 Jill Ford1†
Jill Ford1† Angela Hughson1†Kihong Lim1
Angela Hughson1†Kihong Lim1 Susana V. Bardina2Wuyuan Lu3
Susana V. Bardina2Wuyuan Lu3 Israel F. Charo4
Israel F. Charo4 Jean K. Lim2
Jean K. Lim2 Deborah J. Fowell1*
Deborah J. Fowell1*- 1Department of Microbiology and Immunology, David H. Smith Center for Vaccine Biology and Immunology, Aab Institute of Biomedical Sciences, University of Rochester, Rochester, NY, United States
- 2Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Department of Biochemistry and Molecular Biology, Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD, United States
- 4Department of Medicine, Cardiovascular Research Institute, University of California, San Francisco, San Francisco, CA, United States
The chemokine CCL7 (MCP3) is known to promote the recruitment of many innate immune cell types including monocytes and neutrophils to sites of bacterial and viral infection and eosinophils and basophils to sites of allergic inflammation. CCL7 upregulation has been associated with many inflammatory settings including infection, cardiovascular disease, and the tumor microenvironment. CCL7's pleotropic effects are due in part to its ability to bind numerous chemokine receptors, namely CCR1, CCR2, CCR3, CCR5, and CCR10. CCL7-blockade or CCL7-deficiency is often marked by decreased inflammation and poor pathogen control. In the context of Leishmania major infection, CCL7 is specifically upregulated in the skin one-2 weeks after infection but its role in L. major control is unclear. To determine CCL7's impact on the response to L. major we infected WT and CCL7−/− C57BL/6 mice. L. major infection of CCL7-deficient mice led to an unexpected increase in inflammation in the infected skin 2 weeks post-infection. A broad increase in immune cell subsets was observed but was dominated by enhanced neutrophilic infiltration. Increased neutrophil recruitment was associated with an enhanced IL-17 gene profile in the infected skin. CCL7 was shown to directly antagonize neutrophil migration in vitro and CCL7 add-back in vivo specifically reduced neutrophil influx into the infected skin revealing an unexpected role for CCL7 in limiting neutrophil recruitment during L. major infection. Enhanced neutrophilic infiltration in CCL7-deficient mice changed the balance of L. major infected host cells with an increase in the ratio of infected neutrophils over monocytes/macrophages. To determine the consequence of CCL7 deficiency on L. major control we analyzed parasite load cutaneously at the site of infection and viscerally in the draining LN and spleen. The CCL7−/− mice supported robust cutaneous parasite control similar to their WT C57BL/6 counterparts. In contrast, CCL7-deficiency led to greater parasite dissemination and poor parasite control in the spleen. Our studies reveal a novel role for CCL7 in negatively regulating cutaneous inflammation, specifically neutrophils, early during L. major infection. We propose that CCL7-mediated dampening of the early immune response in the skin may limit the ability of the parasite to disseminate without compromising cutaneous control.
Introduction
The rapid recruitment of innate effector populations such as monocytes, macrophages, and neutrophils to the site of infection provides immediate pathogen control in many infectious settings. Innate recruitment is driven by pathogen-induced tissue expression of a variety of chemokines and cytokines that facilitate innate cell extravasation into the tissue and mobilization from the bone marrow. For pathogens that benefit from residency in the host, the recruited innate cells can also serve as cellular reservoirs of infection. Thus, the recruited monocytes and neutrophils can mediate direct pathogen killing but may also facilitate pathogen persistence and/or spread. It is likely that the magnitude and composition of the early innate infiltrate will influence the balance between pathogen control and persistence.
Members of the monocyte chemoattractant protein (MCP) family play central roles in the recruitment of innate effectors to sites of inflammation (1–4). CCL2 (MCP-1) and its main receptor CCR2 have been widely studied and linked to a host of inflammatory responses including infection, cardiovascular disease, neuro-inflammatory and degenerative disease, autoimmunity, and cancer. CCL7 (MCP-3) mediates effects on a host of innate and adaptive immune cell types through binding to numerous receptors including CCR1, CCR2, CCR3, CCR5, and CCR10 (5). In many settings, CCL7 genetic deletion or CCL7 blockade reduces innate immune cell recruitment, limits inflammation and renders mice more susceptible to infectious challenge (6–11). Correspondingly, CCL7 overexpression in tumors enhances recruitment of macrophages, dendritic cells, neutrophils, and natural killer cells and slows tumor growth (12–14). In contrast to their agonist actions, the function of MCPs are regulated by proteolytic cleavage that can results in antagonist activity (15–17). Matrix metalloproteinase cleaved-CCL7 binds to CCR1, 2, and 3 receptors but does not elicit signaling and dampens inflammation in vivo (16, 18, 19). In addition, high affinity binding of native CCL7 to CCR5 blocks receptor signaling and antagonizes CCR5 activation by CCL4 (MIP-1β) (20). Thus, disease-specific enhancement of CCL7 expression may lead to either agonist or antagonist activity depending on the local milieu.
Cutaneous infection with the protozoan parasite Leishmania major (L. major) is marked by the rapid influx of neutrophils within hours of infection (21, 22). This early neutrophil response is transient and its role in preventing or promoting parasite infection remains controversial (23–25). Neutrophils can harbor live parasites which can be transferred to tissue macrophages through uptake of infected apoptotic neutrophils or by direct parasite uptake after release from lytic neutrophils (26). L. major resists immune control through interference with a number of macrophage and dendritic cell functions in the infected dermis that can limit innate pathogen control and modulate T cell activation (27). Ultimately, the production of cytokines by CD4+ T cells is responsible for local control of infection (28, 29). Production of IFNγ activates macrophages for iNOS-dependent intracellular parasite killing that controls local parasite load and limits dissemination (30). In susceptible mouse strains, the dominance of T cell-derived IL-4 and IL-13, and the paucity of IFNγ- producing Th1 cells, prevents local parasite control and is associated with wide-spread parasite dissemination (31–34).
L. major can actively modulate the availability of chemokine signals for immune cell recruitment to the infection site (35). Specifically, parasite downregulation of CXCR3 chemokines (CXCL9,10) and CCL5 limits the recruitment of leishmaniacidal IFNγ-producing Th1 cells (36). In contrast, CCL7 is one of a limited number of chemokines to be upregulated specifically in the L. major infected dermis compared to other dermal proinflammatory stimuli (35). The role of CCL7 in L. major infection is not known. In humans, elevated expression of CCL7 has been associated with a late cutaneous stage of L. major infection that is dominated by lymphocytes and macrophages while granulocytes are poorly represented (37). Given CCL7s ability to bind to a host of different receptors on numerous immune cell types, and the possibility of acting as a chemokine antagonist, its impact on tissue immunity to infection is likely to be highly context and stage dependent.
In this current study, the use of CCL7-deficient mice (38) reveals a striking role for CCL7 in limiting the magnitude of local cutaneous inflammation. The enhanced dermal inflammation early in infection in the absence of CCL7 was characterized by elevated neutrophils, IL-4, and IL-17. CCL7 add-back specifically limited neutrophil recruitment to the infection site highlighting an unexpected role for CCL7 in controlling neutrophil numbers in the L. major infected dermis. Moreover, despite local control of L. major in the dermis, CCL7 deficient mice had slightly elevated parasite burdens in the dLN early in the infection and a striking increase in parasite load in the spleen late in infection. Thus, CCL7 plays an important role in limiting cutaneous inflammation following L. major infection and appears critical for visceral parasite control.
Materials and Methods
Mice
WT BALB/c and C57BL/6 mice were purchased from National Cancer Institute. CCL7−/− mice were provided by Dr. Israel Charo (UCSF) and further backcrossed to N12 C57BL/6.
Leishmania major Infection
Mice were infected intradermally in the ear with 2 × 105 infectious (PNA-selected) Leishmania major promastigotes (strain WHOM/IR/-/173) in 10 μl PBS as previously described (39). Parasite titres were determined by limiting dilution analysis. Briefly, ear suspensions were resuspended in 450 μl cRPMI. Fifty microliters of the suspension was plated in a 96 well flat bottom plate containing 150 μl of the parasite growth medium, HOSMEM II. 10-fold dilutions were carried out across the wells of the plate and titers were determined at 6 and 10 days post plating. L. major-RFP parasites (DsRed MHOM/IL/80/Friedlin) were provided by Dr. David Sacks (NIAID) (26).
Tissue Processing
Individual ears were split into dorsal and ventral sides and placed in 1 mL of 1 mg/mL collagenase/dispase (Roche) containing 1 mM CaCl2 and incubated for 30 min with agitation at 37°C. After 30 min, EDTA was added to a final concentration of 5 mM and ears were returned to 37° for another 5 min to complete dissociation. Ears were gently pushed through a strainer and washed twice with HBSS/2% FCS. Draining LNs (cervical) were gently pushed through a strainer and cells isolated.
