 Silvia Preite
Silvia Preite Bonnie Huang1,2
Bonnie Huang1,2 Jennifer L. Cannons
Jennifer L. Cannons Dorian B. McGavern
Dorian B. McGavern Pamela L. Schwartzberg
Pamela L. Schwartzberg- 1National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD, United States
- 2National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
- 3National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, United States
T follicular helper (Tfh) cells are a specialized population of CD4+ T cells that provide help to B cells for the formation and maintenance germinal centers, and the production of high affinity class-switched antibodies, long-lived plasma cells, and memory B cells. As such, Tfh cells are essential for the generation of successful long-term humoral immunity and memory responses to vaccination and infection. Conversely, overproduction of Tfh cells has been associated with the generation of autoantibodies and autoimmunity. Data from gene-targeted mice, pharmacological inhibitors, as well as studies of human and mice expressing activating mutants have revealed that PI3Kδ is a key regulator of Tfh cell differentiation, acting downstream of ICOS to facilitate inactivation of FOXO1, repression of Klf2 and induction of Bcl6. Nonetheless, here we show that after acute LCMV infection, WT and activated-PI3Kδ mice (Pik3cdE1020K/+) show comparable ratios of Tfh:Th1 viral specific CD4+ T cells, despite higher polyclonal Tfh cells in Pik3cdE1020K/+ mice. Thus, the idea that PI3K activity primarily drives Tfh cell differentiation may be an oversimplification and PI3K-mediated pathways are likely to integrate multiple signals to promote distinct effector T cell lineages. The consequences of dysregulated Tfh cell generation will be discussed in the context of the human primary immunodeficiency “Activated PI3K-delta Syndrome” (APDS), also known as “p110 delta-activating mutation causing senescent T cells, lymphadenopathy and immunodeficiency” (PASLI). Overall, these data underscore a major role for PI3K signaling in the orchestration of T lymphocyte responses.
Introduction
Naïve CD4+ T helper (Th) cells play pivotal roles in adaptive immunity through the differentiation into distinct cytokine-producing effector subsets that specifically fight a wide range of pathogens and tumors (1). T follicular helper (Tfh) cells provide help to B cells for the formation of germinal centers (GCs) (2–4), a specialized microenvironment where clonal expansion of B cells, immunoglobulin diversification, affinity maturation, and development of memory B and long-lived plasma cells occur in response to immune challenge (5). The outcome of GC reactions requires proper help provided by Tfh cells (6–9). Most successful human vaccines are based on the generation of long-term protective humoral responses derived from the interactions of Tfh and GC B cells; however, Tfh cells can also promote dysregulated responses and autoimmunity (7, 10, 11). It is, therefore, critical to understand factors that promote or limit Tfh cells to elicit tightly controlled GC responses.
Recent data from gene-targeted mice, as well as mice and humans expressing activating mutants of phosphatidylinositol 3-kinase delta (PI3Kδ), suggest that PI3K activity is an essential component of pathways driving Tfh cell and GC formation (12–16). In this review, we discuss PI3Kδ-mediated pathways involved in the generation, maintenance and function of Tfh cells, including cellular receptors that activate PI3K within T cells, molecular pathways activated, and implications for autoimmunity, with a focus on the genetic disease APDS/PASLI.
PI3K Signaling in Immunity
PI3K Signaling
The PI3Ks are a family of heterodimeric lipid kinases that are activated downstream of a variety of receptors, including growth factor, antigen, costimulatory, cytokine, chemokine, and Toll-like receptors (17, 18). Class IA PI3Ks consist of a p85 regulatory and a p110 catalytic subunit that catalyzes the addition of a phosphate to the membrane phospholipid PI(4,5)P2, to generate phosphoinositide 3,4,5-triphosphate (PIP3). PIP3 helps recruit signaling molecules containing pleckstrin homology and other PIP3-binding domains to the plasma membrane to propagate signaling cascades (Figure 1). Mammals express three class IA catalytic isoforms: the broadly expressed p110α and p110β, and p110δ, which is expressed primarily by immune cells (17). Notably, PI3Kδ is activated by a variety of cell-surface receptors that are critical for Tfh cell differentiation, localization and function, including the T-cell receptor, CD28, and ICOS co-receptors, and cytokine receptors (17).
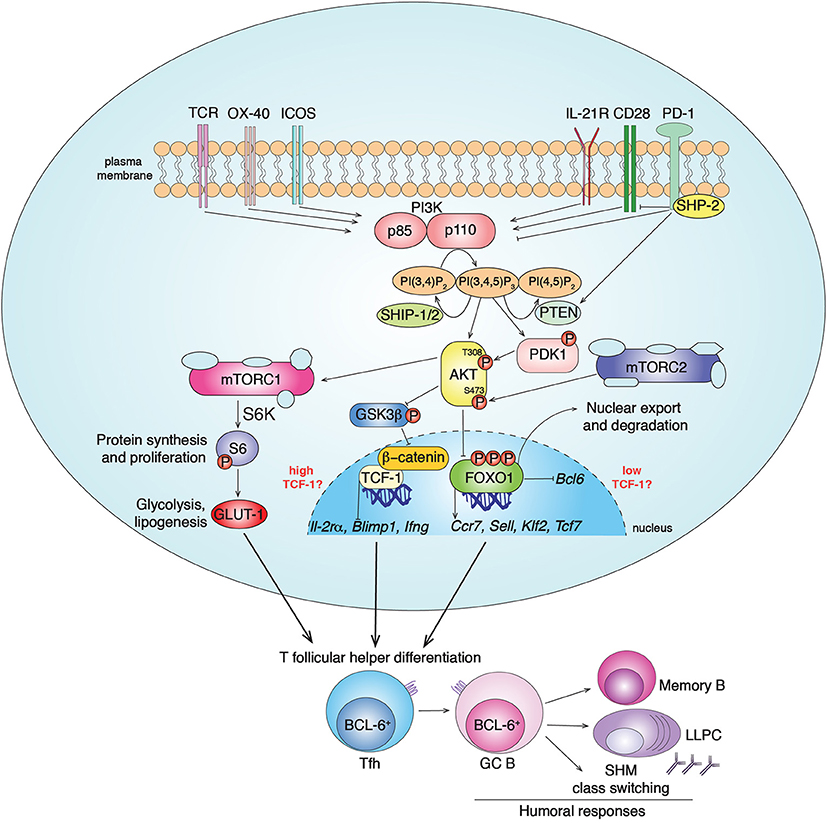
Figure 1. PI3K signaling pathways in Tfh cell differentiation. Class IA PI3Ks (PI3Kα, PI3Kβ, PI3Kδ) are lipid kinases composed by a regulatory (p85) and a catalytic (p110) subunit. Multiple receptors activate PI3K in CD4+ T cells, including TCR, CD28, ICOS, OX-40 and IL-21R, leading to PI3K recruitment to the plasma membrane and conversion of the membrane lipid PI(4,5)P2 to PI(3,4,5)P3. In T cells, chemokine receptors, including CXCR5, preferentially drive the activation of the class IB PI3Kγ (19). PI3K activity is counteracted by the inhibitor receptor PD-1 that blocks CD28 signal transduction through SHP-2 recruitment and PTEN induction. The phosphatases PTEN and SHIP-1/2 counteract PI3K signaling by converting PIP3 to PI(4,5)P2 and PI(3,4)P2, respectively. PIP3 recruits to the plasma membrane proteins containing pleckstrin homology domains, such as AKT and PDK1. The serine/threonine kinase AKT gets