 Giulia Di Lullo1*
Giulia Di Lullo1* Magda Marcatti2Silvia Heltai1Cristina Tresoldi3Anna Maria Paganoni4Claudio Bordignon5
Magda Marcatti2Silvia Heltai1Cristina Tresoldi3Anna Maria Paganoni4Claudio Bordignon5 Fabio Ciceri2,5
Fabio Ciceri2,5 Maria Pia Protti1*
Maria Pia Protti1*- 1Tumor Immunology Unit, Division of Immunology, Transplantation and Infectious Diseases, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) San Raffaele Scientific Institute, Milan, Italy
- 2Hematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Molecular Hematology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 4Laboratory for Modeling and Scientific Computing (MOX), Dipartimento di Matematica, Politecnico di Milano, Milan, Italy
- 5Vita-Salute San Raffaele University, Milan, Italy
Immunomodulatory drugs (IMiDs) are effective therapeutics for multiple myeloma (MM), where in different clinical settings they exert their function both directly on MM cells and indirectly by modulating immune cell subsets, although with not completely defined mechanisms. Here we studied the role of IMiDs in the context of autologous hematopoietic stem cell transplantation on the T cell subset distribution in the bone marrow of newly diagnosed MM patients. We found that after transplantation pro-tumor Th17-Th1 and Th22 cells and their related cytokines were lower in patients treated with IMiDs during induction chemotherapy compared to untreated patients. Of note, lower levels of IL-17, IL-22, and related IL-6, TNF-α, IL-1β, and IL-23 in the bone marrow sera correlated with treatment with IMiDs and favorable clinical outcome. Collectively, our results suggest a novel anti-inflammatory role for IMiDs in MM.
Introduction
Multiple Myeloma (MM) is a plasma cell neoplastic disorder primarily localized in the bone marrow (BM), where interactions between neoplastic cells and cells within the tumor microenvironment (i.e., mesenchymal stromal cells, endothelial cells, osteoblasts, osteoclasts, and immune cells) mediate disease development and progression (1–3).
Th cells play fundamental regulatory function in adaptive immune responses and in antitumor immunity being, according to their secretory cytokine profile, either anti-tumor or pro-tumor (4). In MM imbalanced Th cell polarization and subset distribution in the BM niche largely impact on disease progression (5).
Immunomodulatory drugs (IMiDs), namely thalidomide and its derivatives lenalidomide and pomalidomide, are major therapeutics in the treatment of MM (6), where their efficacy has been demonstrated in newly diagnosed patients either eligible or ineligible for autologous hematopoietic stem cell transplantation (ASCT), in the maintenance setting after ASCT and in refractory/relapsed disease (6).
In MM, in addition to anti-proliferative and pro-apoptotic effects on malignant cells, IMiDs exert immune regulatory function and interfere with tumor microenvironment interactions (7). In vitro IMiDs enhanced T cell proliferation, IL-2 and IFN-γ secretion and NK cell activation (8, 9), lenalidomide improved immune checkpoint blockade-induced immune responses (10) and inhibited T regulatory cell (Treg) proliferation and suppressor function (11). In vivo lenalidomide augmented (i) vaccine responses and endogenous anti-tumor immunity (12), (ii) the number of central and effector memory CD8+ T cells, Tregs and CD14+CD15+ myeloid derived suppressor cells in patients that received lenalidomide as monotherapy or in combination with other treatments (13), (iii) the number of Tregs in patients in consolidation/maintenance therapy (14, 15), (iv) anti-myeloma specific T cell responses in patients that received lenalidomide as consolidation therapy after ASCT (16), and (v) the number of IFN-γ and IL-21 producing cells in the setting of maintenance therapy (17). Treatment with lenalidomide was also associated with impaired long-term thymic reconstitution and decreased number of CD4+ and CD8+ effector terminally differentiated T cells (14, 15) and reduced PD-1 expression on T cells in the maintenance setting (18).
A prospective randomized trial comparing induction regimens prior to ASCT including or not thalidomide (i.e., bortezomib-thalidomide-dexamethasone) vs. bortezomib-cyclophosphamide-dexamethasone) recently reported significantly higher overall clinical response rate in the bortezomib-thalidomide-dexamethasone arm (19).
In this study, we evaluated in newly diagnosed MM patients the impact of IMiDs used in the induction chemotherapy preceding ASCT on the distribution of T cell subsets and related cytokines in the BM after transplantation and whether changes in those immunological parameters correlated with the clinical outcome.
Materials and Methods
Subjects and Samples
Forty-four newly diagnosed MM patients, who had been hospitalized at the Hematology Department of our Institution and received ASCT as first line therapy, were selected for the study. The Institutional Ethics Committee (Comitato Etico Fondazione Centro San Raffaele, Istituto Scientifico Ospedale San Raffaele) had approved the study protocol and written informed consent was obtained from all donors. Clinical data and information on the induction chemotherapy received by each patient are reported in Table 1. BM mononuclear cells were isolated by density gradient centrifugation with Ficoll-PaqueTM Plus (GE Healthcare, Uppsala, Sweden), frozen in fetal bovine serum (Lonza, Milan, Italy) + 10% DMSO (Sigma-Aldrich, Milan, Italy), and stored according to standardized operating procedures by the Institutional Biobank. BM mononuclear cells were used after thawing and viable cell counting. BM sera were taken and collected according to standardized operating procedures by the Institutional Biobank. Briefly, non-heparinized BM blood (5–7 ml) was incubated for 1 h at room temperature to achieve complete clotting. Then samples were centrifuged at 1,600 g without brake for 10 min at 4°C and supernatants transferred in cryovials and stored in liquid nitrogen until use.