Gene Expression
RNA from whole ear homogenates was isolated using the RNeasy Fibrous tissue kit from Qiagen. cDNA was synthesized using the Life Technologie's High Capacity cDNA Reverse Transcription kit. Samples were loaded onto TLDA cards and RT-PCR performed using 7900HT Fast Real-Time PCR System (Applied Biosystems) at the University of Rochester Genomics Center. HPRT and GAPDH were used as endogenous controls to normalize input cDNA and one WT sample used as the calibrator.
Protein Quantification
Individual ears were incubated in 500 μl of collagenase/dispase containing 1 mM CaCl2 as above, gently pushed through a strainer using a minimal volume of PBS to rinse strainer. Samples were centrifuged at 10,000 rpm and supernatants were collected for ELISA. Murine MCP-3 ELISA development Kit was purchased from Peprotech and assay was performed according to manufacturer's protocol.
Flow Cytometry
The following antibodies were used: anti-Ly6G/Ly-6C (RB6-8C5), anti-CD103 (2E7), APC anti-CD11b (M1/70), anti-Ly6C (HK1.4), anti-Ly6G (1A8), anti-F4/80 (BM8), and MHC-II (M5/114.15.2) from Biolegend; anti-CD11c (N418), anti-FcεR1a (MAR-1), anti-CD49b (DX5), and anti-CD4 (RM4-5) from Ebioscience; anti-CD45 (30-F11) from BD Biosciences; Live/Dead Aqua Fixable Dead Cell stain from Life Technologies.
Cytokine Analysis by ELISPOT
Single cell suspensions from ear tissue and draining lymph nodes were cultured on anti-cytokine mAb-coated elispot plates (Millipore) in the presence or absence of soluble Leishmania antigen (SLA) (equivalent to 1 × 106 parasites/ml) overnight. Plates were developed as previously described (39).
In vitro Neutrophil Migration Assay
Neutrophils were freshly isolated from mouse bone marrow using the EasySep Neutrophil enrichment kit (STEMCELL). Cell migration chambers (Millicell EZ slide eight-well glass, Millipore) were coated with 5 μg/ml recombinant mouse ICAM-1 with or without the indicated chemokines. For in vitro live cell imaging, neutrophils were placed in L15 medium (Invitrogen) in the chamber at 37°C, and video microscopy was conducted using TE2000-U microscope (Nikon) with × 20 magnification. Migration analysis was performed using Volocity software (Quorum).
CCL7 Add Back
Synthetic active CCL7 (pCCL7) and control inactive CCL7 (Ctrl) peptides were prepared as described (6). L. major infected WT C57BL/6 or CCL7−/− mice were injected i.v. with 500 ng of synthetic active CCL7 peptide or control peptide 2 times a day on days 9, 10, 11, 12, and 13 post infection for a total of 10 injections (6). Mice were euthanized and tissues harvested on day 14.
Statistical Analysis
Student t-test, Mann Whitney test, one way ANOVA with multiple comparisons or two way ANOVA were performed where indicated: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Ethics Statement
All animal experimentation in this study was reviewed and approved by the University Committee on Animal Resources (UCAR), the University's Institutional Animal Care and Use Committee (IACUC) under protocol “UCAR-2006-145R.” The University of Rochester and its animal research facilities are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International, and adheres to the humane use as dictated by the Animal Welfare Act, and “The Guide For the Care and Use of Laboratory Animals.” PHS Assurance number is D16-00188 (A3292-01).
Results
CCL7 Induction Following L. major Infection
Our previous work assessed the regulation of chemokines in the L. major infected dermis and found that both CCL1 and CCL7 were specifically upregulated by L. major in the infected dermis (35). While CCL1 was upregulated directly in the infected macrophage (35), CCL7 upregulation was detected from mRNA extracts of unfractionated infected skin tissue, but not from mRNA of infected macrophages, suggesting its induction in non-infected immune and/or stromal cells in the infected skin. To dissect the regulation of early host expression of CCL7 we initially determined the kinetics of CCL7 induction. Elevated CCL7 protein expression at the infection site peaked 1 and 2 weeks post-infection (Figure 1A). Basic fractionation of cells in the infected ear, revealed CCL7 was predominantly expressed by CD45- non-hematopoietic cells in the skin (Figure 1B) consistent with observations of cutaneous fibroblasts being a potent source of CCL7 (40, 41). Thus, CCL7 is upregulated within the tissue stroma 1–2 weeks post-Leishmania infection.
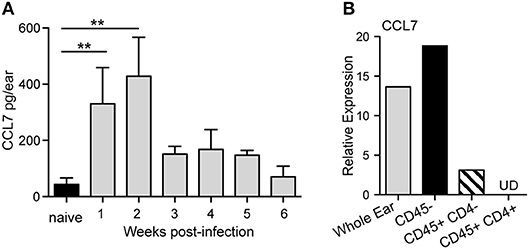
Figure 1. CCL7 expression following L. major infection. WT C57BL/6 mice were infected with L. major intradermally in the ear. (A) Ear tissue was homogenized at indicted times post-infection and the supernatants assayed for CCL7 protein by ELISA. Statistics by Mann Whitney. Four to six mice per group, two independent experiments. (B) Cells were isolated from 2 week L. major-infected ears, sorted into the respective cell populations and mRNA isolated. CCL7 mRNA measured by RT-PCR, normalized to HPRT, and expression levels calibrated to whole ear cell suspensions from uninfected mice. Results from mRNA pooled from 4 to 6 mice, one representative experiment of 2–3 independent comparable experiments. UD, undetected. **p ≤ 0.01.
Enhanced Inflammation in the L. major-Infected Skin in the Absence of CCL7
To determine the role of CCL7 in the immune response to L. major we infected WT and CCL7 deficient (CCL7 KO) C57BL/6 mice (38) with L. major and first analyzed the recruitment of immune cell subsets to the infected skin. Similar to previously published work on CCL7 deficient mice (6, 38), uninfected (steady state) CCL7 KO mice had a specific reduction in the frequency and number of monocytes in circulation, and in the spleen and skin (Supplementary Figure 1), but representation of other major immune cell subsets was similar to WT (6, 38), including neutrophils (Supplementary Figure 1). Steady state CD45 numbers in the uninfected skin were comparable between WT and CCL7 KO (Figure 2A). Two weeks post-infection, when CCL7 expression peaked at the infection site in WT mice (Figure 1A), the CCL7 KO mice exhibited a striking 4- to 5-fold increase in immune infiltration into the infected skin (Figures 2A,B). In contrast, WT and CCL7 KO cell numbers in the draining lymph node (LN) following L. major infection were comparable (Figure 2A, right panel). Phenotypic analysis of the CD45+ cells in the ear indicated that most immune cell types were elevated in the infected ear of CCL7 KO mice (Figures 2B–E). Neutrophils were the dominant immune cell type, with CD4+ T cells and eosinophils also major contributors to the infiltrate (Figures 2B–D). Monocyte/macrophage numbers in the infected ear were not significantly different between WT and CCL7 KO (Figure 2E, left panel). However, consistent with the reduced starting number of monocytes in the CCL7 KO (Supplementary Figure 1), there remained a significant and robust increased fold change in monocyte numbers in the infected ears compared to uninfected ears in the absence of CCL7 (mean fold change in monocyte numbers in the infected skin/uninfected skin: 11.2 for WT BL/6, 36.52 for CCL7 KO BL/6), similar to the other immune cell types (Figure 2E, right panel). Therefore, monocytes/macrophages can be recruited to the L. major infected skin independently of CCL7. The phenotypic distinction between neutrophils (CD11b+ Gr-1hi) and monocytes/macrophage (CD11b+ Gr-1lo/+) was confirmed with additional analysis of expression of Ly6C and Ly6G: neutrophils were CD11b+ Ly6C+ Ly6Ghi while monocytes/macrophage were CD11b+ Ly6C+ Ly6Glo, (Supplementary Figure 2). Moreover, the CD11b+ Gr-1hi neutrophils were negative for F4/80 and MHC Class II expression (Supplementary Figure 2). The heightened inflammatory response was not associated with a paucity of regulatory T cells: similar numbers, and frequency within the CD4 compartment, of Foxp3+ regulatory T cells were found at the infection site of CCL7 KO and WT mice (mean/SEM number/ear: WT 45.46 ± 11.7, CCL7 KO 48.54 ± 17.03). The enhanced accumulation of immune cells at the site of infection in the CCL7 KO was also not associated with a compensatory increase in expression of other monocyte chemoattractant proteins (MCPs) CCL2, CCL8, CCL12 compared to infected WT mice (Supplementary Figure 3). Therefore, CCL7 deficiency results in an unexpected enhancement in inflammation at the L. major infection site, suggesting CCL7 regulates the overall magnitude of the local immune response. These results are counter to the reported early decrease in immune infiltration into the infected brain following West Nile Virus (6) and decreases in inflammation on CCL7-blockade. Indeed, the unexpected augmented inflammatory response appears to be specific to L. major infection as no enhancement was seen in the size of the immune response in the ear on challenge with protein antigen and adjuvant (CFA or Alum) or on infection with C. albicans (Supplementary Figure 4). Kinetic analysis of the local immune response in the L. major infected ear revealed the immune changes to be remarkably transient. The increase in number of CD45+ cells and all respective subset analysis had resolved by week 4 of infection (Figures 2F,G), similar to the kinetics of CCL7 expression in WT mice (Figure 1).
