 Manu Chhabra1†
Manu Chhabra1† Jawaher Alsughayyir1†
Jawaher Alsughayyir1† M. Saeed Qureshi1
M. Saeed Qureshi1 Mekhola Mallik1
Mekhola Mallik1 Jason M. Ali1
Jason M. Ali1 Ivonne Gamper1
Ivonne Gamper1 Ellen L. Moseley2
Ellen L. Moseley2 Sarah Peacock3
Sarah Peacock3 Vasilis Kosmoliaptsis1
Vasilis Kosmoliaptsis1 Martin J. Goddard2
Martin J. Goddard2 Michelle A. Linterman4
Michelle A. Linterman4 Reza Motallebzadeh5,6,7*†
Reza Motallebzadeh5,6,7*† Gavin J. Pettigrew1*†
Gavin J. Pettigrew1*†- 1School of Clinical Medicine, University of Cambridge, Cambridge, United Kingdom
- 2Department of Pathology, Papworth Hospital, Papworth Everard, United Kingdom
- 3Histocompatibility and Immunogenetics Laboratory, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
- 4Laboratory of Lymphocyte Signalling and Development, Babraham Institute, Cambridge, United Kingdom
- 5Division of Surgery and Interventional Science, University College London, London, United Kingdom
- 6Centre for Transplantation, Department of Renal Medicine, University College London, London, United Kingdom
- 7Institute of Immunity and Transplantation, University College London, London, United Kingdom
Different profiles of alloantibody responses are observed in the clinic, with those that persist, often despite targeted treatment, associated with poorer long-term transplant outcomes. Although such responses would suggest an underlying germinal center (GC) response, the relationship to cellular events within the allospecific B cell population is unclear. Here we examine the contribution of germinal center (GC) humoral alloimmunity to chronic antibody mediated rejection (AMR). A murine model of chronic AMR was developed in which T cell deficient (Tcrbd−/−) C57BL/6 recipients were challenged with MHC-mismatched BALB/c heart allografts and T cell help provided by reconstituting with 103 “TCR75” CD4 T cells that recognize self-restricted allopeptide derived from the H-2Kd MHC class I alloantigen. Reconstituted recipients developed Ig-switched anti-Kd alloantibody responses that were slow to develop, but long-lived, with confocal immunofluorescence and flow cytometric characterization of responding H-2Kd-allospecific B cells confirming persistent splenic GC activity. This was associated with T follicular helper (TFH) cell differentiation of the transferred TCR75 CD4 T cells. Heart grafts developed progressive allograft vasculopathy, and were rejected chronically (MST 50 days), with explanted allografts displaying features of humoral vascular rejection. Critically, late alloantibody responses were abolished, and heart grafts survived indefinitely, in recipients reconstituted with Sh2d1a−/− TCR75 CD4 T cells that were genetically incapable of providing TFH cell function. The GC response was associated with affinity maturation of the anti-Kd alloantibody response, and its contribution to progression of allograft vasculopathy related principally to secretion of alloantibody, rather than to enhanced alloreactive T cell priming, because grafts survived long-term when B cells could present alloantigen, but not secrete alloantibody. Similarly, sera sampled at late time points from chronically-rejecting recipients induced more vigorous donor endothelial responses in vitro than sera sampled earlier after transplantation. In summary, our results suggest that chronic AMR and progression of allograft vasculopathy is dependent upon allospecific GC activity, with critical help provided by TFH cells. Clinical strategies that target the TFH cell subset may hold therapeutic potential.
This work is composed of two parts, of which this is Part II. Please read also Part I: Alsughayyir et al., 2019.
Introduction
The contribution of humoral alloimmunity to transplant failure is increasingly recognized (1–3). In addition to acute antibody mediated rejection (AMR) that typically occurs early after transplantation [considered in the companion paper (4)], donor specific alloantibody (DSA) responses are associated with chronic graft dysfunction, progressive allograft vasculopathy and early graft failure (5). Chronic AMR is less well-defined, and not recognized as a distinct clinical entity for some organs (6). Nevertheless, the development of DSA against mismatched MHC antigen is now known to be a major risk factor for early transplant failure of liver (7, 8) kidney (9–12), lung (13, 14), and heart (15, 16) allografts. Chronic AMR is most clearly described following kidney transplantation and presents as transplant glomerulopathy (17–19), with an associated graft loss of 50% at 18 months (10).
The hallmark feature of chronic AMR—progressive allograft vasculopathy—is thought to be a culmination of cycles of insidious injury and repair, and it is notable that the associated alloantibody response usually persists long after its first detection (20, 21). Such long-lived antibody responses are typically mediated by long-lived plasma cells (LLPCs) that are deposited in the bone marrow (22), and that, in turn, are generally considered a product of an affinity-matured germinal center reaction (GC) (23–25). However, somatic hypermutation has been described within extrafollicular foci (26, 27), and, notably, the allospecific GC response has not yet been delineated, because of the difficulty in accessing tissue in human transplant patients, and because murine models of chronic AMR are limited (28, 29). Thus, it is not yet known whether a GC response against MHC alloantigen is essential for the production of long-lived alloantibody that underpins the development of chronic AMR and allograft vasculopathy. Experimental confirmation of a central role for the GC response in chronic rejection would have immediate relevance for clinical transplantation, because the GC reaction is now known to require help for its initiation and maintenance from a specialized subset of PD-1hiCXCR5hi CD4 T follicular helper (TFH) cells (30–33).
Here, we develop a murine cardiac model of chronic AMR to demonstrate that progression of allograft vasculopathy is dependent upon germinal center alloantibody responses that are associated with affinity maturation, and that are critically dependent upon the provision of help from TFH cells that recognize processed target alloantigen via the indirect pathway.
Materials and Methods
Animals
C57BL/6 (BL/6; H-2b) and BALB/c mice (H-2d) were purchased from Charles River Laboratories (Margate, UK) and maintained according to the institutional guidelines of The University of Cambridge. T cell receptor-deficient mice (H-2b, Tcrbd−/−) BL/6.129P2-Tcrbtm1MomTcrdtm1Mom/J were purchased from the Jackson Laboratory (Bar Harbor, ME). C57BL/6 Rag2−/− mice (H-2b) were gifted by Prof T. Rabbitts (Laboratory of Molecular Biology, Cambridge, UK). TCR-transgenic Rag1−/− TCR75 mice (H-2b), specific for I-Ab-restricted H-2K peptide (34) and C57BL/6-Tg(Kd)RPb (BL/6.Kd) mice, which express the full sequence of H-2Kd (35), were gifted by Prof. P. Bucy (University of Alabama, Birmingham, AL). BCR-transgenic SWHEL (VH10 x LC2) mice (H-2b) specific for Hen Egg Lysozyme (HEL) protein (36) and BL/6.mHEL mice (H-2b, KLK3 Tg) that express membrane bound HEL (37) under the H-2Kb promoter, were gifted by Prof R. Brink (Garvan Institute of Medical Research, Darlinghusrt, Australia). BL/6 Rag2−/− SWHEL were generated by crossing SWHEL BCR–transgenic mice crossed onto the Rag2−/− background (38). Sh2d1a−/− (SLAM-associated protein [SAP]-deficient) mice (39) were gifted by Dr S. Crotty (University of California, La Jolla, California). BL/6.Kd mice were crossed with BL/6.mHEL mice to create a BL/6 mHEL-Kd donor strain. Mice were bred and maintained in specific pathogen-free animal facilities and were maintained in individual ventilated cages in specific-pathogen free facilities and fed standard rodent feeds. Mice weighed between 18 and 22 g at the time of their use for in vitro experiments and transplants.
Skin and Heterotopic Cardiac Transplantation
Full-thickness tail skin was sutured as 1 cm2 grafts onto the recipients' back. Vascularized cardiac allografts were transplanted intra-abdominally as previously described (40, 41). See also our companion paper (4).
Histopathology
Heart graft rejection was defined as cessation of palpable myocardial contraction, confirmed at the time of explant. Grafts were excised at predetermined time points after transplantation and stored at −80°C or fixed in 10% buffered formalin. Cardiac allograft vasculopathy was assessed on elastin van Gieson-stained paraffin sections by morphometric analysis as previously described (42). All elastin-positive vessels in each section were evaluated, with approximately 10 vessels/heart analyzed. The severity of parenchymal allograft damage was scored on hematoxylin and eosin (H&E) stained paraffin sections by a cardiac histopathologist (EM and MG), blinded to the study groups, using a scale modified from the International Society for Heart and Lung transplantation (43) as follows: 0, no parenchymal damage; 1, < 30% parenchymal damage; 2, 30–60% parenchymal damage; 3, >60% parenchymal damage.
Assay of Anti-H-2Kd Humoral Immunity
See our companion paper (4).
Immunohistology and Confocal Imaging
Seven micrometer spleen and heart cryostat sections were air-dried and fixed in acetone. Primary mAbs specific for the following mouse epitopes were used for immunohistochemical/fluorescent staining: C4d (clone 16-D2 Abcam, Cambridge, UK), NK1.1 (PK136, Abcam) CD68 (ER-HR3, Abcam), mucosal addressin cell adhesion molecule (MAdCAM-1; clone MECA-367, Abcam), CD31 (Novus Biologicals, CO, USA), α-smooth muscle Actin (Thermo Fisher Scientific), and IgG-FITC (BD Biosciences, San Diego, CA, USA). Splenic GCs were identified by double-labeling sections with rat anti-mouse B220-APC (clone RA3-6B2) and rat anti mouse GL7-FITC (both BD Biosciences). Numbers of GL7+ GCs were expressed as a percentage of total B220+ lymphoid follicles (44). CD4 T cells within GCs were located with rat anti-mouse CD4-biotin (BD Biosciences) & Streptavidin-Alexa Fluor 555 (Thermo Fisher Scientific). Confocal images were captured with a Leica SP5 confocal microscope using LAS AF software, version 2.7.2.9586 (Leica Microsystems, Wetzlar, Germany).
Alloantibody Purification From Serum Samples
IgG antibodies were purified from mouse serum samples using the Thermo Scientific Antibody Purification Kit (Thermo Fisher Scientific). Protein G spin columns were loaded with serum samples and binding buffer (0.1 M phosphate, 0.15 M sodium chloride; pH 7.2), centrifuged at 5,000 g and samples were eluted after addition of neutralization buffer followed by IgG elution buffer. A NanoDrop Microvolume Spectrophotometer was used to determine total IgG antibody concentrations using absorbance values at 280 nm. Samples were subsequently used in analysis of endothelial intracellular signaling.
Endothelial Cell Migration Assay
In vitro wound-healing assay was performed as previously described (45). For endothelial cell culture, 10–14 day old neonatal hearts were digested with collagenase and endothelial cells labeled with biotin-conjugated antibodies against CD31 (clone MEC 13.3, BD Pharmingen), CD105 (clone MJ7/18, BioLegend, San Diego, CA, USA), and Isolectin B4 (clone B-1205, Vector, Burlingame, CA), and then separated using anti-biotin MicroBeads (Mitenyi Biotec) with an AutoMACS™ Separator (Mitenyi Biotec). Endothelial cells were cultured until 80–90% confluent and cells were subsequently incubated with medium lacking growth factors for 24 h to minimize baseline proliferation. A linear lesion was made in the cell monolayer across the diameter of the dish using a sterile 200 μl pipette tip. Cells were incubated with test sera (purified IgG) or control antibody for a further 24–36 h, fixed with paraformaldehyde (BD Cytofix kit, BD Biosciences), and then stained with 0.05% Crystal Violet solution. For each plate, six high power fields along the lesion were analyzed and degree of migration induced by test sera was calculated by determining the proportion of outward migration area in relation to a positive control [pooled hyperimmune anti-H-2Kd IgG serum recovered from BL/6 recipients of BALB/c skin grafts (SG)], normalized to 100%.