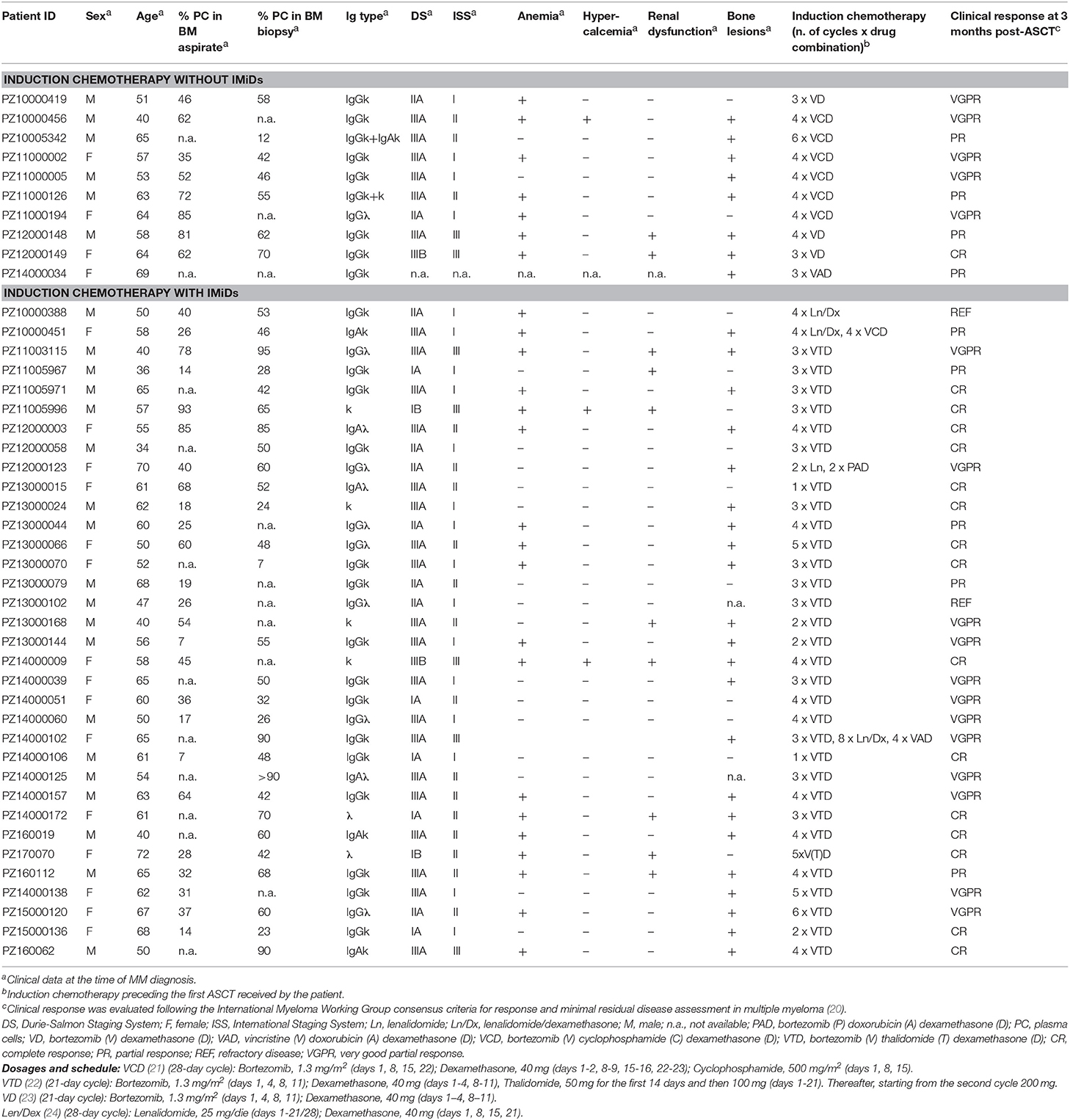
Table 1. Characteristics of the patients.
Flow Cytometry Analysis
The following antibodies were used: Pacific blue-conjugated CD3 (clone UCHT1) from Dako Cytomation (Cernusco sul Naviglio (MI), Italy); PerCP-conjugated CD4 (clone L200), PE-conjugated CD25 (clone 2A3), PE-conjugated IL-13 (clone JES10-5A2), and APC-conjugated IL-5 (clone TRFK5), all from BD Biosciences (Milan, Italy); PE-Cy7-conjugated CD127 (clone R34.34) from Beckman Coulter (Cassina De' Pecchi (MI), Italy); PE-Cy7-coniugated IL-22 (clone 22URTI) from eBioscience (Milan, Italy); FITC-conjugated CD3 (clone UCHT1), FITC-conjugated IFN-γ (clone B27), Alexa Fluor488-conjugated IL-4 (clone MPA-25D2), Alexa Fluor647-conjugated IL-17 (clone BL168), all from Biolegend (Milan, Italy). For the study of the percentage of T cells secreting distinct effector cytokines, thawed BM mononuclear cells were rested at room temperature for 30 min before viable cell counting and then cultured at a cell density of 2 × 106 cells/ml for 5 h in x-vivo-15 medium (Lonza), supplemented with penicillin (100 U/ml), streptomycin (100 U/ml), and 3% human serum type AB (Lonza), in the absence or the presence of 50 ng/ml PMA (phorbol-12-myristate-13-acetate) and 1 μg/ml ionomycin (both from Sigma-Aldrich) to induce cytokine production. After 2 h, 10 μg/ml brefeldin A (Sigma-Aldrich) was added to both unstimulated and stimulated cells. After stimulation, cells were harvested, washed and stained first for the surface markers CD3 and CD4 and then for intracellular cytokines, using a kit from BD Biosciences, following the manufacturer's instructions. In detail, for intracellular cytokine staining cells were fixed, permeabilized with Cytofix/Cytoperm buffer, and incubated with antibodies diluted in Perm/Wash buffer. Percentages of cells positive for intracellular cytokine expression were referred to total CD3+ T cells because a clear-cut distinction between CD4+ and CD4− T cells was hampered by the downregulation of surface CD4 expression on T cells stimulated with PMA + ionomycin (Figure S1). For Treg staining, cells were first stained for the surface antigens CD3, CD4, CD25, and CD127 and then fixed, permeabilized, and stained for the nuclear transcription factor FoxP3 with the dedicated FoxP3/Transcription Factor Staining Buffer from eBioscience, according to the manufacturer's instructions. Percentages of Treg cells, identified as CD3+CD4+CD127−CD25+FoxP3+ cells, were also referred to total CD3+ T cells. Cells were analyzed using a FACSCanto flow cytometer (BD Biosciences) and data were illustrated by FlowJo software (Tree Star Inc., Ashland, OR, USA).
