 Denise van Uden
Denise van Uden Karin Boomars
Karin Boomars Mirjam Kool
Mirjam Kool- Department of Pulmonary Medicine, Erasmus MC, Rotterdam, Netherlands
Pulmonary arterial hypertension (PAH) is a cardiopulmonary disease characterized by an incurable condition of the pulmonary vasculature, leading to increased pulmonary vascular resistance, elevated pulmonary arterial pressure resulting in progressive right ventricular failure and ultimately death. PAH has different underlying causes. In approximately 30–40% of the patients no underlying risk factor or cause can be found, so-called idiopathic PAH (IPAH). Patients with an autoimmune connective tissue disease (CTD) can develop PAH [CTD-associated PAH (CTD-PAH)], suggesting a prominent role of immune cell activation in PAH pathophysiology. This is further supported by the presence of tertiary lymphoid organs (TLOs) near pulmonary blood vessels in IPAH and CTD-PAH. TLOs consist of myeloid cells, like monocytes and dendritic cells (DCs), T-cells, and B-cells. Next to their T-cell activating function, DCs are crucial for the preservation of TLOs. Multiple DC subsets can be found in steady state, such as conventional DCs (cDCs), including type 1 cDCs (cDC1s), and type 2 cDCs (cDC2s), AXL+Siglec6+ DCs (AS-DCs), and plasmacytoid DCs (pDCs). Under inflammatory conditions monocytes can differentiate into monocyte-derived-DCs (mo-DCs). DC subset distribution and activation status play an important role in the pathobiology of autoimmune diseases and most likely in the development of IPAH and CTD-PAH. DCs can contribute to pathology by activating T-cells (production of pro-inflammatory cytokines) and B-cells (pathogenic antibody secretion). In this review we therefore describe the latest knowledge about DC subset distribution, activation status, and effector functions, and polymorphisms involved in DC function in IPAH and CTD-PAH to gain a better understanding of PAH pathology.
Introduction Pulmonary Arterial hypertension
Pulmonary arterial hypertension (PAH) is characterized by a mean pulmonary arterial pressure (PAP) of ≥25 mmHg at rest and a mean capillary wedge pressure of ≤15 mmHg (1). The high PAP causes hypertrophy of the right ventricle (RV) leading eventually to RV dilatation, heart failure, and ultimately death. Particularly small pulmonary arteries (PAs) and arterioles are affected. They show a thickened vascular wall and formation of plexiform lesions due to endothelial dysfunction and proliferation of all three cell layers, the endothelium, smooth muscle cells (SMC), and the adventitia (2).
PAH patients can be subdivided into groups based on associated conditions and risk factors. However, in a substantial proportion of PAH patients no cause or associated condition can be identified: idiopathic PAH (IPAH). In another subgroup of patients, PAH is associated with autoimmune diseases (AD) such as connective tissue disease (CTD). CTD includes systemic sclerosis (SSc), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and mixed connective tissue disease (MCTD). SSc is the most common AD associated with PAH, followed by SLE (3–6). PAH patients have a low 1-year survival rate: only 82% of SSc-PAH patients and 93% of IPAH patients are still alive after 1 year (6).
Role for Immune Activation in the Development of PAH
The presence of PAH in a proportion of autoimmune patients suggests that activated immune cells (or their mediators) directly provoke pulmonary vascular remodeling. Local immune activation is also observed as tertiary lymphoid organs (TLOs or ectopic lymphoid structures) are present in the lungs of IPAH and CTD-associated PAH (CTD-PAH) patients (7, 8). TLOs are organized structures similar to lymph nodes (LNs), including distinct T-cell areas containing dendritic cells (DCs), organized B-cell follicles with germinal centers (GCs), high endothelial venules (HEV), and lymphatics. TLOs most likely develop due to long-lasting local immune activation and are considered a hallmark of chronic disease (9). In lungs of IPAH patients, TLOs are found in the vicinity of PAs, suggesting that they promote vascular remodeling (7). Not surprisingly, as TLOs are characteristic for ongoing/chronic immune activation, they are often found in target organs of several ADs. For instance, in SLE patients TLOs are present in the kidneys, and in SSc-PAH patients TLOs have even been found in the lungs (8, 10, 11). Even though the SSc-PAH patient group used in this study is small, it is conceivable that TLOs are present in the lungs of various CTD-PAH patients. In addition, it is very likely that immune activation in PAH patients will also occur in draining LNs.
During chronic antigenic stimulation, the lymphotoxin (LT)α1β2-LTβ receptor axes is crucial for development of TLOs (12), whereby lymphoid tissue inducer (LTi) cells interact with lymphoid tissue organizer (LTo) cells. Repeated DC injection in the lungs of mice, mimicking chronic activation, provokes TLO development (13). Activated DCs can produce chemokines which attract T-cells and B-cells (e.g., CCL19/21 and CXCL13, respectively), as well as T- and B-cell survival factors (e.g., interleukin (IL)-15 and BAFF/IL-6, respectively) (13–17). They furthermore secrete cytokines creating a pro-inflammatory milieu and promote innate and adaptive responses. This milieu can also induce post-translational modifications of proteins, altering self-antigens into new antigens which could provoke autoimmune responses as seen in SLE (18). Within TLOs and LNs, tissue-migrated DCs present antigens to naïve T-cells, inducing their activation and differentiation. The main T helper (Th)-cell subsets are Th1, Th2, Th17, follicular Th-cells (Tfh), and regulatory T-cells (Tregs). Within the GC reaction in TLOs and LNs, Tfh-cells provide help to B-cells by producing cytokines that induce class switching, survival, proliferation, and antibody production.
The role of DC subsets and their effector function in pathogenesis of IPAH, AD, and CTD-PAH will be discussed in this review and is shown in Table 1.