Analysis of Endothelial Intracellular Signaling by Preparation of Cell Lysates and Western Blot
The ability of column-purified test sera from transplanted mice to induce phosphatidylinositol-3-kinase (PI3K)/Akt signaling of cultured BALB/c endothelial cells was tested as previously described (46) with minor modifications. For endothelial cell culture, 10–14 day old neonatal hearts were digested with collagenase and endothelial cells labeled with biotin-conjugated antibodies against CD31 (clone MEC 13.3, BD Biosciences), CD105 (clone MJ7/18, BioLegend), and Isolectin B4 (clone B-1205, Vector, Burlingame, CA), and then separated using anti-biotin MicroBeads (Mitenyi Biotec) with an AutoMACSTM Separator (Mitenyi Biotec). Endothelial cells were cultured until 80–90% confluent and cells were subsequently incubated with medium lacking growth factors for 24 h to minimize baseline proliferation. Cultured BALB/c endothelial cells that were starved for 24 h in medium containing fetal calf serum without additional growth factors, were subsequently incubated with column-purified serum (see above) of interest (diluted to 1:100) for 30 min at 37°C. The supernatant was removed, cells were then washed and subsequently lysed with CelLytic M reagent (Sigma-Aldrich) containing protease inhibitor cocktail (Roche Life Sciences, Penzberg, Germany). Cell lysate was heated in sodium dodecyl sulfate lysis buffer, electrophoresed on 20% polyacrylamide gel (with approximately 10 μg of cell lysate per lane quantified using the protein quantification kit-rapid [Sigma-Aldrich]), and transferred to a polyvinylidene difluoride membrane. The membrane was blocked in 5% milk in phosphate-buffered saline (PBS) containing 0.1% Tween-20 for 1 h at room temperature, and incubated overnight at 4°C with phosphorylated-Akt (Ser473) mouse mAb (Cell Signaling Technology, Danvers, MA, USA) diluted to 1:1000 in blocking buffer. The blot was washed with PBS containing 2.5% Tween-20 followed by incubation in polyclonal HRP-conjugated rabbit anti-mouse IgG (Abcam) and then developed with chemiluminescence using standard procedure. Anti-GADPH (Abcam) was blotted as a loading control. Data depicted are representative of three independent experiments.
Bio-Layer Interferometry (BLI)
Off-rate screening was performed on RED96 platform instrument ForteBio (Menlo Park, CA, USA). All experiments included biosensor pre-equilibration step in 1 × PBS buffer containing 0.1% BSA, 0.02% Tween-20, and 0.02% NaN3 (pH 7.4); hereafter referred to as kinetics buffer for 10 min, and data were collected at 27°C with agitation at 1,000 rpm. Biotinylated MHC class I Kd antigen (1.25 μg/mL) was captured on streptavidin coated biosensors (ForteBio) at a maximal threshold of 1.5 nm in kinetics buffer. After MHC antigen capture, sensors were blocked for 60 s with naive serum obtained from BL/6 Tcrbd−/− mice. After blocking, sensors were transferred to corresponding wells containing immune serum diluted in kinetics buffer for association over 1,000 s, followed by transfer to dissociation wells containing diluted naïve serum over a period of 1,000 s. Each biological sample was tested at two dilutions: 1:20 and 1:40. Dissociation rate constants for each sample were calculated by applying a 1:1 interaction model (fitting local, partial), and double referenced with both reference biosensor (no biotinylated MHC class I Kd antigen loading) and sample reference (naïve BL/6 serum). Control experiments were performed using non-relevant control anti-HEL IgG antibody (obtained from sera of BL/6 SWHEL recipients challenged with hearts from BL/6.mHEL mice) and showed no binding to sensors loaded with biotinylated Kd antigen. Data analysis was performed using the ForteBio data analysis software 7.0.1.5.
Statistical Analysis
Data is presented as mean ± S.E.M where appropriate, with each animal constituting one biological replicate where indicated. Unpaired t-tests and Mann-Whitney U-tests were used for analysis of parametric data and non-parametric data, respectively. Two-way ANOVA was employed for comparison of anti-H-2Kd IgG alloantibody responses. Graft survival is depicted using Kaplan-Meier analysis and groups compared by log-rank (Mantel-Cox) testing. Analysis was conducted using GraphPad 4 (Graph- Pad Software, San Diego, CA, USA). Values of P < 0.05 were considered significant.
Study Approval
This research has been regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). All surgery was performed under inhalational anesthesia and every effort was made to minimize suffering. Human transmyocardial biopsy samples were obtained through the Papworth Hospital Research Tissue Bank (REC Ref: 08/H0304/56+5), project number T02333: “Visualization of tissue components in cardiac tissue post-transplantation.” All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Results
Development of Model of Chronic Alloantibody Mediated Allograft Rejection
As detailed in the companion paper (4), we developed a murine model of cardiac allograft rejection in which the humoral alloimmune response was likely the sole effector mechanism of graft rejection, because the recipients otherwise lacked CD8 T cells or direct-pathway CD4 T cells with the potential for generating cellular cytotoxicity directed against the graft. Thus, T cell deficient Tcrbd−/− C57BL/6 recipients of a BALB/c heart allograft were reconstituted the day after transplant with TCR75 CD4 T cells (34, 35) that recognize donor MHC class I H-2Kd alloantigen as processed allopeptide via the indirect pathway [Figure 1A; see also companion paper (4)]. Recipient mice reconstituted with as few as 103 TCR75 CD4 T cells generated long-lasting anti-H-2Kd IgG alloantibody, that was slow to develop, but was still evident 15 weeks after transplant (Figure 1B). This alloantibody response was associated with gradual failure of the BALB/c heart allograft [median survival time [MST]−50 days (4)], whereas BALB/c heart allografts survived indefinitely when transplanted into unmodified Tcrbd−/− recipients.
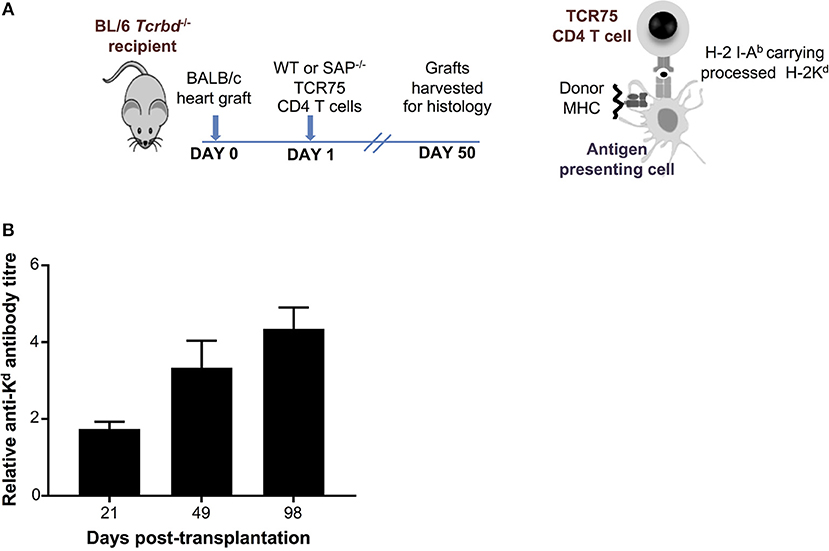
Figure 1. Development and characterization of murine model of antibody-mediated heart allograft rejection. (A) Figure is adapted from our companion paper (4). BL/6 Tcrbd−/− recipients of BALB/c heart allografts were either unmodified (no cells group) or reconstituted the day after with either wild-type or SAP-deficient CD4 T cells from TCR transgenic Rag1−/− TCR75 animals that recognize I-Ab-restricted H-2K peptide. (B) ELISA assays of serum samples obtained from BL/6 Tcrbd−/− recipients of BALB/c hearts and reconstituted with 103 TCR75 CD4 T cells (n = 4) shows progressive increase of anti-H-2Kd IgG alloantibody until day 100.
Explanted heart allografts were assessed for histopathological features of AMR. Allografts, explanted at day 50 from the T cell reconstituted recipients, revealed chronic parenchymal injury, with diffuse myocyte loss and replacement fibrosis (Figure 2A). Areas of damage were associated with concentric proliferative vascular lesions, in keeping with a vasculopathic process causing chronic ischaemic injury (Figure 2B, right). These vascular lesions comprised endothelial and smooth muscle components, and were confined mainly to small intra-myocardial arteries and small/medium veins. Heart allografts transplanted into unmodified Tcrbd−/− recipients remained disease-free (Figures 2A,B, left), confirming that allograft vasculopathy in the reconstituted recipients was mediated by the transferred TCR75 CD4 T cells. The histological features were not suggestive of chronic cellular rejection, in that although inflammatory infiltrates were present within the allografts, these consisted primarily of neutrophils and macrophages, with some plasma cells (Figure 2C); lymphocyte mediated myocyte damage-analogous to the pattern of cellular rejection observed in human heart allografts [“grade 2R” of the ISHLT grading system 43]–was not observed. Instead, a humoral effector component is strongly supported by: strong endothelial staining for C4d complement by-product associated with widespread IgG deposition (Figure 2D); the presence of macrosialinpos (CD68) macrophages within the heart allograft; and by perivascular accumulations of NK cells (Figure 2A).