Cytokine Measurements
Cytokines in BM sera were measured using ELISA kits from Mabtech (Milan, Italy) (i.e., IL-1β, TNF-α, IFN-γ, IL-4, IL-5, IL-6, IL-13, IL-17, IL-22, and IL-23), following the manufacturer's instructions.
Statistical Analysis
Statistical significance was determined with Mann Whitney U test and Wilcoxon Signed-Rank Test, as reported in the figure legends. In addition, to evaluate the equality of variances for the values in the IMiD-treated vs. the IMiD-untreated group of patients we performed the Levene's test with no statistical evidence to support difference between the variance of the distributions. Statistical analyses were performed with an alpha level of 5% using GraphPad Prism version 5.0 for Mac (GraphPad Software), thus values of p < 0.05 were considered significant.
Results
We studied forty-four newly diagnosed MM patients, who had received ASCT as frontline therapy and whose clinical features are detailed in Table 1. Patients were administered an induction chemotherapy, most often consisting of a bortezomib-based poly-chemotherapy (including or not thalidomide or, less often, lenalidomide) followed by stem cell mobilization with cyclophosphamide and G-CSF, high-dose melphalan and a single or tandem ASCT (see Table 1 for dosage and schedule details).
BM mononuclear cells were analyzed by flow cytometry for intracellular cytokine staining of the T cell subset-distinctive cytokine patterns or directly for Treg specific markers within the T cell fraction (see Figure 1 for gating strategy).
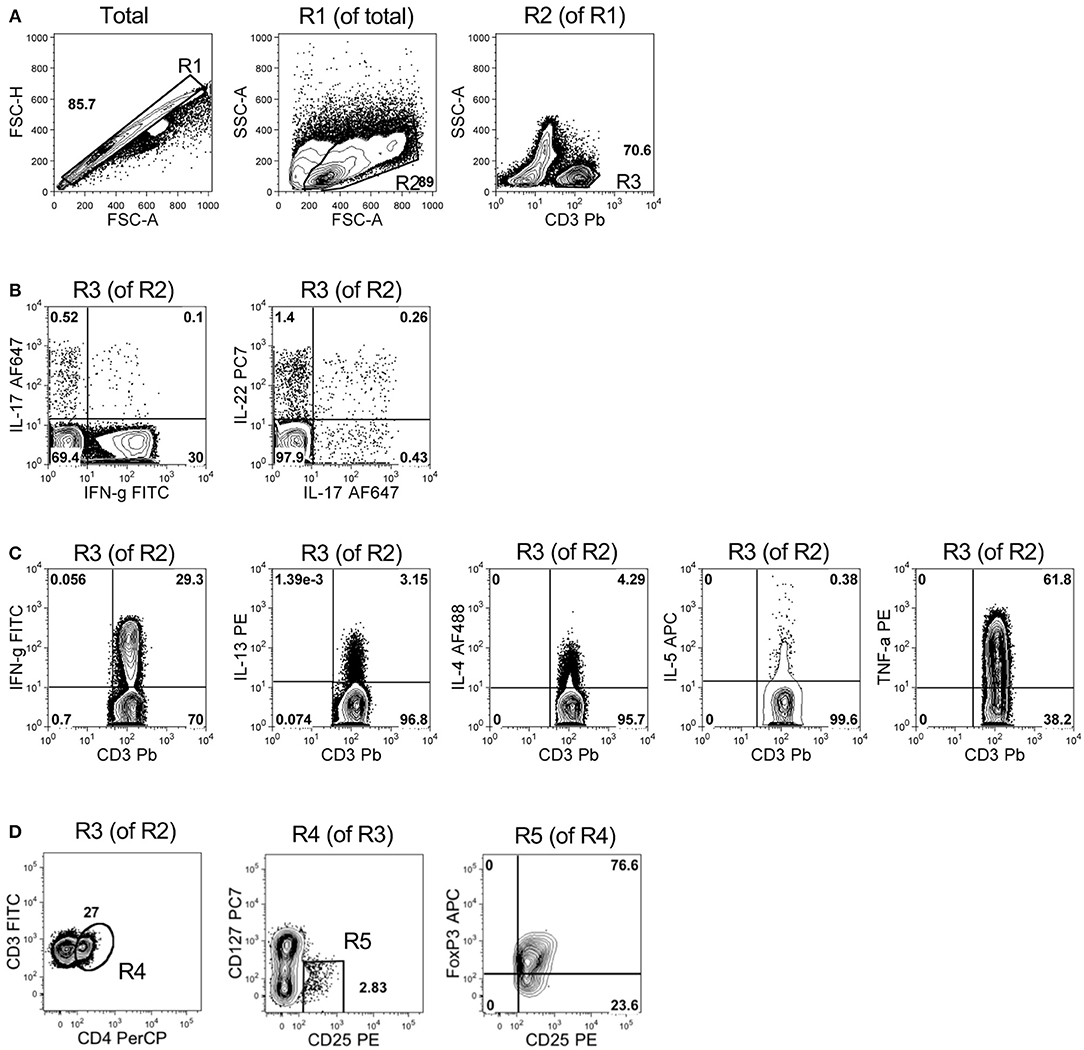
Figure 1. Gating strategy for immunophenotypic analyses in representative samples of BM mononuclear cells. (A) Panels represent, from left to right: the pulse geometry R1 gate (in FSC-A x FSC-H dot plot of all the analyzed cells) used to exclude doublets, the morphology-based R2 gate of leukocytes (in FSC-A x SSC-A dot plot of R1-gated cells) and the R3 gate of T cells (i.e., CD3+ cells of R2-gated leukocytes). (B) Left panel: quadrant gates defining the expression and the percentage of IL-17 and IFN-γ in R3-gated CD3+ T cells; right panel: quadrant gates defining the expression of IL-17 and IL-22 in R3-gated CD3+ T cells. (C) Panels represent, from left to right, the quadrant gates defining expression and the percentage of IFN-γ+, IL-13+, IL-4+, IL-5+, and TNF-α+ cells in R3-gated CD3+ T cells. (D) Dot plots showing the gates used to identify and enumerate the percentage of Treg cells (CD4+CD127−CD25+FoxP3+) in R3-gated CD3+ T cells. From left to right: R4 gate represents the percentage of CD4+ cells among total CD3+ T cells (i.e., R3-gated cells); R5 gate represents the percentage of CD127− cells among total CD4+ T cells (i.e., R4-gated cells); the upper right quadrant gate in the CD25 x FoxP3 dot plot identifies bona fide Treg cells (i.e., CD25+FoxP3+ T cells in R5-gated CD4+CD127− T cells). Numbers within the plots represent percentages of R gates (A,D) and quadrant statistics (B–D).