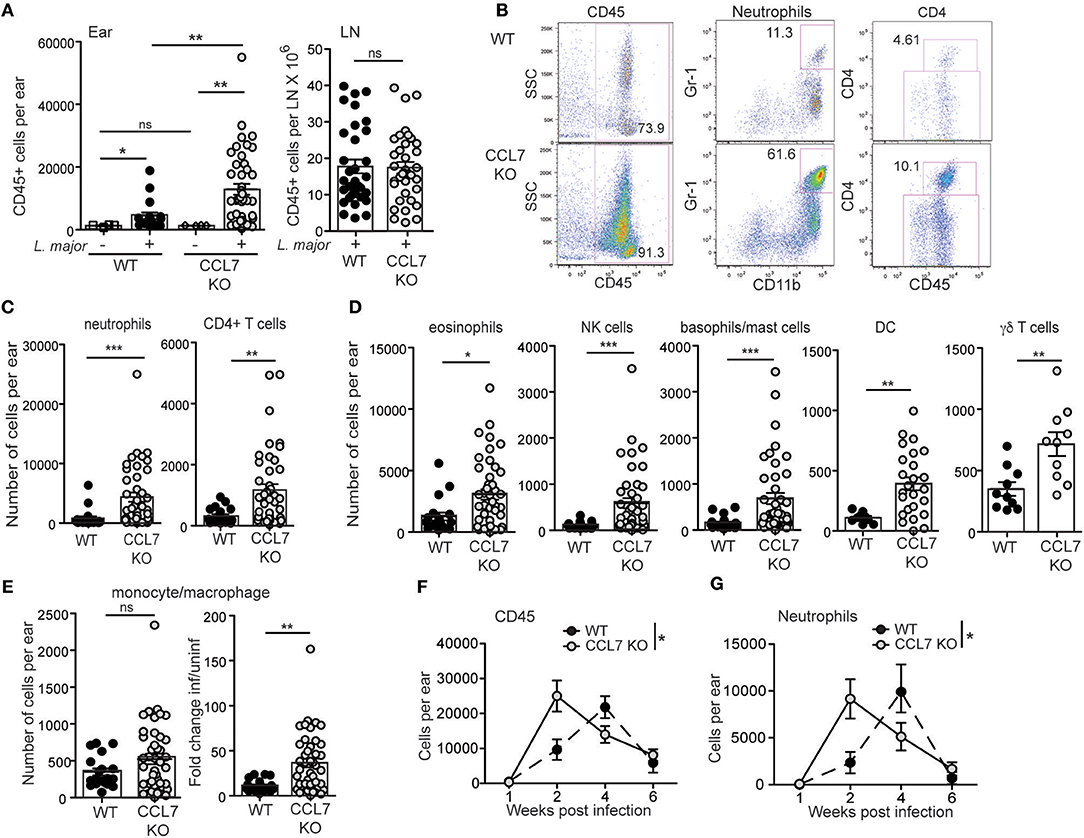
Figure 2. Enhanced inflammation at the L. major infection site in the absence of CCL7. WT and CCL7 KO mice were infected with L. major in the ear and immune cells in the infected tissue 2 weeks post-infection characterized by flow cytometry. (A) Leukocyte counts (CD45+ cells) in the uninfected (– L. major) and infected (+ L. major) ears (left panel) and draining LN (right panel) of C57BL/6 WT and CCL7 KO mice. (B) Representative FACS plots from 2 week-infected ears. Numbers in the FACS plots represent % of total cells for CD45 and % of CD45+ cells for neutrophils and CD4. (C) Neutrophils and CD4 T cell numbers in the ear 2 weeks post-infection. (D) Phenotypic analysis of CD45+ cells types, numbers of cells per ear 2 weeks post-infection. (E) Left, monocyte numbers per ear of 2 week infected mice; right, fold change relative to uninfected. Symbols represent individual mice, 4–6 mice per group, pooled from at least 4 independent experiments. Statistics by Mann Whitney. For all cell types in (C,D) P < 0.05 WT vs. CCL7 KO for additional paired analysis of the means of individual experiments (n = 4) by Wilcoxon matched-pairs rank test. (F,G) Kinetic analysis of inflammation in the ear following L. major infection. (F) Total number of CD45+ cells per ear by flow cytometry and (G) number of neutrophils per ear by flow cytometry. 4–5 mice per group per timepoint, 3 independent experiments. Statistics by two way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
We next determined the cytokine response associated with the elevated inflammation. The 2-week L. major infection timepoint is marked by an early CD4 T cell response that is dominated by IL-4 producing cells, with very few IFNγ producing cells, in both resistant (C57BL/6) and susceptible (BALB/c) mouse strains (35, 42–44). To determine if the loss of CCL7 alters the function of the developing CD4 T cell response to L. major, we performed L. major specific ELISPOTs from the dLN. CCL7-deficiency did not alter the generation, magnitude or cytokine balance of L. major-specific effector T cells in the dLN, with an early IL-4 dominance observed in both WT and CCL7 KO mice (Figure 3A) and a later Th1 protective response with predominant IFNγ production in both B6 and CCL7 KO mice (Figure 3B). However, at the 2 week timepoint of enhanced inflammation, the number of IL-4 producing cells, but not IFNγ producing cells, was significantly enhanced in the infected tissue in the CCL7 KO mice (Figure 3C). Thus, CCL7 deficiency appears to enhance the efficiency of early IL-4-producing effector T cell recruitment and/or retention at the infection site. A similar enhancement in IL-4 was seen in a lung model of fungal infection when CCL7 was neutralized (45). However, IL-4 does not appear to be the driver of the heightened inflammation as blockade of IL-4 (anti-IL-4 mAb, 11B11) did not alter the enhanced leukocytic infiltration (data not shown). At later timepoints after infection, the anti-leishmania T cell response in CCL7 KO C57BL/6 mice resembled that of their WT C57BL/6 counterparts with a predominance of IFNγ producers (Figure 3B) suggesting CCL7 is not required for the priming of a protective anti-leishmania Th1 response.
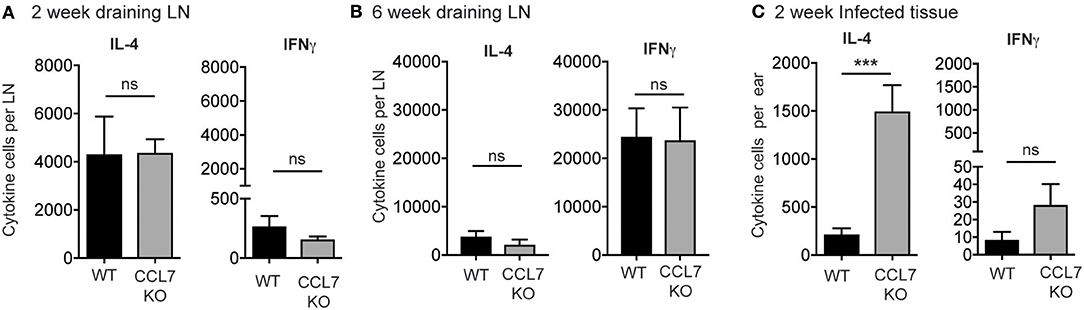
Figure 3. Elevated number of L. major-specific IL-4-producing cells in the infected ear in the absence of CCL7. WT and CCL7 KO mice were infected with L. major in the ear and cytokine-producing cells in the infected ear and draining LN analyzed by L. major-specific ELISPOT. Cytokine producing cells per LN 2 weeks post-infection (A) and 6 weeks post-infection (B). (C) Cytokine producing cells per infected ear 2 weeks post-infection. ***p ≤ 0.001.
Enhanced Neutrophilic Inflammation Is Associated With Increased Expression of IL-17 at the Infection Site
To identify possible mechanisms that facilitate the enhanced inflammation in the absence of CCL7 we analyzed cellular and gene expression changes in whole-ear extracts 10 days after L. major infection, immediately preceding the enhanced inflammatory infiltrate. Flow cytometry, 10 days post-infection, revealed that neutrophils were the only immune population to be significantly elevated in CCL7 KO mice at this time point (mean/SEM number/ear: WT 122.6 ± 44.11, CCL7 KO 501.6 ± 124.5, p ≤ 0.05) suggesting infiltration of neutrophils precedes the more general increase in immune recruitment at 2 weeks. We screened for changes in expression of 94 inflammatory genes (including chemokines, cytokines, and trafficking molecules) between WT and CCL7 KO infected mice using a custom gene array micro-fluidics card (36) (gene list, Supplementary Figure 5). Surprisingly, of the 94 genes tested, just 6 genes were significantly up-regulated and CCL7 was the only significantly downregulated gene in the CCL7 KO environment compared to WT (Figures 4A,B). The up-regulated genes IL-17a, IL-3, IL-6, IL-1β, and CCL3 are all associated with Type 17 inflammation and important drivers of local recruitment and expansion of neutrophils (46). Indeed, genetic ablation of IL-17 has been shown to reduce neutrophilic infiltration in L. major infected tissues (25, 47). IL-4 was also upregulated confirming the elevated IL-4 producing cells on Day 14 as measured by ELISPOT (Figure 3C). Thus, the absence of CCL7 induction early after L. major infection triggers a local IL-17 milieu in the skin that is consistent with the enhanced recruitment of neutrophils.
