that might correspond to Ly49C (56). Nevertheless, in vivo studies indicate that Q9 sensitizes tumors to trigger a NK cell-mediated cytolytic response either by directly killing tumor cells or by stimulating downstream T cell responses through secretion of cytokines and chemokines. Interestingly, the protection conferred by Q9 to the host was stronger that the anti-tumor effect exerted by the expression of its structurally homologous class Ia H2-Kb antigen (57). Further studies demonstrated that Q9 protein is a target for anti-tumor immune surveillance, as CD8+ T-cell-deficient mice as well as β2 microglobulin and CD8-knockout mice were unable to reject Q9-bearing melanoma tumors. Moreover, the display of peptide-loaded Q9 on the surface of melanoma cells induced strong immunological memory (58). The ability of Q9 to act as a restriction element for anti-tumor CTL is not specific of melanoma cells, as Q9 surface expression on 3LLA9F1 Lewis lung carcinoma or RMA T cell lymphoma cell lines also induces a potent anti-tumor CTL response that eliminate tumor cells in syngeneic hosts. Mice challenged with one Q9-expressing tumor develop immunological memory against subsequent challenge with different Q9-bearing tumor cells, suggesting that Q9 has the capacity to present tumor antigens shared by distinct lineages of cancer cells (59). Like Q9, Q8 protein is ubiquitously distributed in mouse normal tissues and is frequently downregulated in tumors (42). Despite Q8 differs significantly from Q9 in the amino acid sequence of α1 and α2 domains involved in peptide binding and T cell recognition, Q8 and Q9 have overlapping binding motifs and exhibit cross-reactive CTL responses recognizing shared tumor-associated antigens from melanoma, Lewis lung carcinoma and T cell lymphoma (61). Whereas, the studies of Chiang and Stroynowski suggest that Qa-2 activates innate and adaptive immune cells to reject tumors, they hold some limitations that should be taken into account. First, these studies come from only one laboratory and should be confirmed by others. Second, the B78H1melanoma cell model used in these experiments is not completely negative for MHC class I molecules, but express low levels of HLA class I antigens (http://www.lollini.it/b78.htm), and the possibility that they were upregulated after TAP-2 transfection was not ruled out experimentally.
We have addressed the role of Qa-2 in tumor growth and progression using the 4T1 mammary carcinoma cell model. 4T1 tumor cells are representative of the highly aggressive triple-negative subtype of human breast cancer (62). In the 4T1 cell line, only a small percentage of cells (about 4%) express Qa-2 on the cell surface, and Qa-2 protein levels were further reduced during in vivo tumor growth and in tumor-derived cultured cells (63), suggesting that Qa-2 expression is downregulated during breast cancer development. Cell lines derived from tumors induced by 4T1 in the back skin, or in the mammary fat pad, of syngeneic Balb/c mice elicited a partial epithelial-mesenchymal transition (EMT) and exhibited increased stem cell characteristics and enhanced tumor-initiating and invasive capacities that correlated with reduced Qa-2 expression. In fact, Qa-2 expression was completely lost in a CD44high/CD24med/low cancer stem cell subpopulation (64) isolated from these cell lines (63). This striking result suggests the possibility that Qa-2 is excluded from cancer stem cells, thus contributing to their evasion from immunosurveillance. Nonetheless, this hypothesis deserves further investigation. Downregulation of Qa-2 in cancer stem cells appears to be mediated, at least partially, by the Src signaling pathway. Indeed, pharmacological inhibition of Src kinase activity enhanced Qa-2 expression and concomitantly reduced stemness of 4T1 tumor-derived cell lines (63). Increased Src activity is a general characteristic of malignancy associated with EMT, stemness, invasion and metastasis (65). In order to confirm the anti-tumor role of Qa-2, we forced the expression of Q7 in 4T1 cells. Q7 is a key member of the Qa-2 family in Balb/c mice (42). 4T1 cells overexpressing Q7 produced tumors that grew slower and were less metastatic than control or parental cells (63).