Firstly, we measured the frequency of distinct effector cytokine-secreting T and Treg cell subsets in paired samples at diagnosis and 3 months after ASCT (i.e., a time point at which immune cells are mostly reconstituted), to evaluate potential changes of the T cell subset distribution occurring early after transplantation as a consequence of the complete therapy received by the patients (i.e., induction regimen including or not IMiDs, stem cell mobilization, chemotherapy and ASCT). We found that T cells secreting Th1 (i.e., IFN-γ and TNF-α) and Th2 (i.e., IL-13, IL-4, and IL-5) cytokines were significantly increased after ASCT compared to those at diagnosis as well as T cells coproducing IL-17 and IFN-γ, whereas T cells secreting IL-17, IL-22, and Tregs (CD4+CD127−CD25+FoxP3+) were not (Figure 2). Secondly, to evaluate the impact of IMiDs used in the induction regimen, we focused on the frequency of the T cell subsets in the post-ASCT time point and performed analyses in patients grouped based on the absence or the presence of IMiDs in the induction chemotherapy. We found that IL-17+IFN-γ+ (i.e., mostly Th17-Th1, see Figure S1) and IL-22+IL-17− (i.e., mostly Th22, see Figure S1) T cells were significantly lower in the IMiD-treated compared to untreated patients (Figure 3A). Remarkably, increased frequency of Th17-Th1 and Th22 cell subsets correlated with development of symptomatic MM and worse prognosis (25, 26), respectively. Indeed, poly-functional Th17-Th1 were found increased in the BM of MM patients compared to those of patients with pre-neoplastic gammopathy (25), and we previously reported increased frequency of Th22 cells in the blood and BM of patients with stage III disease at diagnosis and refractory/relapsed disease compared to those of asymptomatic patients or patients with stage I/II disease (26). Total IL-17+ and IL-22+ T cells were also lower within BM T cells from IMiD-treated compared to untreated patients (Figure 3A). In agreement with previous reports (9, 13), IFN-γ+ secreting cells were higher in the IMiD-treated compared to the untreated group (Figure 3A), whereas T cells secreting the other cytokines (i.e., IL-13, IL-4, IL-5, and TNF-α) and Tregs were comparable between the two groups (Figure 3A).
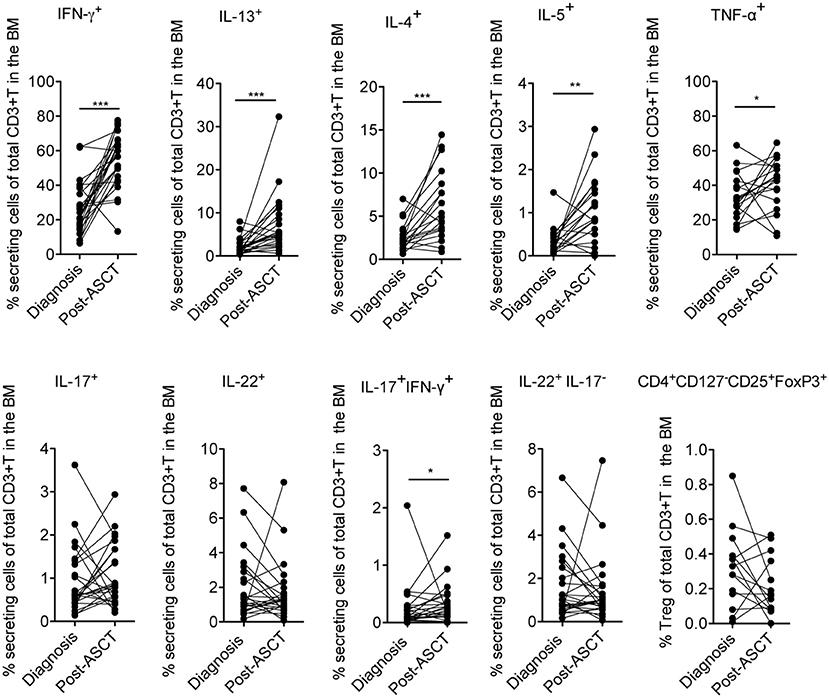
Figure 2. Frequency of distinctive cytokine secreting T cells and Tregs in newly diagnosed MM patients measured in paired BM samples at diagnosis and at 3 months after ASCT. The frequency of CD3+ T cells secreting IFN-γ, IL-13, IL-17, IL-22 (n = 25), IL-4, and IL-5 (n = 19), TNF-α (n = 18) and CD3+ T cells expressing a Treg phenotype (i.e., CD4+CD127−CD25+FoxP3+) (n = 14) are reported. Responses significantly different by Wilcoxon Signed-Rank Test are indicated as: *p < 0.05, **0.001 < p < 0.01 and ***p < 0.001.