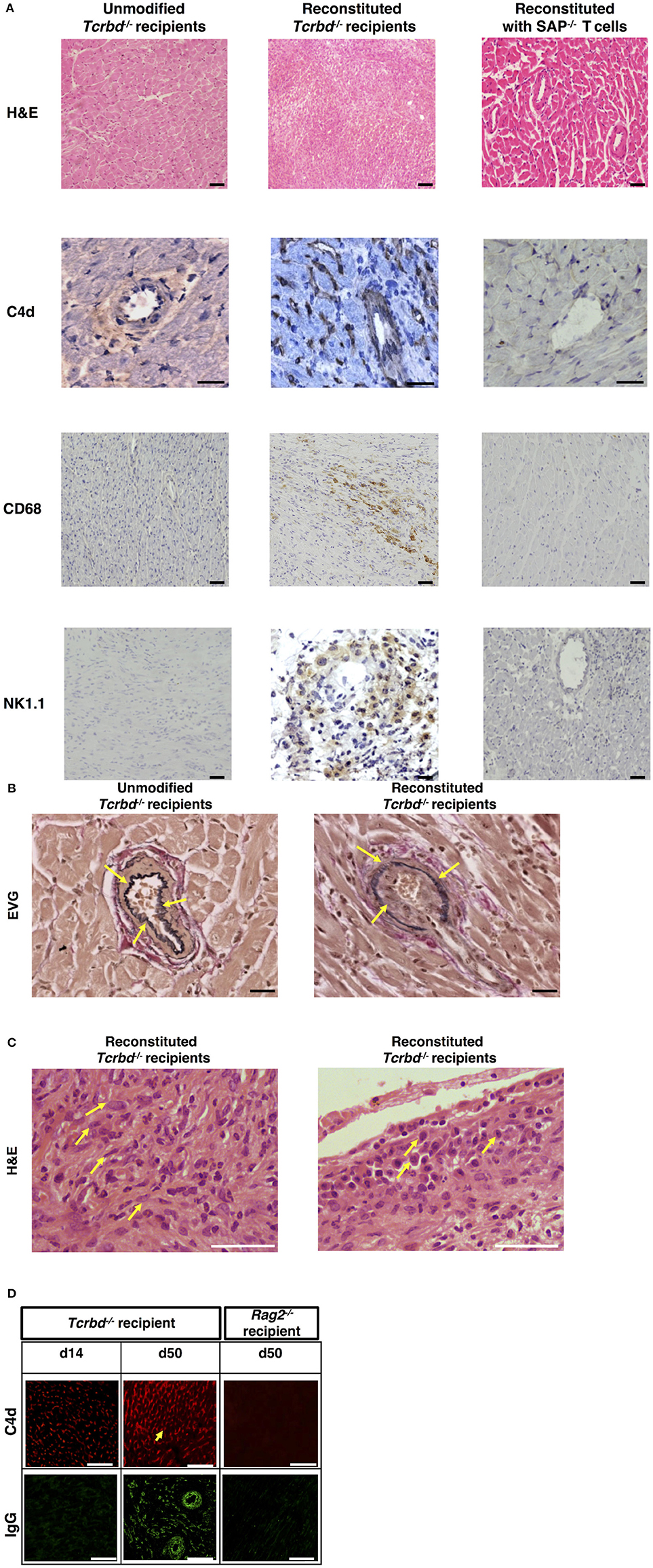
Figure 2. Histopathological confirmation of humoral rejection. (A) Representative photomicrographs of BALB/c hearts explanted at d50 from BL/6 Tcrbd−/− recipients (reconstituted with 103 wild-type [WT] TCR75 CD4 T cells) demonstrate: inflammatory infiltrates with myocyte loss and replacement fibrosis (on H&E staining); endothelial complement C4d deposition and areas of CD68+ (macrophage) and NK1.1+ (NK cell) staining, compared with grossly normal histology and negative staining in equivalent grafts from unmodified (not reconstituted) BL/6 Tcrbd−/− recipients and BL/6 Tcrbd−/− recipients reconstituted with Sh2d1a−/− (SAP−/−) TCR75 CD4 T cells. Representative photomicrographs of (B) elastin van Gieson stained sections of d50 allografts from the wild-type reconstituted group depicting fibroproliferative arterial intimal thickening, compared to control unmodified Tcrbd−/− recipients, and (C) H&E stained sections demonstrating inflammatory infiltrates consisting predominantly of (left) polymorphs and (right) occasional plasma cells (arrows). (D) Representative photomicrographs of immunofluorescence staining showing interstitial capillary/vascular staining (arrowed) for C4d (red) and IgG deposition (green) in BALB/c cardiac allografts explanted at days 14 and 50 from BL/6 Tcrbd−/− recipients reconstituted with 103 WT TCR75 CD4 T cells, and at day 50 from WT reconstituted Rag2−/− recipients. Intensity of C4d and IgG staining increased from day 14 to day 50 in Tcrbd−/− recipients reconstituted with WT TCR75 CD4 T cells. All images are representative of at least 5 animals: scale bars–A: 50 μm (H&E, CD68, and NK1.1 images, except for the NK1.1 image of the reconstituted BL/6 Tcrbd−/− group which is at 75 μm) or 100 μm (C4d images), B: 100 μm, C: 150 μm, and D: top row; scale bar-−50 μm; bottom row; scale bar−100 μm.
Chronic Graft Injury Is Dependent Upon Germinal Center Alloantibody Responses
We sought to confirm whether the long-lasting humoral alloimmunity that underpinned progression of chronic allograft injury was the consequence of a GC response. In this regard, splenic GCs were poorly defined, rudimentary structures at 2 weeks (Figure 3A, left), but distinctly present 7 weeks after transplantation in the T cell-reconstituted recipient group (Figure 3A, right), with approximately half the B cell follicles staining for markers of GC differentiation (Figure 3B). In accordance, bone marrow deposition of H-2Kd-specific plasma cells-an indicator of GC activity–was readily detectable by day 50 after transplantation, and had increased from day 14 (Figure 3C).
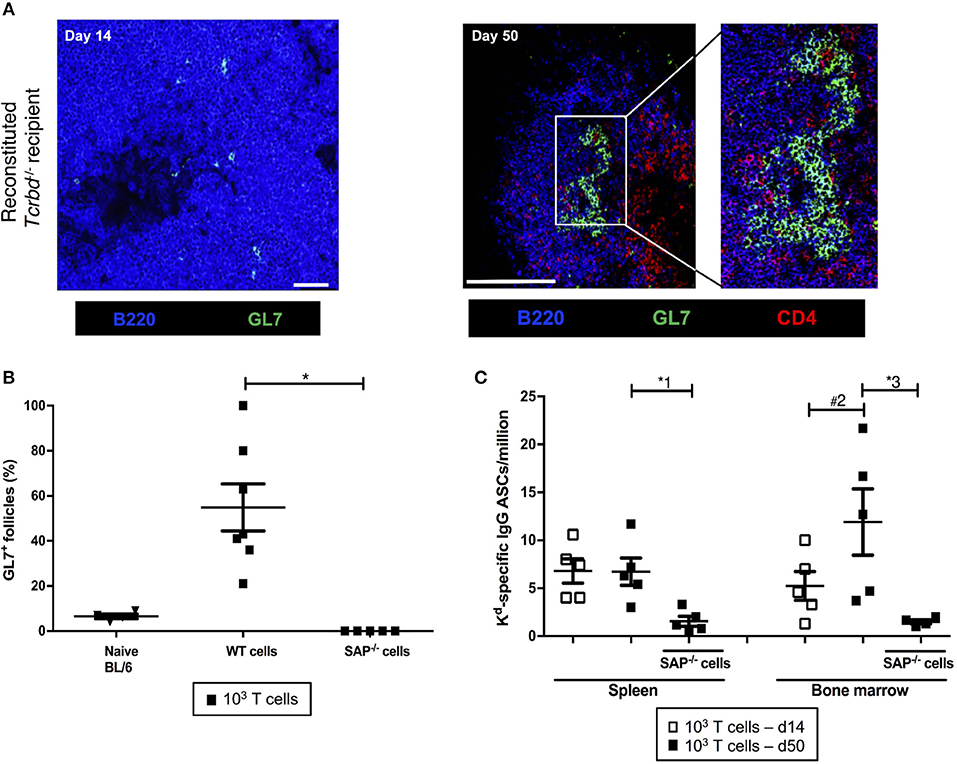
Figure 3. Germinal center responses and TFH cell profile of adoptively transferred CD4 T cells following challenge a BALB/c heart allograft. (A) Representative confocal immunofluorescence photomicrograph of a splenic B cell follicle at d14 (left; scale bar−50 μm) and d50 (right; scale bar−250 μm) from reconstituted BL/6 Tcrbd−/− recipients of BALB/c heart grafts stained with antibodies against B220 (B cells, blue), GL7 (GC B cells, green), and CD4 (T cells, red), demonstrating poorly defined GCs at d14 but by d50 a typical secondary follicle, with TFH cells present within the GC (inset), readily evident. (B) Histogram of secondary follicles expressed as percentage of total follicles within d50 spleens of BL/6 Tcrbd−/− recipients either non-reconstituted (naïve) or adoptively transferred with 1 × 103 WT or Sh2d1a−/− (SAP−/−) TCR75 CD4 T cells. *P = 0.01, Mann-Whitney test. (C) ELISPOT assay of splenic and bone-marrow anti-Kd IgG antibody secreting cells (ASCs) 50 days after transplantation. Numbers of bone marrow ASCs in recipients reconstituted with SAP−/− TCR75 CD4 T cells were not above background. Data represents mean ± S.E.M of a minimum of 5 animals/group, with each dot representing the biological replicate in a distinct animal; *1P = 0.02, #2P = 0.15, *3P = 0.02, Mann–Whitney test.
The GC response was further analyzed by using synthetic MHC class I H-2Kd tetramers to label individual allospecific B cells in recipient mice, as detailed previously (47–49). Flow cytometric characterization of the labeled allospecific B cell population revealed that in recipient mice 7 weeks after transplantation, ~40% of the H-2Kd-specific splenic B cells displayed a GC phenotype (Figures 4A,B). In comparison, in the same group 2 weeks after transplant, numbers of total, and GC H-2Kd-specific B cells were much fewer (Figures 4A,B). This is in keeping with the limited deposition of anti-Kd specific LLPCs in the bone marrow at this stage (Figure 3C).