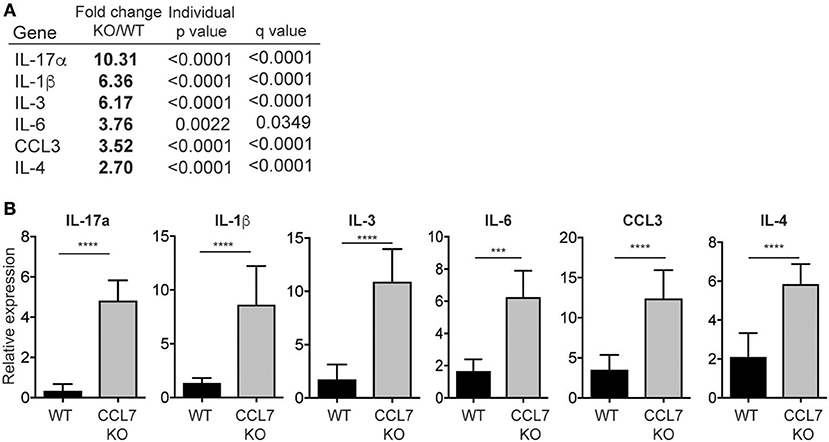
Figure 4. Neutrophil-promoting cytokines and chemokines upregulated following L. major infection in the absence of CCL7. mRNA was isolated from whole ear extracts of WT and CCL7 KO mice 10 days post-infection and expression of 94 inflammatory genes tested using a custom gene array micro-fluidics card (see Supplementary Figure 2 for gene list). (A) Fold change for all genes that were significantly differentially expressed between WT and CCL7 KO mice. Statistics by two way ANOVA, corrected for multiple comparisons with a 5% FDR (Benjamini, Frieger, and Yekutieli). (B) Relative gene expression for all genes significantly up-regulated in the absence of CCL7. Three to four mice per group, representative data from one of two independent experiments. ***p ≤ 0.001, ****p ≤ 0.0001.
CCL7 Negatively Regulates Neutrophil Migration and Recruitment to the L. major Infection Site
CCL7 is often associated with the positive regulation of myeloid cell recruitment to sites of inflammation and infection. In contrast, we found that in the absence of CCL7 there was enhanced recruitment of many immune cell types suggesting CCL7 may directly or indirectly dampen the magnitude of immune cell recruitment in some inflammatory/infectious settings.
In particular, the unexpected increase in neutrophil accumulation within the 2-week L. major infected dermis (Figure 2) resulted in the ratio of neutrophils to monocytes/macrophages being heavily skewed in favor of neutrophils (Figure 5A). This altered balance in neutrophil and monocyte/macrophage accumulation in the infected skin of CCL7 KO mice could be a consequence of the loss of CCL7 as a positive regulator of monocyte/macrophage recruitment or due to the loss of a previously undescribed CCL7-dependent pathway that antagonizes neutrophil recruitment. We focused on the possible role of CCL7 in negatively regulating neutrophil migration/recruitment in vitro and in vivo.
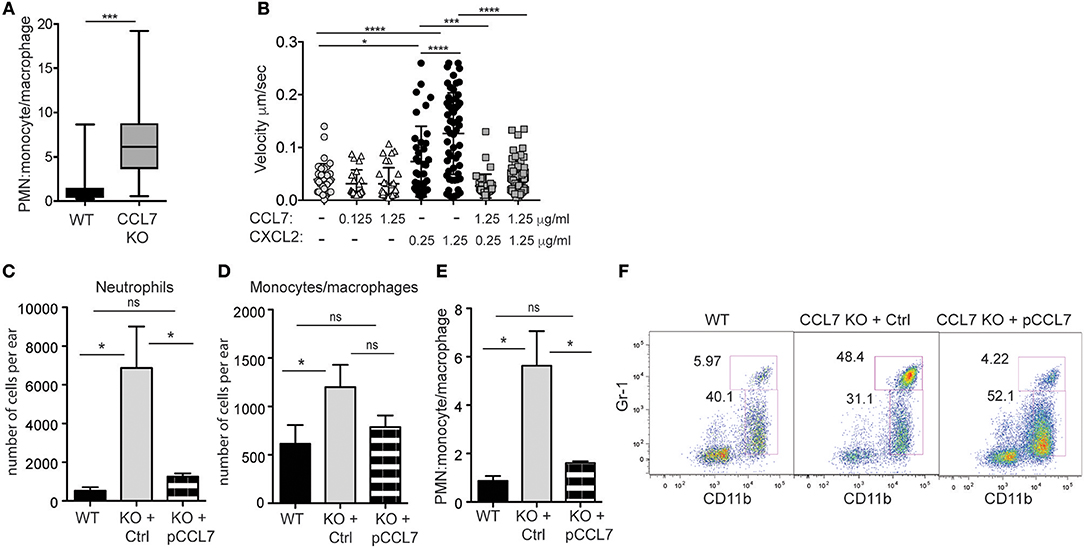
Figure 5. CCL7 negatively regulates neutrophilic accumulation in the L. major-infected dermis. Analysis of neutrophil (PMN) to monocyte/macrophage ratios in the 2 week L. major-infected skin by flow cytometry. (A) PMN:monocyte/macrophage ratio based on cell number. Four to six mice per group mice from at least four independent experiments. Statistics by Mann Whitney. (B) Live imaging of neutrophil migration on ICAM-1 coated-plates in response to indicated chemokines, at designated μg/ml. Three independent experiments. Statistics by one way ANOVA. (C–F) CCL7 add-back. Control, inactive (Ctrl), or active (pCCL7) CCL7 peptides were administered i.v. twice daily for 5 consecutive days starting day 9 post-infection and ear tissue harvested 2 weeks post-infection. Three to four mice per group, representative data from one of two independent experiments. (C) Number of neutrophils in infected ear. (D) Number of monocytes/macrophages in infected ear. (E) PMN:monocyte/macrophage ratio. (C–E) Statistics by one way ANOVA. (F) Representative flow cytometry. Numbers represent % of all CD45+ cells in the respective gates. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
In vitro, we compared neutrophil migration to CCL7 and CXCL2, a canonical chemotactic factor for neutrophils. By live-imaging (48), we examined the migration of freshly isolated neutrophils on ICAM-1 coated surfaces in the presence or absence of immobilized chemokines, alone or in combination. CXCL2 drove neutrophil migration in a dose-dependent manner, while CCL7 was poorly chemotactic (Figure 5B). However, when combined, the presence of CCL7 significantly inhibited neutrophil migration to CXCL2 (Figure 5B). These data suggest that CCL7 may directly alter the migration of neutrophils to sites of inflammation.
In vivo, to directly test the role of CCL7 in shaping the immune infiltrate in the infected tissue, we used synthetic active peptides of CCL7 to add-back CCL7 function to CCL7 KO mice (6). Control or active CCL7 peptides were administered twice daily as described (6) from day 9 to day 13 post infection, around the time of CCL7 induction in WT mice. CCL7 add-back led to an acute reduction in the number of neutrophils at the infection site (Figures 5C,F), with minimal changes in the number of monocytes/macrophages (Figures 5D,F). This change in the number of neutrophils, restored the neutrophil:monocyte/macrophage ratio in the infected ear to that seen in WT mice (Figures 5E,F). Thus, unexpectedly, CCL7 creates a tissue environment that appears to limit neutrophil accumulation in the context of cutaneous L. major. Although the magnitude of the differences between WT BL/6 and CCL7 KO was less in the spleen, neutrophils were also elevated in the spleen on L. major infection, relative to monocytes/macrophages, and the add-back of CCL7 significantly reduced neutrophil accumulation (Supplementary Figure 6), reinforcing a negative regulatory role for CCL7 in neutrophil accumulation in tissues.
Loss of CCL7 Results in Local Cutaneous Parasite Control but Enhanced Visceral Parasite Load
We next wanted to determine the physiological consequence of CCL7-deficiency on parasite control. The cellular distribution of L. major is known to change over time: neutrophils being the dominant host cell type hours after infection and monocyte/macrophages emerging as the predominant host cell type 6–7 days post-infection (26). This switch in cellular host has evoked mechanisms where infected-neutrophils “hand over” parasites to macrophages and/or DC by infected-neutrophil engulfment or direct parasite uptake following neutrophil lytic release (23, 26, 49). To determine the impact of the CCL7-deficiency on L. major host cell distribution, we used fluorescently-tagged L. major-RFP (26). Two weeks post-infection, the RFPhi cells in the infected WT ear were primarily CD11bhi Gr-1lo monocytes/macrophages (Figure 6A), as previously described (26). In contrast, CD11bhi Gr-1hi neutrophils remained the predominant RFP+ cells in the infected ear of CCL7-deficient mice (Figure 6A). We observed a heterogeneous pattern on RFP expression by infected cells, with a population of RFPhi cells (gated in Figure 6A) and a population with a lower RFP signal, RFPint cells. Analysis of both the RFPhi and RFPint populations showed, in the absence of CCL7, the balance of infected host cell types was skewed in favor of the neutrophil (Figure 6B). To determine if this change in L. major host cell type may influence the ability of the CCL7 KO mouse to control parasite growth, we directly compared the neutrophil:monocyte ratio with the splenic parasite load in individual CCL7 KO and WT BL/6 mice. We found a striking positive correlation between enhanced neutrophil:monocyte ratios and the inability to control parasites in the spleen (Figure 6C).