activated by phosphorylation by PDK1 (at Thr308) and mTORC2 (at Ser473). In turn, activated pAKT phosphorylates inhibitors of mTORC1 leading to its activation. mTORC1 phosphorylates several factors including S6-kinase (S6K), that phosphorylates S6, driving protein synthesis and cell proliferation, important events for Tfh cell differentiation. pAKT also phosphorylates the FOXO-1 transcription factor leading to its export outside the nucleus and degradation by binding 14-3-3 proteins. FOXO-1 represses Bcl6, essential for Tfh cells, and promotes expression of multiple genes including Sell, Tcf7, Ccr7, and Klf2. While KLF-2 restrains Tfh cell program through multiple mechanisms, TCF-1 promotes Tfh cell formation by inhibiting Il-2rα, Blimp1, Ifng. At the same time, it has been shown that mTORC2-pAKT may also support TCF-1 activity through the inactivation of GSK3β, an inhibitor of β-catenin and TCF-1. Overall, PI3K pathways drive BCL-6+ Tfh cell differentiation that coordinates GC responses and humoral immunity after infections and vaccination through the generation of memory B cells and long-lived plasma cells (LLPC).
Activated-PI3K coordinates the recruitment of molecules such as PDK1 that phosphorylates and activates the serine/threonine kinase AKT, which in turn phosphorylates multiple targets. Among these are the FOXO transcription factors, which are then sequestrated outside the nucleus by 14-3-3 proteins and degraded. FOXOs regulate transcription of multiple genes involved in lymphocyte development, differentiation and function (17, 20). Another downstream effector of PI3K is the mammalian Target of Rapamycin kinase (mTOR), which forms two complexes, mTORC1 and mTORC2, with different scaffolding partners (21). AKT activates mTORC1, an ancient regulator of metabolism, protein synthesis, and cell growth. mTORC2 is essential to fully phosphorylate and activate AKT, thus contributing to downstream signaling, including FOXO1 inactivation, and actin reorganization (21–23). PI3K is counteracted by the lipid-phosphatases PTEN and SHIP-1/2 that convert PIP3 to PI(4,5)P2 and PI(3,4)P2, respectively, (17) (Figure 1).
The importance of PI3Kδ in lymphocyte function is highlighted by the human primary immunodeficiency APDS/PASLI, in which patients are heterozygous for activating mutations in PIK3CD, the gene encoding p110δ. These patients show immunodeficiency and lymphopenia, as well as lymphoproliferation and autoimmunity (14, 15, 24, 25). Four independent groups, including us, have recently generated mouse models expressing the E1020K activating mutant of p110δ, which recapitulate many features of APDS/PASLI (16, 26–28). Notably, patients and Pik3cdE1020K/+ mice exhibit elevated circulating Tfh cells and GCs associated with autoantibody production (15, 16, 24, 29). Mice that express constitutively active p110α in T cells (30) or have a T cell-specific deletion of PTEN (13) also have elevated Tfh cell frequencies, supporting a more general connection between PI3K activity and Tfh cells. Nonetheless, the observation that p110δ-inactivation in T cells abrogates Tfh cell generation, supports a non-redundant role of p110δ in this process (12, 13). Together, these data provide strong evidence that PI3Kδ is an important component of pathways driving Tfh cell differentiation.
Tfh Cell Differentiation
The generation of Tfh cells is a multistage process that requires the integration of signals from different cell types (31). In the T cell zone of secondary lymphoid organs, antigen-presenting dendritic cells (DCs) activate T cells to initiate the pre-Tfh cell program, leading to induction of the costimulatory molecule ICOS and chemokine receptor CXCR5, as well as downregulation of CCR7, which together permit migration to the T-B cell border zone (32–34). Here, activated B cells receive signals from pre-Tfh cells to differentiate either along extra-follicular or GC pathways (5, 35). Cognate interactions with activated B cells help promote the differentiation into GC-Tfh cells (36), identified as CXCR5hiPD-1hiFoxp3−CD4+ T cells that also express high levels of ICOS, CD40L, and the Tfh-master transcription factor BCL-6, which are all critical for Tfh cell differentiation (37, 38). In turn, Tfh cells provide signals via costimulatory molecules and cytokines that help establish and maintain GCs. Thus, the generation of Tfh cells and GC reactions requires intimate communication between T and B cells involving multiple receptors that activate PI3K.
ICOS-PI3K Pathways in Tfh Cells
One of the key costimulatory receptors expressed by Tfh cells is ICOS, a CD28 family member. CD28 and ICOS both activate PI3Kδ and are required for Tfh cell development and function. CD28-CD80/CD86 interactions are involved in early T cell activation, including initial induction of ICOS, BCL-6, and CXCR5 (39), which are necessary for Tfh cell formation; Cd28−/− mice show a total absence of Tfh cells and thymus-dependent (TD) germinal centers (40–42). ICOS is upregulated on activated T cells shortly after TCR stimulation and interacts with ICOS-ligand (ICOS-L) on antigen presenting cells including DCs and B cells (43). Icos−/− and Icosl−/− mice display severely reduced humoral response to TD-antigens characterized by a lack of immunological memory and defective GC formation (44–49). Patients lacking ICOS display a common variable immunodeficiency (CVID) with reduced circulating Tfh cells (50, 51). Conversely, mutations affecting Roquin, a negative post-transcriptional regulator of ICOS mRNA, increase Tfh cells and drive autoimmunity (52, 53).