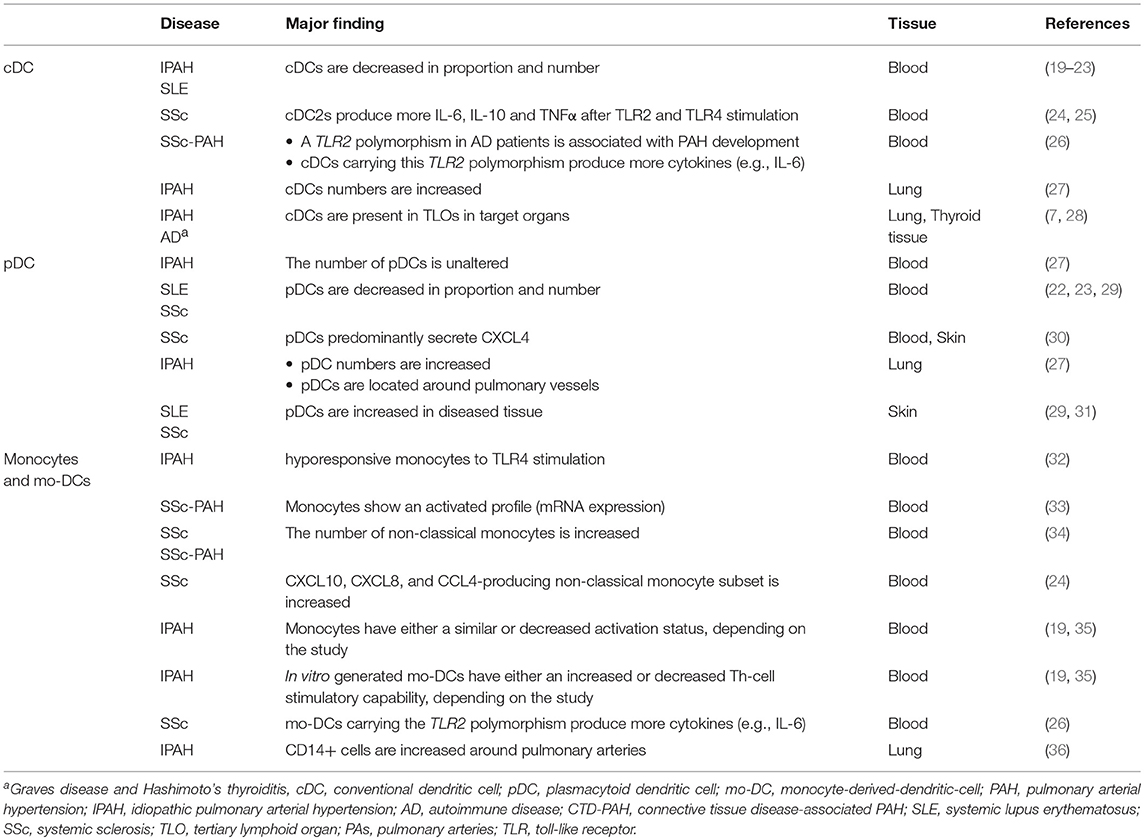
Table 1. Involvement of DCs and monocytes in IPAH, AD, and CTD-PAH.
Dendritic Cells in IPAH, CTD-PAH, and AD
DCs are equipped with pathogen recognition receptors (PRRs) like toll-like receptors (TLR) to sense their surroundings. Antigen recognition leads to DC activation and migration toward LNs. Activated DCs upregulate co-stimulatory molecules like CD86, produce pro-inflammatory cytokines, and present antigen to T-cells using major histocompatibility complex class-II (MHC-II). In TLOs, DCs are mature, indicated by high CD86 expression and IL-12 production (37). The maintenance of TLOs in two lung infection models, has been shown to be dependent on DCs as they disintegrate when DCs are ablated (13, 38). Furthermore, impaired DC migration due to defects in the CCR7-signaling, has been shown to lead to the formation of bronchus-associated lymphoid tissue (39).
Under steady state conditions, several DC subsets with unique functions can be identified (40, 41). Conventional DCs (cDCs), identified by CD11c, and HLA-DR expression in humans, are a major DC subset and can be divided in two subtypes, type 1 cDCs (cDC1s) and type 2 cDCs (cDC2s). cDC1s express IRF8 and CD141 and excel in cross presentation (42). IRF4 and CD1c classify cDC2s, which are potent inducers of Th-cell responses. Plasmacytoid DCs (pDCs) produce interferons (IFN) and do not express CD11c, but express HLA-DR and CD123. Recently, within this HLA-DR+CD123+ population potent Th-cell inducers have been found, which additionally express AXL and Siglec6 (AXL+Siglec6+ (AS)-DCs) (43, 44). Under inflammatory conditions monocytes can differentiate into DCs, giving rise to monocyte-derived-DCs (mo-DCs).
Conventional Dendritic Cells
In IPAH patients, the proportion of circulating cDCs is decreased compared to controls (19). Numbers of circulating cDCs are also altered in several ADs associated with PAH. Both cDC1s and cDC2s are decreased in proportion and number in SLE patients compared to HCs, especially in patients with active disease (20–23). The decrease in circulating cDCs in PAH could indicate an increased cDC migration toward lung TLOs (Figure 1). In support of this idea, DCs can be found in lung TLOs of IPAH patients and cDC numbers were increased in total lung cell suspensions of these patients (7, 27). In IPAH TLOs, DCs are found inside T-cell zones, suggesting that they promote T-cell activation. In patients with ADs, cDCs in TLOs show increased expression of costimulatory molecules and a cDC2 phenotype, since they express CD1c and CD11c (28). Alternatively, the reduction in circulating cDCs might also be caused by alterations in cDC viability or DC progenitors resulting in a decreased output of cDCs from the bone marrow.
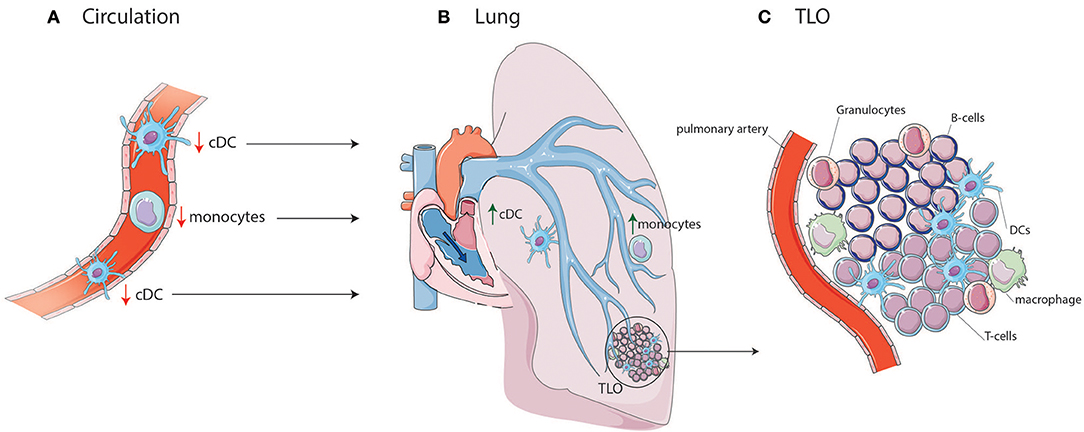
Figure 1. cDC and monocyte migration toward lung TLOs. (A) cDCs and monocytes are decreased in circulation of IPAH patients due to migration to the lungs in which cDCs and monocytes are increased. (B) In the lung they can add to the development of TLOs surrounding PAs. (C) TLOs consist, besides DCs, of different immune cells such as T-cells, B-cells, macrophages, and granulocytes.
