 Alice H. Costain
Alice H. Costain Andrew S. MacDonald
Andrew S. MacDonald Hermelijn H. Smits
Hermelijn H. Smits- 1Department of Parasitology, Leiden University Medical Center, Leiden, Netherlands
- 2Lydia Becker Institute of Immunology and Inflammation, University of Manchester, Manchester, United Kingdom
Many parasitic worms possess complex and intriguing life cycles, and schistosomes are no exception. To exit the human body and progress to their successive snail host, Schistosoma mansoni eggs must migrate from the mesenteric vessels, across the intestinal wall and into the feces. This process is complex and not always successful. A vast proportion of eggs fail to leave their definite host, instead becoming lodged within intestinal or hepatic tissue, where they can evoke potentially life-threatening pathology. Thus, to maximize the likelihood of successful egg passage whilst minimizing host pathology, intriguing egg exit strategies have evolved. Notably, schistosomes actively exert counter-inflammatory influences on the host immune system, discreetly compromise endothelial and epithelial barriers, and modulate granuloma formation around transiting eggs, which is instrumental to their migration. In this review, we discuss new developments in our understanding of schistosome egg migration, with an emphasis on S. mansoni and the intestine, and outline the host-parasite interactions that are thought to make this process possible. In addition, we explore the potential immune implications of egg penetration and discuss the long-term consequences for the host of unsuccessful egg transit, such as fibrosis, co-infection and cancer development.
Introduction
Schistosomiasis is a chronic and potentially lethal tropical disease, mainly caused by the parasitic blood flukes Schistosoma mansoni, S. haematobium, and S. japonicum. Schistosomes have evolved to develop and thrive in their infected hosts, with untreated infections generally persisting for 3–10 years and a minority of infected individuals developing severe, life-threatening pathology (1). Common among parasites, schistosomes possess rather peculiar life cycles. This includes stages within definitive human hosts and secondary snail vectors, transformation through various larval forms, and—importantly—a unique process of egg migration to leave their human host. In this essential life cycle event, schistosome eggs pass from the host vasculature, across intervening tissue and into the environment via host excretions. This enigmatic process, and its pathological/immunological consequences, is the focus of this review, with particular emphasis placed on the intestinal response to S. mansoni.
Adult S. mansoni worms reside deep within the mesenteric veins of the intestine, where they feed on blood and acquire nutrients necessary for growth, development, and egg production (2). Each worm pair produces ~300 eggs daily, which exit the host by moving from the depths of the mesenteric vessels, across the intestinal wall and into the intestine lumen (3, 4). Importantly, as schistosome eggs are not in possession of any obvious motility mechanisms themselves, their expulsion is likely to be heavily reliant on host-driven processes. However, successful egg passage is not guaranteed. Approximately half of all deposited eggs never reach the intestine, but instead are swept to the liver, where they evoke strong granulomatous inflammation, as characterized by the infiltration of alternatively activated (AA) macrophages, eosinophils and T-helper 2 (Th2) cells, with additional fibroblast proliferation and generation of extracellular matrix (3–5). For the remainder of intestinally-bound eggs, success is still not certain. Firstly, eggs remain viable for a mere 2–3 weeks following oviposition, providing them with a relatively short timeframe to make this journey (6, 7). Secondly, due their high antigenicity and continual release of antigens and other metabolites, transiting eggs are easily detected by the host immune system, becoming the focal point of inflammatory granulomatous reactions. If these responses are too extreme, a variety of immune-pathologic sequelae will follow (8).
As our understanding of schistosome immunobiology has increased, it has become increasingly obvious that schistosomes implement a variety of strategies to ensure efficient egg transit. Within the vasculature, egg extravasation is promoted by angiogenesis, endothelial activation, and interactions with blood clotting components (9, 10). In the intestinal tissues, schistosomes exert a variety of immunomodulatory influences to support granuloma formation around transiting eggs, which is an essential process in egg excretion (11–14). Directly related to this, and to prevent overwhelming immunopathology, schistosomes guide the immune response toward a more regulatory phenotype during chronic disease. In this review we discuss the strategies employed by schistosomes to favor egg passage, and outline the potential immune implications and pathological consequences that may follow (Figure 1).
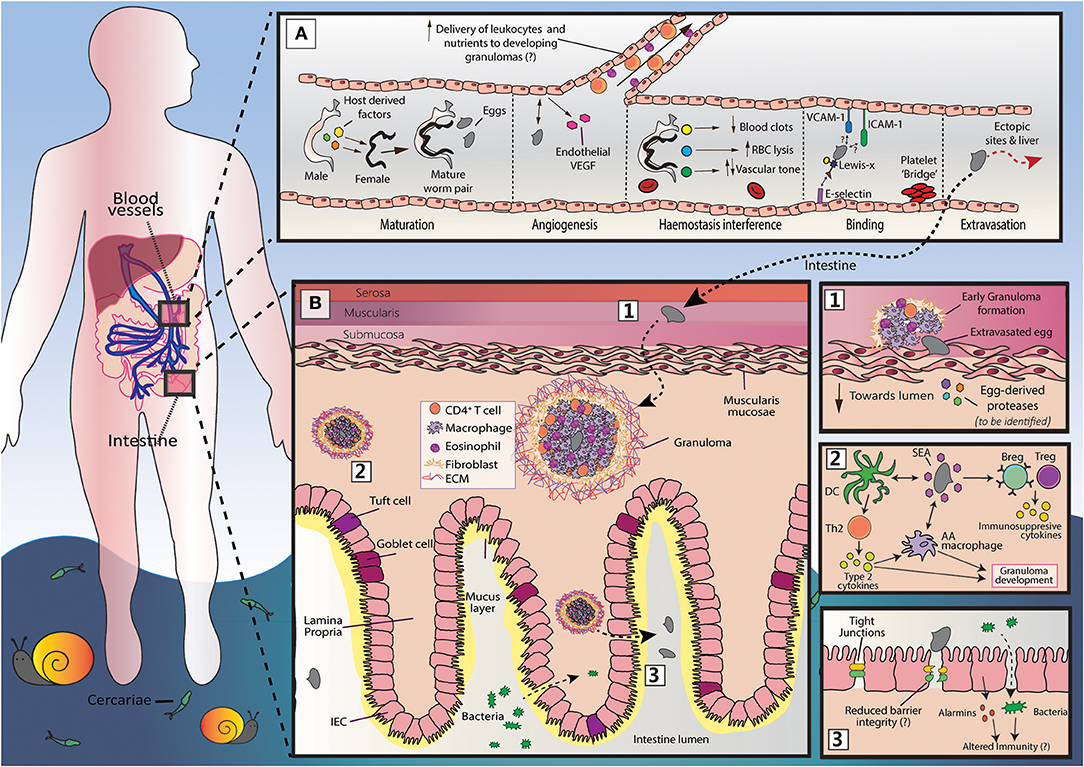
Figure 1. An overview of S. mansoni egg migration. Schistosome egg transit is facilitated by a series of host interactions at the intestinal and vascular interface. (A) The development of schistosomes into sexually mature, egg-producing adults occurs within the portal vein (~3–5 weeks post infection) and requires the transduction of host-derived signals (including those from the innate and adaptive immune system) to the developing worm pair. Once sexual maturity is reached, worm pairs migrate toward the mesenteric vessels, where the females lay approximately 300 eggs per day and actively modulates the intravascular environment to support their long-term survival. The production of eggs at ~5–6 weeks post infection is a milestone event in the schistosome life cycle, that is characterized by induction of a marked Th2 response and angiogenesis. Notably, the generation of a Th2 response by the host is critical for egg passage, and new vessel formation may favor egg transit, promoting the recruitment of immune cells and nutrients to developing granulomas. Freshly deposited eggs cannot move by themselves and must somehow attach and extravasate the endothelium. Although yet to be fully defined, this process may involve E-selectin:-Lewis-x interactions, and participation from platelets, ICAM-1 and VCAM-1. While a large proportion of eggs successfully penetrate the endothelium and reach intestinal tissue, many are swept to the liver or other distal locations (e.g., brain or spinal cord). Since schistosome eggs are unable to transit through these organs, overwhelming tissue pathology and inflammation may ensue. (B) Once schistosome eggs have passed across the host endothelium and out of the vasculature, they must cross the multi-layered intestinal wall. The host immune system responds to transiting eggs via an inflammatory granuloma response, in which individual eggs are encapsulated by immune cells [including alternatively activated (AA) macrophages, Th2 cells and eosinophils] and extracellular matrix (ECM), which protects host tissues from egg-derived toxins, but ultimately leads to formation of fibrotic lesions. For unknown reasons, granulomatous responses need to successfully develop for effective egg excretion from the host. Accordingly, schistosomes and their host have co-evolved a wide range of mechanisms to skew the host immune response toward granuloma-inducing Th2 profile. These include the ability of soluble egg antigens (SEA) to promote alternative activation in macrophages and to condition dendritic cells (DCs) for Th2 polarization. However, to prevent unwanted bystander tissue damage and potentially fatal immunopathology, schistosomes also implement various strategies to dampen host immunity and expanded regulatory networks (Bregs and Tregs). There remain many unknowns surrounding egg migration. This includes the molecules secreted by eggs to disrupt host barriers and modulate immune responses and, importantly, how egg penetration and intestinal ‘leakiness' may influence local and systemic immune reactions.
Endothelial extravasation
Maturation and Mesenteric Migration
Before egg production can commence, schistosomes need to mature to adulthood while navigating from the skin, via the lungs to the mesenteric veins of the intestine (S. mansoni and S. japonicum, causing hepatosplenic disease) or bladder (S. haematobium, causing urinary schistosomiasis) (15–17). Worm maturation occurs in the blood vessels and requires the transduction of host-derived signals from male worms to their female partner (18). Signals from the adaptive and innate immune system are thought to be intimately linked with this process (18). Notably, worm growth and reproductive activity is severely stunted in the absence of CD4+ T cells, but can be sufficiently restored through repeated stimulation of innate immunity via toll-like receptor signaling or inflammasome activation by endogenous danger signals (18–21). The specific immunological factors that guide parasite development remain poorly defined and controversial. For instance, while a functional role for interleukin (IL)-7 in parasite development is generally agreed upon, there is ongoing controversy surrounding the role of TNF, with studies showing that both TNF neutralization and administration can promote egg production (21–23).
At approximately 4–6 weeks post infection, sexually mature worm pairs move to the mesenteric vessels, which is the site of oviposition (17). Although eggs can be found throughout the vasculature, certain sites may be favored (15). In mice, oviposition appears to be concentrated in the Peyer's patches of the small intestine, while in primates and man, eggs are more readily detected in the colon and rectum (15, 16, 24, 25). Why oviposition shows such patterns is subject to speculation. It is possible that worm migration is dictated by host-derived signals (e.g., hormones or digestion products absorbed across the intestine wall) or that worms preferentially exploit regions of low sheer stress and high vascularization to avoid eggs being swept away with the blood stream (15). Alternatively, blood vessel diameter could be the major determinant, with adult worm pairs being relatively large (~0.5 mm in diameter and up to 10 mm long) in comparison to the mesenteric vessels that they reside in Da'dara et al. (26). Furthermore, while S. mansoni eggs are laid diffusely across the intestine and seldom produce bulky, concentrated lesions, S. haematobium and S. japonicum worms tend to deposit eggs in a few areas where a large number of them are concentrated (15, 16). These deposition patterns may reflect the unavoidable clumping of worms within host vessels or, alternatively, worms may be attracted to factors at the site of eggs-induced lesions, including substances released from breached blood vessels (15). Finally, it is possible that egg aggregation supports extravasation, with the build-up of egg-derived proteases creating channels from the intravascular to intraluminal space (27).
Even though migrating worm pairs are clear potential obstructions to blood flow, schistosome infections are not associated with enhanced blood clotting. In fact, individuals with advanced hepatosplenic schistosomiasis have a reduced level of blood coagulation factors, and blood clots are not observed around worms in host vessels (28–31). Experimental studies have also shown schistosome infections to impact blood-coagulation, where blood from 7-week infected mice coagulates more rapidly than control, with faster lysis of the clot formed (26). However, ex vivo studies involving the exposure of adult worms to blood from infected or non-infected mice, demonstrate an anticoagulant effect of the adult parasite (26). Mechanistically, there is strong evidence indicating that that schistosomes directly modulate the host haemostatic system via a variety of bioactive secretory products and molecules on the schistosome's outer-surface (tegument) (9). For instance, schistosomes inhibit blood clot formation and/or promote blood clot lysis through the activities of several tegumental enzymes, including enolase, SmSP2, SmAP and SmCalp1&2, and vascular tone is modified through the activities of SmSP2 and SmPOP (32–36). Altogether, such processes can be viewed as a schistosome survival mechanism in the blood stream, likely promoting residence and movement while preventing unwanted vessel occlusion.