ICOS helps drive multiple stages of Tfh cell differentiation, including the early generation of CXCR5high T cells, modulation of other chemokine and homing receptors through regulation of the KLF2 transcription factor (39, 54), and T:B cell non-cognate interactions that promote T cell motility at the T:B cell border (55). ICOS-ICOS-L interactions are also critical for localization and maintenance of GC-Tfh cells (9, 39, 54).
The essential role of PI3K in ICOS function was highlighted by data showing that mutation of the p85-binding site, which selectively abrogates PI3K recruitment, led to defects in Tfh cell formation similar to ICOS-deficiency (56). Inhibition of p110δ also prevented ICOS-mediated changes in cell migration and morphology in vitro (55). Conversely, we found that activated-PI3Kδ mice show T cell-intrinsic increases in Tfh cell differentiation, even in the presence of blocking anti-ICOS-L antibody, therefore bypassing the requirement for ICOS for Tfh cell development (16). Thus, PI3K appears to be a major effector of ICOS, required for Tfh cell formation and maintenance.
PI3K Signaling Downstream of ICOS
After ICOS ligation, activated-PI3Kδ transduces its signals through several intermediates, including pAKT-mediated inactivation of FOXO1 (20). FOXO1 transcriptionally represses Bcl6, a driver of Tfh cell differentiation (30, 57, 58); strong PI3K activity relieves this repression. FOXO1 also transcriptionally activates Klf2 (59), which restrains Tfh cells and promotes alternative T helper subsets through at least four mechanisms: (1) induction of S1pr1, downregulation of which is essential for Tfh cell retention in GC and efficient polarization; (2) induction of BLIMP1, which negatively regulates Bcl6 and Tfh cell generation; (3) induction of T-bet and GATA3 which drives Th1 and Th2 cell differentiation, respectively; and (4) repression of Cxcr5 (39, 60). Accordingly, Foxo1−/− CD4+ T cells generate increased percentages of pre-Tfh cells (CXCR5intBCL-6int) early post-immunization, even in the presence of anti-ICOS-L (57), similar to cells expressing activated-PI3Kδ (16). These data support the ICOS-PI3Kδ-FOXO1 pathway as critical for Tfh cell development; accordingly, Pik3cdE1020K/+ CD4+ T cells exhibit elevated pFOXO1 upon TCR stimulation, even without further ICOS re-stimulation (16). Furthermore, an AKT-resistant mutant of FOXO1 prevents increased Tfh cells in the presence of activated-PI3Kδ (16). It is also of note that ICOS is a stronger inducer of PI3K than CD28, resulting in greater inhibition of FOXO1; this may account for the inability of CD28 to compensate for ICOS-deficiency in promoting Tfh cells (39, 42, 56, 61, 62). However, Foxo1−/− T cells show defective GC-Tfh (CXCR5highBCL-6high) cell formation (57) which is not observed with activated-PI3Kδ (16). Thus, cells expressing activated-PI3Kd likely still retain some FOXO1 activity. FOXO1 is required for sustained surface ICOS expression (57), providing a possible explanation for this defect. Indeed, chromatin immunoprecipitation and deep sequencing revealed FOXO1 binding sites in multiple genes that influence Tfh cell fate, including Cxcr4, Batf, Irf4, Icos, and Prdm1 (57, 63).
Nonetheless, despite increased GC-Tfh cell differentiation, Pik3cdE1020K/+ mice show disorganized GCs with increased Tfh cell infiltration and impaired class-switched antigen-specific responses to immunization (16, 27, 28). Multiple factors may contribute to these poor responses, including impaired B cell selection due to increased Tfh cells (7) and Tfh cell mis-localization (16), or intrinsic B cell defects. Indeed, although deletion of p110δ in B cells only minimally affected GC formation and T cell-dependent humoral responses after protein immunization (13), activated-PI3Kδ drove B cell-intrinsic increases in GC B and plasma cells, as well as impaired class-switched antibody production (16, 28). Increased GC B cells may in turn further drive expanded Tfh cell numbers, contributing to immune dysregulation. Additionally, increased GCs that fill the follicular dendritic cell network at baseline, may prevent new GC formation as mice age (16). Whether and how FOXO1 contributes to defects in antigen-specific responses or whether additional downstream effectors of PI3K are involved remain intriguing questions. It should also be noted that additional receptors expressed by Tfh cells, including OX-40, and IL-21R activate PI3K (64) and may contribute to expanded Tfh cell populations in these mice; in contrast, PD-1, an inhibitory receptor highly expressed by Tfh cells (65), counteracts PI3K by blocking CD28 signaling and increasing PTEN expression (66–69) (Figure 1).
Context-Dependent Roles For PI3K in T Cell Differentiation
Viral Infection
Although the connection between ICOS and PI3Kδ provides strong evidence for PI3Kδ driving Tfh cells, the view that PI3K exclusively promotes Tfh cells may be a simplification; this is particularly apparent when looking at the differentiation of Tfh vs. Th1 cells during viral infection. In response to viral or strong Th1 polarizing infections, CD4+ T cells undergo an early bifurcation such that up to 50% of viral-specific T cells express BCL-6 and become Tfh cells, while the rest express BLIMP1 and SLAM, and become IFNγ-producing Th1 cells (54). Although activated-PI3Kδ increased percentages of Tfh cells at baseline and in response to immunization (16), as well as polyclonal CXCR5+PD-1+ Tfh cells after LCMV infection (Figure 2A), it did not alter Tfh cell percentages, nor Tfh/Th1 ratios, within viral-specific GP66-tetramer+ CD4+ T cells in the same mice (Figures 2B,C). We also observed increased percentages of circulating CXCR3+ Tfh1 cells in patients with APDS/PASLI compared to controls (16), suggesting that PI3K can drive both Tfh cells and Type 1 immunity. Thus, PI3K activity may promote multiple effector T cell lineages and the effects of PI3K on Tfh cells may depend on the activating stimuli and microenvironment.