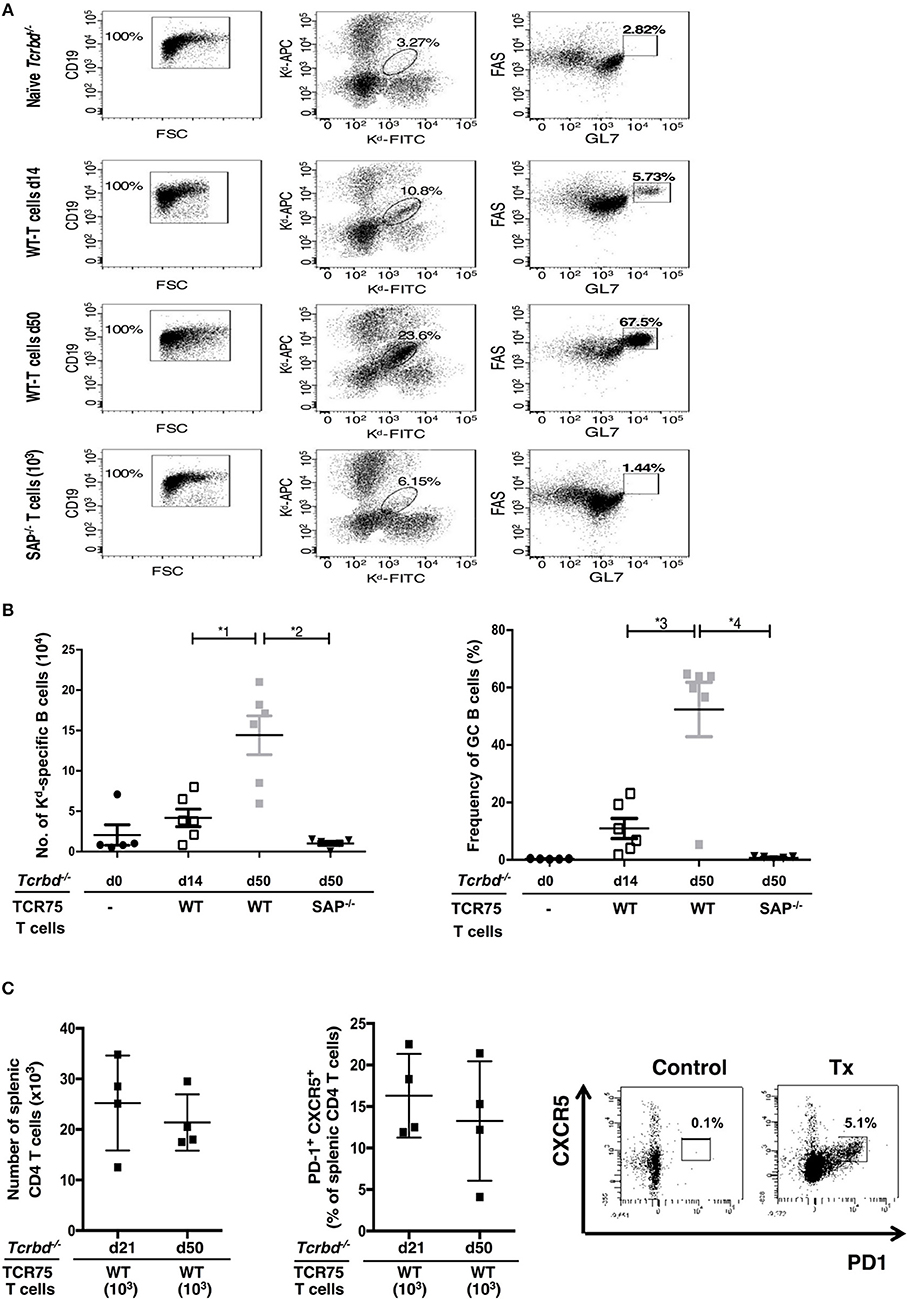
Figure 4. Characterization of allospecific B cells. (A), Splenic H-2Kd-specific B cells were identified by flow cytometric detection for binding of CD19+ve B cells to FITC-conjugated and APC-conjugated synthetic H-2Kd tetramers in: naïve (un-reconstituted) BL/6 Tcrbd−/− mice (n = 5); BL/6 Tcrbd−/− mice, reconstituted with 103 WT TCR75 CD4 T cells, 14 (n = 6) and 50 days (n = 6) after challenge with BALB/c heart allografts; and at 50 days after transplantation with BALB/c hearts and transfer of 103 Sh2d1a−/− TCR75 CD4 T cells (SAP−/−; n = 5). Gated cells in middle column of representative dot plots show percentage of enriched CD19+ve B cells binding H-2Kd tetramer; right column shows percentage of GC-specific tetramer bound CD19+ve B cells. (B) Histogram (left)—absolute numbers of splenic H-2Kd-specific B cells (x 104 cells per mouse) enumerated; histogram (right)—percentage of H-2Kd-specific B cells expressing FAShiGL7+ve GC phenotype. Data represents mean ± S.E.M, with each dot representing the biological replicate in a separate animal; *1P = 0.01, *2P = 0.004, *3P = 0.02 *4P = 0.004 Mann-Whitney test. (C) Total numbers of splenic CD4 T cells (left histogram) and proportion displaying PD1+ CXCR5+ TFH cell phenotype (right histogram) as determined by flow cytometry analysis at indicated time points following transplantation of Tcrbd−/− recipients with a BALB/c heart graft and reconstitution with 103 WT TCR75 CD4 T cells. Representative flow cytometry plot of unchallenged, but adoptively transferred (with 5 × 105 TCR75 CD4 T cells) BL/6 Tcrbd−/− mouse (left) and transplanted WT reconstituted recipient (right).
The TCR-transgenic TCR75 CD4 T cells undergoe only minimal lymphopenia-induced homeostatic proliferation (38, 44), but would be expected to undergo robust antigen-specific expansion upon encounter of target H-2Kd allopeptide in transplanted recipients (50), with presumably, acquisition of TFH cell phenotype. In this respect, flow cytometric analysis of CD4 T cells after transplantation (Figure 4C) confirmed the presence of transferred TCR75 T cells in spleens of recipient BL/6 Tcrbd−/− mice. Reliable assessment of T cell phenotype was not possible at early time points (days 5–7), reflecting the small number of cells viable immediately after transfer, but at later time points, a sub-population that had acquired CXCR5+ PD1+ TFH cell signature-phenotype (51) was evident (Figure 4C), although the precise proportion of TFH cells varied considerably between recipient animals. This TFH cell population presumably corresponded to those CD4 T cells identified within the GCs on immunofluorescent staining (Figure 3A, right panel).
To confirm that GC activity was essential for mediating chronic rejection in this group, BL/6 Tcrbd−/− recipients of BALB/c heart allografts were instead reconstituted with Sh2d1a−/− TCR75 CD4 T cells the day after transplantation. Lacking the Sh2d1a gene, these T cells do not express SLAM-associated protein (SAP) and cannot undergo productive interaction with B cells for formation of TFH cells (52). As expected, recipient BL/6 Tcrbd−/− mice reconstituted with 103 Sh2d1a−/− TCR75 CD4 T cells did not develop splenic GC activity (Figure 3B, Figure 5A) and FAShiGL7+ve H-2Kd-allospecific GC B cells were not detectable (Figures 4A,B). Compared to recipient mice reconstituted with similar numbers of wild-type (WT) TCR75 CD4 T cells, the anti-Kd alloantibody response in mice receiving Sh2d1a−/− TCR75 T cells was markedly abbreviated (Figure 5B), with absence of LLPC deposition in the bone marrow (Figure 3C). Notably, unlike the chronic rejection observed in the WT group, BALB/c heart grafts survived long-term in the SAP−/− recipients (Figure 5C), without evidence of endothelial complement deposition (Figure 2A) and with the development of only minimal allograft vasculopathy, and parenchymal injury (Figure 5D). The absence of GC activity was not due to early death of the transferred Sh2d1a−/− TCR75 T cells, because they were detectable within the spleens of recipient BL/6 Tcrbd−/− mice at late time points after transfer, albeit not as abundantly as following transfer of similar numbers of WT TCR75 CD4 T cells (Figure 5E). This possibly reflects continued proliferation relating to the robust GC response and ongoing graft injury in the recipient group reconstituted with WT TCR75 CD4 T cells (50).
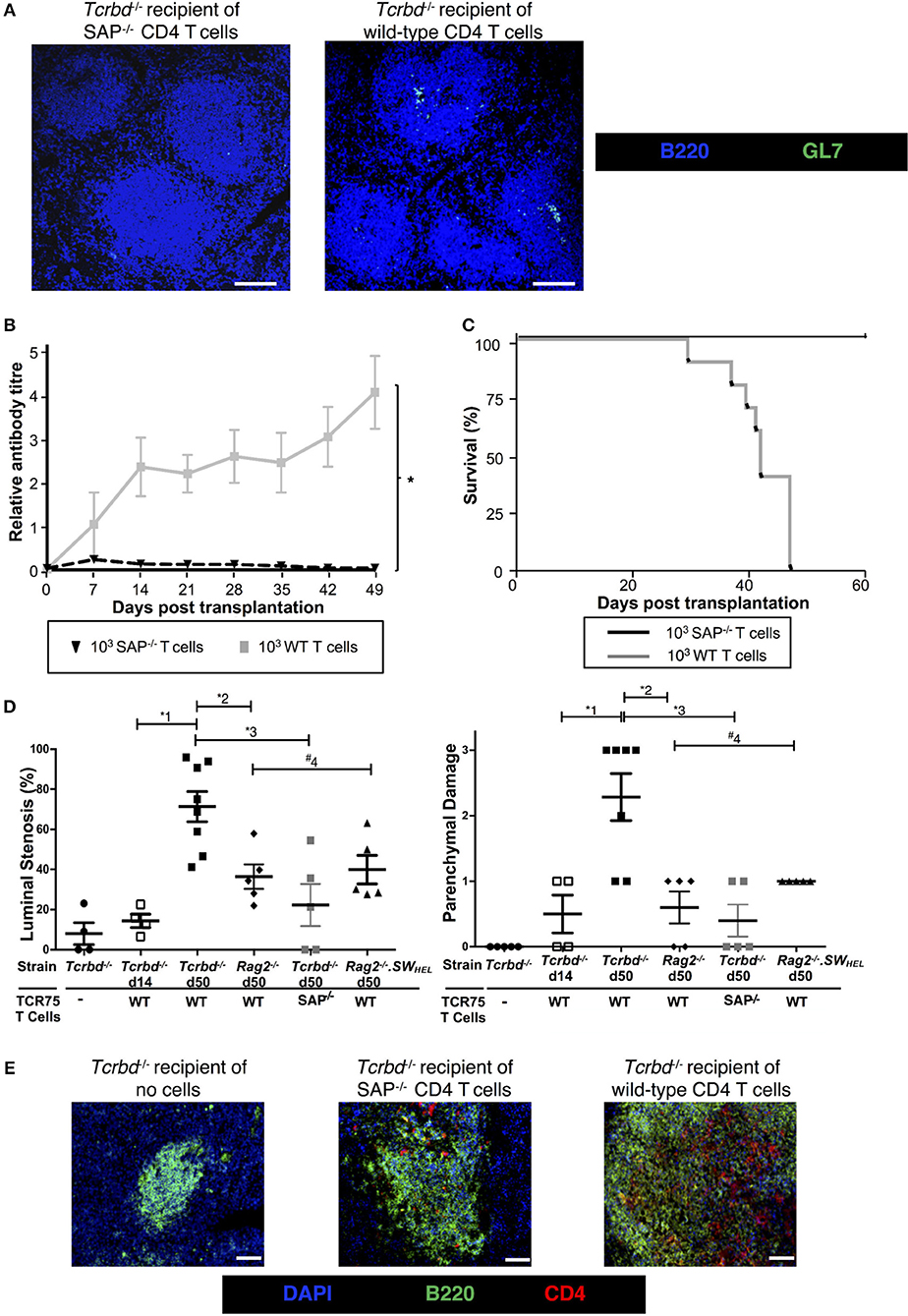
Figure 5. Germinal center alloantibody responses mediate chronic allograft rejection. (A) Representative low magnification confocal immunofluorescence photomicrographs of splenic B cell follicles stained with antibodies against B220 (B cells, blue) and GL7 (GC B cells, green) at d50 from BL/6 Tcrbd−/− recipients of BALB/c heart grafts adoptively transferred with 103 Sh2d1a−/− (left) or wild-type (right) TCR75 CD4 T cells (scale bar – 200 μm). (B) Anti-H-2Kd alloantibody in recipients with 103 SAP−/− TCR75 CD4 T cells (n = 7) was markedly diminished compared to recipients given WT TCR75 CD4 T cells (n = 7); *P < 0.001, two-way ANOVA. (C) Whereas BALB/c heart allografts survived long-term in BL/6 Tcrbd−/− recipients reconstituted with 103 SAP−/− TCR75 CD4 T cells (n = 7), allografts were rejected in recipients given 1 × 103 WT (SAP+/+) TCR75 CD4 T cells (n = 7, MST-50 days; P < 0.001, log-rank test). (D) Severity of vasculopathy (left) and parenchymal damage (right) in BALB/c hearts explanted: from unmodified (non-reconstituted) BL/6 Tcrbd−/− mice (day 50, n = 4); at d14 (n = 4) and d50 (n = 8) from BL/6 Tcrbd−/− recipients reconstituted with WT TCR75 CD4 T cells; at d50 from Rag2−/− recipients reconstituted with WT TCR75 CD4 T cells (n = 5); at d50 from BL/6 Tcrbd−/− recipients reconstituted with 103 SAP−/− TCR75 CD4 T cells (n = 5) and at day 50 from Rag2−/− SWHEL recipients simultaneously transplanted with BALB/c heart and mHEL-Kd skin graft and reconstituted with 103 TCR75 CD4 T cells (n = 5). Each dot represents a single animal; Left: *1P = 0.001, *2P = 0.008, *3P = 0.003, #4P = 0.29, Right: *1P = 0.008, *2P = 0.005, *3P = 0.003, #4P = 0.18; two-tailed Student's t-test for normally distributed and Mann-Whitney tests for non-parametric data. (E) Representative confocal immunofluorescence photomicrographs of splenic cryostat sections stained with antibodies against B220 (B cells, green) and CD4 (red) at day 50 after transplantation of BALB/c heart grafts into BL/6 Tcrbd−/− recipients reconstituted with no cells (left), 103 Sh2d1a−/− (middle) or wild-type (right) TCR75 CD4 T cells (scale bar – 50 μm).