In addition to DC or DC precursors entering the affected tissue from the blood circulation, DCs may accumulate in tissue and contribute to TLO formation as they fail to go to LNs (39). Upon activation, DCs upregulate CCR7. The CCR7 allows the DC to respond to CCL19 and CCL21 expressed by the lymphatic endothelial cells and to enter the lymphatic vessels to migrate to the draining LN. Both CCL19 and CCL21 are expressed by lymphatic vessels in IPAH patients, which could facilitate DC attraction (7). Strikingly, CCR7-deficient mice develop lung TLOs and signs of PH, perhaps due to DC retention in the lungs (39, 45). DCs, amongst other cells, can produce CCL20 and CXCL13, which attract T-cells, B-cells, and immature DCs. CCL20 and CXCL13 mRNA expression are increased in IPAH lungs compared to controls (7), contributing to TLO formation. However, the cell responsible for this increased expression in IPAH is yet unknown.
Research into cDC subset activation is still limited in PAH and ADs. In SSc patients, circulating cDC2s produce more IL-6, IL-10, and TNF-α after TLR2 and TLR4 stimulation (24, 25). These cytokines appear to play a central role in the immunopathology of PAH, as IL-6 and IL-10 are increased in the serum of IPAH patients and correlate with mortality (46). Especially IL-6 appears to be a crucial cytokine in PAH pathobiology, as mice overexpressing IL-6 develop signs of PH, while IL-6-deficient mice do not develop PH after hypoxia (47, 48). At this time, a phase II trial using Tocilizumab, an IL-6 receptor antagonist, is conducted in PAH patients (49).
In conclusion, in both IPAH and ADs circulating cDC proportions are decreased possibly due to migration to target organs, where they can both initiate adaptive immune responses and maintain TLOs (Figure 2B). Currently, only little is known about cDC subset distribution and function in IPAH, CTD-PAH, and ADs.
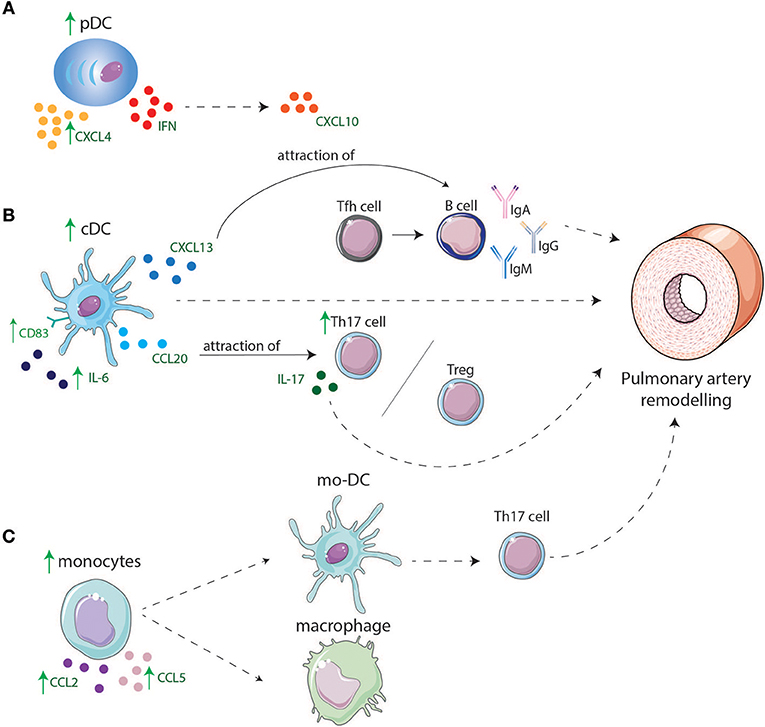
Figure 2. Involvement of DCs and monocyte in lungs of IPAH and CTD-PAH patients. (A) pDCs are increased in lungs and might play a role in IPAH and CTD-PAH pathology by producing higher levels of CXCL4 and CXCL10 that is induced by IFNs. (B) cDC display higher levels of CD83 and have an enhanced cytokine production e.g., IL-6. cDCs are increased in lungs of PAH patients and can directly lead to PA remodeling or indirectly by production of CXCL13 and CCL20. CXCL13 leads to migration of B-cells toward the lungs, B-cells will produce pathogenic antibodies after interaction with Tfh cells, leading to remodeling of PAs. CCL20 attracts T-cells such as Tregs and Th17 cells leading to an increase in Th17 cells in the lung resulting in a Th17/Treg disbalance and by IL-17 production contributes to PA remodeling. (C) Monocytes are increased in the lung and produce CCL2 and CCL5 which might lead to attraction of other monocytes. Monocytes might differentiate in macrophages or mo-DCs. Mo-DCs induce Th17 cells adding to PA remodeling.
Plasmacytoid Dendritic Cells
Plasmacytoid DCs are predominantly found in lymphoid tissues and blood in steady state conditions. During inflammation, pDCs home toward peripheral tissues, produce type I IFNs, and promote activation of immune cells. In IPAH lungs pDC numbers are enhanced and pDCs are specifically located around the pulmonary vessels, while circulating pDC numbers are unaltered (27). In contrast, in SLE and SSc patients, circulating pDC number and frequency are decreased compared to controls, which could be due to emigration into diseased tissues (22, 23, 29, 31). Indeed, pDCs are present in diseased organs of SSc patients (29). Several ADs are associated with the interferon gene signature (IGS), to which different cells contribute. pDCs are major contributors to the IGS through their production of type I IFNs. One of the most strongly upregulated genes in pDCs within the IGS is CXCL10 (50). Augmented serum CXCL10 levels are associated with PAH in SSc patients (51). Likewise, in IPAH patients, serum CXCL10 is elevated and even associated with poor RV function (52), suggesting the possibility of a prominent role for pDCs in disease immunopathology. Next to IFNs, pDCs are also large producers of CXCL4 in SSc (30). CXCL4 can induce an influx of CD45+ cells in target tissues, perhaps leading to tissue remodeling and disease progression.