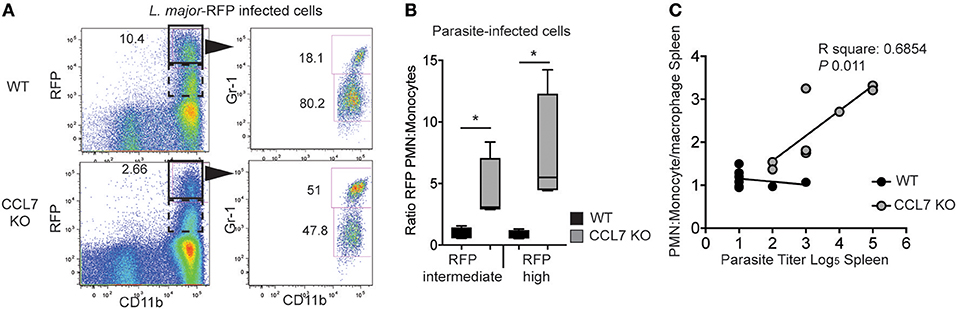
Figure 6. CCL7 deficiency skews the cellular host for L. major in favor of the neutrophil. L. major-RFP were used to phenotype the host cells harboring L. major in the infected ear. WT and CCL7 KO mice were infected intradermally with L. major-RFP and the phenotype of RFP+ host cells determined by flow cytometry 2 weeks post-infection. (A) Representative flow cyometry plots. Left panels, RFP by CD11b expression and gate used to further phenotype RFPhi cells (solid line) and RFPint (dashed line); right panels, Gr-1 and CD11b expression of the RFPhi cells with numbers indicating the frequency of RFPhi cells with these markers. (B) Ratio of the number of RFPint and RFPhi PMN to monocytes/macrophages. Three to four mice per group, data from three independent experiments. Statistics by one way ANOVA. (C) Linear regression of PMN:monocyte/macrophage ratio and parasite titer in the spleen. (R square: 0.077 WT and 0.685 CCL7 KO. P-value: 0.507 WT and 0.011 CCL7 KO). *p ≤ 0.05.
C57BL/6 mice are genetically resistant to L. major infection, locally controlling parasite numbers through a robust IFNγ-dependent immune response, and limiting parasite visceralization as measured in the spleen. In contrast, BALB/c mice are genetically susceptible to L. major infection and fail to control parasite numbers cutaneously and viscerally in the spleen, due to the induction of a dominant IL-4 response to the pathogen (28, 29). Given the robust infiltration of immune cells into the cutaneous site of infection (Figure 2), the appearance of parasites in the spleens of CCL7 deficient mice on the C57BL/6 background was unexpected (Figure 6C). To examine parasite control in more detail, we infected WT and CCL7 deficient (CCL7 KO BL/6) C57BL/6 mice with L. major subcutaneously in the ear and assessed the cutaneous lesion size over time and the parasite load locally in the skin and viscerally in the dLN and spleen. We included susceptible WT BALB/c mice as a positive control for elevated parasite burdens in the skin and spleen. CCL7 KO mice had an increased lesion size compared to WT C57BL/6 control mice 4 weeks after infection that was resolved by 8 weeks (Figure 7A), suggesting a possible delay in the ability to control parasite growth in the skin. However, kinetic analysis of cutaneous parasite loads using limiting dilution analysis showed control of parasite load in the skin of CCL7 KO that was comparable to their WT counterparts (Figures 7B,C). Thus, the increased lesion size at 4 weeks in the absence of CCL7 was not due to enhanced parasite load and may be a consequence of the altered cutaneous inflammatory response. To determine early dissemination of the parasite we analyzed parasite burdens in the skin-draining LN (dLN) over time post infection (Figures 7D–F). Temporal changes in dLN parasite load between CCL7 KO and WT C57BL/6 mice were not significantly different and both KO and WT C57BL/6 mice had controled parasite numbers by 8 weeks compared with BALB/c mice (Figure 7D). On more detailed analysis we found modest but significantly increased parasite numbers (~5-fold) in the dLN (2 week Figure 7E; 6 week Figure 7F) of mice lacking CCL7, suggesting enhanced dissemination from the skin. Parasites were first detected in the spleen at 4 weeks post-infection in all groups (Figure 7G). While WT C57BL/6 mice prevented further parasite growth in the spleen, CCL7 KO mice had significantly more parasites in the spleen at 6 and 8 weeks post-infection compared to WT C57BL/6, with levels approximating those seen for susceptible WT BALB/c mice (Figure 7G). To confirm this striking observation, we infected mice in an alternate subcutaneous site, the footpad. Similar to ear infection, the size of the footpad lesions and parasite loads in the footpad were not significantly different in the CCL7 KO mice compared to WT C57BL/6 mice throughout the 8 week timecourse (data not shown), but the CCL7 KO C57BL/6 mice remained unable to control parasite loads in the spleen (Figure 7H). Therefore, the absence of CCL7 does not alter the ability of mice to control parasite growth cutaneously (Figure 7C), but CCL7 appears critical for parasite control viscerally, as highlighted in the spleen (Figure 7H).
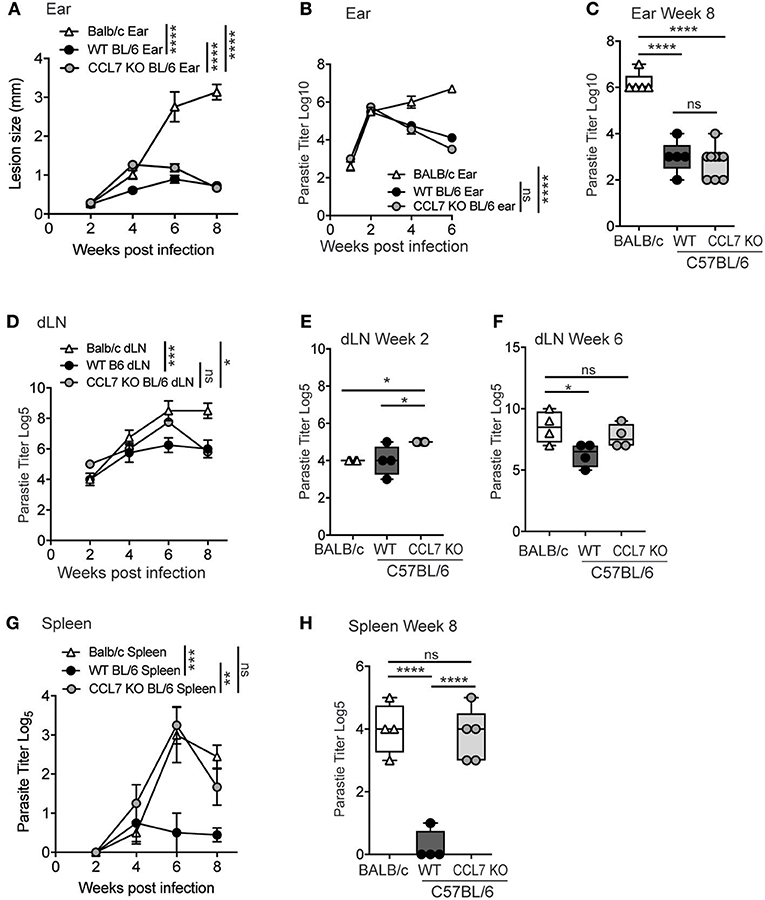
Figure 7. CCL7 deficiency maintains local parasite control but results in enhanced parasite load in the spleen. (A) WT BALB/c, WT C57BL/6 (BL/6), and CCL7 KO C57BL/6 (BL/6) mice were infected with L. major in the ear pinna and the lesion size measured weekly. Statistics by two way ANOVA. (B) Kinetic analysis of parasite load in the infected ear by limiting dilution analysis. Statistics by two way ANOVA. (C) Parasite load in the ear 8 weeks post infection. Statistics by one way ANOVA. (D) Kinetic analysis of parasite numbers in the draining LN by limiting dilution analysis, statistics by two way ANOVA. Parasite load in the dLN at 2 weeks (E) and 6 weeks (F) by limiting dilution analysis, statistics by one way ANOVA. (G) Kinetic analysis of parasite load in the spleen following cutaneous infection in the ear pinna, by limiting dilution analysis. Statistics by two way ANOVA. (H) Parasite load in the spleen 8 weeks post-infection in the footpad. Representative data from 1 of 3 independent experiments, 4–6 mice per group per timepoint per experiment. Statistics by one way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Discussion
We reveal a novel role for CCL7 in limiting cutaneous inflammation in response to infection with the protozoan parasite Leishmania major. In the absence of CCL7, mice exhibited enhanced dermal inflammation 2 weeks post-infection that was associated with an elevated IL-17 gene signature and an increase in neutrophil recruitment. CCL7 add-back reduced the local accumulation of neutrophils at the infection site suggesting a specific role for CCL7 in limiting neutrophil influx into infected tissues. Indeed, in vitro, we found that CCL7 could directly antagonize neutrophil migration. Disrupting CCL7 had important consequences for parasite control: cutaneous parasite control remained intact but there was a failure to contain parasites in the spleen. Thus, host induction of CCL7 in response to L. major infection appears critical for limiting neutrophilic inflammation in the first few weeks of infection and also protects against visceral parasite growth.