Endothelial Adherence
Schistosome eggs are striking structures, encased by a rigid network of cross-linked proteins and, in the case of S. mansoni, characterized by a large protruding lateral spine. Due to the high rigidity and inflexibility of their outer shell, schistosome eggs must rely on external forces to bring them toward to the endothelial lining (37). It has long been suggested that the active migration of endothelial cells over schistosome eggs, brings the eggs into close contact with the vessel lining (38). More recently, video imaging has suggested that female worms prompt egg-endothelial associations via strong muscular contractions at their genital pore (dorsiflexion) that thrusts their eggs into the endothelium (7). Once brought into close contact with the endothelium, eggs likely tether themselves to endothelial surface adhesion molecules, including ICAM-1, VCAM-1 and E-selectin (39, 40). However, while binding to E-selectin is likely mediated by egg-shell components glycosylated with Lewis-x motifs (40), there are no obvious integrin-like structures within the egg-shell identified yet that bind ICAM-1 or VCAM-1. Interestingly, ICAM-1 shows strong upregulation in response to eggs and SEA (39), and soluble ICAM-1 levels are constitutively higher in schistosome-infected individuals and positively correlate with egg excretion rates (41). Additionally, there is evidence to show that ICAM-1 not only mediates egg binding, but also participates in the generation of granulomatous inflammation around parasite eggs, by regulating leukocyte trafficking, vascular permeability and modulating T cell responsiveness to soluble egg antigens (SEA) (39, 41, 42). As later discussed, intact granuloma formation is essential for successful egg expulsion.
Freshly deposited eggs are immediately surrounded by cells and proteins of the haemostatic system, including the plasma proteins von Willebrand factor (VWF), fibrin and fibrinogen (7, 43, 44). In addition to ICAM-1, E-selectin and VCAM-1, schistosome eggs may bind to these haemostatic components to promote their anchorage to the endothelium and to prevent them from being swept away with circulation. Indeed, the administration of platelet inactivating drugs to S.mansoni-infected mice results in significantly diminished egg excretion rates (45). By closely analysing the interactions between eggs and such components, deWalick and colleagues demonstrated that the schistosome egg-shell directly binds to VWF: an adhesive glycoprotein that tethers clotting material (such as platelets) to the activated endothelium (43, 46). This binding is suggested to benefit egg extravasation in two ways. VWF could form a direct bridge between eggs and the extracellular matrix, and/or the binding of VWF to clotting material may induce stable clot formation, making it easier for eggs to adhere to the endothelium.
The role of the schistosome spine in egg migration is not known. Given that S. japiconium eggs are virtually spineless, it is unlikely that the schistosome spine plays a major function. In fact, a recent report comparing egg morphology between Praziquantel resistant and susceptible S. mansoni infection suggests that the spine actually hinders egg transit (47). More specifically, resistant strain eggs were shown to have smaller lateral spines than susceptible strain eggs, and were also more frequently shed into host feces (47). Thus, from an evolutionary standpoint, perhaps S. japiconium has taken advantage of spine absence. However, based on the repeated observation that S.mansoni eggs clump to one another at their spine tip, it is also possible that spine-to-spine clumping enhances the cytotoxicity of freshly deposited eggs, promoting channel formation through host tissues and/or accelerating granuloma development (27, 48).
Eggs and Angiogenesis
Schistosomes not only reside and produce eggs within blood vessels, but also appear to promote their formation (49). Angiogenesis is a complex process in which new vessels develop from pre-existing ones, creating an environment that favors tissue growth and repair (50). This sequential process is guided by pro-angiogenic factors (such as VEGF, angiopoietin and inflammatory cell-derived chemokines) which instruct endothelial cell activation, proliferation and reorganization (50). Evidence for schistosome-induced angiogenesis can be found in both human studies and experimental models. During murine infection, vascularity is significantly enhanced in areas of high egg concentration (including the Peyer's patches) and, when angiogenesis is inhibited, there is a reduction in worm load and hepatic egg deposition (10, 24, 51). In human studies, mucosal biopsies containing S. haematobium eggs are more vascularized than healthy, egg-free control tissue (52). In addition, schistosomiasis patients have significantly higher serum VEGF levels than healthy individuals, or those with active hookworm infections (10) Schistosomes likely promote neovascularization to sustain their life cycle, for several reasons. First, the remodeling of intestinal vasculature may increase the number of worms the blood vessels can accommodate and reduce egg “spill over” into hepatic tissue (24). Second, angiogenic responses could enable the recruitment of leukocytes to developing granulomas, and ensure an adequate supply of oxygen and nutrients at these sites (53). Third, increased vessel density may impair intestinal tissue, making it easier for eggs to disrupt and move through (52). Finally, similar to what has been observed in cancer, growth of new vessels would maintain blood flow in scenarios of vessel occlusion (e.g., by worm pairs and their eggs). Furthermore, conditions created by vessel occlusion such as hypoxia, acidic PH and low glucose concentration, may also contribute to the neovascularization observed (54).
While adult worms have poorly defined roles in the induction of angiogenesis, secretory products of schistosome eggs (soluble egg antigens, or SEA) have been shown to instruct angiogenesis via direct and indirect mechanisms. Investigations using human umbilical vein endothelial cells have shown that SEA can directly encourage endothelial cell proliferation, migration, sprouting and production of VEGF (54, 55). The extent of angiogenic activity can be influenced by host genetics, and lies within the glycoprotein fraction of SEA (56, 57). Indirectly, SEA induces angiogenesis through the actions of alternatively activated (AA) macrophages and hedgehog signaling (58). In this case, SEA stimulates macrophage secretion of biologically active hedgehog ligands, which subsequently activate hedgehog-responsive endothelial cells to proliferate and secrete angiogenic factors. The fact that sprouting blood vessels are more frequently observed around viable ova as opposed to dead or dying calcified ova, strongly suggests that substances actively secreted from eggs are responsible for new blood vessel formation (52).
In addition to angiogenesis, endothelial activation by schistosomes may also support granuloma formation. Schistosome triggered VEGF significantly increases proliferation of and extracellular matrix deposition by hepatic stellate cells, which are the main source of extracellular matrix around schistosome eggs in the liver (59). Whether similar mechanisms are involved in intestinal granuloma formation is unknown, though it is tempting to speculate that endothelial cells lining mesenteric vessels could be activated upon worm encounter to secrete pro-fibrotic factors, such as VEGF, IL-13, TGF-β, or IL-33, to intestinal-resident cells involved in fibrosis. IL-33, for instance, is constitutively expressed by endothelial cells, and has recently been shown to support liver granuloma pathology through the induction of pro-fibrotic AA macrophages (60, 61). In addition, VEGF partially regulates Th2 inflammatory responses in the lung and liver in response to schistosome eggs (62). Given that type 2 immune responses are essential for adequate granuloma formation, it would be interesting to define a role for VEGF in intestinal granulomatous reactions.
Overall, schistosomes employ a variety of strategies within host blood vessels that favor their survival and life cycle propagation. Increased understanding of these interactions could identify new targets for the prevention of severe disease during schistosomiasis. For instance, direct targeting of adult worms could prevent or reduce the production of eggs and their accumulation within host tissues, which is the primary cause of pathogenesis during schistosome infection.
Intestinal Passage
After successfully extravasating the mesenteric vessels schistosome eggs are confronted with a much larger anatomical hurdle: the intestinal wall. This extensive barricade is the host's largest interface with the external environment, its luminal side in constant exposure to the contents of the intestines, including innocuous (food) antigens, commensals and pathogens (63). To efficiently segregate the host from this hazardous environment, the intestinal wall incorporates a dense network of immune cells and an innermost monolayer of tightly aligned intestinal epithelial cells (IECs). IECs and resident immune cells communicate with one another to reinforce barrier integrity, maintain homeostasis and mount robust responses against invading threats—schistosome eggs included. However, despite the host response against them, a large proportion of eggs successfully transit across the intestinal tissues and exit the host body. Crucially, to enable this process while limiting enteric inflammation, schistosomes have evolved several strategies to modulate host immune responses and manipulate host barriers. For example, as illustrated in Figure 2, schistosome eggs are capable of digesting through the intestinal muscular layer (muscularis mucosae) without triggering significant inflammation or immune infiltration. This process likely involves a collaboration between egg-derived proteases (yet to be defined) and immune cells that have been recruited to the serosal tissue and/or mesenteric vessels (24). In particular, the strategies employed by schistosomes to favor migration of eggs through the intestine likely heavily involve Th2 polarization and granuloma maintenance.
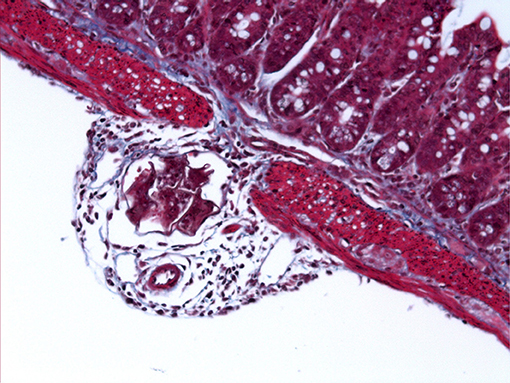
Figure 2. S. mansoni egg penetrating through the intestinal wall. Light microscope image of Masson's Trichrome stained ileal section (5 μm) from a C57BL/6 mouse 6 weeks after percutaneous infection with approximately 40 S. mansoni cercariae.
Moreover, due to infection longevity and the continuous passing of eggs, it is currently unclear, and difficult to assess, which intestinal layer schistosome eggs are predominantly located (i.e., serosa, submucosa, or mucosa). However, H&E images from infected mouse intestine show that eggs tend to penetrate through the muscularis-serosa layer en route to the lamina propria (64).
Promoting Granuloma Formation
S. mansoni eggs are highly antigenic structures that continuously secrete a variety of innocuous, toxic or antigenic substances into host tissues (6). Accordingly, infiltrating eggs are focal points for the host immune system, which mounts a distinct attack and sequestration strategy in response: the granuloma. Granulomas are highly organized, multi-cellular structures that are enriched with a range of immune cells including Th2 cells, macrophages and eosinophils, with mast cell infiltration, accumulation of type 2 cytokines (such as IL-4, IL-13 and IL-5) and additional involvement from stromal cells/fibroblasts. Type-2 cytokines instruct the polarization of macrophages toward an alternatively activated (AA) phenotype, whom are essential to the resolution of egg-induced inflammation and tissue damage (65–67). After reaching maximal responses, granulomatous lesions decline in size and become fibrotic, with IL-13 being the main cytokine responsible for fibrosis, mediating its effects through AA macrophages and fibroblasts (5, 68, 69).
It is important to highlight that although these egg-triggered inflammatory reactions can also be found throughout infected liver, there are distinct differences between intestinal and hepatic granulomas in terms of size, cellular composition and extracellular matrix deposition (7, 70). These finding arguably reflect the higher proportion of dead eggs within infected liver, and so differences in egg secretions between the two tissues. Alternatively, they may suggest that the local tissue environment has a large bearing on granuloma function and development. In the case of intestinal granulomas, it would be very interesting to disentangle intestinal bacterial involvement, as studies on antibiotic-treated mice show only intestinal granulomas, but not hepatic granulomas, to be influenced by antibiotic administration (71). Moreover, it is unknown whether intestinal granulomas are better “designed” for egg excretion than their hepatic counterparts. Furthermore, while the immunology of hepatic granulomas has been studied extensively over past decades, our understanding of intestinal granulomas is considerably less. Accordingly, most of the data discussed in the following section has been obtained from studies on hepatic granulomas, and we cannot rule out the possibility that intestinal granuloma development may differ from their hepatic counterparts.
The schistosome granuloma is both beneficial and detrimental for the host (5). On the one hand, granulomas protect host tissues from egg-derived toxins such as Omega-1 and IPSE/alpha-1, which can cause severe damage (72). On the other hand, the fibrotic sequelae that follows granuloma resolution is the main cause of pathology and lethality in schistosomiasis (5). At the same time, in a remarkable example of co-evolution, granulomas are thought to be essential for schistosomes and continuation of their life cycle. Early studies in immunodeficient mice demonstrated the crucial role of granulomas in egg passage, with egg excretion rates plummeting in mice failing to generate intact granulomas, leading to lethal microvascular damage (11–14). These data also underscored the importance of host T cells in granuloma development, results that were later corroborated by studies in HIV+ infected people, in whom egg excretion rates were found to positively correlate with circulating CD4+ T cell levels (73). Furthermore, Th2 immunity is critical for intact granuloma development and host survival, since mice deficient in IL-4 and IL-13 signaling die during acute disease due to elevated intestinal and hepatic pathology, oxidative damage, endotoxemia and cachexia (65, 68, 74–76). Therefore, it comes as no surprise that schistosomes actively modulate host immune responses to ensure a Th2 bias is achieved.