The associations of pDC with CTD-PAH and the increase in pDCs in lungs of IPAH patients suggest that type-I IFN and chemokine secretion by pDCs not only play an important role in several ADs, but also in CTD-PAH and IPAH pathology (Figure 2A).
Monocytes and Monocyte-Derived DCs
Monocytes are precursors of mo-DCs that arise under inflammatory conditions (40). Monocytes are heterogeneous and can be divided into 3 subsets based on CD14 and CD16 expression (53, 54). Classical monocytes, also called inflammatory monocytes, express CD14 and can infiltrate tissues, produce pro-inflammatory cytokines, and differentiate into inflammatory macrophages. Classical monocytes express several PRRs and are superior in phagocytosis. Monocytes expressing both CD14 and CD16 are termed intermediate monocytes, can also produce pro-inflammatory cytokines (55) and are unique in their ability to produce reactive oxygen species. Their gene expression signature indicates their ability to present antigens and induce T-cell activation (56). Intermediate monocytes specifically promote pro-inflammatory Th17-cell responses, which also contribute to PAH development, as discussed below (55). Finally, non-classical monocytes, expressing CD16, are known to survey the endothelium for danger signals (54). They differentiate into tissue-resident macrophages in steady state or into anti-inflammatory macrophages during inflammation, to repair damaged tissues.
The number of non-classical monocytes is increased in SSc associated with PAH development, whereas there is no difference in the number of classical monocytes (34). The number of CTD-PAH patients in this study was very small, so this should be confirmed in a larger cohort. Increased numbers of CD14+ cells, including classical/intermediate monocytes and macrophages, are observed around PAs of IPAH patients (36). Monocytes might be attracted to the PAs through their expression of CCR2 and CCR5 and an increased expression of their ligands CCL2 and CCL5 in lungs and serum of IPAH patients (57, 58). In SSc and CTD-PAH enhanced CCL2 is also observed in either skin or serum (59–61).
Strikingly, circulating monocytes of IPAH patients are hyporesponsive, as demonstrated by decreased cytokine production upon TLR4 stimulation (32). The local and/or systemic pro-inflammatory milieu in IPAH patients could provoke a feedback mechanism, resulting in hyporesponsive monocytes. However, the underlying mechanism is still unknown and further research is needed. In contrast to IPAH monocytes, monocytes from SSc-PAH patients are activated, as shown by their mRNA expression profile. This profile is even discriminative between SSc-PAH and SSc patients (33). Non-classical monocytes, expressing CXCL10, CXCL8, and CCL4 are involved in SSc pathology, and are found in increased numbers in SSc patients compared to controls (24).
Mo-DCs for in vitro assays, used to model and monitor human DC function, are commonly generated from monocytes. Contradictory results have been found using this model in IPAH. Decreased activation of monocytes together with lower T-cell stimulation (19), as well as a similar activation status with an increased Th-cell stimulatory capability have been observed (35). These opposite findings might be caused by the type of stimulation used to mature mo-DCs and different mo-DC:T-cell ratios in the T-cell stimulation assays.
Taken together, increased pulmonary expression of chemokines may attract monocytes to lungs of IPAH and CTD-PAH patients, where they become activated and alter their gene expression due to the pro-inflammatory environment. These altered monocytes may give rise to mo-DCs, which arise at places of inflammation and can induce T-cell activation (Figure 2C).
Effector Function of DCs in IPAH, CTD-PAH and ADS
T-Cell Responses
DCs excel at antigen presentation to T-cells and together with their costimulatory molecule expression and cytokine production, they are pivotal for the succeeding T-cell response. Specifically, Th17-cells are implicated in the pathogenesis of many ADs and are observed inside mature TLOs of IPAH patients (7). Th17 differentiation from naïve Th-cells occurs in the presence of IL-1β, IL-6, and TGFβ (62), cytokines produced by activated DCs. Both IL-1β and IL-6 are elevated in serum of IPAH patients (46). Th17-cells are the main source of IL-17, IL-21, and IL-22. IL-21+ cells are present in remodeled PAs of IPAH patients (63). In addition, IL-17 may affect structural remodeling observed in PAH, as IL-17 enhances fibroblast proliferation and collagen production in vitro (64). In SSc, IL-17 induces adhesion molecule expression and IL-1/chemokine production on endothelial cells (ECs) (65–67). Additionally, in IPAH PBMCs the IL-17 gene is hypo-methylated, indicating increased IL-17 transcription and supporting a possible role for Th17-cells in the pathology of IPAH (35). Indeed, IL-17 gene expression is enhanced in lungs of both IPAH and SSc-PAH compared to idiopathic pulmonary fibrosis (IPF) and pulmonary fibrosis associated SSc (SSc-PF) (68), this IL-17 may be expressed by cells in TLOs as well as in tissues outside of TLOs.
Furthermore, IL-23, also produced by DCs, stabilizes the phenotype of Th17-cells, but also promotes their pro-inflammatory potential (62). Th17-cells are also highly plastic cells and under the influence of IL-23 start co-expressing cytokines from the Th1-cell lineage. This leads to possibly pathogenic IFNγ-producing Th17-cells, also called Th17.1-cells. Enhanced expression of the IL-23 receptor on Th17(.1)-cells might contribute to their pro-inflammatory pathogenic phenotype (62, 69, 70). IL-23 is increased in exhale breath condensate of SSc patients, so perhaps Th17 plasticity plays a role in SSc pathology (71). Furthermore, IFNγ, IL-12, and TNFα can induce plasticity toward Th17.1-cells (62). Both serum IL-12 and TNFα are enhanced in IPAH patients and mRNA transcripts of these cytokines were increased in lungs rats in a PH model (46, 72). IL-17/IFNγ-double producing Th-cells are observed within the arteries of atherosclerosis patients, where they provoke pro-inflammatory cytokine production (e.g., IL-6, CXCL10) by vascular SMCs (73). This feedback loop could also exist within PAH, since IL-6 is highly produced by pulmonary ECs of IPAH patients. In addition, IL-6 promotes SMC proliferation in a hypoxia-induced PH model (74, 75). Blocking of IL-6 signaling improved PH physiology in a hypoxia-induced PH mouse model and prevented accumulation of Th17-cells (63). IL-6 also converts Th17-cells into IL-17+ Tregs, which are less suppressive than conventional Tregs (76). In SSc, IL-17+ Tregs are observed in the circulation and possibly also in the skin, indicated by IL-17 and FoxP3 positivity (64, 65, 77). The balance between pro-inflammatory Th17-cells and anti-inflammatory Tregs is crucial to control autoimmune features. IL-6 is a key cytokine in Th17/Treg balance, since TGF-β alone polarizes naïve Th-cells to Tregs, while TGF-β together with IL-6 induces Th17-cells (78). Active TGF-β signaling is very prominent in PAH and can be produced by different cells, like monocytes and DCs (79). However, whether DC-derived IL-6 plays a prominent role is unknown yet, as many cells can produce IL-6. In favor of a disturbed balance are the decreased number of Tregs observed in SLE, which correlates with disease severity (66). In CTD-PAH patients Th17-cells and Th17-related cytokines are elevated compared to AD patients without PAH (80). The disturbed Th17/Treg ratio even appears to correlate with PAH severity in APAH patients (80). This demonstrates that Th17-cells and Tregs are implicated not only in ADs but also in PAH (80).