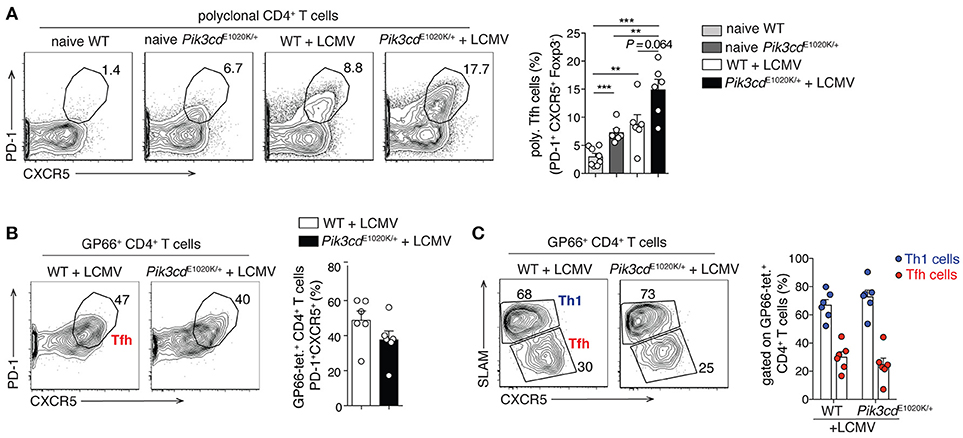
Figure 2. LCMV infection promotes comparable Th1/Tfh cell differentiation within GP66-tetramer+ T cells, despite increased polyclonal Tfh cells in Pik3cdE1020K/+ mice. 2/3-month-old WT and Pik3cdE1020K/+ mice were infected i.v. with LCMV Arm and analyzed in the spleen 7/8 days post infection. Naïve WT and Pik3cdE1020K/+ mice were analyzed as control. (A) Representative contour plots and summary histogram of polyclonal (GP66 tetramer negative) PD-1+CXCR5+Foxp3− Tfh cells (percentage of CD4+B220−GP66− T cells). (B) Representative contour plot and summary histogram of Tfh cells (PD-1+CXCR5+), gated on GP66-tetramer+ CD4+B220−Foxp3− T cells. (C) Representative contour plot and summary histogram of Th1 (SLAMhiCXCR5lo) and Tfh (SLAMloCXCR5hi) cells (gated on GP66-tetramer+CD4+B220−Foxp3− T cells). We were unable to evaluate GP66+ Tfr cells due to the low numbers of GP66+ Foxp3+ cells (70). However, polyclonal Tfr cells were increased after LCMV infection (data not shown), as we have previously reported in naïve mice (16). (A–C), n = 5–8. Data are representative of three independent experiments and are expressed as mean ± SEM with each dot indicating one mouse. Significance analyzed by Mann-Whitney U-test. ** p < 0.01; *** p < 0.001.
IL2 Signaling
Among potential PI3K-mediated signaling pathways that influence Tfh and Th1 cell differentiation are those downstream from the cytokine IL-2. Early data suggested that PI3K is activated by the IL-2R signaling complex (71–73); PI3K inhibitors arrest IL-2 induced CTL growth (74, 75). However, recent reports question the direct connection between IL-2 and PI3K activation (76), as that: (1) certain PI3K inhibitors (such as LY294002) have off-target effects (77); (2) many studies evaluate pAKTS473 and pS6, rather than pAKTT308, which more accurately reflects PI3K activity (78); and (3) IL-2 can promote mTORC1 activation independent of PI3K (79). Indeed, IL-2 potently inhibits Tfh cell generation via STAT5-mediated induction of BLIMP1 (80–82); BLIMP1+ Th1 cells express high levels of the high-affinity IL-2 receptor, CD25, and pSTAT5. As that IL-2 activates multiple signaling pathways, the integration, kinetics, and balance of these and other signals elicited in response to multiple receptors, may ultimately help determine T helper cell fates.
Metabolic Pathways in Tfh vs. Th1 Cells
Other PI3K-mediated signaling pathways that may influence both Tfh and Th1 cells are those involving mTORC1 and mTORC2. During acute LCMV infection, Th1 cells appear more proliferative and bio-energetically demanding with greater glucose metabolism and metabolic respiration than Tfh cells (83). Data suggest that these Th1 cells were more dependent on the IL-2-PI3K-AKT-mTORC1 axis, which preferentially promoted BLIMP1+ Th1 cells at the expense of BCL-6+ Tfh cells and humoral immunity (83, 84). However, other studies have demonstrated requirements for mTORC1 and mTORC2 in driving Tfh cells in Peyer's Patches at steady state and in the periphery after LCMV infection and immunization (30, 85). Mechanistically, Tfh cells were supported by mTORC1-promotion of pS6, GLUT1 expression, glycolysis, lipogenesis and overall proliferation; and by mTORC2-pAKT, which decreased FOXO1 activity (30).
While these studies provide conflicting conclusions on the requirements for PI3K and downstream effectors for Tfh cells, this may result from different experimental systems (knockdown vs. knockout) as well as bio-energetic demands during immune challenges. However, there is also evidence that mTOR may be activated independently of PI3K via pathways involving nutrient sensing that may also affect T helper cell differentiation (22, 79, 86, 87).
PI3K-TCF-1 Cross-Talk
Several recent studies revealed that the transcription factor TCF-1 is expressed at high levels in Tfh cells after viral infection and plays an essential role in their generation and maintenance, via repression of Il2ra, and Prdm1 (which encodes BLIMP1), promotion of Bcl6 (55, 88–90), and possibly repression of Ifng (91). Intriguingly, PI3K has been implicated both positively and negatively in TCF-1 regulation (92, 93). In CD8+ T cells, Tcf7, which encodes for TCF-1, is induced by FOXO1 (94), and both are required for memory T cell formation (95–98). Strong PI3K signaling would therefore be expected to decrease TCF-1 levels (25), as observed in studies of asymmetric cell division (92, 93). Conversely, a positive link between PI3K/AKT and TCF-1 has been proposed via β-catenin (85), a coactivator of TCF-1 that is negatively regulated by phosphorylation by Glycogen Synthase Kinase 3β (GSK3β), which is inactivated by pAKT (Figure 1). mTORC2-deficient T cells, which do not fully activate AKT, show reduced β-catenin and TCF-1 (85). Nonetheless, most studies implicating TCF-1 in Tfh cell generation have been done in the context of strong Th1-inducing infections (88–90), and how these findings relate to Tfh cells in other contexts remains unknown. Thus, the relationship between PI3K and TCF-1, how they affect Tfh cell differentiation, and involvement in possible feedback loops remain intriguing questions.