Distinguishing Alloantibody Production From Antigen Presentation in GC-Mediated Allograft Injury
The humoral features in explanted heart allografts, coupled to the absence of overt lymphocytic infiltrates, suggest that the role of GC humoral alloimmunity in mediating chronic rejection relates to production of effector alloantibody. However, a number of additional experiments were performed to confirm the contribution of B cells to rejection. Firstly, T and B cell deficient Rag2−/− C57BL/6 mice were reconstituted with 103 TCR75 CD4 T cells following transplantation with a BALB/c heart allograft. Heart allografts transplanted into the reconstituted Rag2−/− recipients [which, as expected, did not mount an alloantibody response (not shown)] continued to beat until explant, and when compared to the reconstituted Tcrbd−/− recipient group recipients, developed less severe vasculopathy and less extensive parenchymal damage (Figures 5D, 6A). Endothelial C4d deposition was not detectable in explanted heart allografts, nor was macrophage or NK cell infiltration (Figures 6A, 2D), suggesting these features are a direct consequence of alloantibody production. Nevertheless, BALB/c heart allografts in the reconstituted Rag2−/− recipients displayed some features of chronic damage, with sparse intravascular lymphocytic infiltrates in medium and large epicardial arteries and diffuse myocyte loss and replacement fibrosis evident at late time points (Figures 5D, 6A). These features presumably represent low-grade cellular rejection.
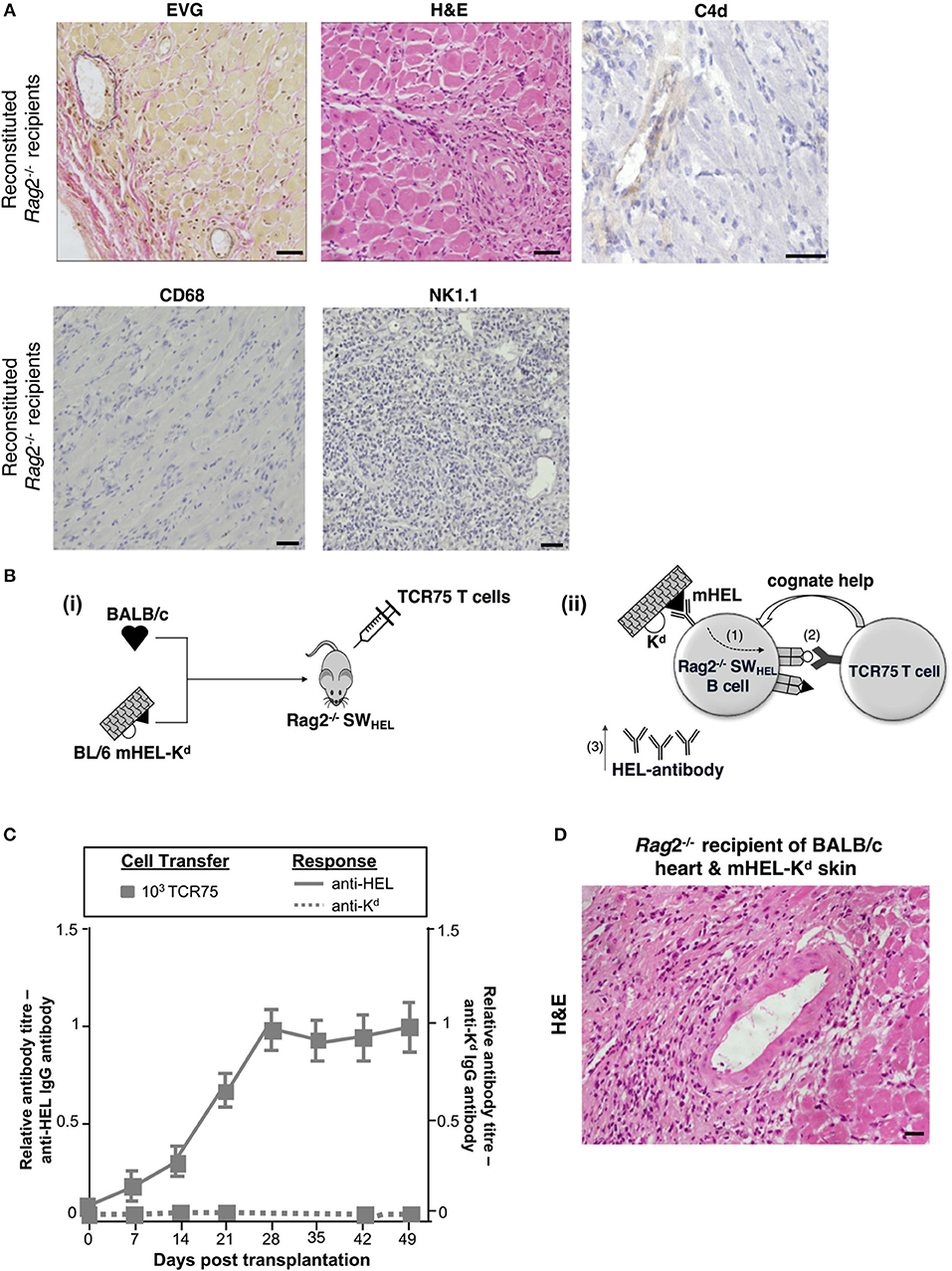
Figure 6. Contribution of alloantibody to allograft rejection. (A) Elastin van Gieson and H&E stained sections of d50 BALB/c heart allografts from reconstituted Rag2−/− recipients with 103 TCR75 CD4 T cells demonstrate focal areas of mild concentric vasculopathy and lymphocytic infiltrates (EVG) within well-preserved vasculature and myocardium (H&E), with absence of capillary C4d staining and NK cell and macrophage infiltration. Scale bars: H&E, CD68, and NK1.1–50 μm; EVG−65 μm; C4d−100 μm. (B) Model to distinguish alloantibody production from alloantigen presentation. (i) BALB/c heart graft and BL/6 mHEL-Kd skin graft which co-express membrane-bound Hen Egg Lysosyme (HEL, black triangle) and MHC class I H-2Kd (white semicircle) were engrafted to a Rag2−/− SWHEL recipient, simultaneously reconstituted with TCR75 CD4 T cells. Critically, on the Rag2−/− background, VDJ recombination is not possible and all B cells express a single HEL-specific BCR. (ii) Following challenge with a BL/6 mHEL-Kd skin graft, SWHEL B cell recognition of mHEL target antigen on the skin graft is expected to result in additional internalization and processing of the H-2Kd alloantigen co-expressed on the surface of donor cells (step 1). CD4 T cell help is limited to the adoptively-transferred TCR75 CD4 T cells that recognize processed H-2Kd allopeptide. Hence, generation of an Ig-switched anti-HEL antibody response is dependent upon presentation of co-internalized H-2Kd antigen by the SWHEL B cell for receipt of cognate help from the TCR75 CD4 T cell (step 2). This results in generation of class-switched anti-HEL alloantibody (step 3). (C) Rag2−/−SWHEL recipients reconstituted with 103 TCR75 CD4 T cells and simultaneously transplanted with a BALB/c heart and mHEL-Kd skin graft developed strong anti-HEL IgG (continuous lines), and undetectable anti-H-2Kd IgG (dotted lines). Data represent mean and S.E.M of n = 6 mice/group. [(D) Representative photomicrographs of H&E stained sections of d50 heart allografts [in (C)] depicting only mild vasculopathy and parenchymal injury (scale bar: 50 μm)].
The requirement for B cells in mediating chronic heart allograft rejection could conceivably reflect a principal role for B cells as antigen presenters for allospecific T cell activation, rather than as producers of alloantibody. To distinguish alloantibody production from alloantigen presentation, we devised a system whereby the B cell antibody output would not recognize determinants expressed on a heart graft, but nevertheless, the B cells would receive cognate help and activation from indirect-pathway CD4 T cells that recognize processed alloantigen from that heart graft (Figure 6B). BL/6 Rag2−/− SWHEL recipients, which lack all T cells, but contain a monoclonal population of B cells specific for HEL protein (36), were reconstituted with limiting numbers (103) of TCR75 CD4 T cells and challenged simultaneously with a BALB/c heart graft and a BL/6 mHEL-Kd skin graft (that co-expresses membrane-bound HEL protein and MHC class I Kd antigen). We have shown previously that allospecific B cells can present allopeptide derived from graft alloantigens that are distinct from their target alloantigen, most likely because the additional alloantigen is internalized bound to target antigen via the B cell receptor (38). Hence, as indicated by the strong anti-HEL IgG antibody responses generated to the mHEL-Kd skin graft (Figure 6C), we reasoned that the SWHEL B cells presented H-2Kd allopeptide both for activation of, and receipt of help from, the adoptively-transferred TCR75 CD4 T cells. This was associated with rapid rejection of the mHEL-Kd skin graft (MST 16 days). As control, without T cell reconstitution, anti-HEL antibody was not generated in the BL/6 Rag2−/− SWHEL recipients and skin and heart grafts survived indefinitely (not shown). Of note, no anti-Kd alloantibody was detected in the reconstituted BL/6 Rag2−/− SWHEL recipients (Figure 6C) and the simultaneously-transplanted BALB/c heart allografts survived until day 100, with development of mild vasculopathy and parenchymal injury, comparable in severity to that observed in similarly-reconstituted Rag2−/− recipients (Figures 5D, 6D). These experiments thus provide further evidence that chronic AMR is mediated by a GC reaction, and principally through production of alloantibody.
Alloantibody as an Effector of Graft Damage
A direct effector role for alloantibody on the allograft was then examined by testing the ability of sera from chronically-rejecting animals to activate cultured allogeneic endothelium, using an in vitro endothelial cell migration assay (45). Whereas test serum pooled from chronically-rejecting recipients sampled 7 weeks after transplant did not alter motility of syngeneic BL/6 endothelial cells, its addition to cultured BALB/c endothelial cells provoked migration similar to that observed following addition of positive control anti-H-2Kd IgG hyperimmune serum (Figure 7A). In contrast, only minimal BALB/c endothelial cell migration was observed upon addition of sera sampled from the same recipients at 2 weeks after transplantation (Figure 7A), even though a moderately strong anti-Kd IgG alloantibody response had developed by this time (Figure 1B). The endothelial cell migration observed possibly relates to signaling via the phosphoinositide 3-kinase (PI3K)/Akt pathway; a pro-survival and proliferative signaling cascade reportedly triggered by alloantibody binding to endothelial MHC class I (46, 53). Western blot analysis of the expression of phosphorylated Akt Ser473 in cultured BALB/c endothelial cells upon addition of test sera revealed similar signaling patterns; with strong Akt signaling observed upon addition of week 7 sera from chronically-rejecting recipients, but little or no signaling observed upon addition of either week 2 sera from the same recipients, or week 7 sera from the Sh2d1a−/− reconstituted recipients (Figure 7B).