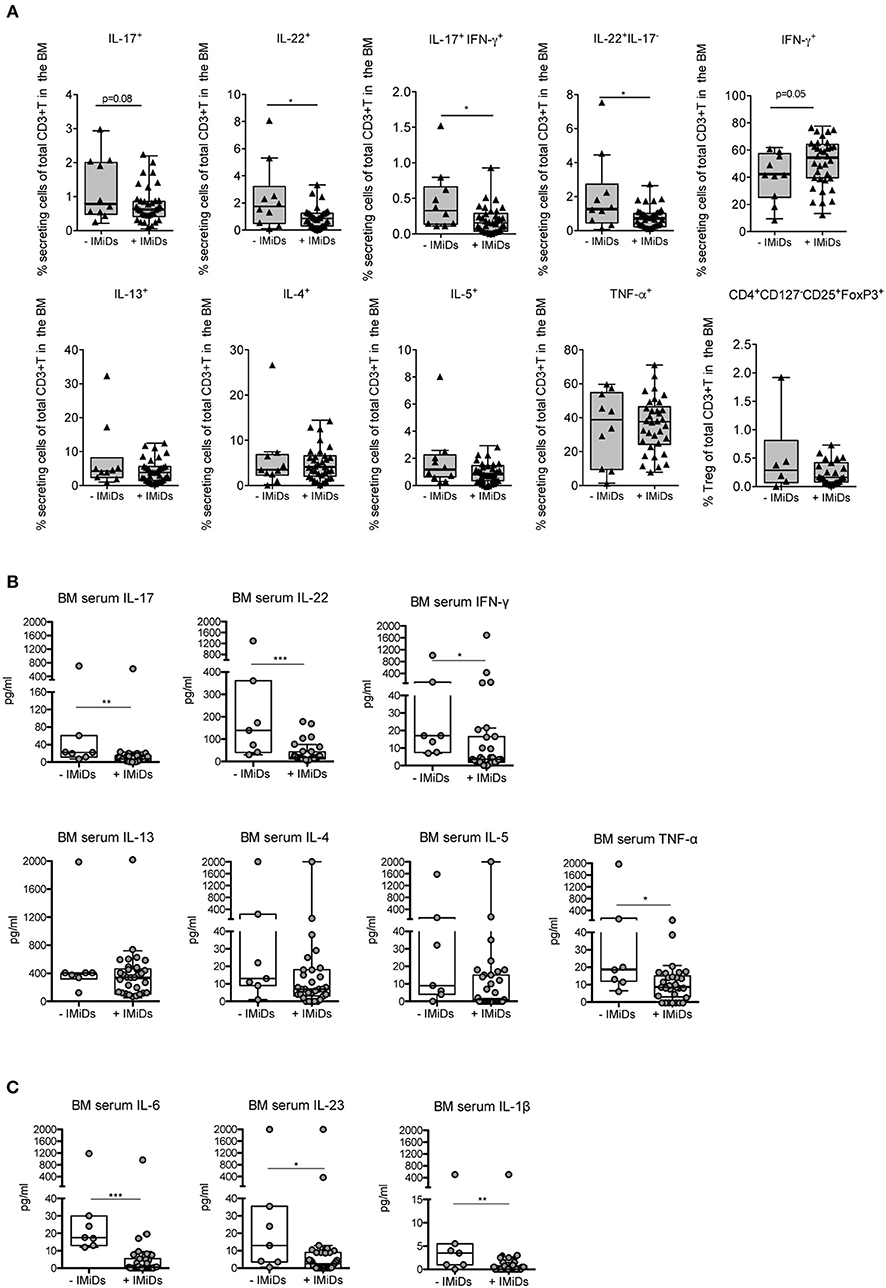
Figure 3. Effects of IMiD treatment on ex-vivo distribution of T cell subsets and related cytokines in the BM of MM patients at 3 months after ASCT. (A) Percentages of CD3+ T cells secreting the indicated cytokines and CD3+ T cells expressing a Treg phenotype (i.e., CD4+CD127−CD25+FoxP3+), as assessed by surface and intracellular staining analyses, in the BM of patients grouped based on the absence (–IMiDs, n = 10 for T cells and n = 6 for Tregs) or the presence (+IMiDs, n = 34 for T cells and n = 23 for Tregs) of IMiDs in the induction chemotherapy. Data from each patient are represented as black-filled triangles. (B,C) BM serum levels of the indicated cytokines in MM patients grouped as above (-IMiDs, n = 7; +IMiDs, n = 31). Data from each patient are represented as gray-filled circles. Responses significantly different by Mann-Whitney test are indicated as: *p < 0.05, **0.001 < p < 0.01 and ***p < 0.001.
We then measured the levels of the same cytokines in the BM sera. We found that, as a result of the complete therapy received by the patients, in paired samples at diagnosis and at 3 months after ASCT IFN-γ, IL-13, IL-4, IL-5, TNF-α, IL-17, and IL-22 were all significantly increased at 3 months after transplantation (Figure S2A). In agreement with the T cell data, when we considered the levels of the cytokines in the BM sera after ASCT in patients grouped based on the absence or the presence of IMiDs in the induction chemotherapy, we found that the levels of IL-17 and IL-22 were significantly lower in the BM sera of IMiD-treated vs. untreated patients, and those of Th2 cytokines (i.e., IL-13, IL-4 and IL-5) were not (Figure 3B). On the contrary, the levels of IFN-γ and TNF-α were both significantly lower in IMiD-treated patients (Figure 3B), suggesting that the total amount of these cytokines in the BM sera after transplantation possibly depends on immune cells, other than T cells, which may be differentially targeted by the drugs used in the two induction chemotherapy regimens.
Next, we measured the BM serum levels of cytokines implicated in Th17-Th1 and Th22 cell polarization/expansion, namely, in addition to TNF-α, IL-6, IL-1β, and IL-23 (27). We did not find any significant change in the level of these cytokines when comparing paired BM sera at diagnosis and at 3 months after ASCT (Figure S2B). Interestingly, these cytokines were all significantly reduced in IMiD-treated vs. untreated patients in the BM sera after ASCT (Figure 3C).
Together, these data show that IMiD-including therapies are associated with a lower frequency in the BM at 3 months after transplantation of pro-tumor Th17-Th1 and Th22 cells and of the serum levels of their distinctive cytokines and cytokines implicated in their polarization/expansion.