Interestingly, Servín-Blanco and colleagues have reported that Qa-2-derived peptides elicited an anti-tumor immune response against carcinomas induced by 4T1 cells in Balb/c mice resulting in a significant inhibition of tumor growth and a reduction in the number of metastatic lesions (66). This possibility of immunotherapy targeting Qa-2 is shared by HLA-G, as HLA-G-derived peptides were able to induce a CTL response against HLA-G-expressing human renal carcinoma cells (67).
Concluding Remarks
In summary, Qa-2 has been found to have a different role in cancer with respect to its putative human homolog HLA-G. Whereas, HLA-G seems to help tumor cells to escape from immunosurveillance by directly interacting with inhibitory receptors that halt innate and adaptive immune cells, Qa-2 appears to activate NK and CD8+ CTLs to reject tumors. Thus, despite similarities in structure and function during embryogenesis and autoimmunity, both families of HLA class lb proteins act in an opposite manner in the context of tumors (Table 1). Therefore, downregulation of Qa-2 and upregulation of HLA-G expression are major immune evasive mechanisms used by murine and human tumors, respectively. Nevertheless, in contrast to HLA-G, only a handful of reports have addressed the functional implication of Qa-2 in tumors, and mainly using a limited number of cell lines. Thus, studies on the role of Qa-2 in cancer should be extended. Specially, in vivo models of carcinogenesis with normal or genetically engineered mice should be used in order to ascertain more accurately the mechanisms of tumor suppression exhibited by these molecules. Moreover, it would be of great interest to clarify the apparent inverse relationship between Qa-2 and cancer stem cells using both in vitro and in vivo models of cancer. Particularly relevant to understand the different roles of HLA-G and Qa-2 in cancer is to identify the specific receptors for Qa-2 in different immune cells, either inhibitory or stimulatory, and the regulatory signals triggered upon its binding.

Table 1. Functional characteristics of HLA-G and Qa-2.
Author Contributions
IdS and MQ wrote the manuscript. LM-M and EF reviewed it.
Funding
The research developed in our laboratories is supported by grants SAF2017-84183-R from the Spanish Ministry of Science, Innovation and Universities to MQ, and APQ-04269-10 and RED-00011-14 from the Foundation for Research Support of the State of Minas Gerais (FAPEMIG) to EF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Javier Pérez for his help with Figure 1. We apologize with authors whose excellent work was not cited in this article due to space limitations.
References
1. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Ann Rev Immunol. (2011) 29:235–71. doi: 10.1146/annurev-immunol-031210-101324
2. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. (2013) 1284:1–5. doi: 10.1111/nyas.12105
3. Morandi F, Rizzo R, Fainardi E, Rouas-Freiss N, Pistoia V. Recent advances in our understanding of HLA-G biology: lessons from a wide spectrum of human diseases. J Immunol Res. (2016) 2016:4326495. doi: 10.1155/2016/4326495
4. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: an immune checkpoint molecule. Adv Immunol. (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001
5. Rizzo R, Bortolotti D, Bolzani S, Fainardi E. HLA-G molecules in autoimmune diseases and infections. Front Immunol. (2014) 5:592. doi: 10.3389/fimmu.2014.00592