Infection with S.mansoni follows a predictable immunological pattern, with a mixed, low level Th1/Th2 profile prevailing in the initial 4–5 weeks, then Th2 responses dominating from the point of egg production, with parallel expansion of regulatory cell networks that exert their influence most dramatically during chronic infection (77). Although many innate cell types help establish the Th2 milieu, dendritic cells (DCs) are essential (78) (79). Using their vast array of pattern-recognition receptors (PRRs), DCs sense, process and present schistosome egg-derived antigens to naïve CD4+ T cells, instructing Th2 development by yet to be fully defined mechanism (80).
SEA is a complex mixture of immune-stimulatory antigens, that is well known for its capacity to condition DCs for Th2 priming (81–84). Omega-1, a glycosylated T2 RNase, is a single component of SEA and potentially, the most powerful inducer of Th2 responses (85, 86). Omega-1 is taken up through mannose receptors on DCs and enhances Th2 polarization by degrading mRNA and ribosomal RNA, leading to reduced co-stimulatory molecules expression (CD83 and CD86) and reduced production of the Th1 promoting cytokine IL-12 (85, 86). Somewhat unexpectedly, SEA and Omega-1 can also stimulate DC production of Type-I IFN (normally associated with anti-viral responses), which enables their initiation of Th2 responses (87, 88). However, it is important to note that Omega-1 is not the only SEA component involved in DC conditioning, as SEA depleted of Omega-1 and S.mansoni eggs with specific Omega-1 knock-down still retain some of their Th2 polarizing potential (85, 89). In addition, Kaisar and colleagues very recently identified a novel pathway in Th2 polarization that entails Dectin1/2 signaling on DCs and works independently of the actions of Omega-1 (90). Overall, the impact of egg antigens on host innate immune cells is complex and multifactorial, often flouting convention, but with the cumulative effect being initiation of a strong Th2 response.
It is important to realize that although Th2 skewing is primarily ascribed to eggs, adult worms are also capable of instructing a Th2 environment before the onset of egg production (91). In fact, by comparing granuloma formation between models of natural infection and portal vein egg injection, Leptak and colleagues demonstrate that granuloma development requires both worm- and egg-derived signals (92). Notably, while the inflammatory reaction toward portal vein injected eggs is minimal in terms of reaction volume, cellular infiltration and collagen deposition, these reactions can be sufficiently restored if eggs are injected into mice previously infected with single-sex male cercariae or exposed to adult worm homogenates (92). Mechanistically, this ‘priming' effect could be explained by the sharing of antigens between adult worms and eggs (92). Further, cytokines produced in response to adult worms may be key to granuloma induction (23, 42, 92). In particular, the cytokine TNF is thought to be exclusively produced in response to adult worms, and may be able to restore granuloma formation in naïve mice injected with eggs alone (92). Interestingly, it has been suggested that the production of TNF may be linked to phagocytosis of worm-regurgitation products by local macrophage populations (92, 93).
The schistosome granuloma is hallmarked by the accumulation of AA macrophages, which predominantly arise from the recruitment of Ly6CHi monocytes, as opposed to the proliferation of local macrophage populations (94, 95). AA macrophages are activated by type-2 cytokines (IL-4/IL-13), characterized by the expression of various signature genes (such as arginase 1, Ym1, Relmα) and are thought to prevent potentially lethal host pathology via a variety of mechanisms (67, 96). This includes coordinating the recruitment of collagen and cells to developing granulomas, regulating T-cell proliferation, facilitating tissue repair and regeneration, and inhibiting the differentiation of classically activated macrophages (CAMs), which generate pro-inflammatory cytokines and cause oxidative tissue damage (67, 97, 98). Crucially, thanks to the wound healing capacities of AA macrophages, intestinal barrier integrity is sufficiently maintained during egg migration and the host is shielded from enteric bacteria (67). Indeed, in mice deficient in IL-4/IL-13 signaling specifically on macrophages and neutrophils [LysM(Cre)IL-4Ralpha(-/flox) mice], AA macrophages do not develop and mice are extremely susceptible to S. mansoni infection (100% mortality during acute disease) (65). Importantly, acute mortality was associated with exaggerated Th1 pathology, severe hepatic and intestinal damage, impaired egg excretion and sepsis (65). In more recent studies, AA macrophages have also shown to maintain tissue integrity in models of urogenital schistosomiasis (66), and their protective function has been partially attributed to the enzyme arginase-1 and resistin-like molecule (RELM)-α (98–101). Notably, arginase-1 contributes to the long-term survival of schistosomiasis by restricting Th1 and Th17 associated immunopathology, and RELM-α acts as a negative regulator of Th2 responses (98–101). Moreover, while YM-1 is an additional phenotypic marker of AA macrophages, its contribution to Th2 responses during schistosomiasis remains undefined. However, various reports suggest a role for YM-1 in Th2 differentiation (102, 103). Of note, our understanding of the complexity of ‘M2' macrophages, and the range of cytokines that can promote aspects of M2 polarization, is continually growing, with IL-10, TGF-β and IL-33 also shown capable of instructing alternative activation (61, 104–106).
Schistosomes not only rely on cytokines to instruct AA macrophages induction, but also play a direct role in their polarization. Schistosome-derived molecules that may contribute to alternative activation include SEA, peroxiredoxin, lysophosphatidylcholine and hemozoin (101, 107–110). SEA, for instance, appears able to directly interact with macrophages to instruct an AA phenotype, or indirectly elicits an AA profile via IL-33 and its receptor ST2 (61, 109). Interestingly, in a model of intravenous S.mansoni egg injection, mice deficient in ST2 demonstrated impaired production of Th2 cytokines and primary granuloma development (111). However, this study did not assess the influence of ST2 deficiency on macrophage function or polarization. Another molecule that influences macrophage polarization is hemozoin. Hemozoin is a neutralized version of heme that schistosomes regurgitate into the bloodstream following the digestion of host erythrocytes (110). Hemozoin crystals spontaneously aggregate in the liver, where their consumption by patrolling macrophages promotes an alternatively activated phenotype, including the expression of the Th2 negative regulator RELM-α (99–101). Moreover, the uptake of these worm-regurgitation products by antigen-presenting cells has been suggested to induce the production TNF, which has previously shown a key role in granuloma formation (23, 42, 92).
In terms of other myeloid cells, eosinophils and mast cells are cardinal features of the schistosome granuloma, but their function there remains elusive. While older studies suggest that eosinophils favor egg passage by digesting the epithelial basal membrane, more recent models of eosinophil ablation found no obvious role for them in this process or for granuloma formation, fibrosis or worm burden (112–114). In this context, mast cell function also remains a mystery. During Trichinella spiralis infections, mast cells mediate parasite expulsion and disrupt epithelial barrier function through the release of mast cell protease 1 (mMCP-1), a serine protease that degrades TJ proteins (115). These observations prompted investigations into the role of mMCP-1 in barrier integrity during schistosome infections, but no functional role was found in this setting (116). However, mast cell secretion of pro-angiogenic factors could support egg extravasation (117). Additionally, the strong Th2 milieu may simply promote the recruitment of eosinophils and mast cells to the area, where it is possible that both cells types represent (minor) innate sources of Th2 associated cytokines, and so help sustain granulomas.
The alarmins IL-33, IL-25 and TSLP (thymic stromal lymphopoietin) are important initiators of type 2 immune reactions that are released from the epithelium, endothelium and other stromal compartments upon damage and stress (118). Although yet to be formally demonstrated, their release could be triggered by egg migration. While individual ablation of TSLP, IL-25, or IL-33 has no discernible impact on hepatic granuloma formation and fibrosis during chronic S. mansoni infection, simultaneous blockade of all three signaling pathways results in a modest decrease in Th2 associated pathology by 9 weeks of infection, including a reduction in granuloma-associated eosinophilia, hepatic fibrosis and IL-13 producing type 2 innate lymphoid cells (ILC2s) (119, 120). However, such effects were transient in nature and no longer significant by 12 weeks post infection, suggesting that these alarmins are dispensable for the development and maintenance of egg-induced pathology (120). Similarly, the observed reduction in ILC2 activity appeared to be compensated for by enhanced CD4+ T cell responses, indicating that ILC2s also have no major function in the maintenance of type 2 inflammation in this particular setting. Considering the essential role of Th2 responses in granuloma formation and egg expulsion, it makes sense that compensatory mechanisms are at play, and that schistosomes are not overly reliant on the unregulated release of stromal mediators for development or type 2 immunity. However, given that ILC2s have been shown to be important initiators of Th2 responses during gastrointestinal helminth infections, and to significantly contribute to wound repair responses at mucosal surfaces, further studies are required to reach a formal conclusion on the involvement of ILC2s in schistosome-associated Th2 inflammation (121–123). In addition, although the aforementioned study signposts the redundancy of alarmins in type 2 immunity during schistosome infection, there remains the possibility that they contribute to regulatory cell expansion during chronic stages of disease (124).
Exiting the Epithelium
Tight junctions (TJs) are the final hurdle that schistosome eggs must overcome to transit through the intestinal wall and exit the host. TJs are multi-protein complexes that tightly link adjacent IECs at their apical membranes, creating virtually impermeable seals. In this manner, TJs regulate para-cellular permeability (the movement of substances between adjacent cells) and are essential for maintenance of intestinal barrier function (125). In scenarios of TJ disruption, intestinal barrier function is impaired, allowing for enhanced permeation of luminal substances (such as bacteria, antigens, toxins and metabolites) into mucosal tissues and the systemic circulation (125). Both experimental models and human infection studies hint toward increased intestinal permeability during schistosome infections. Mice experimentally infected with S. mansoni demonstrate reduced ileal integrity from 8 weeks post infection, and S. mansoni infections in humans are associated with extremely high levels of endotoxin in the bloodstream (116, 126). In addition, the lethality of experimental schistosomiasis in scenarios of AA macrophage impairment is partially attributed to mass disruption of the intestinal wall and sepsis, with a common gastrointestinal symptom of intestinal schistosomiasis being blood in stool (65, 127).
One future goal is to better define schistosome-epithelial interactions. Namely, is intestinal epithelial disruption solely driven by damage caused by egg migration and/or, do schistosomes actively modulate components of the epithelial barrier to favor egg migration? In a unique model of urogenital schistosomiasis, in which S. haematobium eggs are directly introduced to the bladder wall, eggs were shown to suppress the transcription of multiple genes implicated in urothelial barrier maintenance, including junctional adhesion molecules claudin and uroplakin (128). These data indicate that schistosome eggs not only physically disrupt TJs as they pierce through the intestinal lining, but also suppress the transcription of various TJ related genes, which could facilitate egg transit. However, in a separate study, S. japonicum eggs were instead found to reinforce intestinal epithelial barrier function and relieve inflammation in the TNBS colitis model (129). Crucially, prevention of experimental colitis was accompanied by reduced bacterial translocation and enhanced levels of tight junction molecules (Zo-1 and occludin). This study suggests that eggs can fortify the intestinal barricade to prevent potentially lethal bacterial translocation, and thereby enhance host survival. Perhaps the discrepancies between these two studies reflect the manner of egg introduction: direct egg injection into the bladder wall vs. injection into the peritoneal cavity. More specifically, could the introduction of eggs into the peritoneal cavity influence the function of intestinal cell types? Direct injection of eggs into intestinal tissue, as very recently described (130), may provide a novel approach to study the impact of S. mansoni eggs on intestinal barrier integrity.
In addition to direct effects of schistosome eggs, intestinal permeability can be influenced by cytokines such as IFN-y, TNF and IL-13, as well as epithelial cell apoptosis and exogenous factors such as diet and non-steroidal anti-inflammatory drugs (125, 131). With regards to cytokine-mediated barrier dysfunction, it was recently demonstrated that schistosome eggs are incapable of penetrating through epithelial cells themselves, but require pro-inflammatory mediators TNF and/or IFN-γ to disrupt tight junctions and reduce intestinal epithelial resistance (71). Schistosome eggs appear not to stimulate production of pro-inflammatory cytokines by DCs, which suggests that other local mediators or cell types may be involved (71, 80). Alternatively, the tissue damage caused by egg transit, compounded by involvement of intestinal bacteria, could conceivably stimulate production of such cytokines (71). Furthermore, IL-13 has shown to induce epithelial apoptosis and increase the expression of the pore forming TJ molecule claudin-2, indicating that Th2 responses themselves can be instrumental to decreased barrier integrity (131).