Our results are counter to a number of studies where CCL7 blockade mitigates inflammation and suggest that in certain settings in vivo CCL7 induction can be associated with antagonist activity that dampens inflammation. Most inflammatory and infectious studies have linked CCL7 to the promotion of inflammation with positive effects on recruitment of many innate immune cell types including neutrophils, NK cells, monocytes and DC (6, 10, 13, 50–53), and basophils and eosinophils (11, 54, 55). The balance of CCL7 agonist and antagonist activity with respect to leukocyte recruitment will likely depend on a variety of conspiring events in the local inflammatory milieu (56–59). The degree of pathogen or tissue leukocyte-driven proteolytic cleavage will regulate the degree of degradation vs. liberation of cleaved CCL7 with antagonist activity (16, 18). Indeed, the Leishmania metalloprotease GP63 is an extensively studied parasite virulence factor (60) and could directly alter chemokine activity. Competition between chemokines in the milieu for binding chemokine receptors and/or glycosaminoglycans (GAGs) may also regulate net chemotactic activity in the infected tissue. Differences between CCL7's monomeric binding to GAGs compared to oligomerizing CCL2 may change relative availability/duration and/or agonist activity (61). A number of natural chemokine antagonists have been described (56), including CCL7 that can bind to CCR5 and block CCL4 activity (20). Here we show that CCL7 can also antagonize neutrophil migration in response to CXCL2, despite no evidence for CCL7 binding to the CXCL2 receptor, CXCR2. How CCL7 mediates this effect will require additional mechanistic studies.
It is unclear how CCL7 negatively regulates cutaneous inflammation in the context of L. major. Our preliminary characterization of the cell types involved suggests the main source of CCL7 in the L. major-infected skin is non-hematopoietic stromal cells (Figure 1) (35) that are not directly L. major-infected suggesting CCL7 induction is indirect, triggered by other host factors in response to infection, possibly through IL-1β or IFNs (62, 63). CCL7 has also been associated with Type 2 responses (64) being linked to basophil and eosinophil recruitment and/or activation in allergic inflammation and has recently been shown to be induced by IL-33 (65). CCL7 induction early in L. major infection when IL-4 dominates the infected cutaneous milieu in both genetically susceptible and resistant mouse strains (35, 42–44) is consistent with such Type 2 instruction. However, what is unusual about our studies is the apparent role CCL7 plays in reducing the magnitude of inflammation in the L. major infected skin. This could be mediated through a direct negative effect on neutrophils themselves through active antagonism of chemokine receptors on infiltrating neutrophils (as seen in vitro). CCL7 may also work indirectly, by promoting the accumulation of myeloid cells to the infection site (66, 67) that might compete for, or inhibit, signals that enhance neutrophil accumulation. For example, in response to mucosal infection with Toxoplasma gondii incoming inflammatory monocytes directly inhibited neutrophil accumulation and activation at the site of infection and blocking monocyte recruitment led to severe neutrophil-dependent pathology (68). Additional indirect exacerbation of neutrophil accumulation in the absence of CCL7 could come from impaired neutrophil clearance due to reduced number or function of recruited monocytes/macrophage (69).
CCL7-deficiency reveals an unusual disconnect between robust parasite control in the skin and a failure to control parasite load in the spleen. At present, we are unclear if these two events are linked. The increased parasite load in the spleen in absence of CCL7 could arise due to increased dissemination from the skin and/or could be due to an enhanced ability of the parasites to proliferate in the spleen once there. Our analysis of parasite load in the draining LN suggests that initial dissemination to the LN may be enhanced in the absence of CCL7 and this effect might be subsequently amplified in the spleen. We failed to detect differences in L. major numbers in the spleen of CCL7 KO mice early after cutaneous infection, despite using an additional sensitive qPCR-based approach, suggesting that early seeding of the spleen does not appear to explain the elevated numbers late in infection. CCL7 may drive the recruitment of distinct monocyte subsets and/or numbers to the two tissues and these monocytes may differ in their ability to support or attenuate parasite growth as host cells or as drivers of protective Th1 immunity (70). In response to infection with a highly viscerally-prone Leishmania strain such as L. donovani, infection induces emergency hematopoiesis with the expansion of non-classical myeloid progenitors. These myeloid cells have a regulatory phenotype, are more permissive to infection and appear to facilitate parasite persistence in the spleen (71, 72). Indeed, we cannot rule out the possibility of our neutrophil or monocyte/macrophage populations containing myeloid-derived suppressor cells, indistinguishable by phenotypic analysis (73), which could account for the impaired parasite control in the spleen. Alternatively, CCL7 may recruit the same monocyte populations to both sites but differ in the negative regulation of neutrophil recruitment to specific tissues, where neutrophils can act as a “safe haven” for parasites (74, 75). Thus, there may be CCL7-dependent location-specific differences in immune requirements for clearance or in permissiveness of parasite growth (25). Future characterization of the splenic innate compartment following L. major infection in the presence and absence of CCL7 should shed light on local immune changes that are unique to the spleen.
In addition to possible CCL7 requirements specifically in the spleen, it is worth speculating on whether the enhanced cutaneous inflammation seen in the absence of CCL7 may play a direct role in enhancing the transport of L. major out of the skin to visceral sites. Despite intensive study of the host response to L. major in the infected dermis, the mechanisms whereby L. major exits the dermis and disseminates to visceral organs are poorly understood. Parasites could disseminate via lymphatic drainage or be carried out by motile host cells. The transient increase in the magnitude of cutaneous immune infiltration seen in the absence of CCL7, dominated by neutrophils, raises the possibility that this milieu provides increased cellular targets that aid in leishmania dissemination. Indeed, susceptibility to infection is reduced when neutrophil influx is limited via IL-17 deficiency (47). As suggested from in vivo imaging studies (26) and early immunohistological studies (21), neutrophils contain live parasites and appear to contribute little to the direct elimination of L. major. Rather, neutrophils may act as tissue reservoirs for L. major with lytic release of parasites promoting local lymphatic drainage and/or hand-off to other immune cells in the skin such as macrophages and DCs (25, 26, 49, 74, 76). Interestingly, recent measures of neutrophil life-span, particularly in inflammatory settings, have challenged the notion that neutrophils are short-lived (77), raising the possibility that neutrophils could represent a more long-term harbor for L. major and/or act as “slow release” vehicles at the infection site extending the timeframe for possible dissemination. In addition, neutrophil reverse migration back out of tissues, either via lymphatics (78–81) or retrograde transendothelial entry back into the bloodstream (82–84), provides a potential mechanism whereby neutrophils can directly transport L. major between tissues. Despite this attractive idea, we have been unable to detect parasite-containing cells in the peripheral blood of L. major infected WT or CCL7 KO mice. Nevertheless, host induction of CCL7 may limit the magnitude of the cutaneous immune response to protect against visceral disease by reducing the number of cellular “pathogen-transporters.”
Chemokines are inheritantly promiscuous and often play redundant roles in leukocyte recruitment to damaged or infected tissues. Characterization of disease-specific chemokine profiles have helped to define context-specific chemokine milieus, yet the functional contribution of single (or combinations of) chemokine players is unclear (56). Recently, CXCR2 deficient mice were shown to have a counterintuitive exaggerated inflammatory response linked to imbalanced myeloid cell populations (85). Our studies also highlight that a chemokine such as CCL7 that is usually associated with enhancing inflammation has an unexpected regulatory role in limiting cutaneous inflammation in the context of L. major infection, and specifically in negatively regulating neutrophil tissue accumulation. Limiting cutaneous inflammation following L. major infection via CCL7 may have potential therapeutic application for locally controlling the inflammation at the infection site and attenuating visceral parasite growth.