PI3K Pathways in Tfr Cells
A subset of thymic derived T regulatory cells, defined as T follicular regulatory (Tfr) cells, are localized at the T-B cell border and inside the GC area (99), and directly control the activation and differentiation of Tfh and GC B cells, including the development of autoimmunity (100). Tfr cells are phenotypically similar to Tfh cells and express BCL-6 (101–103), yet lack expression of B-cell-helper molecules, such as CD40L, and express inhibitory molecules CTLA-4, GITR, IL-10, and granzymes (104). Although excessive PI3K-mTOR activity is detrimental for induced-Treg cell differentiation (105–107), Pik3cdE1020K/+ mice show increased Treg and Tfr cells at steady state (16, 26). Indeed, although Tfr cells derive from Tregs, Tfr have different requirements for their differentiation and function. For example, IL-2 is necessary for Treg development and suppressive capability (108), yet prevents Tfr cell differentiation in a BLIMP1-dependent manner, similar to IL-2's effects on Tfh cells (109). Tfr cells also display high mTORC1 activity that promotes differentiation and STAT3 phosphorylation, which induces Tcf7 and Bcl6 (110); increased AKT-mTOR activity in Roquin-deficient Treg cells upregulates Tfh cell gene signatures that drive Tfr cell differentiation (111). How PI3K affects ratios of Tfh:Tfr cells, which are important for regulating humoral responses and autoimmunity (104), is less clear; notably, Pik3cdE1020K/+ mice have parallel increases in both cell populations (16).
Dysregulated PI3K Pathways in Autoimmunity
In addition to helping antigen-specific humoral responses to vaccination and infection, Tfh cells have been linked to autoimmunity in both animals and humans (10, 112). Correlations between circulating Tfh (cTfh) cells and disease have been reported in systemic lupus erythematosus (SLE) (113), rheumatoid arthritis (RA) and Sjögren's syndrome (112). Similarly, APDS/PASLI patients have high cTfh cells (16), and autoimmune manifestations including autoantibodies, cytopenias and glomerulonephritis (15, 29). In parallel, we found that Pik3cdE1020K/+ mice develop autoantibodies against a wide range of self-antigens. Indeed, PIDs caused by mutations affecting PI3K signaling cascades, or “immune TOR-opathies,” often display a combination of defective immune-responses and autoimmunity (114); animal models demonstrate that PI3K activity in B cells, T helper and regulatory T cells contributes to autoimmune manifestations (16, 115–121). Additionally, increased PI3K activity has been observed in several autoimmune diseases (121), and inhibitors of PI3Kδ and PI3Kγ are currently being explored in pre-clinical models of RA and SLE, and clinically for psoriasis (NCT02303509) and Sjögren's (NCT02775916) (121, 122).
Recent data suggest that certain autoantibodies cross-react with gut microbiota, supporting links between the microbiome and autoimmunity (123). Interestingly, we found increased local and systemic immune responses against gut commensals in Pik3cdE1020K/+ mice, with evidence for cross-reactivity between anti-self and anti-bacterial antibodies. Furthermore, autoantibodies could be prevented by systemic antibiotic treatment (16). Such data highlight roles for PI3Kδ in modulating T and B lymphocyte activation, including that induced by the microbiota, which can lead to autoimmunity.
Concluding Remarks
Together, a growing body of evidence supports a connection between PI3Kδ and Tfh cell differentiation, raising the possibility that altered PI3K pathways may contribute to both immunodeficiency and autoimmunity. Nonetheless, results during viral infection suggest that effects of PI3K on Tfh cell differentiation may be context-dependent and that PI3K may promote multiple effector cell lineages. A recent report demonstrated that treatment of APDS/PASLI patients with leniolisib, a selective PI3Kδ inhibitor, showed promising improvements in cellular dysfunction and lympho-proliferation (124) Notably, selective PI3Kδ inhibition reduced serum IgM in vivo (124), while increasing IgG class-switching in vitro (16, 27); however, effects on Tfh cells and autoantibodies have not yet been reported. Our results further suggest that evaluation of microbiota composition and systemic responses to gut commensals in APDS/PASLI may provide new opportunities, possibly in association with leniolisib, for managing this and other conditions where Tfh cells and autoantibodies contribute to pathogenesis. Such approaches may also be relevant for autoimmunity induced by checkpoint-blockade therapy, where PI3Kδ-inhibition may provide a selective control of immune responses. Finally, PI3Kδ activation may help improve vaccine responses, although this would have to be carefully assessed. Thus, a more comprehensive understanding of PI3K regulation and signaling in T and B cells is of crucial importance to more effectively improve humoral immune responses while minimizing autoimmunity.
Methods
Animal Care and Ethics
Control (C57Bl/6J) and Pik3cdE1020K/+ mice (16) were maintained and treated under specific pathogen-free (SPF) conditions under protocols reviewed and approved by the NINDS (protocol 1295-12) and NHGRI (protocol G98-3) Animal Care and Use Committees at the NIH.