Once eggs rupture past IECs they are met by a stratified layer of mucus, whose nature and function in schistosomiasis remains undefined. Mucus secretion is likely enhanced during schistosome infections, given that the IL-4/IL-13 signaling axis is critical to goblet-cell hyperplasia and mucus secretion (132, 133). Whether such alterations influence egg expulsion is unclear. In gastrointestinal nematode infections, mucus mediates the rapid expulsion of these parasites by limiting their motility and preventing their establishment within the gastrointestinal tract (132). Applying these observations to schistosome infections, it is possible that increased mucus generation leads to accelerated expulsion of eggs into the environment. In addition, since intestinal mucins are capable of instructing important pro-inflammatory functions in DCs, it is tempting to suggest that mucins may contribute to modulation of the inflammatory environment during schistosomiasis (134). Directly related to this, the exposure of DCs to mucins may be increased by egg-induced disruption of the intestinal barrier.
While larval and adult gastrointestinal nematodes likely interact with IECs for prolonged periods of time, schistosome eggs transiently move past or through them during transit. It is unknown whether these different exposure times influence how IECs sense and respond to the parasite infections. Very recently, intestinal tuft cells (a rare IEC population) were shown to instigate type 2 immunity in response to gastrontestinal nematodes infections (135–138). As tuft cell research is still in its infancy, defining the interactions between tuft cells and schistosome eggs remains an open and interesting point of further study.
Microbial Mediated Migration
In the face of the complexity of the immunopathology they generate, schistosome eggs ultimately transit from the mesenteric vessels via the serosa, the muscularis, the epithelium and the mucosa into the intestinal lumen, where a dense and vibrant microbiota surrounds them. As discussed above, successful egg penetration requires carefully coordinated interactions between host and parasite. These interactions can be extended to a third partner: the intestinal microbiome (herein, defined as commensal bacteria, viruses and fungi).
Helminths and the microbiome have co-evolved with their mammalian hosts over millennia, so extensive interactions exist between the three parties (139). Many helminths favor the establishment of defined microbial communities to support their own infectious life cycle and improve the overall wellbeing of the host. For instance, Trichuris muris uses intestinal bacteria as environmental hatching cues and Heligmosomoides polygyrus infections alter cecal microbiome composition, leading to a greater availability of short chain fatty acids (SCFAs) that dampen allergic responses (140, 141). While the interface between host, microbiota and intestinal-dwelling nematodes is beginning to be understood, there is currently a paucity of information on this topic about schistosomes, and whether they also remodel gut microbiome composition to support their own life cycle and lessen host pathology.
In mice, experimental S. mansoni infections are accompanied by profound changes in microbiota composition from at the point of egg production, including the expansion of Akkermansia muciniphila and bacterial populations from the Family Lactobacillaceae (142). The expansion of these bacterial communities may favor chronic schistosome infection by elevating the frequency of regulatory cell populations, or repairing egg-induced damage to the intestinal wall (143, 144). Further, a recent study using a broad spectrum of antibiotics showed that bacterial depletion reduces fecal egg excretion and intestinal granuloma development during murine infection with S. mansoni (71). In contrast, worm fecundity and liver pathology was not influenced by antibiotic administration, indicating that intestinal granuloma development and egg transit is particularly dependent on local, bacterially-derived factors. These factors could include bacterially-induced cytokines, such as IFN-y and TNF, that are needed for tight junction severance (71, 125). Complementing these studies, metabolic analysis of feces from mice experimentally infected with S. mansoni reveals several alterations in gut-bacteria related metabolites from day 41 of infection, including a greater availability of the SCFA, propionate (145). Additionally, differences in intestinal microbiome structure have been reported between children with or without S. haematobium infection (146). As S. haematobium is a urogenital parasite, this observation indicates that the schistosomes have a systemic influence on microbiome composition. Similar to that observed in Trichuris infection, it is possible that the schistosome-induced Th2 responses promote the establishment of a particular microbiome (147). Moreover, as signals from the gut microbiome are known to influence immune responses at both local and distal locations (e.g., the lung), it is possible that schistosome-induced alterations to the intestinal microbiome influence the development and/or protection against a range of diseases (148, 149).
The Immune Implications of Egg Penetration
In mice, granulomatous responses are maximal by approximately 8–10 weeks post infection, before gradually declining in magnitude (150, 151). This decline corresponds with a reduction in lymphocyte proliferation and responsiveness, and represents the transition from acute to chronic disease (77, 152, 153). Chronic schistosomiasis is characterized by high circulating levels of anti-inflammatory IL-10 and/or TGF-β and the profound expansion of regulatory T cells (Tregs) and B cells (Bregs), which function to supress potentially deleterious activities of T-helper cells and limit granulomatous pathology (77). Schistosome eggs are continually produced during this chronic period, but thanks to the immune-modulatory capacities of these regulatory cell networks, the majority of chronically infected individuals do not develop lethal pathology.
Schistosomes drive regulatory cell induction via a variety of immunomodulatory influences (154). The egg-derived glycoprotein IPSE-1/ alpha-1,was recently shown to promote B cell IL-10 and equip B cells with Treg inducing capacities (155). In the case of worm-derived products, both Cyclophilin A and schistosomal lysophosphatidylserine (lyso-PS) can modulate DC function, leading to preferential expansion of IL-10 producing Tregs (156, 157). Small exosome-like extracellular vehicles (EVs) from schistosomes could also represent untapped and important sources of immunomodulation (158, 159). In addition to such direct effects of worm and egg-derived products on inflammation and the immunological environment, it is tempting to speculate that egg penetration itself will shape the immunoregulatory landscape, perhaps in part via facilitating the systemic spread of immune-modulating luminal molecules.
Such molecules could include LPS and bacterially-derived metabolites. SCFAs, for instance, are known to increase Treg responses, mediate the dampening of allergic airway inflammation in the context of H. polygyrus bakeri infection, and down-regulate the pro-inflammatory effector functions of intestinal macrophages (140, 149, 160, 161). A role for SCFAs in schistosome-driven immune regulation has not yet been demonstrated, but this would make for an interesting point of further study. Intriguingly, although reported endotoxemia levels are 10 times higher in sera from individuals with human schistosomiasis than that observed in lethal toxic shock, study participants have neither poor health nor systemic inflammation (126). This observation has been suggested to reflect the type of LPS in the blood stream, where the structure of LPS (specifically Lipid A acylation) dictates whether it has an antagonistic or agonistic effect (126, 162). As the structure of LPS varies between different bacterial communities, potential differences in LPS immunogenicity could reflect schistosome-induced alterations to the gut microbiome (142, 162). Alternatively, it is possible that schistosomes modulate innate cell responsiveness to TLR ligands and/or reprogram innate cell function to acquire a more regulatory phenotype (71, 156, 163, 164). In support of these hypotheses, schistosome-derived lysophosphatidylserine has been shown to be capable of modifying TLR2 signaling pathways in DCs, leading to altered maturation and enhanced induction of Tregs (156). In addition, S. japonicum infections can increase the survival rate of mice with LPS-induced sepsis, in a mechanism that likely entails the polarization of macrophages to an M2 phenotype (165, 166). Molecules that may mediate this effect include schistosome-derived cysteine proteases, which were recently shown to protect against sepsis challenge and lower the production of pro-inflammatory cytokines and NO from LPS-stimulated macrophages (167). Interestingly, similar protective mechanisms appear to be induced during infections with the filarial worm Litomosoides sigmodontis (163) and the trematode Fasciola hepatica (168). Notably, F. hepatica reduces the release of inflammatory mediators from macrophages via mechanisms of molecular mimicry, and L. sigmodontis protects from bacterial sepsis by down-regulating the expression of macrophages genes involved in TLR signaling (163, 168).
The translocation of bacterial ligands into host tissues likely has immunomodulatory consequences. In the absence of Myd88 signaling, which is central to bacterial-innate cell interactions, schistosome infected mice demonstrate impaired Th1 responses and reduced granulomatous responses (169). The regulatory environment may also be influenced by microbial translocation, where translocated LPS could combine with SEA to trigger the release of inflammasome-derived IL-1β, which partakes in Breg induction (170, 171). Finally, there remains the possibility that bacterial leakage influences the dynamics of macrophage proliferation and alternative activation at the site of inflammation. For example, during experimental schistosomiasis, it is possible that intestinal bacteria act as a trigger for monocyte recruitment from the bone marrow and resultant macrophage accumulation, as opposed to the in situ proliferation of resident macrophage populations (95, 172).
By strategically expanding regulatory cell populations, schistosomes not only limit egg-induced pathology, but can also suppress the development of various inflammatory diseases including allergy (173). Indeed, many epidemiological studies have found inverse correlations between schistosomiasis and allergies, and in experimental models schistosome infection provides relief against allergic airway inflammation and OVA-induced airway hyper-responsiveness, with optimal protection achieved during chronic but not acute disease stages (174–178). There is also evidence to show that schistosome eggs can protect from allergic asthma in the absence of adult worms, despite the strong Th2 responses they evoke (179, 180). A current research goal is to better define the immunomodulatory mechanisms employed by schistosomes, where this new-found knowledge could potentially be used to reengineer and recover regulatory cell function in allergic individuals.
Eggs that Fail
Despite the multiple strategies employed, many eggs fail to exit their mammalian host. Experimental infections indicate that only 20–55% of eggs are successful excreted, while the remainder inevitably become trapped within host tissues (3, 4). Although egg deposition is targeted for the urinary bladder (S. haematobium) and the intestine (S. mansoni and S. japonicum), eggs swept systemically through the blood stream can readily be detected at various other locations including the eyes, skin, kidney, spleen and central nervous system (CNS) (4, 16). Egg deposition at intended and unintended sites can have serious pathological consequences for the host. The more severe disease complications generally manifest many years after infection, reflecting gradual egg accumulation in host tissues and the resolution of granulomas by fibrosis and calcification (127).
This aspect of schistosome infection is perhaps most evident in liver pathology during S. mansoni and S. japonicum infection. Granuloma formation around hepatic egg deposits leads to severe fibrosis, blood flow obstruction and, subsequently, the formation of ascites and blood vessels that bypass the liver (portosystemic shunting) (127). If these vessels rupture, life-threatening bleeding may follow. Pathology in the intestine is generally less severe than in the liver, and can be characterized by pseudopolyposis, ulceration and stricture formation. In S. haematobium infections, egg entrapment within pelvic organs (urinary bladder, ureters, cervix, vagina, prostate gland and seminal vesicles) can also result in severe pathology, including obstructive bladder and ureteral fibrosis, bladder cancer, kidney failure and the formation of gross genital sores (127, 181). In females, these lesions may give rise to infertility, ectopic pregnancies and menstrual irregularities, while in men, higher rates of sperm apoptosis have been reported (181, 182).
Importantly, reduced integrity of the genital epithelium constitutes a significant risk factor for the acquisition of HIV and other pathogens, including oncogenic viruses and bacteria (183, 184). Indeed, a recent longitudinal study in Tanzania demonstrated a clear gender bias in schistosomiasis and HIV acquisition, with schistosome-infected women demonstrating a 3 fold higher chance of acquiring HIV than uninfected women, whereas odds remained the same for men with or without infection (185). The increased risk of HIV acquisition in females very likely reflects the local physical changes caused by schistosome eggs at the female genitalia (183). The sequestration of eggs within the mucosal tissue of the vagina and cervix leads to ulceration, erosion and the formation of tiny cervical abnormalities (yellow and/or grainy sandy patches) that are surrounded by an irregular network of blood vessels. These blood vessels are believed to represent egg-induced angiogenesis and could accelerate the transmission of HIV during intercourse (52, 183). In contrast, S. haematobium infected men are at lesser risk of acquiring HIV because eggs do not infiltrate male genital organs that are exposed to the virus. However, other studies have shown mild associations between male urogenital schistosomiasis and HIV-1 acquisition, and even though S. mansoni eggs do not localize to the genitalia, S. mansoni-infected individuals also appear more susceptible to HIV (183, 186, 187). These associations are believed to be a consequence of schistosome-associated immunomodulation, as opposed to local tissue damage caused by eggs (183). In particular, the co-receptors for HIV are more highly expressed on the surface of CD4+ T cells from schistosome-infected individuals, allowing for greater viral uptake (186).
Similar to HIV, there is a clear link between urogenital schistosomiasis and bladder cancer, with promotion of carcinogenesis likely due to S. haematobium enhancing entry sites for oncolytic viruses and bacteria (184, 188). Alternatively, egg-driven inflammation may have a bystander effect on host cells, or immunosuppressive cytokines (produced during chronic stages) may reduce host ability to clear oncolytic viral and bacterial infections. In addition, S. haematobium eggs have shown to directly influence the transcription of carcinogenesis-associated genes within the bladder wall (128). This effect could potentially be mediated by schistosome-derived estrogenic molecules, albeit their carcinogenic activity has yet to be defined (189).