6. Curigliano G, Criscitiello C, Gelao L, Goldhirsch A. Molecular pathways: human leukocyte antigen G (HLA-G). Clin Cancer Res. (2013) 19:5564–71. doi: 10.1158/1078-0432.CCR-12-3697
7. Gomes AQ, Correia DV, Silva-Santos B. Non-classical major histocompatibility complex proteins as determinants of tumour immunosurveillance. EMBO Rep. (2007) 8:1024–30. doi: 10.1038/sj.embor.7401090
8. Rajagopalan S, Fu J, Long EO. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol. (2001) 167:1877–81. doi: 10.4049/jimmunol.167.4.1877
9. Kikuchi-Maki A, Yusa S, Catina TL, Campbell KS. KIR2DL4 is an IL-2-regulated NK cell receptor that exhibits limited expression in humans but triggers strong IFN-gamma production. J Immunol. (2003) 171:3415–25. doi: 10.4049/jimmunol.171.7.3415
10. Le Page ME, Goodridge JP, John E, Christiansen FT, Witt CS. Killer Ig-like receptor 2DL4 does not mediate NK cell IFN-gamma responses to soluble HLA-G preparations. J Immunol. (2014) 192:732–40. doi: 10.4049/jimmunol.1301748
11. Le Page ME, Goodridge JP, John E, Christiansen FT, Witt CS. Response to comment on “killer Ig-like receptor 2DL4 does not mediate NK cell IFN-gamma responses to soluble HLA-G preparations”. J Immunol. (2014) 192:4003–4. doi: 10.4049/jimmunol.1400492
12. LeMaoult J, Krawice-Radanne I, Dausset J, Carosella ED. HLA-G1-expressing antigen-presenting cells induce immunosuppressive CD4+ T cells. Proc Natl Acad Sci USA. (2004) 101:7064–9. doi: 10.1073/pnas.0401922101
13. Hviid TV. HLA-G in human reproduction: aspects of genetics, function and pregnancy complications. Hum Reprod Update (2006) 12:209–32. doi: 10.1093/humupd/dmi048
14. Diehl M, Munz C, Keilholz W, Stevanovic S, Holmes N, Loke YW, et al. Nonclassical HLA-G molecules are classical peptide presenters. Curr Biol. (1996) 6:305–14. doi: 10.1016/S0960-9822(02)00481-5
15. Di Marco M, Schuster H, Backert L, Ghosh M, Rammensee HG, Stevanovic S. Unveiling the peptide motifs of HLA-C and HLA-G from naturally presented peptides and generation of binding prediction matrices. J Immunol. (2017) 199:2639–51. doi: 10.4049/jimmunol.1700938
16. de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol. (2010) 185:7452–9. doi: 10.4049/jimmunol.1002629
17. Yie SM, Hu Z. Human leukocyte antigen-G (HLA-G) as a marker for diagnosis, prognosis and tumor immune escape in human malignancies. Histol Histopathol. (2011) 26:409–20. doi: 10.14670/HH-26.409
18. Ferguson R, Ramanakumar AV, Koushik A, Coutlee F, Franco E, Roger M. Human leukocyte antigen G polymorphism is associated with an increased risk of invasive cancer of the uterine cervix. Int J Cancer (2012) 131:E312–9. doi: 10.1002/ijc.27356
19. Zidi I, Ben Amor N. Nanoparticles targeting HLA-G for gene therapy in cancer. Med Oncol. (2012) 29:1384–90. doi: 10.1007/s12032-011-9942-8
20. Martinez-Canales S, Cifuentes F, Lopez De Rodas Gregorio M, Serrano-Oviedo L, Galan-Moya EM, Amir E, et al. Transcriptomic immunologic signature associated with favorable clinical outcome in basal-like breast tumors. PLoS ONE (2017) 12:e0175128. doi: 10.1371/journal.pone.0175128
21. Bahri R, Hirsch F, Josse A, Rouas-Freiss N, Bidere N, Vasquez A, et al. Soluble HLA-G inhibits cell cycle progression in human alloreactive T lymphocytes. J Immunol. (2006) 176:1331–9. doi: 10.4049/jimmunol.176.3.1331