Author Contributions
JF, AH, KL, and DF designed the experiments. JF, AH, and KL performed the experiments. SB, WL, IC, JL, and KL provided reagents and advice on experimental design and data interpretation. JF and DF wrote the manuscript. AH, SB, JL, and DF edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Fowell lab for helpful comments on the manuscript. The L. major-RFP parasites were generously provided by Dr. David Sacks (NIAID). We thank Dr. Melanie Wellington (University of Rochester) for help with the C. albicans infections. The work was supported by NIH grants R01 AI072690 and P01 AI02851 to DF and T32 HL066988 to JF.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.03063/full#supplementary-material
References
1. Van Coillie E, Van Damme J, Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. (1999) 10:61–86. doi: 10.1016/S1359-6101(99)00005-2
2. Cheng JW, Sadeghi Z, Levine AD, Penn MS, Von Recum HA, Caplan AI, et al. The role of CXCL12 and CCL7 chemokines in immune regulation, embryonic development, and tissue regeneration. Cytokine (2014) 69:277–83. doi: 10.1016/j.cyto.2014.06.007
3. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. (2008) 26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326
4. Bianconi V, Sahebkar A, Atkin SL, Pirro M. The regulation and importance of monocyte chemoattractant protein-1. Curr Opin Hematol. (2018) 25:44–51. doi: 10.1097/MOH.0000000000000389
5. Ben-Baruch A, Xu L, Young PR, Bengali K, Oppenheim JJ, Wang JM. Monocyte chemotactic protein-3 (MCP3) interacts with multiple leukocyte receptors. C-C CKR1, a receptor for macrophage inflammatory protein-1 alpha/Rantes, is also a functional receptor for MCP3. J Biol Chem. (1995) 270:22123–8. doi: 10.1074/jbc.270.38.22123
6. Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, Charo IF, et al. Differential roles of chemokines CCL2 and CCL7 in monocytosis and leukocyte migration during West Nile virus infection. J Immunol. (2015) 195:4306–18. doi: 10.4049/jimmunol.1500352
7. Shang XZ, Chiu BC, Stolberg V, Lukacs NW, Kunkel SL, Murphy HS, et al. Eosinophil recruitment in type-2 hypersensitivity pulmonary granulomas: source and contribution of monocyte chemotactic protein-3 (CCL7). Am J Pathol. (2002) 161:257–66. doi: 10.1016/S0002-9440(10)64177-6
8. Girkin J, Hatchwell L, Foster P, Johnston SL, Bartlett N, Collison A, et al. CCL7 and IRF-7 mediate hallmark inflammatory and IFN responses following rhinovirus 1B infection. J Immunol. (2015) 194:4924–30. doi: 10.4049/jimmunol.1401362
9. Jose RJ, Williams AE, Mercer PF, Sulikowski MG, Brown JS, Chambers RC. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol. (2015) 194:6024–34. doi: 10.4049/jimmunol.1500124
10. Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, et al. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. (2008) 180:6846–53. doi: 10.4049/jimmunol.180.10.6846
11. Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol. (2010) 11:608–17. doi: 10.1038/ni.1883
12. Wetzel K, Menten P, Opdenakker G, Van Damme J, Grone HJ, Giese N, et al. Transduction of human MCP-3 by a parvoviral vector induces leukocyte infiltration and reduces growth of human cervical carcinoma cell xenografts. J Gene Med. (2001) 3:326–37. doi: 10.1002/jgm.191
13. Fioretti F, Fradelizi D, Stoppacciaro A, Ramponi S, Ruco L, Minty A, et al. Reduced tumorigenicity and augmented leukocyte infiltration after monocyte chemotactic protein-3 (MCP-3) gene transfer: perivascular accumulation of dendritic cells in peritumoral tissue and neutrophil recruitment within the tumor. J Immunol. (1998) 161:342–6.
14. Wetzel K, Struyf S, Van Damme J, Kayser T, Vecchi A, Sozzani S, et al. MCP-3 (CCL7) delivered by parvovirus MVMp reduces tumorigenicity of mouse melanoma cells through activation of T lymphocytes and NK cells. Int J Cancer (2007) 120:1364–71. doi: 10.1002/ijc.22421
15. Proost P, Struyf S, Couvreur M, Lenaerts JP, Conings R, Menten P, et al. Posttranslational modifications affect the activity of the human monocyte chemotactic proteins MCP-1 and MCP-2: identification of MCP-2(6-76) as a natural chemokine inhibitor. J Immunol. (1998) 160:4034–41.
16. McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood (2002) 100:1160–7.
17. Struyf S, Proost P, Vandercappellen J, Dempe S, Noyens B, Nelissen S, et al. Synergistic up-regulation of MCP-2/CCL8 activity is counteracted by chemokine cleavage, limiting its inflammatory and anti-tumoral effects. Eur J Immunol. (2009) 39:843–57. doi: 10.1002/eji.200838660
18. McQuibban GA, Gong JH, Tam EM, Mcculloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science (2000) 289:1202–6. doi: 10.1126/science.289.5482.1202
19. Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2,−7,−8, and−13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood (2008) 112:3455–64. doi: 10.1182/blood-2007-12-129080
20. Blanpain C, Migeotte I, Lee B, Vakili J, Doranz BJ, Govaerts C, et al. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood (1999) 94:1899–905.
21. Beil WJ, Meinardus-Hager G, Neugebauer DC, Sorg C. Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leukoc Biol. (1992) 52:135–42. doi: 10.1002/jlb.52.2.135
22. Muller K, van Zandbergen G, Hansen B, Laufs H, Jahnke N, Solbach W, et al. Chemokines, natural killer cells and granulocytes in the early course of Leishmania major infection in mice. Med Microbiol Immunol. (2001) 190:73–6. doi: 10.1007/s004300100084
23. Ribeiro-Gomes FL, Sacks D. The influence of early neutrophil-Leishmania interactions on the host immune response to infection. Front Cell Infect Microbiol. (2012) 2:59. doi: 10.3389/fcimb.2012.00059
24. Ritter U, Frischknecht F, van Zandbergen G. Are neutrophils important host cells for Leishmania parasites? Trends Parasitol. (2009) 25:505–10. doi: 10.1016/j.pt.2009.08.003
25. Goncalves-de-Albuquerque SDC, Pessoa ESR, Trajano-Silva LAM, De Goes TC, De Morais RCS, Da COCN, et al. The equivocal role of Th17 cells and neutrophils on immunopathogenesis of leishmaniasis. Front Immunol. (2017) 8:1437. doi: 10.3389/fimmu.2017.01437
26. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (2008) 321:970–4. doi: 10.1126/science.1159194
27. Liu D, Uzonna JE. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol. (2012) 2:83. doi: 10.3389/fcimb.2012.00083
28. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. (2002) 2:845–58. doi: 10.1038/nri933
29. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol. (2016) 16:581–92. doi: 10.1038/nri.2016.72
30. Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (1995) 375:408–11. doi: 10.1038/375408a0
31. Kopf M, Brombacher F, Kohler G, Kienzle G, Widmann KH, Lefrang K, et al. IL-4-deficient Balb/c mice resist infection with Leishmania major. J Exp Med. (1996) 184:1127–36. doi: 10.1084/jem.184.3.1127
32. Matthews DJ, Emson CL, Mckenzie GJ, Jolin HE, Blackwell JM, Mckenzie AN. IL-13 is a susceptibility factor for Leishmania major infection. J Immunol. (2000) 164:1458–62. doi: 10.4049/jimmunol.164.3.1458
33. Mohrs M, Ledermann B, Kohler G, Dorfmuller A, Gessner A, Brombacher F. Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J Immunol. (1999) 162:7302–8.
34. Stamm LM, Raisanen-Sokolowski A, Okano M, Russell ME, David JR, Satoskar AR. Mice with STAT6-targeted gene disruption develop a Th1 response and control cutaneous leishmaniasis. J Immunol. (1998) 161:6180–8.