Curiously, while a clear association between S. haematobium and bladder cancer has been established, there is limited and controversial evidence to implicate S. japonicum and S. mansoni in intestinal and liver cancer (184, 188, 190). The reason for these strain-specific differences is unclear, but it has been suggested this reflects the site of egg deposition and/or the contribution of environmental carcinogens, including tobacco smoke or industrial and agricultural dyes (184, 188).
The spreading of eggs to the CNS (neuroschistosomaisis) is one of the more devastating outcomes of schistosome infection that can result in seizure or paralysis, depending on brain or spinal cord involvement. While S. japonicum eggs are more frequently found in the brain, eggs from S. mansoni and S. haematobium appear to have a predilection toward the spinal cord (127). These differences could reflect the smaller size of S. japonicum eggs and/or their absence of a protruding spine (127). Moreover, given that schistosome eggs disperse to many ectopic locations, it is possible that they affect co-infections encountered at these sites too. At a local and mechanistic level, the inflammatory environment established by eggs may exacerbate already established pathology or create an environment that favors other pathogen survival. At an immunological level, schistosome co-infections may suppress immune responses toward viral and bacterial antigens, leading to ineffective pathogen clearance and chronicity (191, 192).
Conclusion and Outlook
How schistosome eggs successfully exit the host body has long been a focus of many researchers. Very early studies revealed the severity of egg-driven pathogenesis and the curious migration patterns taken by adult worms to reach favored sites of oviposition (3, 16, 17, 51, 181). In more recent years, research has shed light on the molecular and immunological mechanisms that govern this process. Within host blood vessels, schistosomes establish a suitable environment for maturation, movement and egg extravasation by interfering with host haemostasis and angiogenesis (9, 10). For intestinal egg passage and granuloma formation, schistosomes actively tamper with host immune responses, to achieve a delicate balance between immune effector and immune regulatory activity, and to limit bystander tissue damage. However, despite the exit strategies employed, egg transit is not a certainty and lethal pathology may follow egg entrapment.
Our understanding of the interactions between schistosomes and the mammalian host is continually growing, with many interesting avenues still open for exploration (see Box 1). For example, while schistosome infection influences gut microbiome composition, we are yet to define how these fluctuations impact egg migration, disease pathogenesis, immunomodulation and long-term host/parasite survival (142). With powerful sequencing techniques emerging and our knowledge of the intestinal microbiome constantly expanding, we anticipate that the relationship between the mammalian host, schistosomes and the microbiome will soon be much better understood. Moreover, we are optimistic that the study of schistosomes will continue to increase fundamental understanding of the mechanisms governing immune-regulation and type 2 immunity. In turn, this could lead to the identification and generation of new vaccine candidates and targets for the treatment of egg-induced pathogenesis in schistosomiasis, as well as other type 2 inflammatory diseases.
Box 1. Outstanding questions
• Recent studies show schistosome infections to influence the composition of the gut microbiota. Do these changes reflect bacterial adaptations to the inflammatory environment established by schistosomes infections, or do schistosomes actively promote the colonization of select bacterial communities? Furthermore, do these microbiome fluctuations influence (i) egg excretion, (ii) pathogenesis, (iii) the immune landscape, and/or (iv) schistosome long-term survival?
• Which exact molecules mediate egg binding to the vascular endothelium, and which process(s) ultimately allow for egg extravasation?
• The effects of schistosome infections on cancer require further attention. Notably, why is the association between carcinogenesis and schistosome infection stronger for S. haematobium than for S. mansoni and S. japonicum?
• Many cells of the intestinal immune system have undefined roles in the instigation and maintenance of type 2 immunity during schistosome infection. Notably, what is the actual function of eosinophils and mast cells in the granuloma? Are chemosensory tuft cells involved in egg detection and the initiation of type 2 responses? Which intestinal macrophage subsets are involved? Do ILC2s participate in these immune reactions?
• Do worms and/or eggs secrete molecules that influence host intestinal barrier integrity? Does the secretion of such molecules promote egg penetration through host tissues?
• Which co-infections/pathologies in the gut are worsened by the inflammatory environment established by eggs?
• Does egg migration trigger the release of alarmins (e.g., IL-25, IL-33, and TSLP) in the intestine, and do these molecules influence the development of Th2 or regulatory responses in the gut?
• Serosal immune responses are poorly characterized. Do serosa-resident immune populations play a role in egg migration and how do eggs transit through these tissues?
• Mucins - Do they play a functional role in intestinal egg-migration or local immune modulation
Author Contributions
AC wrote the manuscript, AM and HS critically reviewed and modified the manuscript.
Funding
HS was supported by a ZonMW-VIDI grant (91714352) from the Netherlands Organisation for Scientific Research. AM was supported by MRC (MR/N013751/1) and the BBSRC (BB/P504543/1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The image in Figure 2 was captured by Angela Marley during her PhD studies in the MacDonald laboratory.
References
1. Warren KS, Mahmoud AA, Cummings P, Murphy DJ, Houser HB. Schistosomiasis mansoni in Yemeni in California: duration of infection, presence of disease, therapeutic management. Am J Trop Med Hyg. (1974) 23:902–9. doi: 10.4269/ajtmh.1974.23.902
2. Skelly PJ, Da'dara AA, Li XH, Castro-Borges W, Wilson RA. Schistosome feeding and regurgitation. PLoS Pathog. (2014) 10:e1004246. doi: 10.1371/journal.ppat.1004246
3. Moore DV, Sandground JH. The relative egg producing capacity of Schistosoma mansoni and Schistosoma japonicum. Am J Trop Med Hyg. (1956) 5:831–40. doi: 10.4269/ajtmh.1956.5.831
4. Fan PC, Kang YC. Egg production capacity of one-pair worms of Schistosoma japonicum in albino mice. Southeast Asian J Trop Med Public Health (2003) 34:708–12.
5. Hams E, Aviello G, Fallon PG. The Schistosoma granuloma: friend or foe? Front Immunol. (2013) 4:89. doi: 10.3389/fimmu.2013.00089
6. Ashton PD, Harrop R, Shah B, Wilson RA. The schistosome egg: development and secretions. Parasitology (2001) 122:329–38. doi: 10.1017/S0031182001007351
8. Wilson MS, Mentink-Kane MM, Pesce JT, Ramalingam TR, Thompson R, Wynn TA. Immunopathology of schistosomiasis. Immunol Cell Biol. (2007) 85:148–54. doi: 10.1038/sj.icb.7100014
9. Mebius MM, Van Genderen PJJ, Urbanus RT, Tielens AGM, De Groot PG, Van Hellemond JJ. Interference with the host haemostatic system by schistosomes. PLoS Pathog. (2013) 9:e1003781. doi: 10.1371/journal.ppat.1003781
10. Shariati F, Perez-Arellano JL, Carranza C, Lopez-Aban J, Vicente B, Arefi M, et al. Evaluation of the role of angiogenic factors in the pathogenesis of schistosomiasis. Exp Parasitol. (2011) 128:44–9. doi: 10.1016/j.exppara.2011.01.016
11. Doenhoff MJ, Pearson S, Dunne DW, Bickle Q, Lucas S, Bain J, et al. Immunological control of hepatotoxicity and parasite egg excretion in Schistosoma mansoni infections: stage specificity of the reactivity of immune serum in T-cell deprived mice. Trans R Soc Trop Med Hyg. (1981) 75:41–53. doi: 10.1016/0035-9203(81)90012-2
12. Phillips SM, Diconza JJ, Gold JA, Reid WA. Schistosomiasis in the congenitally athymic (nude) mouse. I Thymic dependency of eosinophilia, granuloma formation, and host morbidity. J Immunol. (1977) 118:594–9.
13. Mathew RC, Boros DL. Anti-L3T4 antibody treatment suppresses hepatic granuloma formation and abrogates antigen-induced interleukin-2 production in Schistosoma mansoni infection. Infect Immun. (1986) 54:820–6.
14. Byram JE, Von Lichtenberg F. Altered schistosome granuloma formation in nude mice. Am J Trop Med Hyg. (1977) 26:944–56. doi: 10.4269/ajtmh.1977.26.944
15. Cheever AW, Duvall RH. Schistosoma japonicum: migration of adult worm pairs within the mesenteric veins of mice. Trans R Soc Trop Med Hyg. (1982) 76:641–5. doi: 10.1016/0035-9203(82)90231-0
16. Cheever AW, Kamel IA, Elwi AM, Mosimann JE, Danner R. Schistosoma mansoni and S. haematobium infections in Egypt II Quantitative parasitological findings at necropsy. Am J Trop Med Hyg. (1977) 26:702–16. doi: 10.4269/ajtmh.1977.26.702
17. Kamel IA, Cheever AW, Elwi MA, Mosimann J, Ray D. Schistosoma Mansoni and S. Haematobium infections in Egypt: I. Evaluation of techniques for recovery of worms and eggs at necropsy*. Am J Trop Med Hyg. (1977) 26:696–701. doi: 10.4269/ajtmh.1977.26.696
18. Hernandez DC, Lim KC, Mckerrow JH, Davies SJ. Schistosoma mansoni: sex-specific modulation of parasite growth by host immune signals. Exp Parasitol. (2004) 106:59–61. doi: 10.1016/j.exppara.2004.01.003
19. Davies SJ, Grogan JL, Blank RB, Lim KC, Locksley RM, Mckerrow JH. Modulation of blood fluke development in the liver by hepatic CD4+ lymphocytes. Science (2001) 294:1358–61. doi: 10.1126/science.1064462
20. Harrison RA, Doenhoff MJ. Retarded development of Schistosoma mansoni in immunosuppressed mice. Parasitology (2009) 86:429–38. doi: 10.1017/S0031182000050629
21. Riner DK, Ferragine CE, Maynard SK, Davies SJ. Regulation of innate responses during pre-patent schistosome infection provides an immune environment permissive for parasite development. PLOS Pathog. (2013) 9:e1003708. doi: 10.1371/journal.ppat.1003708
22. Wolowczuk I, Nutten S, Roye O, Delacre M, Capron M, Murray RM, et al. Infection of mice lacking interleukin-7 (IL-7) reveals an unexpected role for IL-7 in the development of the parasite Schistosoma mansoni. Infect Immun. (1999) 67:4183–90.
23. Amiri P, Locksley RM, Parslow TG, Sadick M, Rector E, Ritter D, et al. Tumour necrosis factor alpha restores granulomas and induces parasite egg-laying in schistosome-infected SCID mice. Nature (1992) 356:604–7. doi: 10.1038/356604a0
24. Turner JD, Narang P, Coles MC, Mountford AP. Blood flukes exploit peyer's patch lymphoid tissue to facilitate transmission from the mammalian host. PLoS Pathog. (2012) 8:e1003063. doi: 10.1371/journal.ppat.1003063
25. Cao J, Liu WJ, Xu XY, Zou XP. Endoscopic findings and clinicopathologic characteristics of colonic schistosomiasis: a report of 46 cases. World J Gastroenterol. (2010) 16:723–7. doi: 10.3748/wjg.v16.i6.723
26. Da'dara AA, De Laforcade AM, Skelly PJ. The impact of schistosomes and schistosomiasis on murine blood coagulation and fibrinolysis as determined by thromboelastography (TEG). J Thromb Thrombol. (2016) 41:671–7. doi: 10.1007/s11239-015-1298-z