Lastly, to assess potential correlations between the immunological changes observed, the clinical status and the use of IMiDs in the induction chemotherapy, we analyzed the levels of cytokines in patients grouped according to their clinical status at 3 months after ASCT (Table 1). We found that IL-17, IL-22, TNF-α, IL-6, IL-23, and IL-1β were all significantly lower in patients with complete response compared to patients with refractory disease or partial response, whereas IFN-γ did not significantly change (Figure 4A). IL-6, IL-23, and IL-1β were also significantly lower in patients with complete response compared to those with very good partial response (Figure 4A).
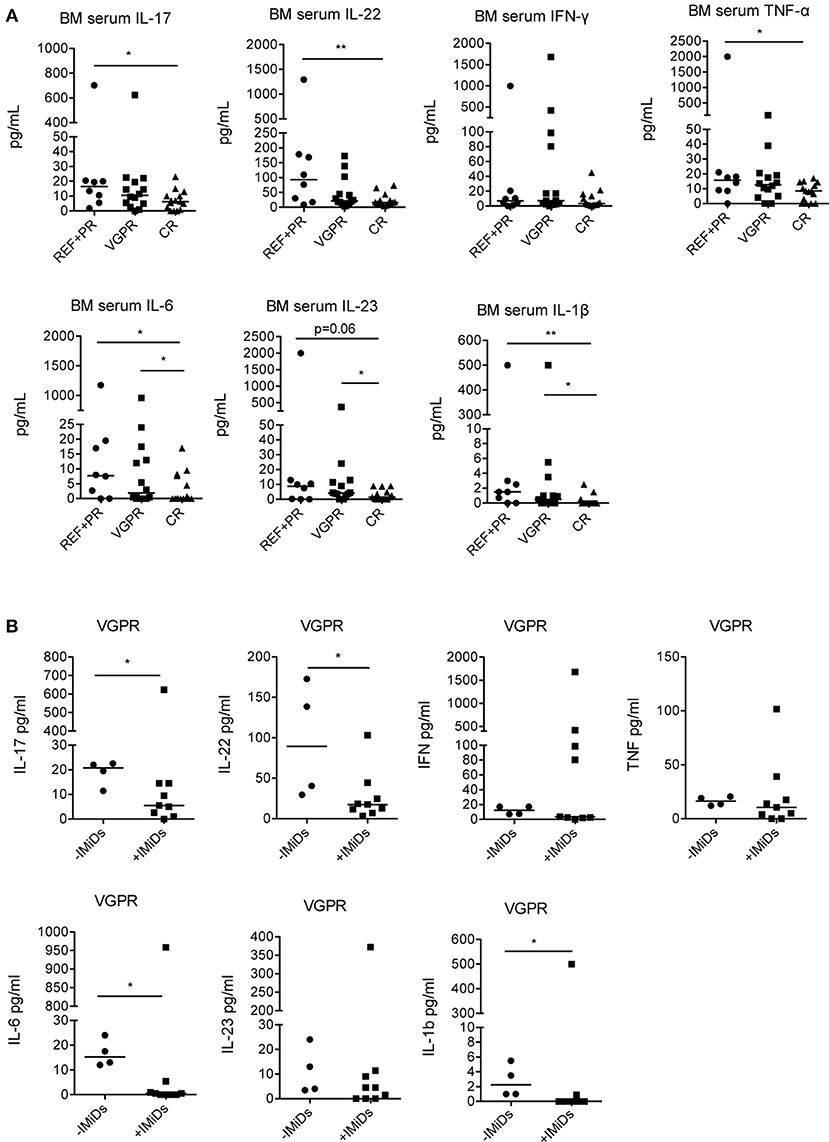
Figure 4. Correlation between cytokine levels in the BM at 3 months after ASCT, clinical outcome and induction chemotherapy regimens. (A) MM patients (n = 37) were grouped based on the clinical status: REF, refractory disease (n = 2); PR: partial response (n = 6); VGPR, very good partial response (n = 14); CR, complete response (n = 15). (B) MM patients with VGPR (n = 13) were grouped based on the absence (–IMiDs) (n = 4) or the presence (+IMiDs) (n = 9) of IMiDs in the induction chemotherapy. Responses significantly different by Mann-Whitney test are indicated as: *p < 0.05 and **0.001 < p < 0.01.
As the cohort of patients that received IMiDs had better clinical outcome (complete response in 44 vs. 10% patients, respectively, see Table 1), to exclude that the immunological changes observed were simply the consequence of reduced tumor burden and not associated to IMiD-driven immune modulation, we compared the cytokine profile in IMiD-treated vs. untreated patients within patients with very good partial response (i.e., patients who had achieved very deep responses to therapy, yet still have some residual markers of disease). Although the number of patients compared is relatively small, we found that IL-17, IL-22, IL-6, and IL-1β were significantly lower in IMiD-treated compared to untreated patients (Figure 4B).
Together, these data suggest that the better clinical responses observed in IMiD-treated patients depend not only on the well-known anti-proliferative effects of IMiDs on myeloma cells (6) but possibly also on immune regulatory functions, among which the down-modulation of pro-tumor Th17-Th1, Th22 cells and their related cytokines.
Conclusions
In this study we report that (i) pro-tumor Th17-Th1 and Th22 cells and their related cytokines in the BM after transplantation are lower in patients treated with IMiDs during induction chemotherapy compared to untreated patients and (ii) the levels of the same cytokines are lower in patients with favorable clinical outcome (i.e., complete response) compared to patients with persistent disease (i.e., refractory disease and partial response).
In agreement with previous reports (9, 13) we found higher numbers of IFN-γ secreting T cells in the BM of patients treated with IMiDs in the induction chemotherapy: however, even though the levels of IFN-γ in the BM sera were increased after transplantation compared with those at diagnosis, we did not find a significant correlation between the levels of IFN-γ and the clinical outcome after transplantation.