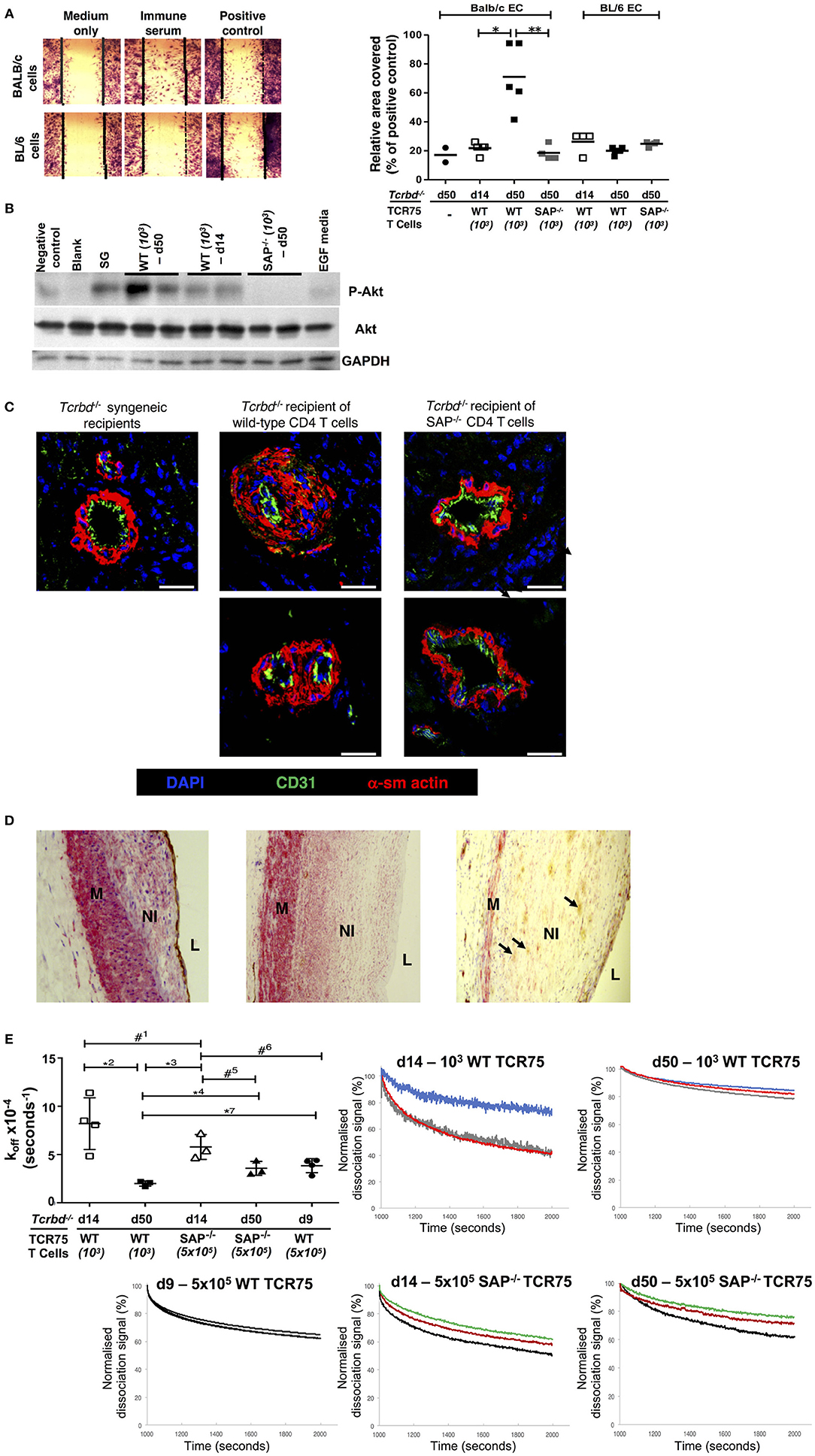
Figure 7. Alloantibody mediates endothelial activation and proliferative vasculopathy. (A) Representative photomicrograph of scratch-wound assay (left) and histogram (right) demonstrating enhanced migration of BALB/c endothelial cells (ECs) upon incubation with day 50 serum sampled from BL/6 Tcrbd−/− reconstituted with wild-type (WT) or SAP−/− TCR75 CD4 T cells, whereas BL/6 EC migration was not above background (mean values, with each dot representing an average of six high power fields analyzed per biological replicate). Migration of BALB/c ECs in response to sera from d14 WT or d50 SAP−/− was significantly weaker compared to serum from d50 WT recipients; *P = 0.01 and **P = 0.02, Mann–Whitney tests. (B) Quiescent BALB/c endothelial cells stimulated with media containing epidermal growth factor (EGF) or column-purified sera from: d50 non-reconstituted BL/6 Tcrbd−/− recipients of BALB/c grafts (negative control), d14 WT, d50 WT, d50 SAP−/− recipients, and pooled hyperimmune anti-H-2Kd IgG serum (BALB/c skin grafts [SG] to BL/6 mice, positive control). Cell lysates were separated and immunoblotted with anti-Akt Ser473, anti-phospho-Akt Ser473, and GAPDH (control) mAb. Displayed Western blot represents three independent experiments. (C) Representative photomicrographs of coronary arteries from allografts explanted at day 50 and double immunolabeled with endothelial marker CD31 (green) and α-smooth muscle actin (α-sm actin, red); scale bar−200 μm. Increased neointimal lesions in chronically rejecting BALB/c allografts containing a large number of α-actin–positive smooth muscle cells were observed in BL/6 Tcrbd−/− recipients reconstituted with 103 TCR75 T cells (middle column) but not with SAP−/− TCR75 CD4 T cells (right); Tcrbd−/− syngeneic grafts shown as comparison (left). (D) Representative photomicrographs of endomyocardial biopsies of human heart allografts in recipients with antibody-mediated rejection and double-immunostained with the endothelial marker CD31 (brown) and α-smooth muscle actin (red); magnification x25. Pronounced neointimal (NI) lesions, composed of dense smooth muscle cells (pink), lying between the media (M) and arterial lumen (L). Remnants of endothelial cell layer seen within neointima (arrows, right image). Images representative of staining patterns observed in three individual patients. (E) Ranking anti-H-2Kd alloantibody dissociation kinetics. Left: dot plot histogram comparing anti-H-2Kd alloantibody dissociation constants (koff) among BL/6 Tcrbd−/− recipients of BALB/c hearts reconstituted with either WT or SAP−/− TCR75 CD4 T cells (at d9, 14, and 50 as depicted). Each value (mean ± SEM) represents the koff after global fit of two serum dilutions of the same biological replicate (n = 3); #1P = 0.21, *2P = 0.01, *3P = 0.008, *4P = 0.02, #5P = 0.06, #6P = 0.05, #7P = 0.01; two-tailed Student's t-test. Right: graphs depict sensograms of anti-H-2Kd antibody dissociation rates in sera from BL/6 Tcrbd−/− recipients of BALB/c hearts reconstituted as shown, with each curve representing individual recipient mice.
Although the mechanisms by which alloantibody provoke development of arterial disease in clinical solid-organ transplantation remain unclear (54), the allograft vasculopathy described is generally considered a disorder of neointimal proliferation. The in vitro endothelial responses observed in our model may therefore mimic the dysregulated endothelial activation and proliferative responses that are triggered by alloantibody binding in human transplant recipients (45, 46, 55–57). In this regard, staining for α-actin–positive smooth muscle cells revealed marked expansion of the smooth muscle cell component of the cardiac allograft microvasculature within the arterial neointima in hearts from chronically-rejecting recipients reconstituted with WT TCR75 CD4 T cells, but not in hearts transplanted into recipients adoptively transferred with Sh2d1a−/− TCR75 CD4 T cells. (Figure 7C). Thus, GC humoral alloimmunity is associated with intra-allograft smooth muscle cell accumulation and proliferation resulting in intimal expansion. Of note, a similar staining pattern was present in heart allografts from human patients with a diagnosis of AMR (Figure 7D).
The Germinal Center as a Source of Somatically-Mutated, High-Affinity Alloantibody
The above experiments detailing markedly augmented endothelial cell responses upon addition of sera sampled at late time points after rejection, even though seemingly robust titres of alloantibody were present at earlier time-points, suggests that the “quality” of the alloantibody response had possibly been improved by somatic hypermutation and affinity maturation. To examine this further, bio-layer interferometry (BLI) was performed, to rank H-2Kd-specific antibody in the test sera according to binding strength (dissociation rate ranking). Because the precise concentration of H-2Kd-specific antibody in the sera was not known, a koff ranking method was employed (the kinetic rate of dissociation is concentration independent), with serum as the analyte and synthetic H-2Kd antigen immobilized on the sensor (58). As can be seen from Figure 7E (and see also Figure S1), test sera obtained from the week 7 chronically-rejecting group bound more strongly to target H-2Kd alloantigen (significantly lower dissociation constant koff) than sera obtained from the same animals at week 2, with less variability between individual recipient mice at the late time point. This decrease in the kinetic rate of dissociation observed over time is in keeping with GC-mediated affinity maturation of (allo)antibody responses (59). The minimal levels of alloantibody present at late time points in the recipients reconstituted with 103 Sh2d1a−/− TCR75 T cells prevented a similar BLI analysis, but day 14 sera from recipients reconstituted with 5 × 105 Sh2d1a−/− TCR75 T cells [that generated very robust, early extrafollicular responses (4)] bound with similar affinity to target H-2Kd antigen as sera sampled at the same time point from recipients reconstituted with 103 WT TCR75 CD4 T cells (Figure 7E).
Discussion
Although the durable nature of the alloantibody responses frequently observed in clinical transplantation is suggestive of an underlying GC response, this has not been demonstrated, largely because of difficulties in sampling the appropriate recipient tissue. The use of a murine model allowed us to demonstrate a critical role for the allospecific GC in the progression of allograft vasculopathy. Given the increasing emphasis on the contribution of alloantibody to chronic graft rejection, our results suggest that strategies that specifically target the GC humoral alloimmune response may improve long-term outcomes.