35. Katzman SD, Fowell DJ. Pathogen-imposed skewing of mouse chemokine and cytokine expression at the infected tissue site. J Clin Invest. (2008) 118:801–11. doi: 10.1172/JCI33174
36. Lazarski CA, Ford J, Katzman SD, Rosenberg AF, Fowell DJ. IL-4 attenuates Th1-associated chemokine expression and Th1 trafficking to inflamed tissues and limits pathogen clearance. PLoS ONE (2013) 8:e71949. doi: 10.1371/journal.pone.0071949
37. Campanelli AP, Brodskyn CI, Boaventura V, Silva C, Roselino AM, Costa J, et al. Chemokines and chemokine receptors coordinate the inflammatory immune response in human cutaneous leishmaniasis. Hum Immunol. (2010) 71:1220–7. doi: 10.1016/j.humimm.2010.09.002
38. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. (2007) 117:902–9. doi: 10.1172/JCI29919
39. Fowell DJ, Magram J, Turck CW, Killeen N, Locksley RM. Impaired Th2 subset development in the absence of CD4. Immunity (1997) 6:559–69. doi: 10.1016/S1074-7613(00)80344-1
40. Malhotra D, Fletcher AL, Astarita J, Lukacs-Kornek V, Tayalia P, Gonzalez SF, et al. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol. (2012) 13:499–510. doi: 10.1038/ni.2262
41. Ong VH, Evans LA, Shiwen X, Fisher IB, Rajkumar V, Abraham DJ, et al. Monocyte chemoattractant protein 3 as a mediator of fibrosis: overexpression in systemic sclerosis and the type 1 tight-skin mouse. Arthritis Rheum. (2003) 48:1979–91. doi: 10.1002/art.11164
42. Belkaid Y, Mendez S, Lira R, Kadambi N, Milon G, Sacks D. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J Immunol. (2000) 165:969–77. doi: 10.4049/jimmunol.165.2.969
43. Scott P, Eaton A, Gause WC, Di Zhou X, Hondowicz B. Early IL-4 production does not predict susceptibility to Leishmania major. Exp Parasitol. (1996) 84:178–87. doi: 10.1006/expr.1996.0103
44. Stetson DB, Mohrs M, Mallet-Designe V, Teyton L, Locksley RM. Rapid expansion and IL-4 expression by Leishmania-specific naive helper T cells in vivo. Immunity (2002) 17:191–200. doi: 10.1016/S1074-7613(02)00363-1
45. Szymczak WA, Deepe GS Jr. The CCL7-CCL2-CCR2 axis regulates IL-4 production in lungs and fungal immunity. J Immunol. (2009) 183:1964–74. doi: 10.4049/jimmunol.0901316
46. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
47. Lopez Kostka S, Dinges S, Griewank K, Iwakura Y, Udey MC, Von Stebut E. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J Immunol. (2009) 182:3039–46. doi: 10.4049/jimmunol.0713598
48. Lim K, Hyun YM, Lambert-Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science (2015) 349:aaa4352. doi: 10.1126/science.aaa4352
49. Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog. (2012) 8:e1002536. doi: 10.1371/journal.ppat.1002536
50. Gonzalez J, Mouttalib S, Delage C, Calise D, Maoret JJ, Pradere JP, et al. Dual effect of chemokine CCL7/MCP-3 in the development of renal tubulointerstitial fibrosis. Biochem Biophys Res Commun. (2013) 438:257–63. doi: 10.1016/j.bbrc.2013.07.025
51. Mercer PF, Williams AE, Scotton CJ, Jose RJ, Sulikowski M, Moffatt JD, et al. Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am J Respir Cell Mol Biol. (2014) 50:144–57. doi: 10.1165/rcmb.2013-0142oc
52. Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, et al. CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J Immunol. (2002) 168:846–52. doi: 10.4049/jimmunol.168.2.846
53. Qiu Y, Zeltzer S, Zhang Y, Wang F, Chen GH, Dayrit J, et al. Early induction of CCL7 downstream of TLR9 signaling promotes the development of robust immunity to cryptococcal infection. J Immunol. (2012) 188:3940–8. doi: 10.4049/jimmunol.1103053
54. Dahinden CA, Geiser T, Brunner T, Von Tscharner V, Caput D, Ferrara P, et al. Monocyte chemotactic protein 3 is a most effective basophil- and eosinophil-activating chemokine. J Exp Med. (1994) 179:751–6. doi: 10.1084/jem.179.2.751
55. Walsh ER, Sahu N, Kearley J, Benjamin E, Kang BH, Humbles A, et al. Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma. J Exp Med. (2008) 205:1285–92. doi: 10.1084/jem.20071836
56. Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. (2004) 25:75–84. doi: 10.1016/j.it.2003.12.005
57. Proudfoot AEI, Johnson Z, Bonvin P, Handel TM. Glycosaminoglycan interactions with chemokines add complexity to a complex system. Pharmaceuticals (2017) 10:E70. doi: 10.3390/ph10030070
58. Stone MJ, Hayward JA, Huang C, E Huma Z, Sanchez J. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. (2017) 18:E342. doi: 10.3390/ijms18020342
59. Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. (2006) 6:907–18. doi: 10.1038/nri1964
60. Murase LS, De Souza JVP, De Lima Neto QA, De Mello TFP, Cardoso BM, Lera-Nonose D, et al. The role of metalloproteases in Leishmania species infection in the New World: a systematic review. Parasitology (2018) 145:1499–509. doi: 10.1017/S0031182018000367
61. Salanga CL, Dyer DP, Kiselar JG, Gupta S, Chance MR, Handel TM. Multiple glycosaminoglycan-binding epitopes of monocyte chemoattractant protein-3/CCL7 enable it to function as a non-oligomerizing chemokine. J Biol Chem. (2014) 289:14896–912. doi: 10.1074/jbc.M114.547737
62. Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. (2009) 175:2023–33. doi: 10.2353/ajpath.2009.090328
63. Menten P, Proost P, Struyf S, Van Coillie E, Put W, Lenaerts JP, et al. Differential induction of monocyte chemotactic protein-3 in mononuclear leukocytes and fibroblasts by interferon-alpha/beta and interferon-gamma reveals MCP-3 heterogeneity. Eur J Immunol. (1999) 29:678–85.
64. Romagnani S. Cytokines and chemoattractants in allergic inflammation. Mol Immunol. (2002) 38:881–5. doi: 10.1016/S0161-5890(02)00013-5
65. Kim J, Kim W, Le HT, Moon UJ, Tran VG, Kim HJ, et al. IL-33-induced hematopoietic stem and progenitor cell mobilization depends upon CCR2. J Immunol. (2014) 193:3792–802. doi: 10.4049/jimmunol.1400176
66. Pereira WF, Ribeiro-Gomes FL, Guillermo LV, Vellozo NS, Montalvao F, Dosreis GA, et al. Myeloid-derived suppressor cells help protective immunity to Leishmania major infection despite suppressed T cell responses. J Leukoc Biol. (2011) 90:1191–7. doi: 10.1189/jlb.1110608
67. Schmid M, Zimara N, Wege AK, Ritter U. Myeloid-derived suppressor cell functionality and interaction with Leishmania major parasites differ in C57BL/6 and BALB/c mice. Eur J Immunol. (2014) 44:3295–306. doi: 10.1002/eji.201344335
68. Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, et al. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med. (2013) 19:713–21. doi: 10.1038/nm.3189
69. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA. (1997) 94:12053–8. doi: 10.1073/pnas.94.22.12053
70. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity (2007) 26:519–31. doi: 10.1016/j.immuni.2007.01.017
71. Abidin BM, Hammami A, Stager S, Heinonen KM. Infection-adapted emergency hematopoiesis promotes visceral leishmaniasis. PLoS Pathog. (2017) 13:e1006422. doi: 10.1371/journal.ppat.1006422
72. Hammami A, Abidin BM, Charpentier T, Fabie A, Duguay AP, Heinonen KM, et al. HIF-1alpha is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PLoS Pathog. (2017) 13:e1006616. doi: 10.1371/journal.ppat.1006616
73. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
74. Laskay T, van Zandbergen G, Solbach W. Neutrophil granulocytes—Trojan horses for Leishmania major and other intracellular microbes? Trends Microbiol. (2003) 11:210–4. doi: 10.1016/S0966-842X(03)00075-1
75. Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, et al. Divergent roles for Ly6C+CCR2+CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PLoS Pathog. (2017) 13:e1006479. doi: 10.1371/journal.ppat.1006479
76. van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, et al. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. (2004) 173:6521–5. doi: 10.4049/jimmunol.173.11.6521
77. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What's your age again? Determination of human neutrophil half-lives revisited. J Leukoc Biol. (2013) 94:595–601. doi: 10.1189/jlb.1112571
78. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood (2005) 106:1843–50. doi: 10.1182/blood-2005-03-1281
79. Gorlino CV, Ranocchia RP, Harman MF, Garcia IA, Crespo MI, Moron G, et al. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J Immunol. (2014) 193:1966–74. doi: 10.4049/jimmunol.1301791
80. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun. (2015) 6:7139. doi: 10.1038/ncomms8139
81. Rigby DA, Ferguson DJ, Johnson LA, Jackson DG. Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin-dependent proteolysis and lipoxin-induced junctional retraction. J Leukoc Biol. (2015) 98:897–912. doi: 10.1189/jlb.1HI0415-149R
82. Buckley CD, Ross EA, Mcgettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
83. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
84. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. (2006) 80:1281–8. doi: 10.1189/jlb.0506346
Keywords: chemokine, CCL7, skin, inflammation, neutrophils, macrophage, parasite, Leishmania major
Citation: Ford J, Hughson A, Lim K, Bardina SV, Lu W, Charo IF, Lim JK and Fowell DJ (2019) CCL7 Is a Negative Regulator of Cutaneous Inflammation Following Leishmania major Infection. Front. Immunol. 9:3063. doi: 10.3389/fimmu.2018.03063
Received: 31 July 2018; Accepted: 11 December 2018;
Published: 08 January 2019.
Edited by:
Sofie Struyf, KU Leuven, BelgiumReviewed by:
Peter Clay Melby, The University of Texas Medical Branch at Galveston, United StatesJoao Santana Silva, University of São Paulo, Brazil
Copyright © 2019 Ford, Hughson, Lim, Bardina, Lu, Charo, Lim and Fowell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah J. Fowell, RGVib3JhaF9Gb3dlbGxAdXJtYy5yb2NoZXN0ZXIuZWR1
†These authors have contributed equally to this work