22. Xu YF, Lu Y, Cheng H, Jiang J, Xu J, Long J, et al. High expression of human leukocyte antigen-G is associated with a poor prognosis in patients with PDAC. Curr Mol Med. (2015) 15:360–7. doi: 10.2174/1566524015666150401102218
23. Cao M, Yie SM, Liu J, Ye SR, Xia D, Gao E. Plasma soluble HLA-G is a potential biomarker for diagnosis of colorectal, gastric, esophageal and lung cancer. Tissue Antigens (2011) 78:120–8. doi: 10.1111/j.1399-0039.2011.01716.x
24. Johansen LL, Lock-Andersen J, Hviid TV. The pathophysiological impact of HLA class Ia and HLA-G expression and regulatory T cells in malignant melanoma: a review. J Immunol Res. (2016) 2016:6829283. doi: 10.1155/2016/6829283
25. Riteau B, Faure F, Menier C, Viel S, Carosella ED, Amigorena S, et al. Exosomes bearing HLA-G are released by melanoma cells. Hum Immunol. (2003) 64:1064–72. doi: 10.1016/j.humimm.2003.08.344
26. Kshirsagar SK, Alam SM, Jasti S, Hodes H, Nauser T, Gilliam M, et al. Immunomodulatory molecules are released from the first trimester and term placenta via exosomes. Placenta (2012) 33:982–90. doi: 10.1016/j.placenta.2012.10.005
27. Weiss EH, Golden L, Fahrner K, Mellor AL, Devlin JJ, Bullman H, et al. Organization and evolution of the class I gene family in the major histocompatibility complex of the C57BL/10 mouse. Nature (1984) 310:650–5. doi: 10.1038/310650a0
28. Stroynowski I, Tabaczewski P. Multiple products of class Ib Qa-2 genes which ones are functional? Res Immunol. (1996) 147:290–301. doi: 10.1016/0923-2494(96)89642-8
29. Comiskey M, Warner CM. Spatio-temporal localization of membrane lipid rafts in mouse oocytes and cleaving preimplantation embryos. Dev Biol. (2007) 303:727–39. doi: 10.1016/j.ydbio.2006.12.009
30. Ulker N, Lewis KD, Hood LE, Stroynowski I. Activated T cells transcribe an alternatively spliced mRNA encoding a soluble form of Qa-2 antigen. EMBO J. (1990) 9:3839–47. doi: 10.1002/j.1460-2075.1990.tb07602.x
31. Tian Z, Xu Y, Warner CM. Removal of Qa-2 antigen alters the Ped gene phenotype of preimplantation mouse embryos. Biol Reprod. (1992) 47:271–6. doi: 10.1095/biolreprod47.2.271
32. He X, Tabaczewski P, Ho J, Stroynowski I, Garcia KC. Promiscuous antigen presentation by the nonclassical MHC Ib Qa-2 is enabled by a shallow, hydrophobic groove and self-stabilized peptide conformation. Structure (2001) 9:1213–24. doi: 10.1016/S0969-2126(01)00689-X
33. Tabaczewski P, Stroynowski I. Expression of secreted and glycosylphosphatidylinositol-bound Qa-2 molecules is dependent on functional TAP-2 peptide transporter. J Immunol. (1994) 152:5268–74.
34. Tabaczewski P, Chiang E, Henson M, Stroynowski I. Alternative peptide binding motifs of Qa-2 class Ib molecules define rules for binding of self and nonself peptides. J Immunol. (1997) 159:2771–81.
35. Shang S, Siddiqui S, Bian Y, Zhao J, Wang CR. Nonclassical MHC Ib-restricted CD8+ T cells recognize mycobacterium tuberculosis-derived protein antigens and contribute to protection against infection. PLoS Pathog. (2016) 12:e1005688. doi: 10.1371/journal.ppat.1005688
36. Swanson PA II, Pack CD, Hadley A, Wang CR, Stroynowski I, Jensen PE, et al. An MHC class Ib-restricted CD8 T cell response confers antiviral immunity. J Exp Med. (2008) 205:1647–57. doi: 10.1084/jem.20080570
37. Hofstetter AR, Ford ML, Sullivan LC, Wilson JJ, Hadley A, Brooks AG, et al. MHC class Ib-restricted CD8 T cells differ in dependence on CD4 T cell help and CD28 costimulation over the course of mouse polyomavirus infection. J Immunol. (2012) 188:3071–9. doi: 10.4049/jimmunol.1103554