The critical role of the GC in progression of allograft vasculopathy in our model most likely reflects its role in producing long-lasting alloantibody responses (60), and it is notable that the LLPC subset (that can potentially produce antibody for the life of the individual) was evident in the bone marrow of the SAP-replete group, but not in the SAP-deficient group. Similarly, in a related piece of work using a different model of chronic rejection, we show the contribution of GC-mediated humoral autoimmunity to allograft vasculopathy independently of conventional alloimmunity, through a mechanism by which donor T cells initiate auto-reactive GC reactions that are then maintained by host TFH cells (61, 62). However, it is possible that at least part of the GC contribution is ultimately T cell mediated, reflecting augmented allopeptide presentation and indirect-pathway CD4 T cell activation secondary to long-lived alloantibody production. Our experiments were not designed to exclude this possibility, and our findings using Rag2−/− recipients highlight that cellular mechanisms can autonomously effect a degree of allograft vasculopathy and parenchymal damage. This would accord with Zeng et al. (63), who recently reported the contribution of B cells, independent of alloantibody production, in promoting allograft rejection by acting as antigen presenting cells for augmenting T cell activation. Notwithstanding, a number of our experiments support a direct effector role for alloantibody in the development of late parenchymal injury and allograft vasculopathy: allograft histology revealing clear evidence of humoral, but a lack of overt cellular, rejection; the association of alloantibody production and endothelial complement deposition; Akt signaling induced in allogeneic endothelial cells by exposure to sera from transplanted mice; and restoration of rejection by passive transfer of alloantibody into T and B cell deficient Rag2−/− recipients [see also (4)]. Furthermore, it is notable that in our experiments involving recipients harboring a monoclonal population of B cells, in which a long-lasting antibody response was generated against irrelevant antigen, but in which B cell presentation to alloreactive CD4 T cells was required for its development, graft rejection was not observed, and the severity of allograft damage mirrored that observed in Rag2−/− recipients. Our findings therefore accord with Hancock et al. (64) who described the development of murine cardiac allograft vasculopathy upon passive transfer of commercial anti-MHC class I alloantibody to T and B cell deficient recipients. We should, however, stress that the mechanisms by which the binding of alloantibody to allograft endothelium results in the neointimal proliferation and medial smooth muscle accumulations that typify allograft vasculopathy remain unclear, and will be the subject of ongoing investigation.
Labeling with synthetic MHC class I tetramer confirmed that transfer of SAP-replete helper CD4 T cells was associated with marked expansion in numbers of alloantigen-specific B cells, with approximately half of these cells expressing a GC phenotype. It is perhaps surprising that robust splenic GC activity was found at late time points after transplantation, in that GC responses against model protein antigen typically involute after about 3 weeks. Their continued presence implies ongoing delivery of help from TFH cells; in support, confocal imaging confirmed CD4 T cells within secondary follicles at these late time points. The TFH population differs from typical Th1 / Th2 subsets in that TFH cells are relatively long-lived and their division limited, but their presence within the GC at 2 months from transplant presumably reflects ongoing differentiation from naïve precursors in response to the continued presence of target alloantigen (50). To what extent these late GC responses contribute to graft rejection is not clear; our experiments were principally designed to distinguish the GC from the extrafollicular response. As discussed above, the deposition of LLPCs in the bone marrow, one of the cardinal characteristics of the GC response, is likely a major factor in the progression of graft injury in our model. Such LLPC deposition would be expected to occur early in the GC response and it is conceivable that even a short-lived GC reaction would lead to eventual graft failure from the durable alloantibody production that ensues. Against this, as demonstrated in our model, as the GC response progresses, SHM produces alloantibody of increasing affinity for target alloantigen. This affinity maturation may be required for binding to target alloantigen on allograft endothelium with sufficiently strong affinity to trigger the proliferative response that is thought to underpin progression of allograft vasculopathy. In support, test sera sampled at late time points after transplantation in the help-limited group provoked pronounced Akt signaling. Hence, our findings suggest that late GC responses are necessary for progression of allograft vasculopathy, most likely reflecting the continual deposition of plasma cells that secrete alloantibody with increasing affinity for target alloantigen. Plasma cells that are produced from the GC at late time points would be expected to out-compete those already established in the bone marrow niche from earlier in the response (22), but it is also possible that they contribute to progressive allograft injury as short-lived cells.
Although the models developed for this study could be criticized for being artificially refined, we felt a reductionist approach, in which additional and potentially confounding effector mechanisms were excluded, was necessary to enable a definitive assessment of the role of the GC in chronic rejection. As such, this has allowed us to conclusively demonstrate the pivotal role of the TFH cell subset in mediating progression of allograft vasculopathy by providing help for GC humoral immunity as well as confirming that the histological manifestations in the heart allografts that resemble those seen in clinical transplantation were a direct consequence of the alloantibody response. Our findings may therefore help to cement definitions of chronic AMR for clinical transplantation. The use of a monoclonal CD4 T cell population may, nevertheless, nullify any potential impact of either conventional regulatory, or follicular regulatory, T cells that would otherwise be present within wild-type, polyclonal populations. Against that, we have previously shown that only a small proportion of transferred TCR75 CD4 T cells adopt a TFH phenotype (44) and our ongoing work has confirmed that other helper T cell subsets, including FoxP3-expressing regulatory CD4 T cells, differentiate within the transferred TCR75 T cell population. Likewise, Aloulou et al have recently described the development of T follicular regulatory cells within a monoclonal population of TCR-transgenic CD4 T cells (65). Our findings will therefore likely apply to wild-type recipients that harbor a polyclonal T cell repertoire. In support, we have recently reported that chronic rejection of “bm12.Kd.IE” heart grafts is associated with late splenic GC responses in wild-type C57BL/6 recipients (50, 66). Similarly, our ongoing investigations have confirmed that GC activity is present at late time points in C57BL/6 recipients of chronically-rejecting MHC class II-mismatched “bm12” heart allografts and in C57BL/6 recipients of BALB/c heart grafts in which acute rejection is prevented by blocking co-stimulation signaling at the time of transplant. Possibly, the chronic alloresponse favors polarization to a TFH cell phenotype, as has been reported for exhausted CD4 T cells responding to chronic viral infection (67). This perhaps explains the late development of alloantibody that is observed in human transplant patients.
We believe there are several aspects to our findings that have implications for clinical transplantation. Most obviously, our description of the role of GC humoral alloimmunity in the progression of allograft vasculopathy appears to mirror the development of durable donor specific alloantibody responses late (months or years) after solid-organ transplantation, and which are increasingly associated with poor graft outcomes (11, 68). Our results suggest these responses are likely to be outputs of the GC response, and further highlight the potential for developing strategies that specifically target the TFH subset as a means of preventing late alloantibody-mediated allograft rejection. Because the TFH cell subset represents only a small proportion of the total CD4 T cell population, this approach offers the potential advantage of preserving general immunocompetence, and could possibly be achieved, for example, by transfer of either allopeptide-specific (69). or donor derived regulatory CD4 T cells (70).
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
All authors contributed extensively to the work presented in this paper. MC and JA jointly conceived the study with RM and GP, designed and implemented the cardiac allograft rejection model with contributions from MQ, MM, JMA, IG, and ML contributed to characterization of germinal center responses, and VK analyzed the BLI data. EM and MG performed cardiac allograft histopathological characterization. GP and RM prepared the manuscript, with contributions from MC, JA, VK, and ML.
Funding
This work was supported by a British Heart Foundation project grant, the National Institute for Health Research Cambridge Biomedical Research Centre and the National Institute for Health Research Blood and Transplant Research Unit. MC was supported by the Agency for Science Technology and Research (A*STAR), Singapore. MQ was supported by a British Heart Foundation Clinical Research Training Fellowship (reference FS/12/87/29899). JA was supported by a grant from King Saud University, Kingdom of Saudi Arabia. VK was supported by an Evelyn Trust grant (reference 14/25) and an Academy of Medical Sciences starter grant (reference RG77106). JMA was supported by a Wellcome Trust Clinical Research Training Fellowship (reference 096207/Z/11/Z) and Raymond and Beverly Sackler Scholarship. RM was supported by a European Society of Organ Transplantation Junior Basic Science Grant. ML is supported by the Bioscience and Biotechnology Research Council.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.03038/full#supplementary-material
References
1. Baldwin WM III, Valujskikh A, Fairchild RL. Mechanisms of antibody-mediated acute and chronic rejection of kidney allografts. Curr Opin Organ Transplant. (2016) 21:7–14. doi: 10.1097/MOT.0000000000000262
2. Garces JC, Giusti S, Staffeld-Coit C, Bohorquez H, Cohen AJ, Loss GE. Antibody-mediated rejection: a review. Ochsner J. (2017) 17:46–55.
3. Montgomery RA, Loupy A, Segev DL. Antibody-mediated rejection: new approaches in prevention and management. Am J Transplant. (2018) 18 (Suppl. 3):3–17. doi: 10.1111/ajt.14584
4. Alsughayyir J, Chhabra M, Qureshi MS, Mallik M, Ali JM, Gamper I, et al. Relative frequencies of alloantigen-specific helper CD4 T cells and B cells determine mode of antibody-mediated allograft rejection. Front Immunol. (2019) 9:3039. doi: 10.3389/fimmu.2018.03039
5. Montgomery RA, Cozzi E, West LJ, Warren DS. Humoral immunity and antibody-mediated rejection in solid organ transplantation. Semin Immunol. (2011) 23:224–34. doi: 10.1016/j.smim.2011.08.021
6. Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest. (2017) 127:2492–504. doi: 10.1172/JCI90597
7. Kaneku H, O'leary JG, Banuelos N, Jennings LW, Susskind BM, Klintmalm GB, et al. De novo donor-specific HLA antibodies decrease patient and graft survival in liver transplant recipients. Am J Transplant. (2013) 13:1541–8. doi: 10.1111/ajt.12212
8. Demetris AJ, Bellamy C, Hubscher SG, O'leary J, Randhawa PS, Feng S, et al. 2016 Comprehensive update of the banff working group on liver allograft pathology: introduction of antibody-mediated rejection. Am J Transplant. (2016) 16:2816–35. doi: 10.1111/ajt.13909
9. Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, et al. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. (2009) 9:2520–31. doi: 10.1111/j.1600-6143.2009.02799.x
10. Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R, Cosio FC, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation (2010) 90:68–74. doi: 10.1097/TP.0b013e3181e065de
11. Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong Van Huyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. (2013) 369:1215–26. doi: 10.1056/NEJMoa1302506
12. Aubert O, Loupy A, Hidalgo L, Duong Van Huyen JP, Higgins S, Viglietti D, et al. Antibody-mediated rejection due to preexisting versus de novo donor-specific antibodies in kidney allograft recipients. J Am Soc Nephrol. (2017) 28:1912–23. doi: 10.1681/ASN.2016070797
13. Snyder LD, Wang Z, Chen DF, Reinsmoen NL, Finlen-Copeland CA, Davis WA, et al. Implications for human leukocyte antigen antibodies after lung transplantation: a 10-year experience in 441 patients. Chest (2013) 144:226–33. doi: 10.1378/chest.12-0587
14. Levine DJ, Glanville AR, Aboyoun C, Belperio J, Benden C, Berry GJ, et al. Antibody-mediated rejection of the lung: a consensus report of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. (2016) 35:397–406. doi: 10.1016/j.healun.2016.01.1223
15. Smith JD, Banner NR, Hamour IM, Ozawa M, Goh A, Robinson D, et al. de novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant. (2011) 11:312–9. doi: 10.1111/j.1600-6143.2010.03383.x
16. Loupy A, Toquet C, Rouvier P, Beuscart T, Bories MC, Varnous S, et al. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. Am J Transplant. (2016) 16:111–20. doi: 10.1111/ajt.13529
17. Regele H, Böhmig GA, Habicht A, Gollowitzer D, Schillinger M, Rockenschaub S, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. (2002) 13:2371–80. doi: 10.1097/01.ASN.0000025780.03790.0F
18. Loupy A, Haas M, Solez K, Racusen L, Glotz D, Seron D, et al. The Banff 2015 kidney meeting report: current challenges in rejection classification and prospects for adopting molecular pathology. Am J Transplant (2017) 17:28–41. doi: 10.1111/ajt.14107
19. Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al. The Banff 2017 kidney meeting report: revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. (2018) 18:293–307. doi: 10.1111/ajt.14625
20. Banasik M, Boratynska M, Koscielska-Kasprzak K, Mazanowska O, Krajewska M, Zabinska M, et al. The impact of de novo donor-specific anti-human leukocyte antigen antibodies on 5-year renal transplant outcome. Transplant Proc. (2013) 45:1449–52. doi: 10.1016/j.transproceed.2012.12.026
21. Bailly E, Anglicheau D, Blancho G, Gatault P, Vuiblet V, Chatelet V, et al. Prognostic value of the persistence of C1q-Binding Anti-HLA antibodies in acute antibody-mediated rejection in kidney transplantation. Transplantation (2018) 102:688–98. doi: 10.1097/TP.0000000000002002
22. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. (2006) 6:741–50. doi: 10.1038/nri1886
23. Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. (1997) 16:2996–3006. doi: 10.1093/emboj/16.11.2996
24. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315:528–31. doi: 10.1126/science.1136736
25. Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol. (2017) 45:21–30. doi: 10.1016/j.coi.2016.12.004
26. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science (2002) 297:2066–70. doi: 10.1126/science.1073924
27. Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, et al. Salmonella infection drives promiscuous b cell activation followed by extrafollicular affinity maturation. Immunity (2015) 43:120–31. doi: 10.1016/j.immuni.2015.06.013
28. Nozaki T, Amano H, Bickerstaff A, Orosz CG, Novick AC, Tanabe K, et al. Antibody-mediated rejection of cardiac allografts in CCR5-deficient recipients. J Immunol. (2007) 179:5238–45. doi: 10.4049/jimmunol.179.8.5238
29. Kohei N, Tanaka T, Tanabe K, Masumori N, Dvorina N, Valujskikh A, et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int. (2016) 89:1293–306. doi: 10.1016/j.kint.2016.02.030
30. Crotty S. follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
31. Deenick EK, Ma CS, Brink R, Tangye SG. Regulation of T follicular helper cell formation and function by antigen presenting cells. Curr Opin Immunol. (2011) 23:111–8. doi: 10.1016/j.coi.2010.10.007
32. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. (2015) 15:185–9. doi: 10.1038/nri3803
33. Vinuesa CG, Linterman MA, Yu D, Maclennan IC. Follicular helper T cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
34. Honjo K, Xu X, Bucy RP. CD4+ T-cell receptor transgenic T cells alone can reject vascularized heart transplants through the indirect pathway of alloantigen recognition. Transplantation (2004) 77:452–5. doi: 10.1097/01.TP.0000112937.12491.42
35. Honjo K, Yan Xu X, Kapp JA, Bucy RP. Evidence for cooperativity in the rejection of cardiac grafts mediated by CD4 TCR Tg T cells specific for a defined allopeptide. Am J Transplant. (2004) 4:1762–8. doi: 10.1046/j.1600-6143.2004.00596.x
36. Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, et al. B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J Exp Med. (2003) 197:845–60. doi: 10.1084/jem.20022144
37. Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature (1991) 353:765–9. doi: 10.1038/353765a0
38. Conlon TM, Cole JL, Motallebzadeh R, Harper I, Callaghan CJ, Bolton EM, et al. Unlinked memory helper responses promote long-lasting humoral alloimmunity. J Immunol. (2012) 189:5703–12. doi: 10.4049/jimmunol.1202257
39. Czar MJ, Kersh EN, Mijares LA, Lanier G, Lewis J, Yap G, et al. Altered lymphocyte responses and cytokine production in mice deficient in the X-linked lymphoproliferative disease gene SH2D1A/DSHP/SAP. Proc Natl Acad Sci USA. (2001) 98:7449–54. doi: 10.1073/pnas.131193098
40. Corry RJ, Winn HJ, Russell PS. Heart transplantation in congenic strains of mice. Transplant Proc. (1973) 5:733–5.
41. Harper SJ, Ali JM, Wlodek E, Negus MC, Harper IG, Chhabra M, et al. CD8 T-cell recognition of acquired alloantigen promotes acute allograft rejection. Proc Natl Acad Sci USA. (2015) 112:12788–93. doi: 10.1073/pnas.1513533112
42. Callaghan CJ, Win TS, Motallebzadeh R, Conlon TM, Chhabra M, Harper I, et al. Regulation of allograft survival by inhibitory FcgammaRIIb signaling. J Immunol. (2012) 189:5694–702. doi: 10.4049/jimmunol.1202084
43. Stewart S, Winters GL, Fishbein MC, Tazelaar HD, Kobashigawa J, Abrams J, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant. (2005) 24:1710–20. doi: 10.1016/j.healun.2005.03.019
44. Conlon TM, Saeb-Parsy K, Cole JL, Motallebzadeh R, Qureshi MS, Rehakova S, et al. Germinal center alloantibody responses are mediated exclusively by indirect-pathway CD4 T follicular helper cells. J Immunol. (2012) 188:2643–52. doi: 10.4049/jimmunol.1102830
45. Jin YP, Valenzuela NM, Ziegler ME, Rozengurt E, Reed EF. Everolimus inhibits anti-HLA I antibody-mediated endothelial cell signaling, migration and proliferation more potently than sirolimus. Am J Transplant. (2014) 14:806–19. doi: 10.1111/ajt.12669
46. Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, et al. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol. (2004) 65:291–302. doi: 10.1016/j.humimm.2004.01.002
47. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science (2011) 331:1203–7. doi: 10.1126/science.1201730
48. Chen J, Yin H, Xu J, Wang Q, Edelblum KL, Sciammas R, et al. Reversing endogenous alloreactive B cell GC responses with anti-CD154 or CTLA-4Ig. Am J Transplant. (2013) 13:2280–92. doi: 10.1111/ajt.12350
49. Chen J, Wang Q, Yin D, Vu V, Sciammas R, Chong AS. Cutting Edge: CTLA-4Ig inhibits memory B Cell responses and promotes allograft survival in sensitized recipients. J Immunol. (2015) 195:4069–73. doi: 10.4049/jimmunol.1500940
50. Ali JM, Negus MC, Conlon TM, Harper IG, Qureshi MS, Motallebzadeh R, et al. Diversity of the CD4 T Cell alloresponse: The short and the long of it. Cell Rep. (2016) 14:1232–45. doi: 10.1016/j.celrep.2015.12.099
51. Johnston RJ, Poholek AC, Ditoro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325:1006–10. doi: 10.1126/science.1175870
52. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity (2010) 32:253–65. doi: 10.1016/j.immuni.2010.01.010
53. Coupel S, Leboeuf F, Boulday G, Soulillou JP, Charreau B. RhoA activation mediates phosphatidylinositol 3-kinase-dependent proliferation of human vascular endothelial cells: an alloimmune mechanism of chronic allograft nephropathy. J Am Soc Nephrol. (2004) 15:2429–39. doi: 10.1097/01.ASN.0000138237.42675.45
54. Mitchell RN. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. (2009) 4:19–47. doi: 10.1146/annurev.pathol.3.121806.151449
55. Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with integrin β4 to stimulate endothelial cell proliferation and migration. Sci Signal. (2010) 3:ra85. doi: 10.1126/scisignal.2001158
56. Tible M, Loupy A, Vernerey D, Suberbielle C, Beuscart T, Cazes A, et al. Pathologic classification of antibody-mediated rejection correlateswith donor-specific antibodies and endothelial cell activation. J Heart Lung Transplant. (2013) 32:769–76. doi: 10.1016/j.healun.2013.05.012
57. Hickey MJ, Valenzuela NM, Reed EF. Alloantibody generation and effector function following sensitization to human leukocyte antigen. Front Immunol. (2016) 7:30. doi: 10.3389/fimmu.2016.00030
58. Ylera F, Harth S, Waldherr D, Frisch C, Knappik A. Off-rate screening for selection of high-affinity anti-drug antibodies. Anal Biochem. (2013) 441:208–13. doi: 10.1016/j.ab.2013.07.025
59. England P, Nageotte R, Renard M, Page AL, Bedouelle H. Functional characterization of the somatic hypermutation process leading to antibody D1.3, a high affinity antibody directed against lysozyme. J Immunol. (1999) 162:2129–36.
60. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
61. Qureshi MS, Alsughayyir J, Chhabra MC, Ali JM, Goddard M, Devine C., et al. Data regarding transplant induced germinal center humoral autoimmunity. J Autoimmun. (2018). doi: 10.1016/j.dib.2018.12.078
62. Qureshi MS, Alsughayyir J, Chhabra MC, Ali JM, Goddard M, Devine C, et al. Germinal center humoral autoimmunity independently mediates progression of allograft vasculopathy. J Autoimmun. (2018). doi: 10.1016/j.jaut.2018.11.006
63. Zeng Q, Ng YH, Singh T, Jiang K, Sheriff KA, Ippolito R, et al. B cells mediate chronic allograft rejection independently of antibody production. J Clin Invest. (2014) 124:1052–6. doi: 10.1172/JCI70084
64. Hancock WW, Buelow R, Sayegh MH, Turka LA. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nat Med. (1998) 4:1392–6. doi: 10.1038/3982
65. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun. (2016) 7:10579. doi: 10.1038/ncomms10579
66. Harper IG, Ali JM, Harper SJ, Wlodek E, Alsughayyir J, Negus MC, et al. Augmentation of recipient adaptive alloimmunity by donor passenger lymphocytes within the transplant. Cell Rep. (2016) 15:1214–27. doi: 10.1016/j.celrep.2016.04.009
67. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, Mcgavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. (2011) 208:987–99. doi: 10.1084/jem.20101773
68. Hidalgo LG, Campbell PM, Sis B, Einecke G, Mengel M, Chang J, et al. de novo donor-specific antibody at the time of kidney transplant biopsy associates with microvascular pathology and late graft failure. Am J Transplant (2009) 9:2532–41. doi: 10.1111/j.1600-6143.2009.02800.x
69. Callaghan CJ, Rouhani FJ, Negus MC, Curry AJ, Bolton EM, Bradley JA, et al. Abrogation of antibody-mediated allograft rejection by regulatory CD4 T cells with indirect allospecificity. J Immunol. (2007) 178:2221–8. doi: 10.4049/jimmunol.178.4.2221
Keywords: allograft, humoral allograft rejection, germinal center (GC), extrafollicular B cell response, transplantation
Citation: Chhabra M, Alsughayyir J, Qureshi MS, Mallik M, Ali JM, Gamper I, Moseley EL, Peacock S, Kosmoliaptsis V, Goddard MJ, Linterman MA, Motallebzadeh R and Pettigrew GJ (2019) Germinal Center Alloantibody Responses Mediate Progression of Chronic Allograft Injury. Front. Immunol. 9:3038. doi: 10.3389/fimmu.2018.03038
Received: 03 July 2018; Accepted: 07 December 2018;
Published: 23 January 2019.
Edited by:
Frans Claas, Leiden University, NetherlandsReviewed by:
Federica Casiraghi, Istituto Di Ricerche Farmacologiche Mario Negri, ItalyLuiza Guilherme, University of São Paulo, Brazil
Copyright © 2019 Chhabra, Alsughayyir, Qureshi, Mallik, Ali, Gamper, Moseley, Peacock, Kosmoliaptsis, Goddard, Linterman, Motallebzadeh and Pettigrew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reza Motallebzadeh, ci5tb3RhbGxlYnphZGVoQHVjbC5hYy51aw==
Gavin J. Pettigrew, Z2pwMjVAY2FtLmFjLnVr
†These authors have contributed equally to this work