Therefore, Th17 plasticity and Th17/Treg balance may contribute to ADs and PAH, potentially in part by modulating vascular remodeling.
Humoral Immune Response
Apart from their interaction with Th17-cells, DCs can induce (immature) Tfh-cells, which develop under the influence of IL-21, IL-6, IL-12, and IL-27 (78). In mature TLOs containing GCs, Tfh-cells interact with B-cells, leading to either antibody-producing plasma cells or memory B-cells. There is clear evidence for B-cell dysregulation in IPAH and CTD-PAH (81, 82). In IPAH patients circulating B-cells have an increased expression of genes involved in inflammatory mechanisms, host defense, and endothelial dysfunction, suggesting increased activation of B-cells (82). Also numbers of circulating plasmablasts are elevated in IPAH patients (83). Anomalies in B-cell homeostasis were also observed in SSc-PAH patients, with increased circulating IgD+ B-cell proportions (81). Tfh-cell numbers crucially control the development of auto-reactive B-cells, since an increase in Tfh-cell number can lead to increased autoantibody production (84, 85). In several ADs, Tfh-cells are increased in blood and target organs (86–89). Serum IgG, IgM, and IgA antibodies are elevated in IPAH patients, and EC-specific IgA promotes cytokine production and upregulation of adhesion molecules (83, 90–92). IgG and IgM antibodies directed against EC-surface antigens are also found in ADs and CTD-PAH, being most prevalent in SSc-PAH patients, followed by IPAH patients and SSc patients without PAH (92). IgG antibodies in SSc and SLE were directed against microvascular ECs antigens, while IgG in SSc, IPAH, and CTD-PAH recognized microvascular dermal and lung EC antigens, and vascular SMCs (90, 91, 93–95). Auto-reactive IgG provoked EC dysfunction, induced pro-inflammatory signals, and increased adhesiveness of T-cells to ECs, which also modulated migration and proliferation of SMC. These autoantibodies from SSc or CTD-PAH patients can directly cause signs of PH when injected into healthy mice (96). It is unknown where the autoantibodies found in IPAH and CTD-PAH patients are produced. TLO might be a likely location since Tfh-cells and B-cells, and perhaps antigens, are present in these TLOs. However, these autoantibodies can also be produced in the draining LNs.
In brief, pathogenic autoantibodies in CTD-PAH and IPAH might be produced by dysregulated B-cells that interact with Tfh-cells in TLOs. These autoantibodies recognize protein epitopes expressed by ECs, leading to endothelial dysfunction and vascular remodeling. So far, the role of Tfh-cells in IPAH is unknown and further research is needed.
Genetics
Increased activation of the immune system in PAH is also supported by different polymorphisms observed in genome wide association studies. A polymorphism in TLR2 of SSc patients is associated with PAH development (26). Functional analysis of mo-DCs and cDCs carrying the TLR2 polymorphism showed enhanced cytokine production, including IL-6, compared to control DCs. As discussed above, IL-6 plays a prominent role in PAH pathology. Strikingly, a decreased IL-6 serum level was observed in healthy individuals and patients with a single nucleotide polymorphism in the promotor region of the IL-6 gene, IL-6-572C/G, which correlated with decreased risk to develop IPAH (97). SNPs might not only be useful to determine disease susceptibility but also to determine disease onset or activity, as is seen for a specific SNP in TGFB gene in heritable PAH patients carrying a BMPR2 mutation (98). Another genetic association found in both PAH and SSc involving immune activation is a SNP in the TNFAIP3 gene (99). TNFAIP3 encodes for the ubiquitinating enzyme A20, which is crucial for down-regulation of the nuclear factor-kappa B (NF-κB) signaling pathway and thereby cell activation (100). Macrophages, pulmonary arterial ECs, and pulmonary arterial SMCs in end-stage IPAH patients showed an increased expression in NF-κB (101), suggesting an important role for the NF-κB pathway in IPAH.
This demonstrates that several SNPs and genes that are involved in DC function are present in PAH patients.
Future Directions
In conclusion, different DC subsets are involved not only in the pathobiology of ADs but appear to play a role in the pathobiology of IPAH and CTD-PAH as well. However, the exact role of these DCs in PAH development has not been fully elucidated. The increasing knowledge on DC biology obtained by advanced immunological techniques has led to a more unified method to identify DC subsets and the discovery of new DC subsets. Determining the role of all currently known DC populations, including AS-DCs, as well as their specific functions may help to unravel the pathobiology of PAH. This might lead to new opportunities for therapies targeting specific DC subsets, their activation, and/or their effector function.
Author Contributions
DvU and MK wrote the manuscript. KB contributed to the review of the manuscript. All authors approved the manuscript for publication.
Funding
This work was supported by a grant of the Dutch Heart Foundation (2016T052).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank O.B.J. Corneth for critically reading the manuscript.