38. Goldbard SB, Verbanac KM, Warner CM. Role of the H-2 complex in preimplantation mouse embryo development. Biol Reprod. (1982) 26:591–6. doi: 10.1095/biolreprod26.4.591
39. Wu L, Feng H, Warner CM. Identification of two major histocompatibility complex class Ib genes, Q9 and Q9, as the Ped gene in the mouse. Biol Reprod. (1999) 60:1114–9. doi: 10.1095/biolreprod60.5.1114
40. Warner CM, Brenner CA. Genetic regulation of preimplantation embryo survival. Curr Topics Dev Biol. (2001) 52:151–92. doi: 10.1016/S0070-2153(01)52011-6
41. Watkins A, Wilkins A, Osmond C, Warner CM, Comiskey M, Hanson M, et al. The influence of mouse Ped gene expression on postnatal development. J Physiol. (2006) 571(Pt 1):211–20. doi: 10.1113/jphysiol.2005.099192
42. Ungchusri T, Chiang EY, Brown G, Chen M, Tabaczewski P, Timares L, et al. Widespread expression of the nonclassical class I Qa-2 antigens in hemopoietic and nonhemopoietic cells. Immunogenetics (2001) 53:455–67. doi: 10.1007/s002510100347
43. Hahn AB, Tian H, Wiegand G, Soloski MJ. Signals delivered via the Qa-2 molecule can synergize with limiting anti-CD3-induced signals to cause T lymphocyte activation. Immunol Invest. (1992) 21:203–17. doi: 10.3109/08820139209072259
44. Teitell M, Holcombe H, Cheroutre H, Aldrich CJ, Stroynowski I, Forman J, et al. The alpha 3 domain of the Qa-2 molecule is defective for CD8 binding and cytotoxic T lymphocyte activation. J Exp Med. (1993) 178:2139–45. doi: 10.1084/jem.178.6.2139
45. De Fazio SR, Warner CM. Activation of T cells by cross-linking Qa-2, the ped gene product, requires Fyn. Am J Reprod Immunol. (2007) 58:315–24. doi: 10.1111/j.1600-0897.2007.00503.x
46. Das G, Gould DS, Augustine MM, Fragoso G, Sciutto E, Stroynowski I, et al. Qa-2-dependent selection of CD8alpha/alpha T cell receptor alpha/beta(+) cells in murine intestinal intraepithelial lymphocytes. J Exp Med. (2000) 192:1521–8. doi: 10.1084/jem.192.10.1521
47. Shimizu T, Ehrlich GE, Inaba G, Hayashi K. Behcet disease (Behcet syndrome). Semin Arthritis Rheum. (1979) 8:223–60. doi: 10.1016/0049-0172(79)90004-0
48. Lee M, Choi B, Kwon HJ, Shim JA, Park KS, Lee ES, et al. The role of Qa-2, the functional homolog of HLA-G, in a Behcet's disease-like mouse model induced by the herpes virus simplex. J Inflamm. (2010) 7:31. doi: 10.1186/1476-9255-7-31
49. Hartono C, Muthukumar T, Suthanthiran M. Noninvasive diagnosis of acute rejection of renal allografts. Curr Opin Organ Transplant. (2010) 15:35–41. doi: 10.1097/MOT.0b013e3283342728
50. Rouas-Freiss N, LeMaoult J, Moreau P, Dausset J, Carosella ED. HLA-G in transplantation: a relevant molecule for inhibition of graft rejection? Am J Transplant. (2003) 3:11–6. doi: 10.1034/j.1600-6143.2003.30103.x
51. Qiu J, Terasaki PI, Miller J, Mizutani K, Cai J, Carosella ED. Soluble HLA-G expression and renal graft acceptance. Am J Transplant. (2006) 6:2152–6. doi: 10.1111/j.1600-6143.2006.01417.x
52. Lu N, Wang C, Yang X, Zhao S, Li X, Jiang H, et al. Dynamic expression of Qa-2 during acute graft rejection. Mol Med. (2011) 17:248–55. doi: 10.2119/molmed.2010.00133
53. Takai T, Nakamura A, Endo S. Role of PIR-B in autoimmune glomerulonephritis. J Biomed Biotechnol. (2011) 2011:275302. doi: 10.1155/2011/275302
54. Liang S, Baibakov B, Horuzsko A. HLA-G inhibits the functions of murine dendritic cells via the PIR-B immune inhibitory receptor. Euro J Immunol. (2002) 32:2418–26. doi: 10.1002/1521-4141(200209)32:9<2418::AID-IMMU2418>3.0.CO;2-L