Conflicting results are reported concerning the role of IMiDs on Tregs: while in vitro lenalidomide reduced the number of Tregs (11), in vivo studies showed an increase in the number of Tregs in different clinical settings (13–15). In our study we did not find significant changes in the numbers of Tregs neither between diagnosis and after transplantation nor between IMiD-treated and untreated patients. Factors such as the clinical setting in different studies (i.e., consolidation and/or maintenance vs. induction chemotherapy) and the site of investigation (i.e., peripheral vs. BM blood) may account for these differences.
Our findings are particularly relevant because of the pro-tumor role of IL-17 and IL-22 in MM. IL-17 promotes MM cell growth/survival by interaction with the IL-17 receptor expressed on MM cells, and inhibits immune cell functions (28). In addition, expansion of Th17 cells correlates with development of bone lesions (29, 30), possibly through IL-17-induced upregulation in BM cells of RANKL, which is a differentiation factor for osteoclasts [rev. in (31)]. A reduction in the RANKL/osteoprotegerin ratio and bone osteolysis was reported in MM patients with relapsed/refractory disease treated with thalidomide-containing regimen (32, 33). In agreement with this report, we found that the percentage of nuclear magnetic resonance and/or positron emission tomography negative patients as from evaluation after transplantation was superior within the IMiD-treated compared to the untreated group (data not shown). Whether the effect on bone disease following treatment with IMiDs might at least partially depend on IL-17 modulation through the RANKL signaling as well as other mechanisms of bone remodeling [i.e., Wnt (34) and LIGHT/TNFSF14 (35) signaling pathways] might warrant further ad hoc investigation. Concerning IL-22, we previously reported that this cytokine directly increases MM cell growth and resistance to drug-induced cell death by binding to its IL22RA1 receptor, which is aberrantly expressed on a fraction of primary MM cells (26).
The immune regulation exerted by IMiDs has actually two sides: they co-stimulate activation of immune responses, as in hematologic cancers (6), but they can also dampen inflammatory reactions, as in autoimmune and inflammatory disorders (36). Such dual role conceivably depends on different immune cell types targeted, such as T and NK cells and antigen presenting cells, respectively (36). It is tempting to speculate that the use of IMiDs during induction chemotherapy might have favored the development and persistence of an anti-inflammatory BM milieu responsible for the reduced frequency of Th17-Th1 and Th22 cells, possibly through modulation of BM resident cells (i.e., mesenchymal stromal cells) and/or immune cells persisting in the BM or transferred within the autologous progenitor cell reinfusion.
In summary, our results contribute to the characterization of the immunomodulatory effects exerted by IMiDs in MM and should be taken into consideration for the implementation of new therapeutic strategies targeting IL-17 and IL-22 to be combined with drugs already used in MM treatment, especially in refractory/relapsed disease stages.
Author Contributions
GDL designed research, performed research, analyzed data and wrote the manuscript. MM and FC contributed to patient selection and clinical data collection. SH performed research. CT contributed sample collection and storage. AMP contributed to statistical analysis design. CB contributed to data analysis. MPP designed research, analyzed data and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Italian Association for Cancer Research (AIRC IG-11353 and AIRC IG-15452). GDL is recipient of a fellowship from the Fondazione Umberto Veronesi. The authors thank all the patients participating in the study and Dr. Francesca Lorentino for support in clinical data retrieval.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.03171/full#supplementary-material
Abbreviations
ASCT, autologous stem cell transplantation; BM, bone marrow; IMiDs, immunomodulatory drugs; MM, multiple myeloma; Treg, T regulatory cells.
References
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. (2011) 364:1046–60. doi: 10.1056/NEJMra1011442
2. Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia (2009) 23:10–24 doi: 10.1038/leu.2008.259
3. Noonan K, Borrello I. The immune microenvironment of myeloma. Cancer Microenviron. (2011) 4:313–23. doi: 10.1007/s12307-011-0086-3
4. Protti MP, Monte LD, Lullo GD. Tumor antigen-specific CD4(+) T cells in cancer immunity: from antigen identification to tumor prognosis and development of therapeutic strategies. Tissue Antigens (2014) 83:237–46. doi: 10.1111/tan.12329
5. Guillerey C, Nakamura K, Vuckovic S, Hill GR, Smyth MJ. Immune responses in multiple myeloma: role of the natural immune surveillance and potential of immunotherapies. Cell Mol Life Sci. (2016) 73:1569–89. doi: 10.1007/s00018-016-2135-z
6. Holstein SA, McCarthy PL. Immunomodulatory drugs in multiple myeloma: mechanisms of action and clinical experience. Drugs (2017) 77:505–20. doi: 10.1007/s40265-017-0689-1
7. Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia (2010) 24:22–32. doi: 10.1038/leu.2009.236
8. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood (2001) 98:210–6. doi: 10.1182/blood.V98.1.210
9. Luptakova K, Rosenblatt J, Glotzbecker B, Mills H, Stroopinsky D, Kufe T, et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol Immunother (2013) 62:39–49. doi: 10.1007/s00262-012-1308-3
10. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. (2015) 21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200
11. Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother (2009) 58:1033–45. doi: 10.1007/s00262-008-0620-4
12. Noonan K, Rudraraju L, Ferguson A, Emerling A, Pasetti MF, Huff CA, et al. Lenalidomide-induced immunomodulation in multiple myeloma: impact on vaccines and antitumor responses. Clin Cancer Res. (2012) 18:1426–34. doi: 10.1158/1078-0432