Abbreviations
PAH, Pulmonary arterial hypertension; PAP, pulmonary arterial pressure; RV, right ventricle; PAs, pulmonary arteries; SMC, smooth muscle cell; IPAH, idiopathic PAH; CTD, connective tissue diseases; CTD-PAH, CTD-associated PAH; AD, autoimmune disease; SSc, systemic sclerosis; SLE, systemic lupus erythematosus; RA, rheumatoid arthritis; MCTD, mixed connective tissue disease; SSc-PAH, Systemic sclerosis-PAH; TLOs, tertiary lymphoid organs; LNs, lymph nodes; DCs, dendritic cells; GCs, germinal centers; HEV, high endothelial venules; PH, pulmonary hypertensions; LT, lymphotoxin; LTi, lymphoid tissue inducers; LTo, lymphoid tissue organizer; Th, T helper; IL, interleukin; Tfh, follicular Th-cells; Tregs, regulatory T-cells; PRRs, pathogen recognition receptors; TLR, toll-like receptor; MHC-II, major histocompatibility complex class II; cDCs, conventional DCs; cDC1s, type 1 cDCs; cDC2s, type 2 cDCs; pDCs, Plasmacytoid DCs; IFN, interferons; AS–DCs, AXL+Siglec6+ DCs; mo-DCs, monocyte-derived dendritic cells; BM, bone marrow; IGS, interferon gene signature; PBMCs, peripheral blood mononuclear cells; LPS, lipopolysaccharide; ECs, endothelial cells; IPF, idiopathic pulmonary fibrosis; SSc-PF, pulmonary fibrosis associated SSc; NF-kB, nuclear factor-kappa B.
References
1. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. (2016) 37:67–119. doi: 10.1093/eurheartj/ehv317
2. Humbert M, Montani D, Perros F, Dorfmüller P, Adnot S, Eddahibi S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol. (2008) 49:113–8. doi: 10.1016/j.vph.2008.06.003
3. McGoon MD, Miller DP. REVEAL: a contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. (2012) 21:8–18. doi: 10.1183/09059180.00008211
4. Dhala A. Pulmonary arterial hypertension in systemic lupus erythematosus: current status and future direction. Clin Dev Immunol. (2012) 2012:854941. doi: 10.1155/2012/854941
5. Tselios K, Gladman DD, Urowitz MB. Systemic lupus erythematosus and pulmonary arterial hypertension: links, risks, and management strategies. Open Access Rheumatol. (2017) 9:1–9. doi: 10.2147/OARRR.S123549
6. Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest (2010) 138:1383–94. doi: 10.1378/chest.10-0260
7. Perros F, Dorfmüller P, Montani D, Hammad H, Waelput W, Girerd B, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 185:311–21. doi: 10.1164/rccm.201105-0927OC
8. Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. (1997) 28:434–42. doi: 10.1016/S0046-8177(97)90032-0
9. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. (2012) 33:297–305. doi: 10.1016/j.it.2012.04.006
10. Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. (2008) 74:448–57. doi: 10.1038/ki.2008.191
11. Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. (2011) 186:1849–60. doi: 10.4049/jimmunol.1001983
12. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. (2010) 10:664–74. doi: 10.1038/nri2832
13. GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med. (2009) 206:2339–49. doi: 10.1084/jem.20090410
14. McDonald KG, McDonough JS, Dieckgraefe BK, Newberry RD. Dendritic cells produce CXCL13 and participate in the development of murine small intestine lymphoid tissues. Am J Pathol. (2010) 176:2367–77. doi: 10.2353/ajpath.2010.090723
15. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, Kool M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity (2011) 35:82–96. doi: 10.1016/j.immuni.2011.05.013
16. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. (2001) 167:1179–87. doi: 10.4049/jimmunol.167.3.1179
17. Gasparini C, Foxwell BM, Feldmann M. RelB/p50 regulates CCL19 production, but fails to promote human DC maturation. Eur J Immunol. (2009) 39:2215–23. doi: 10.1002/eji.200939209
18. Doyle HA, Yang ML, Raycroft MT, Gee RJ, Mamula MJ. Autoantigens: novel forms and presentation to the immune system. Autoimmunity (2014) 47:220–33. doi: 10.3109/08916934.2013.850495
19. Wang W, Yan H, Zhu W, Cui Y, Chen J, Wang X, et al. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J Clin Immunol. (2009) 29:705–13. doi: 10.1007/s10875-009-9322-8
20. Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus (2008) 17:654–62. doi: 10.1177/0961203308089410
21. Khan SA, Nowatzky J, Jiménez-Branda S, Greenberg JD, Clancy R, Buyon J, et al. Active systemic lupus erythematosus is associated with decreased blood conventional dendritic cells. Exp Mol Pathol. (2013) 95:121–3. doi: 10.1016/j.yexmp.2013.06.003
22. Migita K, Miyashita T, Maeda Y, Kimura H, Nakamura M, Yatsuhashi H, et al. Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin Exp Immunol. (2005) 142:84–91. doi: 10.1111/j.1365-2249.2005.02897.x
23. Blomberg S, Eloranta ML, Magnusson M, Alm GV, Rönnblom L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-alpha by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. (2003) 48:2524–32. doi: 10.1002/art.11225
24. Carvalheiro T, Horta S, van Roon JAG, Santiago M, Salvador MJ, Trindade H, et al. Increased frequencies of circulating CXCL10-, CXCL8- and CCL4-producing monocytes and Siglec-3-expressing myeloid dendritic cells in systemic sclerosis patients. Inflamm Res. (2018) 67:169–77. doi: 10.1007/s00011-017-1106-7
25. van Bon L, Popa C, Huijbens R, Vonk M, York M, Simms R, et al. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann Rheum Dis. (2010) 69:1539–47. doi: 10.1136/ard.2009.128207
26. Broen JC, Bossini-Castillo L, van Bon L, Vonk MC, Knaapen H, Beretta L, et al. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. (2012) 64:264–71. doi: 10.1002/art.33325
27. Marsh LM, Jandl K, Grünig G, Foris V, Bashir M, Ghanim B, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. (2018) 51:1701214. doi: 10.1183/13993003.01214-2017
28. Roura-Mir C, Catálfamo M, Cheng TY, Marqusee E, Besra GS, Jaraquemada D, et al. CD1a and CD1c activate intrathyroidal T cells during Graves' disease and Hashimoto's thyroiditis. J Immunol. (2005) 174:3773–80. doi: 10.4049/jimmunol.174.6.3773
29. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med. (2018) 10: eaam8458. doi: 10.1126/scitranslmed.aam8458
30. van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. (2014) 370:433–43. doi: 10.1056/NEJMoa1114576
31. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. (2001) 159:237–43. doi: 10.1016/S0002-9440(10)61689-6