55. Loumagne L, Baudhuin J, Favier B, Montespan F, Carosella ED, Rouas-Freiss N. In vivo evidence that secretion of HLA-G by immunogenic tumor cells allows their evasion from immunosurveillance. Int J Cancer (2014) 135:2107–17. doi: 10.1002/ijc.28845
56. Goodall KJ, Nguyen A, Sullivan LC, Andrews DM. The expanding role of murine class Ib MHC in the development and activation of Natural Killer cells. Mol Immunol. (2018). doi: 10.1016/j.molimm.2018.05.001
57. Chiang EY, Henson M, Stroynowski I. Correction of defects responsible for impaired Qa-2 class Ib MHC expression on melanoma cells protects mice from tumor growth. J Immunol. (2003) 170:4515–23. doi: 10.4049/jimmunol.170.9.4515
58. Chiang EY, Stroynowski I. A nonclassical MHC class I molecule restricts CTL-mediated rejection of a syngeneic melanoma tumor. J Immunol. (2004) 173:4394–401. doi: 10.4049/jimmunol.173.7.4394
59. Chiang EY, Stroynowski I. Protective immunity against disparate tumors is mediated by a nonpolymorphic MHC class I molecule. J Immunol. (2005) 174:5367–74. doi: 10.4049/jimmunol.174.9.5367
60. Chiang EY, Henson M, Stroynowski I. The nonclassical major histocompatibility complex molecule Qa-2 protects tumor cells from NK cell- and lymphokine-activated killer cell-mediated cytolysis. J Immunol. (2002) 168:2200–11. doi: 10.4049/jimmunol.168.5.2200
61. Chiang EY, Stroynowski I. The role of structurally conserved class I MHC in tumor rejection: contribution of the Q8 locus. J Immunol. (2006) 177:2123–30. doi: 10.4049/jimmunol.177.4.2123
62. Kaur P, Nagaraja GM, Zheng H, Gizachew D, Galukande M, Krishnan S, et al. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer (2012) 12:120. doi: 10.1186/1471-2407-12-120
63. da Silva IL, Montero-Montero L, Martin-Villar E, Martin-Perez J, Sainz B, Renart J, et al. Reduced expression of the murine HLA-G homolog Qa-2 is associated with malignancy, epithelial-mesenchymal transition and stemness in breast cancer cells. Sci Rep. (2017) 7:6276. doi: 10.1038/s41598-017-06528-x
64. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
65. Patel A, Sabbineni H, Clarke A, Somanath PR. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. (2016) 157:52–61. doi: 10.1016/j.lfs.2016.05.036
66. Servin-Blanco R, Chavaro-Ortiz RM, Zamora-Alvarado R, Martinez-Cortes F, Gevorkian G, Manoutcharian K. Generation of cancer vaccine immunogens derived from major histocompatibility complex (MHC) class I molecules using variable epitope libraries. Immunol Lett. (2018) 204:47–54. doi: 10.1016/j.imlet.2018.10.008
Keywords: Qa-2, HLA-G, MHC-I, immunosurveillance, cancer
Citation: da Silva IL, Montero-Montero L, Ferreira E and Quintanilla M (2018) New Insights Into the Role of Qa-2 and HLA-G Non-classical MHC-I Complexes in Malignancy. Front. Immunol. 9:2894. doi: 10.3389/fimmu.2018.02894
Received: 30 August 2018; Accepted: 26 November 2018;
Published: 06 December 2018.
Edited by:
Jin S. Im, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Wei-Hua Yan, Wenzhou Medical University, ChinaSilvia Gregori, Istituto San Raffaele Telethon per la Terapia Genica (SR-Tiget), Italy
Jörg Wischhusen, Universität Würzburg, Germany
Copyright © 2018 da Silva, Montero-Montero, Ferreira and Quintanilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Quintanilla, bXF1aW50YW5pbGxhQGlpYi51YW0uZXM=