27. Thors CLE. Clustering of Schistosoma mansoni eggs produced in in vitro culture. Ann Clin Pathol. (2016) 4:1099.
28. Omran SA, Amer AM, El-Kaliouby AH, Eldin AA. Study of contact activation in endemic hepatosplenomegaly. Blood Coagul Fibrinolysis (1991) 2:659–62. doi: 10.1097/00001721-199110000-00012
29. Souza MR, Toledo CF, Borges DR. Thrombocytemia as a predictor of portal hypertension in schistosomiasis. Dig Dis Sci. (2000) 45:1964–70. doi: 10.1023/A:1005535808464
30. El-Bassiouni NE, El Bassiouny AE, Hussein NA, El-Sayed HH, Ibrahim IM, Lotfy MG, et al. The coagulation profile in hepatosplenic schistosomiasis. Blood Coagul Fibrinolysis (1998) 9:189–94. doi: 10.1097/00001721-199803000-00011
31. Keating JH, Wilson RA, Skelly PJ. No overt cellular inflammation around intravascular schistosomes in vivo. J Parasitol. (2006) 92:1365–9. doi: 10.1645/GE-864R.1
32. Fajtova P, Stefanic S, Hradilek M, Dvorak J, Vondrasek J, Jilkova A, et al. Prolyl oligopeptidase from the blood fluke schistosoma mansoni: from functional analysis to anti-schistosomal inhibitors. PLoS Negl Trop Dis. (2015) 9:e0003827. doi: 10.1371/journal.pntd.0003827
33. Figueiredo BC, Da'dara AA, Oliveira SC, Skelly PJ. Schistosomes enhance plasminogen activation: the role of tegumental enolase. PLoS Pathog. (2015) 11:e1005335. doi: 10.1371/journal.ppat.1005335
34. Wang Q, Da'dara AA, Skelly PJ. The human blood parasite Schistosoma mansoni expresses extracellular tegumental calpains that cleave the blood clotting protein fibronectin. Sci Rep. (2017) 7:12912. doi: 10.1038/s41598-017-13141-5
35. Leontovyč A, Ulrychová L, O'donoghue AJ, Vondrášek J, Marešová L, Hubálek M, et al. SmSP2: A serine protease secreted by the blood fluke pathogen Schistosoma mansoni with anti-hemostatic properties. PLoS Negl Trop Dis. (2018) 12:e0006446. doi: 10.1371/journal.pntd.0006446
36. Elzoheiry M, Da'dara AA, Bhardwaj R, Wang Q, Azab MS, El-Kholy ESI, et al. Intravascular Schistosoma mansoni cleave the host immune and hemostatic signaling molecule sphingosine-1-phosphate via tegumental alkaline phosphatase. Front. Immunol. (2018) 9:1746. doi: 10.3389/fimmu.2018.01746
37. Dewalick S, Tielens AGM, Van Hellemond JJ. Schistosoma mansoni: the egg, biosynthesis of the shell and interaction with the host. Exp Parasitol. (2012) 132:7–13. doi: 10.1016/j.exppara.2011.07.018
38. File S. Interaction of schistosome eggs with vascular endothelium. J. Parasitol. (1995) 81:234–8. doi: 10.2307/3283925
39. Ritter DM, Mckerrow JH. Intercellular adhesion molecule 1 is the major adhesion molecule expressed during schistosome granuloma formation. Infect Immun. (1996) 64:4706–13.
40. Lejoly-Boisseau H, Appriou M, Seigneur M, Pruvost A, Tribouley-Duret J, Tribouley J. Schistosoma mansoni: in vitro adhesion of parasite eggs to the vascular endothelium. Subsequent inhibition by a monoclonal antibody directed to a carbohydrate epitope. Exp Parasitol. (1999) 91:20–9. doi: 10.1006/expr.1999.4348
41. Secor WE, Dos Reis MG, Ramos EA, Matos EP, Reis EA, Do Carmo TM, et al. Soluble intercellular adhesion molecules in human schistosomiasis: correlations with disease severity and decreased responsiveness to egg antigens. Infect Immun. (1994) 62:2695–701.
42. Lukacs NW, Chensue SW, Strieter RM, Warmington K, Kunkel SL. Inflammatory granuloma formation is mediated by TNF-alpha-inducible intercellular adhesion molecule-1. J Immunol. (1994) 152:5883–9.
43. Dewalick S, Hensbergen PJ, Bexkens ML, Grosserichter-Wagener C, Hokke CH, Deelder AM, et al. Binding of von Willebrand factor and plasma proteins to the eggshell of Schistosoma mansoni. Int J Parasitol. (2014) 44:263–8. doi: 10.1016/j.ijpara.2013.12.006
44. Wu YP, Lenting PJ, Tielens AG, De Groot PG, Van Hellemond JJ. Differential platelet adhesion to distinct life-cycle stages of the parasitic helminth Schistosoma mansoni. J Thromb Haemost. (2007) 5:2146–8. doi: 10.1111/j.1538-7836.2007.02725.x
45. Ngaiza JR, Doenhoff MJ. Blood platelets and schistosome egg excretion. Proc Soc Exp Biol Med. (1990) 193:73–9. doi: 10.3181/00379727-193-42993
46. Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. (1998) 67:395–424. doi: 10.1146/annurev.biochem.67.1.395
47. Pinto-Almeida A, Mendes T, De Oliveira RN, Corrêa SDAP, Allegretti SM, Belo S, et al. Morphological characteristics of schistosoma mansoni PZQ-resistant and -susceptible strains are different in presence of praziquantel. Front Microbiol. (2016) 7:594. doi: 10.1590/0074-0276108022013011
48. Michaels RM, Prata A. Evolution and characteristics of Schistosoma mansoni eggs laid in vitro. J Parasitol. (1968) 54:921–30. doi: 10.2307/3277120
49. Baptista AP, Andrade ZA. Angiogenesis and schistosomal granuloma formation. Memorias Do Instituto Oswaldo Cruz (2005) 100:183–5. doi: 10.1590/S0074-02762005000200012
50. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature (2000) 407:249. doi: 10.1038/35025220
51. Cheever A. Hepatic vascular lesions in mice infected with Schistosoma mansoni. Arch Pathol. (1961) 72:648–57.
52. Jourdan PM, Roald B, Poggensee G, Gundersen SG, Kjetland EF. Increased vascularity in cervicovaginal mucosa with Schistosoma haematobium infection. PLoS Negl Trop Dis. (2011) 5:e1170. doi: 10.1371/journal.pntd.0001170
53. Osherov N, Ben-Ami R. Modulation of host angiogenesis as a microbial survival strategy and therapeutic target. PLoS Pathog. (2016) 12:e1005479. doi: 10.1371/journal.ppat.1005479
54. Loeffler DA, Lundy SK, Singh KP, Gerard HC, Hudson AP, Boros DL. Soluble egg antigens from schistosoma mansoni induce angiogenesis-related processes by up-regulating vascular endothelial growth factor in human endothelial cells. J Infect Dis. (2002) 185:1650–6. doi: 10.1086/340416
55. Freedman DO, Ottesen EA. Eggs of Schistosoma mansoni stimulate endothelial cell proliferation in vitro. J Infect Dis. (1988) 158:556–62. doi: 10.1093/infdis/158.3.556
56. Kanse SM, Liang O, Schubert U, Haas H, Preissner KT, Doenhoff MJ, et al. Characterisation and partial purification of Schistosoma mansoni egg-derived pro-angiogenic factor. Mol Biochem Parasitol. (2005) 144:76–85. doi: 10.1016/j.molbiopara.2005.08.001
57. Van De Vijver KK, Colpaert CG, Jacobs W, Kuypers K, Hokke CH, Deelder AM, et al. The host's genetic background determines the extent of angiogenesis induced by schistosome egg antigens. Acta Trop. (2006) 99:243–51. doi: 10.1016/j.actatropica.2006.08.011
58. Pereira TA, Xie G, Choi SS Syn WK, Voieta I, Lu J, et al. Macrophage-derived hedgehog ligands promotes fibrogenic and angiogenic responses in human Schistosomiasis mansoni. Liver Int. (2013) 33:149–61. doi: 10.1111/liv.12016
59. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. (2008) 88:125–72. doi: 10.1152/physrev.00013.2007
60. Moussion C, Ortega N, Girard J-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin'? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
61. Peng H, Zhang QX, Li XJ, Liu Z, Shen J, Sun R, et al. IL-33 contributes to Schistosoma japonicum-induced hepatic pathology through induction of M2 macrophages (vol 6, 29844, 2016). Sci Rep. 7:29844. doi: 10.1038/srep29844
62. Chabon JJ, Gebreab L, Kumar R, Debella E, Tanaka T, Koyanagi D, et al. Role of vascular endothelial growth factor signaling in Schistosoma-induced experimental pulmonary hypertension. Pulmonary Circul. (2014) 4:289–99. doi: 10.1086/675992
63. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. (2014) 14:667–85. doi: 10.1038/nri3738
64. Domingo EO, Warren KS. Pathology and pathophysiology of the small intestine in murine schistosomiasis mansoni, including a review of the literature. Gastroenterology (1969) 56:231–40.
65. Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity (2004) 20:623–35. doi: 10.1016/S1074-7613(04)00107-4
66. Fu CL, Odegaard JI, Hsieh MH. Macrophages are required for host survival in experimental urogenital schistosomiasis. FASEB J. (2015) 29:193–207. doi: 10.1096/fj.14-259572
67. Barron L, Wynn TA. Macrophage activation governs schistosomiasis-induced inflammation and fibrosis. Eur J Immunol. (2011) 41:2509–14. doi: 10.1002/eji.201141869
68. Fallon PG, Richardson EJ, Mckenzie GJ, Mckenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. (2000) 164:2585–91. doi: 10.4049/jimmunol.164.5.2585
69. Al Adnani MS. Concomitant immunohistochemical localization of fibronectin and collagen in schistosome granulomata. J Pathol. (1985) 147:77–85. doi: 10.1002/path.1711470202
70. Weinstock JV, Boros DL. Organ-dependent differences in composition and function observed in hepatic and intestinal granulomas isolated from mice with Schistosomiasis mansoni. J Immunol. (1983) 130:418–22.
71. Holzscheiter M, Layland LE, Loffredo-Verde E, Mair K, Vogelmann R, Langer R, et al. Lack of host gut microbiota alters immune responses and intestinal granuloma formation during schistosomiasis. Clin Exp Immunol. (2014) 175:246–57. doi: 10.1111/cei.12230
72. Dunne DW, Jones FM, Doenhoff MJ. The purification, characterization, serological activity and hepatotoxic properties of two cationic glycoproteins (alpha 1 and omega 1) from Schistosoma mansoni eggs. Parasitology (1991) 103(Pt 2), 225–236. doi: 10.1017/S0031182000059503
73. Karanja DM, Colley DG, Nahlen BL, Ouma JH, Secor WE. Studies on schistosomiasis in western Kenya: I. Evidence for immune-facilitated excretion of schistosome eggs from patients with Schistosoma mansoni and human immunodeficiency virus coinfections. Am J Trop Med Hyg. (1997) 56:515–21. doi: 10.4269/ajtmh.1997.56.515
74. Hoffmann KF, Cheever AW, Wynn TA. IL-10 and the dangers of immune polarization: excessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J Immunol. (2000) 164:6406–16. doi: 10.4049/jimmunol.164.12.6406
75. Jankovic D, Kullberg MC, Noben-Trauth N, Caspar P, Ward JM, Cheever AW, et al. Schistosome-infected IL-4 receptor knockout (KO) mice, in contrast to IL-4 KO mice, fail to develop granulomatous pathology while maintaining the same lymphokine expression profile. J Immunol. (1999) 163:337–42.
76. Brunet LR, Finkelman FD, Cheever AW, Kopf MA, Pearce EJ. IL-4 protects against TNF-alpha-mediated cachexia and death during acute schistosomiasis. J Immunol. (1997) 159:777–85.
77. Lundy SK, Lukacs NW. Chronic schistosome infection leads to modulation of granuloma formation and systemic immune suppression. Front Immunol. (2013) 4:39. doi: 10.3389/fimmu.2013.00039
78. Phythian-Adams AT, Cook PC, Lundie RJ, Jones LH, Smith KA, Barr TA, et al. CD11c depletion severely disrupts Th2 induction and development in vivo. J Exp Med. (2010) 207:2089–96. doi: 10.1084/jem.20100734
79. Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M, et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. (2010) 207:2097–111. doi: 10.1084/jem.20101563
80. Macdonald AS, Maizels RM. Alarming dendritic cells for Th2 induction. J Exp Med. (2008) 205:13–7. doi: 10.1084/jem.20072665
81. Jankovic D, Steinfelder S, Kullberg MC, Sher A. Mechanisms underlying helminth- induced Th2 polarization: default, negative or positive pathways? Chem Immunol Allergy (2006) 90:65–81.
82. Vella AT, Pearce EJ. CD4+ Th2 response induced by Schistosoma mansoni eggs develops rapidly, through an early, transient, Th0-like stage. J Immunol. (1992) 148:2283–90.