32. Raychaudhuri B, Bonfield TL, Malur A, Hague K, Kavuru MS, Arroliga AC, et al. Circulating monocytes from patients with primary pulmonary hypertension are hyporesponsive. Clin Immunol. (2002) 104:191–8. doi: 10.1006/clim.2002.5253
33. Pendergrass SA, Hayes E, Farina G, Lemaire R, Farber HW, Whitfield ML, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE (2010) 5:e12106. doi: 10.1371/journal.pone.0012106
34. Lescoat A, Lecureur V, Roussel M, Sunnaram BL, Ballerie A, Coiffier G, et al. CD16-positive circulating monocytes and fibrotic manifestations of systemic sclerosis. Clin Rheumatol. (2017) 36:1649–54. doi: 10.1007/s10067-017-3597-6
35. Hautefort A, Girerd B, Montani D, Cohen-Kaminsky S, Price L, Lambrecht BN, et al. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest (2015) 147:1610–20. doi: 10.1378/chest.14-1678
36. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:897–908. doi: 10.1164/rccm.201202-0335OC
37. Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am J Pathol. (2006) 168:1227–40. doi: 10.2353/ajpath.2006.050125
38. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J Exp Med. (2009) 206:2593–601. doi: 10.1084/jem.20091472
39. Fleige H, Bosnjak B, Permanyer M, Ristenpart J, Bubke A, Willenzon S, et al. Manifold roles of CCR7 and its ligands in the induction and maintenance of bronchus-associated lymphoid tissue. Cell Rep. (2018) 23:783–95. doi: 10.1016/j.celrep.2018.03.072
40. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Guilliams M, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
41. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology (2018) 154:3–20. doi: 10.1111/imm.12888
42. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity (2016) 45:669–84. doi: 10.1016/j.immuni.2016.08.015
43. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (2017) 356:eaah4573. doi: 10.1126/science.aah4573
44. Alcántara-Hernández M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity (2017) 47:1037–1050 e6. doi: 10.1016/j.immuni.2017.11.001
45. Larsen KO, Yndestad A, Sjaastad I, Løberg EM, Goverud IL, Halvorsen B, et al. Lack of CCR7 induces pulmonary hypertension involving perivascular leukocyte infiltration and inflammation. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L50–9. doi: 10.1152/ajplung.00048.2010
46. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation (2010) 122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762
47. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. (2009) 104:236–44:28p following 244. doi: 10.1161/CIRCRESAHA.108.182014
48. Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. (2009) 10:6. doi: 10.1186/1465-9921-10-6
49. Hernández-Sánchez J, Harlow L, Church C, Gaine S, Knightbridge E, Bunclark K, et al. Clinical trial protocol for TRANSFORM-UK: a therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm Circ. (2018) 8:2045893217735820. doi: 10.1177/2045893217735820
50. Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum. (2013) 65:226–35. doi: 10.1002/art.37742
51. Eloranta ML, Franck-Larsson K, Lövgren T, Kalamajski S, Rönnblom A, Rubin K, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis. (2010) 69:1396–402. doi: 10.1136/ard.2009.121400
52. Yang T, Li ZN, Chen G, Gu Q, Ni XH, Zhao ZH, et al. Increased levels of plasma CXC-Chemokine Ligand 10:12 and 16 are associated with right ventricular function in patients with idiopathic pulmonary arterial hypertension. Heart Lung (2014) 43:322–7. doi: 10.1016/j.hrtlng.2014.04.016
53. Ziegler-Heitbrock L. Blood monocytes and their subsets: established features and open questions. Front Immunol. (2015) 6:423. doi: 10.3389/fimmu.2015.00423
54. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. (2012) 53:41–57. doi: 10.1007/s12026-012-8297-3
55. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum. (2012) 64:671–7. doi: 10.1002/art.33418
56. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood (2011) 118:e16–31. doi: 10.1182/blood-2010-12-326355
57. Dorfmüller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. (2002) 165:534–9. doi: 10.1164/ajrccm.165.4.2012112
58. Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, et al. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology (2006) 11:158–63. doi: 10.1111/j.1440-1843.2006.00821.x
59. Yamamoto T, Eckes B, Hartmann K, Krieg T. Expression of monocyte chemoattractant protein-1 in the lesional skin of systemic sclerosis. J Dermatol Sci. (2001) 26:133–9. doi: 10.1016/S0923-1811(00)00169-9
60. Distler O, Pap T, Kowal-Bielecka O, Meyringer R, Guiducci S, Landthaler M, et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: role of platelet-derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum. (2001) 44:2665–78. doi: 10.1002/1529-0131(200111)44:11<2665::AID-ART446>3.0.CO;2-S
61. Cella G, Vianello F, Cozzi F, Marotta H, Tona F, Saggiorato G, et al. Effect of bosentan on plasma markers of endothelial cell activity in patients with secondary pulmonary hypertension related to connective tissue diseases. J Rheumatol. (2009) 36:760–7. doi: 10.3899/jrheum.080542
62. van Hamburg JP, Tas SW. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J Autoimmun. (2018) 87:69–81. doi: 10.1016/j.jaut.2017.12.006
63. Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci USA (2015) 112:E2677–86. doi: 10.1073/pnas.1424774112
64. Yang X, Yang J, Xing X, Wan L, Li M. Increased frequency of Th17 cells in systemic sclerosis is related to disease activity and collagen overproduction. Arthritis Res Ther. (2014) 16:R4. doi: 10.1186/ar4430
65. Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. (2000) 43:2455–63. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K
66. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. (2009) 60:1472–83. doi: 10.1002/art.24499
67. Xing X, Yang J, Yang X, Wei Y, Zhu L, Gao D, et al. IL-17A induces endothelial inflammation in systemic sclerosis via the ERK signaling pathway. PLoS ONE (2013) 8:e85032. doi: 10.1371/journal.pone.0085032
68. Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. (2011) 63:783–94. doi: 10.1002/art.30159
69. Awasthi A, Riol-Blanco L, Jäger A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. (2009) 182:5904–8. doi: 10.4049/jimmunol.0900732
70. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell (2015) 163:1400–12. doi: 10.1016/j.cell.2015.11.009
71. Rolla G, Fusaro E, Nicola S, Bucca C, Peroni C, Parisi S, et al. Th-17 cytokines and interstitial lung involvement in systemic sclerosis. J Breath Res. (2016) 10:046013. doi: 10.1088/1752-7155/10/4/046013