13. Busch A, Zeh D, Janzen V, Mugge LO, Wolf D, Fingerhut L, et al. Treatment with lenalidomide induces immunoactivating and counter-regulatory immunosuppressive changes in myeloma patients. Clin Exp Immunol. (2014) 177:439–53. doi: 10.1111/cei.12343
14. Clave E, Douay C, Coman T, Busson M, Bompoint C, Moins-Teisserenc H, et al. Lenalidomide consolidation and maintenance therapy after autologous stem cell transplant for multiple myeloma induces persistent changes in T-cell homeostasis. Leuk Lymphoma (2014) 55:1788–95. doi: 10.3109/10428194.2013.865182
15. Fostier K, Caers J, Meuleman N, Broos K, Corthals J, Thielemans K, et al. Impact of lenalidomide maintenance on the immune environment of multiple myeloma patients with low tumor burden after autologous stem cell transplantation. Oncotarget (2018) 9:20476–89. doi: 10.18632/oncotarget.24944
16. Kramer I, Engelhardt M, Fichtner S, Neuber B, Medenhoff S, Bertsch U, et al. Lenalidomide enhances myeloma-specific T-cell responses in vivo and in vitro. Oncoimmunology (2016) 5:e1139662. doi: 10.1080/2162402X.2016.1139662
17. Brissot E, Clavert A, Blin N, Roland V, Guillaume T, Dubruille V, et al. Impact of lenalidomide on immune functions in the setting of maintenance therapy for multiple myeloma. Leukemia (2015) 29:2098–100. doi: 10.1038/leu.2015.64
18. Danhof S, Schreder M, Knop S, Rasche L, Strifler S, Loffler C, et al. Expression of programmed death-1 on lymphocytes in myeloma patients is lowered during lenalidomide maintenance. Haematologica (2018) 103:e126–e9. doi: 10.3324/haematol.2017.178947
19. Moreau P, Hulin C, Macro M, Caillot D, Chaleteix C, Roussel M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood (2016) 127:2569–74. doi: 10.1182/blood-2016-01-693580
20. Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. (2016) 17:e328–46. doi: 10.1016/S1470-2045(16)30206-6
21. Reeder CB, Reece DE, Kukreti V, Chen C, Trudel S, Hentz J, et al. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: high response rates in a phase II clinical trial. Leukemia (2009) 23:1337–41. doi: 10.1038/leu.2009.26
22. Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, et al. GIMEMA Italian Myeloma Network. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomized phase 3 study. Lancet (2010) 376:2075–85. doi: 10.1016/S0140-6736(10)61424-9
23. Harousseau JL, Attal M, Avet–Loiseau H, Marit G, Caillot D, Mohty M, et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. J Clin Oncol. (2010) 28:4621–9. doi: 10.1200/JCO.2009.27.9158
24. Palumbo A, Cavallo F, Gay F, Di Raimondo F, Yehuda DB, Petrucci MT, et al. Autologous transplantation and maintenance therapy in Multiple Myeloma. New Engl J Med. (2014) 371:895-905. doi: 10.1056/NEJMoa14028
25. Dhodapkar KM, Barbuto S, Matthews P, Kukreja A, Mazumder A, Vesole D, et al. Dendritic cells mediate the induction of polyfunctional human IL17-producing cells (Th17-1 cells) enriched in the bone marrow of patients with myeloma. Blood (2008) 112:2878–85. doi: 10.1182/blood-2008-03-143222
26. Di Lullo G, Marcatti M, Heltai S, Brunetto E, Tresoldi C, Bondanza A, et al. Th22 cells increase in poor prognosis multiple myeloma and promote tumor cell growth and survival. Oncoimmunology (2015) 4:e1005460. doi: 10.1080/2162402X.2015.1005460
27. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63 doi: 10.1038/ni.1767
28. Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood (2010) 115:5385–92. doi: 10.1182/blood-2009-10-246660
29. Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood (2010) 116:3554–63. doi: 10.1182/blood-2010-05-283895
30. Di Lullo G, Marcatti M, Protti MP. Non-redundant roles for Th17 and Th22 cells in multiple myeloma clinical correlates. Oncoimmunology (2016) 5:e1093278. doi: 10.1080/2162402X.2015.1093278
31. Mori G, D'Amelio P, Faccio R, Brunetti G. Bone-immune cell crosstalk: bone diseases. J Immunol Res. (2015) 2015:108451. doi: 10.1155/2015/108451
32. Terpos E, Mihou D, Szydlo R, Tsimirika K, Karkantaris C, Politou M, et al. The combination of intermediate doses of thalidomide with dexamethasone is an effective treatment for patients with refractory/relapsed multiple myeloma and normalizes abnormal bone remodeling, through the reduction of sRANKL/osteoprotegerin ratio. Leukemia (2005) 19:1969–76. doi: 10.1038/sj.leu.2403890
33. Breitkreutz I, Raab MS, Vallet S, Hideshima T, Raje N, Mitsiades C, et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia (2008) 22:1925–32. doi: 10.1038/leu.2008.174
34. Uluckan O, Jimenez M, Karbach S, Jeschke A, Grana O, Keller J, et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts. Sci Transl Med. (2016) 8:330ra37. doi: 10.1126/scitranslmed.aad8996
35. Brunetti G, Rizzi R, Storlino G, Bortolotti S, Colaianni G, Sanesi L, et al. LIGHT/TNFSF14 as a new biomarker of bone disease in multiple myeloma patients experiencing therapeutic regimens. Front Immunol. (2018) 9:2459. doi: 10.3389/fimmu.2018.02459
Keywords: multiple myeloma, immunomodulatory drugs, autologous hematopoietic stem cell transplantation, bone marrow, anti-tumor and pro-tumor T cell subsets
Citation: Di Lullo G, Marcatti M, Heltai S, Tresoldi C, Paganoni AM, Bordignon C, Ciceri F and Protti MP (2019) Immunomodulatory Drugs in the Context of Autologous Hematopoietic Stem Cell Transplantation Associate With Reduced Pro-tumor T Cell Subsets in Multiple Myeloma. Front. Immunol. 9:3171. doi: 10.3389/fimmu.2018.03171
Received: 21 September 2018; Accepted: 24 December 2018;
Published: 21 January 2019.
Edited by:
Lionel Apetoh, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Frederique Vegran, INSERM U1231 Lipides, Nutrition, Cancer (LNC), FranceRomain Boidot, Centre Georges François Leclerc, France
Giacomina Brunetti, Università degli Studi di Bari, Italy
Copyright © 2019 Di Lullo, Marcatti, Heltai, Tresoldi, Paganoni, Bordignon, Ciceri and Protti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Di Lullo, ZGlsdWxsby5naXVsaWFAaHNyLml0
Maria Pia Protti, cHJvdHRpLm1hcmlhcGlhQGhzci5pdA==