83. De Jong EC, Vieira PL, Kalinski P, Schuitemaker JH, Tanaka Y, Wierenga EA, et al. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse th cell-polarizing signals. J Immunol. (2002) 168:1704–9. doi: 10.4049/jimmunol.168.4.1704
84. Macdonald AS, Straw AD, Bauman B, Pearce EJ. CD8- dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol. (2001) 167:1982–8. doi: 10.4049/jimmunol.167.4.1982
85. Everts B, Perona-Wright G, Smits HH, Hokke CH, Van Der Ham AJ, Fitzsimmons CM, et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. (2009) 206:1673–80. doi: 10.1084/jem.20082460
86. Everts B, Hussaarts L, Driessen NN, Meevissen MHJ, Schramm G, Van Der Ham AJ, et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med. (2012) 209:1753. doi: 10.1084/jem.20111381
87. Webb LM, Lundie RJ, Borger JG, Brown SL, Connor LM, Cartwright ANR, et al. Type I interferon is required for T helper (Th) 2 induction by dendritic cells. EMBO J. (2017) 36:2404–18. doi: 10.15252/embj.201695345
88. Trottein F, Pavelka N, Vizzardelli C, Angeli V, Zouain CS, Pelizzola M, et al. A Type I IFN-Dependent Pathway Induced by <em>Schistosoma mansoni</em> Eggs in Mouse Myeloid Dendritic Cells Generates an Inflammatory Signature. J Immunol. (2004) 172:3011. doi: 10.4049/jimmunol.172.5.3011
89. Hagen J, Young ND, Every AL, Pagel CN, Schnoeller C, Scheerlinck JP, et al. Omega-1 knockdown in Schistosoma mansoni eggs by lentivirus transduction reduces granuloma size in vivo. Nat Commun. (2014) 5:5375. doi: 10.1038/ncomms6375
90. Kaisar MMM, Ritter M, Del Fresno C, Jónasdóttir HS, Van Der Ham AJ, Pelgrom LR, et al. Dectin-1/2–induced autocrine PGE(2) signaling licenses dendritic cells to prime Th2 responses. PLoS Biol. (2018) 16:e2005504.
91. De Oliveira Fraga LA, Torrero MN, Tocheva AS, Mitre E, Davies SJ. Induction of Type 2 Responses by Schistosome Worms during Prepatent Infection. J Infect Dis. (2010) 201:464–72. doi: 10.1086/649841
92. Leptak CL, Mckerrow JH. Schistosome egg granulomas and hepatic expression of TNF-alpha are dependent on immune priming during parasite maturation. J Immunol. (1997) 158:301–7.
93. Pichyangkul S, Saengkrai P, Webster HK. Plasmodium falciparum pigment induces monocytes to release high levels of tumor necrosis factor-alpha and interleukin-1 beta. Am J Trop Med Hyg. (1994) 51:430–5. doi: 10.4269/ajtmh.1994.51.430
94. Girgis NM, Gundra UM, Ward LN, Cabrera M, Frevert U, Loke PN. Ly6Chigh monocytes become alternatively activated macrophages in schistosome granulomas with help from CD4+ cells. PLoS Pathog. (2014) 10:e1004080. doi: 10.1371/journal.ppat.1004080
95. Nascimento M, Huang SC, Smith A, Everts B, Lam W, Bassity E, et al. Ly6Chi monocyte recruitment is responsible for Th2 associated host-protective macrophage accumulation in liver inflammation due to schistosomiasis. PLoS Pathog. (2014) 10:e1004282. doi: 10.1371/journal.ppat.1004282
96. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol. (2001) 167:6533–44. doi: 10.4049/jimmunol.167.11.6533
97. La Flamme AC, Patton EA, Bauman B, Pearce EJ. IL-4 plays a crucial role in regulating oxidative damage in the liver during schistosomiasis. J Immunol. (2001) 166:1903–11. doi: 10.4049/jimmunol.166.3.1903
98. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. (2009a) 5:e1000371. doi: 10.1371/journal.ppat.1000371
99. Pesce JT, Ramalingam TR, Wilson MS, Mentink-Kane MM, Thompson RW, Cheever AW, et al. Retnla (Relmα/Fizz1) Suppresses Helminth-Induced Th2-Type Immunity. PLoS Pathog. (2009b) 5:e1000393. doi: 10.1371/journal.ppat.1000393
100. Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, et al. Alternatively activated macrophage-derived RELM-α is a negative regulator of type 2 inflammation in the lung. J Exp Med. (2009) 206:937–52. doi: 10.1084/jem.2008204841009c
101. Truscott M, Evans DA, Gunn M, Hoffmann KF. Schistosoma mansoni hemozoin modulates alternative activation of macrophages via specific suppression of retnla expression and secretion. Infect Immun. (2013) 81:133–42. doi: 10.1128/IAI.00701-12
102. Arora M, Chen L, Paglia M, Gallagher I, Allen JE, Vyas YM, et al. Simvastatin promotes Th2-type responses through the induction of the chitinase family member Ym1 in dendritic cells. Proc Natl Acad Sci U.S.A. (2006) 103:7777–82. doi: 10.1073/pnas.0508492103
103. Atochina O, Da'dara AA, Walker M, Harn DA. The immunomodulatory glycan LNFPIII initiates alternative activation of murine macrophages in vivo. Immunology (2008) 125:111–21. doi: 10.1111/j.1365-2567.2008.02826.x
104. Dewals BG, Marillier RG, Hoving JC, Leeto M, Schwegmann A, Brombacher F. IL-4Ralpha-independent expression of mannose receptor and Ym1 by macrophages depends on their IL-10 responsiveness. PLoS Negl Trop Dis. (2010) 4:e689. doi: 10.1371/journal.pntd.0000689
105. Gong D, Shi W, Yi S-J, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. (2012) 13:31. doi: 10.1186/1471-2172-13-31
106. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
107. Assunção LS, Magalhães KG, Carneiro AB, Molinaro R, Almeida PE, Atella GC, et al. Schistosomal-derived lysophosphatidylcholine triggers M2 polarization of macrophages through PPARγ dependent mechanisms. Biochim Biophys ca Acta (2017) 1862:246–54.
108. Donnelly S, Stack CM, O'neill SM, Sayed AA, Williams DL, Dalton JP. Helminth 2-Cys peroxiredoxin drives Th2 responses through a mechanism involving alternatively activated macrophages. FASEB J (2008) 22:4022–32. doi: 10.1096/fj.08-106278
109. Zhu J, Xu Z, Chen X, Zhou S, Zhang W, Chi Y, et al. Parasitic antigens alter macrophage polarization during Schistosoma japonicum infection in mice. Parasit Vect. (2014) 7:122. doi: 10.1186/1756-3305-7-122
110. Oliveira MF, D'avila JC, Torres CR, Oliveira PL, Tempone AJ, Rumjanek FD, et al. Haemozoin in Schistosoma mansoni. Mol Biochem Parasitol. (2000) 111:217–21. doi: 10.1016/S0166-6851(00)00299-1
111. Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, Mckenzie ANJ. T1/St2-deficient mice demonstrate the importance of T1/St2 in developing primary T helper cell type 2 responses. J Exp Med. (2000) 191:1069–76. doi: 10.1084/jem.191.6.1069
112. Swartz JM, Dyer KD, Cheever AW, Ramalingam T, Pesnicak L, Domachowske JB, et al. Schistosoma mansoni infection in eosinophil lineage–ablated mice. Blood (2006) 108:2420–7. doi: 10.1182/blood-2006-04-015933
113. Sher A, Coffman RL, Hieny S, Scott P, Cheever AW. Interleukin 5 is required for the blood and tissue eosinophilia but not granuloma formation induced by infection with Schistosoma mansoni. Proc Natl Acad Sci USA (1990) 87:61–5. doi: 10.1073/pnas.87.1.61
114. Lenzi HL, Lenzi JA, Sobral AC. Eosinophils favor the passage of eggs to the intestinal lumen in schistosomiasis. Brazil J Med Biol Res. (1987) 20:433–5.
115. Mcdermott JR, Bartram RE, Knight PA, Miller HRP, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci USA. (2003) 100:7761–6. doi: 10.1073/pnas.1231488100
116. Rychter JW, Van Nassauw L, Brown JK, Van Marck E, Knight PA, Miller HR, et al. Impairment of intestinal barrier and secretory function as well as egg excretion during intestinal schistosomiasis occur independently of mouse mast cell protease-1. Parasite Immunol. (2010) 32:221–31. doi: 10.1111/j.1365-3024.2009.01182.x
117. Corliss BA, Azimi MS, Munson J, Peirce SM, Murfee WL. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation (2016) 23:95–121. doi: 10.1111/micc.12259
118. Camelo A, Rosignoli G, Ohne Y, Stewart RA, Overed-Sayer C, Sleeman MA, et al. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. (2017) 1:577–89. doi: 10.1182/bloodadvances.2016002352
119. Ramalingam TR, Pesce JT, Mentink-Kane MM, Madala S, Cheever AW, Comeau MR, et al. Regulation of helminth-induced Th2 responses by thymic stromal lymphopoietin. J Immunol. (2009) 182:6452–9. doi: 10.4049/jimmunol.0900181
120. Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med. (2016) 8:337ra65. doi: 10.1126/scitranslmed.aaf1938
121. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
122. Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J Exp Med. (2017) 215:2507–21. doi: 10.1084/jem.20170051
123. Turner JE., Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. Il-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951. doi: 10.1084/jem.20130071
124. Sattler S, Ling GS, Xu D, Hussaarts L, Romaine A, Zhao H, et al. IL-10-producing regulatory B cells induced by IL-33 (Breg(IL-33)) effectively attenuate mucosal inflammatory responses in the gut(). J Autoimmunity (2014) 50:107–22. doi: 10.1016/j.jaut.2014.01.032
125. Lee SH. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res. (2015) 13:11–8. doi: 10.5217/ir.2015.13.1.11
126. Onguru D, Liang Y, Griffith Q, Nikolajczyk B, Mwinzi P, Ganley-Leal L. Human schistosomiasis is associated with endotoxemia and toll-like receptor 2- and 4-bearing B cells. Am J Trop Med Hyg. (2011) 84:321–4. doi: 10.4269/ajtmh.2011.10-0397
127. Barsoum RS, Esmat G, El-Baz T. Human schistosomiasis: clinical perspective: review. J Adv Res. (2013) 4:433–44. doi: 10.1016/j.jare.2013.01.005
128. Ray D, Nelson TA, Fu CL, Patel S, Gong DN, Odegaard JI, et al. Transcriptional profiling of the bladder in urogenital schistosomiasis reveals pathways of inflammatory fibrosis and urothelial compromise. PLoS Negl Trop Dis. (2012) 6:e1912. doi: 10.1371/journal.pntd.0001912
129. Xia CM, Zhao Y, Jiang L, Jiang J, Zhang SC. Schistosoma japonicum ova maintains epithelial barrier function during experimental colitis. World J Gastroenterol. (2011) 17:4810–6. doi: 10.3748/wjg.v17.i43.4810
130. Mayer JU, Demiri M, Agace WW, Macdonald AS, Svensson-Frej M, Milling SW. Different populations of CD11b+ dendritic cells drive Th2 responses in the small intestine and colon. Nat Commun. (2017) 8:15820. doi: 10.1038/ncomms15820
131. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129:550–64. doi: 10.1016/j.gastro.2005.05.002
132. Sharpe C, Thornton DJ. A sticky end for gastrointestinal helminths; the role of the mucus Barrier Parasite Immunol. (2018) 40:e12517. doi: 10.1111/pim.12517
133. Mckenzie GJ, Bancroft A, Grencis RK, Mckenzie AN. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr Biol. (1998) 8:339–42. doi: 10.1016/S0960-9822(98)70134-4
134. Melo-Gonzalez F, Fenton TM, Forss C, Smedley C, Goenka A, Macdonald AS, et al. Intestinal mucin activates human dendritic cells and IL-8 production in a glycan-specific manner. J Biol Chem. (2018) 293:8543–53. doi: 10.1074/jbc.M117.789305
135. Grencis RK, Worthington JJ. Tuft Cells: a new flavor in innate epithelial immunity. Trends Parasitol. (2016) 32:583–5. doi: 10.1016/j.pt.2016.04.016
136. Von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature (2015) 529:221. doi: 10.1038/nature16161
137. Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature (2016) 529:226. doi: 10.1038/nature16527
138. Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (2016) 351:1329–33. doi: 10.1126/science.aaf1648
139. Rapin A, Harris NL. Helminth–Bacterial interactions: cause and consequence. Trends Immunol. (2018) 39:724–33. doi: 10.1016/j.it.2018.06.002
140. Zaiss MM, Rapin A, Lebon L, Dubey LK, Mosconi I, Sarter K, et al. The intestinal microbiota contributes to the ability of helminths to modulate allergic inflammation. Immunity (2015) 43:998–1010. doi: 10.1016/j.immuni.2015.09.012
141. Hayes KS, Bancroft AJ, Goldrick M, Portsmouth C, Roberts IS, Grencis RK. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science (2010) 328:1391–4. doi: 10.1126/science.1187703
142. Jenkins TP, Peachey LE, Ajami NJ, Macdonald AS, Hsieh MH, Brindley PJ, et al. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci Rep. (2018) 8:12072. doi: 10.1038/s41598-018-30412-x
143. Chelakkot C, Choi Y, Kim D-K, Park HT, Ghim J, Kwon Y, et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Amp Mol Med. (2018) 50:e450. doi: 10.1038/emm.2017.282
144. Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, et al. Commensal-pathogen interactions in the intestinal tract: Lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microb. (2014) 5:522–32. doi: 10.4161/gmic.32155
145. Li JV, Saric J, Wang Y, Keiser J, Utzinger J, Holmes E. Chemometric analysis of biofluids from mice experimentally infected with Schistosoma mansoni. Parasites Vectors (2011) 4:179. doi: 10.1186/1756-3305-4-179
146. Kay GL, Millard A, Sergeant MJ, Midzi N, Gwisai R, Mduluza T, et al. Differences in the faecal microbiome in schistosoma haematobium infected children vs. uninfected children. PLoS Negl Trop Dis. (2015) 9:e0003861. doi: 10.1371/journal.pntd.0003861
147. Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, et al. Helminth infection promotes colonization resistance via type 2 immunity. Science (2016) 352:608–12. doi: 10.1126/science.aaf3229
148. Budden KF, Gellatly SL, Wood DLA, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut–lung axis. Nat Rev Microbiol. (2016) 15:55. doi: 10.1038/nrmicro.2016.142
149. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. (2014) 20:159–66. doi: 10.1038/nm.3444
150. Boros DL, Pelley RP, Warren KS. Spontaneous modulation of granulomatous hypersensitivity in Schistosomiasis mansoni. J Immunol. (1975) 114:1437–41.