72. Huang WC, Ke MW, Cheng CC, Chiou SH, Wann SR, Shu CW, et al. Therapeutic benefits of induced pluripotent stem cells in monocrotaline-induced pulmonary arterial hypertension. PLoS ONE (2016) 11:e0142476. doi: 10.1371/journal.pone.0142476
73. Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation (2009) 119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618
74. Maston LD, Jones DT, Giermakowska W, Resta TC, Ramiro-Diaz J, Howard TA, et al. Interleukin-6 trans-signaling contributes to chronic hypoxia-induced pulmonary hypertension. Pulm Circ. (2018) 8:2045894018780734. doi: 10.1177/2045894018780734
75. Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. role of the macrophage migration inhibitory factor/CD74 complex. Am J Respir Crit Care Med. (2015) 192:983–97. doi: 10.1164/rccm.201402-0322OC
76. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) 16:448–57. doi: 10.1038/ni.3153
77. Radstake TR, van Bon L, Broen J, Hussiani A, Hesselstrand R, Wuttge DM, et al. The pronounced Th17 profile in systemic sclerosis (SSc) together with intracellular expression of TGFbeta and IFNgamma distinguishes SSc phenotypes. PLoS ONE (2009) 4:e5903. doi: 10.1371/journal.pone.0005903
78. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. (2018) 87:1–15. doi: 10.1016/j.jaut.2017.12.007
79. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. (2012) 122:4306–13. doi: 10.1172/JCI60658
80. Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, et al. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm. (2014) 2014:247372. doi: 10.1155/2014/247372
81. de Bourcy CFA, Dekker CL, Davis MM, Nicolls MR, Quake SR. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci Immunol. (2017) 2:eaan8289. doi: 10.1126/sciimmunol.aan8289
82. Ulrich S, Taraseviciene-Stewart L, Huber LC, Speich R, Voelkel N. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: a cross sectional study. Respir Res. (2008) 9:20. doi: 10.1186/1465-9921-9-20
83. Blum LK, Cao RRL, Sweatt AJ, Bill M, Lahey LJ, Hsi AC, et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur J Immunol. (2018) 48:874–84. doi: 10.1002/eji.201747460
84. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. (2009) 206:561–76. doi: 10.1084/jem.20081886
85. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi: 10.1038/nature03555
86. Taylor DK, Mittereder N, Kuta E, Delaney T, Burwell T, Dacosta K, et al. T follicular helper-like cells contribute to skin fibrosis. Sci Transl Med. (2018) 10:aaf5307. doi: 10.1126/scitranslmed.aaf5307
87. Zhu C, Ma J, Liu Y, Tong J, Tian J, Chen J, et al. Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J Clin Endocrinol Metab. (2012) 97:943–50. doi: 10.1210/jc.2011-2003
88. Zhang X, Lindwall E, Gauthier C, Lyman J, Spencer N, Alarakhia A, et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus (2015) 24:909–17. doi: 10.1177/0961203314567750
89. Armengol MP, Juan M, Lucas-Martín A, Fernández-Figueras MT, Jaraquemada D, Gallart T, et al. Thyroid autoimmune disease: demonstration of thyroid antigen-specific B cells and recombination-activating gene expression in chemokine-containing active intrathyroidal germinal centers. Am J Pathol. (2001) 159:861–73. doi: 10.1016/S0002-9440(10)61762-2
90. Dib H, Tamby MC, Bussone G, Regent A, Berezné A, Lafine C, et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J. (2012) 39:1405–14. doi: 10.1183/09031936.00181410
91. Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax (2005) 60:765–72. doi: 10.1136/thx.2004.029082
92. Arends SJ, Damoiseaux J, Duijvestijn A, Debrus-Palmans L, Boomars K, Broers B, et al. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur Respir J. (2010) 35:923–5. doi: 10.1183/09031936.00164209
93. Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Vroomen M, Boomars KA, et al. Immunoglobulin G anti-endothelial cell antibodies: inducers of endothelial cell apoptosis in pulmonary arterial hypertension? Clin Exp Immunol. (2013) 174:433–40. doi: 10.1111/cei.12166
94. Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Boomars KA, Brunner-La Rocca HP, et al. Functional implications of IgG anti-endothelial cell antibodies in pulmonary arterial hypertension. Autoimmunity (2013) 46:463–70. doi: 10.3109/08916934.2013.812080
95. Bussone G, Tamby MC, Calzas C, Kherbeck N, Sahbatou Y, Sanson C, et al. IgG from patients with pulmonary arterial hypertension and/or systemic sclerosis binds to vascular smooth muscle cells and induces cell contraction. Ann Rheum Dis. (2012) 71:596–605. doi: 10.1136/annrheumdis-2011-200195
96. Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. (2014) 190:808–17. doi: 10.1164/rccm.201403-0442OC
97. Fang M, Huang Y, Zhang Y, Ning Z, Zhu L, Li X. Interleukin-6−572C/G polymorphism is associated with serum interleukin-6 levels and risk of idiopathic pulmonary arterial hypertension. J Am Soc Hypertens (2017) 11:171–77. doi: 10.1016/j.jash.2017.01.011
98. Phillips JA, Poling JS, Phillips CA, Stanton KC, Austin ED, Cogan JD, et al. Synergistic heterozygosity for TGFbeta1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet Med. (2008) 10:359–65. doi: 10.1097/GIM.0b013e318172dcdf
99. Dieudé P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, et al. Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis. (2010) 69:1958–64. doi: 10.1136/ard.2009.127928
100. Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. (2009) 284:8217–21. doi: 10.1074/jbc.R800032200
Keywords: dendritic cell, dendritic cell subsets, pulmonary arterial hypertension, idiopathic pulmonary arterial hypertension, autoimmune disease, dendritic cell effector function, connective tissue disease
Citation: van Uden D, Boomars K and Kool M (2019) Dendritic Cell Subsets and Effector Function in Idiopathic and Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Front. Immunol. 10:11. doi: 10.3389/fimmu.2019.00011
Received: 28 August 2018; Accepted: 04 January 2019;
Published: 22 January 2019.
Edited by:
Diana Dudziak, Universitätsklinikum Erlangen, GermanyReviewed by:
Veronika Lukacs-Kornek, Saarland University, GermanyTheresa T. Lu, Hospital for Special Surgery, United States
Copyright © 2019 van Uden, Boomars and Kool. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Kool, bS5rb29sQGVyYXNtdXNtYy5ubA==