LCMV Infection and Flow-Cytometry
Mice were injected intravenously (i.v.) with 2*105 plaque-forming units (PFUs) of LCMV Armstrong, kindly provided by Dorian McGavern Lab, grown as previously described (88). Day+7/8 post infection, single cell suspensions were prepared from spleen in MACS buffer (PBS with 2% FBS and 2 μM EDTA). GP66 tetramer [I-A(b) QVYSLIRPNENPAHK PE] was obtained from NIH tetramer facility (Emory University); staining was performed at 37°C for 1 h in RPMI with 10% serum. CXCR5 staining was performed using: CXCR5-purified (2G8, BD Biosciences), followed by Biotin-SP AffiniPure Fab Fragment Goat Anti-Rat IgG (H+L) (Jackson ImmunoResearch), and Streptavidin (BioLegend) as previously described (88). The following antibodies (obtained from BioLegend, BD Biosciences, eBioscience) were incubated with spleen cells for 45/60 min on ice: CD4 (RM4-5), B220 (RA3-6B2), PD-1 (RMP1-30), SLAM (TC15-12F12.2). Intracellular staining of Foxp3 (FJK-16s) was performed using the Foxp3-staining buffer (eBioscience). Cells were gated according to FSC-A/SSC-A, doublet exclusion (SSC-H/SSC-W and FSC-H/FSC-W), live cells (negative for LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit, Life Technologies), followed by gating strategies indicated in figure legend. Flow cytometry was performed on a LSRII (BD Biosciences) and data analyzed using FlowJo 9.9 software (TreeStar).
Statistical Analysis
Data were analyzed via Prism 6 (GraphPad Software) using non-parametric unpaired Mann-Whitney U-test. Graphs show the mean ± SEM. *P < 0.05; ** P < 0.01; *** P < 0.001. If not indicated, the P-values were not significant (>0.05).
Author Contributions
SP designed and performed experiments, analyzed and interpreted data, wrote the manuscript and prepared the figures. BH and JLC performed experiments, edited the manuscript, and contributed to discussions. DBM contributed essential reagents and advice. PLS conceived the project, wrote the manuscript, and provided overall direction of the study. All authors concur with this submission.
Funding
This work was supported in part by funds from the intramural programs of the National Human Genome Research Institute, National Institute of Allergy and Infectious Diseases and National Institute of Neurological Disorders and Stroke, NIH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the PLS and DBM labs for their assistance with this work. GP66 tetramer [I-A(b) QVYSLIRPNENPAHK PE] was obtained from the NIH tetramer facility (Emory University).
References
1. Sallusto F. Heterogeneity of human CD4(+) T cells against microbes. Annu Rev Immunol. (2016) 34:317–34. doi: 10.1146/annurev-immunol-032414-112056
2. Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev. (2010) 237:72–89. doi: 10.1111/j.1600-065X.2010.00937.x
3. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
4. Cannons JL, Lu KT, Schwartzberg PL. T follicular helper cell diversity and plasticity. Trends Immunol. (2013) 34:200–7. doi: 10.1016/j.it.2013.01.001
5. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45:471–482. doi: 10.1016/j.immuni.2016.09.001
6. Pratama A, Vinuesa CG. Control of TFH cell numbers: why and how? Immunol Cell Biol. (2014) 92:40–8. doi: 10.1038/icb.2013.69
7. Preite S, Baumjohann D, Foglierini M, Basso C, Ronchi F, Fernandez Rodriguez BM, et al. Somatic mutations and affinity maturation are impaired by excessive numbers of T follicular helper cells and restored by Treg cells or memory T cells. Eur J Immunol. (2015) 45:3010–21. doi: 10.1002/eji.201545920
8. Brink R, Phan TG. Self-reactive B cells in the germinal center reaction. Annu Rev Immunol. (2018) 36:339–57. doi: 10.1146/annurev-immunol-051116-052510
9. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38:596–605. doi: 10.1016/j.immuni.2012.11.020
10. Ueno H. T follicular helper cells in human autoimmunity. Curr Opin Immunol. (2016) 43:24–31. doi: 10.1016/j.coi.2016.08.003
11. Brink R. The imperfect control of self-reactive germinal center B cells. Curr Opin Immunol. (2014) 28:97–101. doi: 10.1016/j.coi.2014.03.001
12. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B, and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science (2002) 297:1031–4. doi: 10.1126/science.1073560
13. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol. (2010) 185:4042–52. doi: 10.4049/jimmunol.1001730
14. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi: 10.1126/science.1243292
15. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. (2014) 15:88–97. doi: 10.1038/ni.2771
16. Preite S, Cannons JL, Radtke AJ, Vujkovic-Cvijin I, Gomez-Rodriguez J, Volpi S, et al. Hyperactivated PI3Kdelta promotes self and commensal reactivity at the expense of optimal humoral immunity. Nat Immunol. (2018) 19:986–1000. doi: 10.1038/s41590-018-0182-3
17. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. (2003) 3:317–30. doi: 10.1038/nri1056
18. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
19. Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG. Cutting edge: differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. (2004) 173:2236–40. doi: 10.4049/jimmunol.173.4.2236
20. Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. (2012) 12:649–61. doi: 10.1038/nri3278
21. Saxton RA, Sabatini DM. mTOR Signaling in growth, metabolism, and disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
22. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
23. Blenis, J. TOR, the gateway to cellular metabolism, cell growth, and disease. Cell (2017) 171:10–13. doi: 10.1016/j.cell.2017.08.019
24. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol. (2014) 34:272–6. doi: 10.1007/s10875-014-0012-9
25. Cannons JL, Preite S, Kapnick SM, Uzel G, Schwartzberg PL. Genetic defects in phosphoinositide 3-kinase delta influence CD8(+) T cell survival, differentiation, and function. Front Immunol. (2018) 9:1758. doi: 10.3389/fimmu.2018.01758
26. Stark AK, Chandra A, Chakraborty K, Alam R, Carbonaro V, Clark J, et al. PI3Kdelta hyper-activation promotes development of B cells that exacerbate Streptococcus pneumoniae infection in an antibody-independent manner. Nat Commun. (2018) 9:3174. doi: 10.1038/s41467-018-05674-8
27. Avery DT, Kane A, Nguyen T, Lau A, Nguyen A, Lenthall H, et al. Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med. (2018) 215:2073–95. doi: 10.1084/jem.20180010
28. Wray-Dutra MN, Al Qureshah F, Metzler G, Oukka M, James RG, Rawlings DJ. Activated PIK3CD drives innate B cell expansion yet limits B cell-intrinsic immune responses. J Exp Med. (2018) 215:2485–96. doi: 10.1084/jem.20180617
29. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606 e4. doi: 10.1016/j.jaci.2016.06.021
30. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45:540–54. doi: 10.1016/j.immuni.2016.08.017
31. Qi H. T follicular helper cells in space-time. Nat Rev Immunol. (2016) 16:612–25. doi: 10.1038/nri.2016.94
32. Goenka R, Barnett LG, Silver JS, O'Neill PJ, Hunter CA, Cancro MP, et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. (2011) 187:1091–5. doi: 10.4049/jimmunol.1100853
33. Baumjohann D, Okada T, Ansel KM. Cutting edge: distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. (2011) 187:2089–92. doi: 10.4049/jimmunol.1101393
34. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943–52. doi: 10.1038/ni1100
35. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular antibody responses. Immunol Rev. (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
36. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. (2012) 209:1241–53. doi: 10.1084/jem.20120994
37. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
38. Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol. (2011) 29:665–705. doi: 10.1146/annurev-immunol-030409-101302
39. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med. (2015) 212:217–33. doi: 10.1084/jem.20141432
40. Mittrucker HW, Kohler A, Mak TW, Kaufmann SH. Critical role of CD28 in protective immunity against Salmonella typhimurium. J Immunol. (1999) 163:6769–76.
41. Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science (1993) 261:609–12. doi: 10.1126/science.7688139
42. Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, et al. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity (2009) 30:228–41. doi: 10.1016/j.immuni.2008.12.015
43. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397:263–6. doi: 10.1038/16717
44. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature (2001) 409:105–9. doi: 10.1038/35051113
45. Mak TW, Shahinian A, Yoshinaga SK, Wakeham A, Boucher LM, Pintilie M, et al. Costimulation through the inducible costimulator ligand is essential for both T helper and B cell functions in T cell-dependent B cell responses. Nat Immunol. (2003) 4:765–72. doi: 10.1038/ni947
46. Gonzalo JA, Tian J, Delaney T, Corcoran J, Rottman JB, Lora J, et al. ICOS is critical for T helper cell-mediated lung mucosal inflammatory responses. Nat Immunol. (2001) 2:597–604. doi: 10.1038/89739
47. Dong C, Temann UA, Flavell RA. Cutting edge: critical role of inducible costimulator in germinal center reactions. J Immunol. (2001) 166:3659–62. doi: 10.4049/jimmunol.166.6.3659
48. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature (2001) 409:102–5. doi: 10.1038/35051107
49. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. (2005) 175:2340–8. doi: 10.4049/jimmunol.175.4.2340
50. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
51. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. (2015) 136:993–1006 e1. doi: 10.1016/j.jaci.2015.05.036
52. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature (2007) 450:299–303. doi: 10.1038/nature06253
53. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi: 10.1038/nature03555
54. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
55. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496:523–7. doi: 10.1038/nature12058
56. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci USA (2009) 106:20371–6. doi: 10.1073/pnas.0911573106
57. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity (2015) 42:239–51. doi: 10.1016/j.immuni.2015.01.017
58. Xiao N, Eto D, Elly C, Peng G, Crotty S, Liu YC. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol. (2014) 15: 657–66. doi: 10.1038/ni.2912
59. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. (2009) 10: 176–84. doi: 10.1038/ni.1689
60. Lee JY, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, et al. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity (2015) 42:252–64. doi: 10.1016/j.immuni.2015.01.013
61. Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. (2003) 171:166–74. doi: 10.4049/jimmunol.171.1.166
62. Arimura Y, Kato H, Dianzani U, Okamoto T, Kamekura S, Buonfiglio D, et al. A co-stimulatory molecule on activated T cells, H4/ICOS, delivers specific signals in T(h) cells and regulates their responses. Int Immunol. (2002) 14:555–66. doi: 10.1093/intimm/dxf022
63. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature (2012) 491:554–9. doi: 10.1038/nature11581
64. Rolf J, Fairfax K, Turner M. Signaling pathways in T follicular helper cells. J Immunol. (2010) 184:6563–8. doi: 10.4049/jimmunol.1000202
65. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. (2007) 179:5099–108. doi: 10.4049/jimmunol.179.8.5099
66. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
67. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
68. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
69. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls follicular T helper cell positioning and function. Immunity (2018) 49:264–74.e4. doi: 10.1016/j.immuni.2018.06.012
70. Tian Y, Mollo SB, Harrington LE, Zajac AJ. IL-10 Regulates Memory T Cell Development and the balance between Th1 and follicular Th cell responses during an acute viral infection. J Immunol. (2016) 197:1308–21. doi: 10.4049/jimmunol.1502481
71. Migone TS, Rodig S, Cacalano NA, Berg M, Schreiber RD, Leonard WJ. Functional cooperation of the interleukin-2 receptor beta chain and Jak1 in phosphatidylinositol 3-kinase recruitment and phosphorylation. Mol Cell Biol. (1998) 18:6416–22. doi: 10.1128/MCB.18.11.6416
72. Truitt KE, Mills GB, Turck CW, Imboden JB. SH2-dependent association of phosphatidylinositol 3'-kinase 85-kDa regulatory subunit with the interleukin-2 receptor beta chain. J Biol Chem. (1994) 269:5937–43.
73. Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, et al. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol Cell Biol. (2000) 20:7109–20. doi: 10.1128/MCB.20.19.7109-7120.2000
74. Cornish GH, Sinclair LV, Cantrell DA. Differential regulation of T-cell growth by IL-2 and IL-15. Blood (2006) 108:600–8. doi: 10.1182/blood-2005-12-4827
75. Karnitz LM, Burns LA, Sutor SL, Blenis J, Abraham RT. Interleukin-2 triggers a novel phosphatidylinositol 3-kinase-dependent MEK activation pathway. Mol Cell Biol. (1995) 15:3049–57. doi: 10.1128/MCB.15.6.3049
76. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
77. Jacobs MD, Black J, Futer O, Swenson L, Hare B, Fleming M, et al. Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J Biol Chem. (2005) 280:13728–34. doi: 10.1074/jbc.M413155200
78. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. (2010) 11:9–22. doi: 10.1038/nrm2822
79. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. (2012) 209:2441–53. doi: 10.1084/jem.20112607
80. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. (2012) 209:243–50. doi: 10.1084/jem.20111174
81. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
82. Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. (2012) 13:405–11. doi: 10.1038/ni.2242
83. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The Interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi: 10.1016/j.immuni.2015.08.017
84. Ye L, Lee J, Xu L, Mohammed AU, Li W, Hale JS, et al. mTOR Promotes Antiviral Humoral Immunity by Differentially Regulating CD4 Helper T Cell and B Cell Responses. J Virol. (2017) 91:e01653–16. doi: 10.1128/JVI.01653-16
85. Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, et al. Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. Elife (2016) 5:e17936. doi: 10.7554/eLife.17936
86. Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity (2011) 34:224–36. doi: 10.1016/j.immuni.2011.01.012
87. Grzes KM, Swamy M, Hukelmann JL, Emslie E, Sinclair LV, Cantrell DA. Control of amino acid transport coordinates metabolic reprogramming in T-cell malignancy. Leukemia (2017) 31:2771–9. doi: 10.1038/leu.2017.160
88. Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell Rep. (2015) 12:2099–110. doi: 10.1016/j.celrep.2015.08.049
89. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. (2015) 16:980–90. doi: 10.1038/ni.3226
90. Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. (2015) 16:991–9. doi: 10.1038/ni.3229
91. Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. (2009) 10:992–9. doi: 10.1038/ni.1762
92. Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, et al. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. (2015) 13:2203–18. doi: 10.1016/j.celrep.2015.10.072
93. Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, et al. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J Exp Med. (2017) 214:39–47. doi: 10.1084/jem.20161046
94. Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity (2013) 39:286–97. doi: 10.1016/j.immuni.2013.07.013
95. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. (2016) 1:eaai8593. doi: 10.1126/sciimmunol.aai8593
96. Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. (2013) 210:1189–200. doi: 10.1084/jem.20130392
97. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. (2010) 107:9777–82. doi: 10.1073/pnas.0914127107
98. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
99. Sayin I, Radtke AJ, Vella LA, Jin W, Wherry EJ, Buggert M, et al. Spatial distribution and function of T follicular regulatory cells in human lymph nodes. J Exp Med. (2018) 215: jem.20171940. doi: 10.1084/jem.20171940
100. Fu W, Liu X, Lin X, Feng H, Sun L, Li S, et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J Exp Med. (2018) 215:815–25. doi: 10.1084/jem.20170901
101. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
102. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983–8. doi: 10.1038/nm.2426
103. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
104. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. (2016) 271:246–59. doi: 10.1111/imr.12411
105. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
106. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. (2008) 205:565–74. doi: 10.1084/jem.20071477
107. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
108. Malek TR. The biology of interleukin-2. Annu Rev Immunol. (2008) 26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357
109. Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol. (2017) 18:1249–60. doi: 10.1038/ni.3837
110. Xu L, Huang Q, Wang H, Hao Y, Bai Q, Hu J, et al. The Kinase mTORC1 promotes the generation and suppressive function of follicular regulatory T cells. Immunity (2017) 47:538–51 e5. doi: 10.1016/j.immuni.2017.08.011
111. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of treg to Tfr cells. Immunity (2017) 47:1067–1082 e12. doi: 10.1016/j.immuni.2017.11.00
112. Shekhar S, Yang X. The darker side of follicular helper T cells: from autoimmunity to immunodeficiency. Cell Mol Immunol. (2012) 9:380–5. doi: 10.1038/cmi.2012.26
113. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. (2010) 62:234–44. doi: 10.1002/art.25032
114. Jung S, Gamez-Diaz L, Proietti M, Grimbacher B. “Immune TOR-opathies,” a Novel Disease Entity in Clinical Immunology. Front Immunol (2018) 9: 966. doi: 10.3389/fimmu.2018.00966
115. Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. (2009) 228:253–72. doi: 10.1111/j.1600-065X.2008.00750.x
116. Haylock-Jacobs S, Comerford I, Bunting M, Kara E, Townley S, Klingler-Hoffmann M, et al. PI3Kdelta drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation. J Autoimmun. (2011) 36:278–87. doi: 10.1016/j.jaut.2011.02.006
117. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. (2015) 16:178–87. doi: 10.1038/ni.3076
118. Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, et al. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med. (2003) 197:657–67. doi: 10.1084/jem.20021101
119. Durand CA, Hartvigsen K, Fogelstrand L, Kim S, Iritani S, Vanhaesebroeck B, et al. Phosphoinositide 3-kinase p110 delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol. (2009) 183:5673–84. doi: 10.4049/jimmunol.0900432
120. Jayachandran N, Mejia EM, Sheikholeslami K, Sher AA, Hou S, Hatch GM, et al. TAPP adaptors control B cell metabolism by modulating the phosphatidylinositol 3-kinase signaling pathway: a novel regulatory circuit preventing autoimmunity. J Immunol. (2018) 201:406–16. doi: 10.4049/jimmunol.1701440
121. Banham-Hall E, Clatworthy MR, Okkenhaug K. The therapeutic potential for pi3k inhibitors in autoimmune rheumatic diseases. Open Rheumatol J. (2012) 6:245–58. doi: 10.2174/1874312901206010245
122. Stark AK, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol. (2015) 23:82–91. doi: 10.1016/j.coph.2015.05.017
123. Zarate-Blades CR, Horai R, Caspi RR. Regulation of autoimmunity by the microbiome. DNA Cell Biol. (2016) 35:455–8. doi: 10.1089/dna.2016.3432
Keywords: Tfh cells, Tfr, ICOS, PI3K, APDS, PASLI, autoimmunity
Citation: Preite S, Huang B, Cannons JL, McGavern DB and Schwartzberg PL (2019) PI3K Orchestrates T Follicular Helper Cell Differentiation in a Context Dependent Manner: Implications for Autoimmunity. Front. Immunol. 9:3079. doi: 10.3389/fimmu.2018.03079
Received: 13 September 2018; Accepted: 12 December 2018;
Published: 07 January 2019.
Edited by:
Shahram Salek-Ardakani, Pfizer, United StatesReviewed by:
Georges Abboud, University of Florida, United StatesDavid A. Fruman, University of California, Irvine, United States
Laurence Morel, University of Florida, United States
Copyright © 2019 Preite, Huang, Cannons, McGavern and Schwartzberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela L. Schwartzberg, cGFtc0BuaWguZ292