151. Turner JD, Jenkins GR, Hogg KG, Aynsley SA, Paveley RA, Cook PC, et al. CD4+CD25+ regulatory cells contribute to the regulation of colonic Th2 granulomatous pathology caused by schistosome infection. PLoS Negl Trop Dis. (2011) 5:e1269. doi: 10.1371/journal.pntd.0001269
152. Pearce EJ, Macdonald AS. The immunobiology of schistosomiasis. Nat Rev Immunol. (2002) 2:499–511. doi: 10.1038/nri843
153. Taylor JJ, Krawczyk CM, Mohrs M, Pearce EJ. Th2 cell hyporesponsiveness during chronic murine schistosomiasis is cell intrinsic and linked to GRAIL expression. J Clin Invest. (2009) 119:1019–28. doi: 10.1172/JCI36534
154. Kahl J, Brattig N, Liebau E. The untapped pharmacopeic potential of helminths. Trends Parasitol. (2018) 34:828–42. doi: 10.1016/j.pt.2018.05.011
155. Haeberlein S, Obieglo K, Ozir-Fazalalikhan A, Chayé MAM, Veninga H, Van Der Vlugt, LEPM et al. Schistosome egg antigens, including the glycoprotein IPSE/alpha-1, trigger the development of regulatory B cells. PLoS Pathog. (2017) 13:e1006539. doi: 10.1371/journal.ppat.1006539.
156. Van Der Kleij D, Latz E, Brouwers JF, Kruize YC, Schmitz M, Kurt-Jones EA, et al. (2002). A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune polarization. J Biol Chem. 277:48122–9. doi: 10.1074/jbc.M206941200
157. Floudas A, Cluxton CD, Fahel J, Khan AR, Saunders SP, Amu S, et al. Composition of the Schistosoma mansoni worm secretome: identification of immune modulatory cyclophilin A. PLoS Negl Trop Dis. (2017) 11:e0006012. doi: 10.1371/journal.pntd.0006012
158. Eichenberger RM, Ryan S, Jones L, Buitrago G, Polster R, Montes De Oca M, et al. Hookworm secreted extracellular vesicles interact with host cells and prevent inducible colitis in mice. Front Immunol. (2018) 9:850. doi: 10.3389/fimmu.2018.00850
159. Nowacki FC, Swain MT, Klychnikov OI, Niazi U, Ivens A, Quintana JF, et al. Protein and small non-coding RNA-enriched extracellular vesicles are released by the pathogenic blood fluke Schistosoma mansoni. J Extracell Vesicles (2015) 4:10.3402/jev.v4.28665. doi: 10.3402/jev.v4.28665
160. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
161. Nastasi C, Candela M, Bonefeld CM, Geisler C, Hansen M, Krejsgaard T, et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci Rep. (2015) 5:16148. doi: 10.1038/srep16148
162. Vatanen T, Kostic AD, D'hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
163. Gondorf F, Berbudi A, Buerfent BC, Ajendra J, Bloemker D, Specht S, et al. Chronic filarial infection provides protection against bacterial sepsis by functionally reprogramming macrophages. PLoS Pathog. (2015) 11:e1004616. doi: 10.1371/journal.ppat.1004616
164. Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. (2003) 171:4984–9. doi: 10.4049/jimmunol.171.10.4984
165. Du LL, Liang YB, Tang H, Chen ZB Li ZY, Wu JG, et al. The protective effect of chronic schistosoma japonica infestation against sepsis in mice. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue (2011) 23:290–3.
166. Tang H, Liang YB, Chen ZB, Du LL, Zeng LJ, Wu JG, et al. Soluble egg antigen activates M2 macrophages via the STAT6 and PI3K pathways, and schistosoma japonicum alternatively activates macrophage polarization to improve the survival rate of septic mice. J Cell Biochem. (2017) 118:4230–9. doi: 10.1002/jcb.26073
167. Li H, Wang S, Zhan B, He W, Chu L, Qiu D, et al. Therapeutic effect of Schistosoma japonicum cystatin on bacterial sepsis in mice. Parasites Vectors (2017) 10:222. doi: 10.1186/s13071-017-2162-0
168. Robinson MW, Donnelly S, Hutchinson AT, To J, Taylor NL, Norton RS, et al. A family of helminth molecules that modulate innate cell responses via molecular mimicry of host antimicrobial peptides. PLoS Pathog. (2011) 7:e1002042. doi: 10.1371/journal.ppat.1002042
169. Layland LE, Wagner H, Da Costa CU. Lack of antigen-specific Th1 response alters granuloma formation and composition in Schistosoma mansoni-infected MyD88-/- mice. Eur J Immunol. (2005) 35:3248–57. doi: 10.1002/eji.200526273
170. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1beta and interleukin-6 production. Nat Med. (2014) 20:1334–9. doi: 10.1038/nm.3680
171. Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, et al. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci USA. (2010) 107:20459–64. doi: 10.1073/pnas.1010337107
172. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, Van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of Th2 inflammation. Science (2011) 332:1284–8. doi: 10.1126/science.1204351
173. Mcsorley HJ, Chayé MAM, Smits HH. Worms: pernicious parasites or allies against allergies? Arasite Immunol. (2018). doi: 10.1111/pim.12574. [Epub ahead of print].
174. Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol. (2010) 125:1114–24.e8. doi: 10.1016/j.jaci.2010.01.018
175. Van Der Vlugt LEPM, Labuda LA, Ozir-Fazalalikhan A, Lievers E, Gloudemans AK, Liu K-Y, et al. Schistosomes induce regulatory features in human and mouse CD1dhi B cells: inhibition of allergic inflammation by IL-10 and regulatory T cells. PLoS ONE (2012) 7:e30883. doi: 10.1371/journal.pone.0030883
176. Liu JY Li LY, Yang XZ Li J, Zhong G, Wang J, et al. Adoptive transfer of dendritic cells isolated from helminth-infected mice enhanced T regulatory cell responses in airway allergic inflammation. Parasite Immunol. (2011) 33:525–34. doi: 10.1111/j.1365-3024.2011.01308.x
177. Smits HH, Hammad H, Van Nimwegen M, Soullie T, Willart MA, Lievers E, et al. Protective effect of Schistosoma mansoni infection on allergic airway inflammation depends on the intensity and chronicity of infection. J Allergy Clin Immunol. (2007) 120:932–40. doi: 10.1016/j.jaci.2007.06.009
178. Van Der Vlugt LE, Mlejnek E, Ozir-Fazalalikhan A, Janssen Bonas M, Dijksman TR, Labuda LA, et al. CD24(hi)CD27(+) B cells from patients with allergic asthma have impaired regulatory activity in response to lipopolysaccharide. Clin Exp Allergy (2014) 44:517–28. doi: 10.1111/cea.12238
179. Obieglo K, Schuijs MJ, Ozir-Fazalalikhan A, Otto F, Van Wijck Y, Boon L, et al. Isolated Schistosoma mansoni eggs prevent allergic airway inflammation. Parasite Immunol. (2018) 40:e12579. doi: 10.1111/pim.12579
180. Layland LE, Straubinger K, Ritter M, Loffredo-Verde E, Garn H, Sparwasser T, et al. Schistosoma mansoni-mediated suppression of allergic airway inflammation requires patency and Foxp3+ treg cells. PLoS Negl Trop Dis. (2013) 7:e2379. doi: 10.1371/journal.pntd.0002379
181. Edington GM, Nwabuebo I, Junaid TA. The pathology of schistosomiasis in Ibadan, Nigeria with special reference to the appendix, brain, pancreas and genital organs. Trans R Soc Trop Med Hyg. (1975) 69:153–6. doi: 10.1016/0035-9203(75)90027-9
182. Leutscher PDC, Høst E, Reimert CM. Semen quality in Schistosoma haematobium infected men in Madagascar. Acta Trop. (2009) 109:41–4. doi: 10.1016/j.actatropica.2008.09.010
183. Mbabazi PS, Andan O, Fitzgerald DW, Chitsulo L, Engels D, Downs JA. Examining the relationship between urogenital schistosomiasis and HIV infection. PLoS Negl Trop Dis. (2011) 5:e1396. doi: 10.1371/journal.pntd.0001396
184. Honeycutt J, Hammam O, Fu CL, Hsieh MH. Controversies and challenges in research on urogenital schistosomiasis-associated bladder cancer. Trends Parasitol. (2014) 30:324–32. doi: 10.1016/j.pt.2014.05.004
185. Downs JA, Dupnik KM, Van Dam GJ, Urassa M, Lutonja P, Kornelis D, et al. Effects of schistosomiasis on susceptibility to HIV-1 infection and HIV-1 viral load at HIV-1 seroconversion: a nested case-control study. PLoS Negl Trop Dis. (2017b) 11:e0005968. doi: 10.1371/journal.pntd.0005968
186. Secor WE, Shah A, Mwinzi PM, Ndenga BA, Watta CO, Karanja DM. Increased density of human immunodeficiency virus type 1 coreceptors CCR5 and CXCR4 on the surfaces of CD4(+) T cells and monocytes of patients with Schistosoma mansoni infection. Infect Immun. (2003) 71:6668–71. doi: 10.1128/IAI.71.11.6668-6671.2003
187. Downs JA, De Dood CJ, Dee HE, Mcgeehan M, Khan H, Marenga A, et al. Schistosomiasis and human immunodeficiency virus in men in tanzania. Am J Trop Med Hyg. (2017a) 96:856–62. doi: 10.4269/ajtmh.16-0897
188. Brindley PJ, Da Costa JMC, Sripa B. Why does infection with some helminths cause cancer? Trends cancer (2015) 1:174–82. doi: 10.1016/j.trecan.2015.08.011
189. Botelho MC, Alves H, Barros A, Rinaldi G, Brindley PJ, Sousa M. The role of estrogens and estrogen receptor signaling pathways in cancer and infertility: the case of schistosomes. Trends Parasitol. (2015) 31:246–50. doi: 10.1016/j.pt.2015.03.005
190. Toda KS, Kikuchi L, Chagas AL, Tanigawa RY, Paranaguá-Vezozzo DC, Pfiffer T, et al. Hepatocellular carcinoma related to schistosoma mansoni infection: case series and literature review. J Clin Transl Hepatol. (2015) 3:260–4. doi: 10.14218/JCTH.2015.00027
191. Kamal SM, Graham CS, He Q, Bianchi L, Tawil AA, Rasenack JW, et al. Kinetics of intrahepatic hepatitis C virus (HCV)-specific CD4+ T cell responses in HCV and Schistosoma mansoni coinfection: relation to progression of liver fibrosis. J Infect Dis. (2004) 189:1140–50. doi: 10.1086/382278
Keywords: Schistosoma mansoni, intestine, endothelium, type 2 immunity, immune modulation
Citation: Costain AH, MacDonald AS and Smits HH (2018) Schistosome Egg Migration: Mechanisms, Pathogenesis and Host Immune Responses. Front. Immunol. 9:3042. doi: 10.3389/fimmu.2018.03042
Received: 13 September 2018; Accepted: 10 December 2018;
Published: 20 December 2018.
Edited by:
Thiago Almeida Pereira, Stanford University, United StatesReviewed by:
Margaret M. Mentink-Kane, Biomedical Research Institute, United StatesFausto Edmundo Lima Pereira, Universidade Vila Velha, Brazil
Copyright © 2018 Costain, MacDonald and Smits. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hermelijn H. Smits, aC5oLnNtaXRzQGx1bWMubmw=