 Romy E. Hoeppli
Romy E. Hoeppli Anne M. Pesenacker
Anne M. Pesenacker- 1Department of Surgery, British Columbia Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada
- 2Division of Infection and Immunity, Institute of Immunity and Transplantation, University College London, London, United Kingdom
Regulatory T cells (Tregs) are believed to be dysfunctional in autoimmunity. Juvenile idiopathic arthritis (JIA) and juvenile dermatomyositis (JDM) result from a loss of normal immune regulation in specific tissues such as joints or muscle and skin, respectively. Here, we discuss recent findings in regard to Treg biology in oligo-/polyarticular JIA and JDM, as well as what we can learn about Treg-related disease mechanism, treatment and biomarkers in JIA/JDM from studies of other diseases. We explore the potential use of Treg immunoregulatory markers and gene signatures as biomarkers for disease course and/or treatment success. Further, we discuss how Tregs are affected by several treatment strategies already employed in the therapy of JIA and JDM and by alternative immunotherapies such as anti-cytokine or co-receptor targeting. Finally, we review recent successes in using Tregs as a treatment target with low-dose IL-2 or cellular immunotherapy. Thus, this mini review will highlight our current understanding and identify open questions in regard to Treg biology, and how recent findings may advance biomarkers and new therapies for JIA and JDM.
Introduction
CD4+FOXP3+ regulatory T cells (Tregs) are a subset of CD4+ T helper cells present in lymphoid and non-lymphoid tissues, and are crucial for mediating tolerance to self, preventing allergies and controlling immune reactions after infections (1). They develop in the thymus or are induced in the periphery and exhibit contact-dependent and -independent mechanisms of action (1). Inactivating mutations in FOXP3 lead to multi-organ autoimmune disease [immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX)], highlighting the importance of Tregs (2). Importantly, it is becoming clear that the local microenvironment affects the phenotype and function of tissue-localized Tregs, which also have additional roles in repair and regeneration (3).
Treg–tissue interaction might be particular important in autoimmunity with tissue-specific presentation, such as juvenile idiopathic arthritis (JIA) and juvenile dermatomyositis (JDM). While JIA is the most common inflammatory rheumatic disease in children, JDM is rare. JIA is characterized by persistent arthritis and subtype-dependent symptoms [reviewed in (4)]. Here, we focus on polyarticular and oligoarticular JIA, which present without involvement of systemic organs or skin. JDM is characterized by inflammation of muscles and skin, resulting in muscle weakness and rashes [reviewed in (5)]. Interestingly, for both conditions researchers may take advantage of clinical sample collection from the site of inflammation: synovial fluid (SF) drained during therapeutic joint injection (JIA) and biopsies (mostly muscle, JDM). While some patients respond to therapy, others do not and studying the underlying differences may lead to better understanding and treatments.
Here, we discuss recent advances in the understanding of Treg biology in oligo-/ polyarticular JIA and JDM, and what we can learn about Treg-related disease mechanisms, treatments and biomarkers from other diseases.
Altered Tregs in JIA and JDM
The phenotype of CD4+FOXP3+ Tregs in JIA has been considerably characterized in the past (6) with the molecular roles of FOXP3 in JIA reviewed by Copland and Bending in this special collection (7). It is now clear that the Treg TCR (T cell receptor) repertoire is highly restricted in JIA, both at the site of inflammation (8–11) and in circulation (10, 12). Interestingly, in blood only Tregs but not conventional CD4+ non-Treg cells (Tconv) are more clonal (10, 12). Some suggest that the TCR repertoires of Tregs from SF and peripheral blood (PB) significantly overlap (8), while others only found a very small overlap (9, 11). These differences might be explained by different sequencing depth and analysis strategies and/or by different Treg subsets studied: total (11) or effector Tregs defined by HLA-DR (8) or CD161 expression (9). Further, one study found that SF Tregs, but not Tconv, share specificity at an amino acid sequence level among different patients (10), suggesting disease-associated Treg clones might foster JIA.
Besides a restricted TCR repertoire, Tregs from the JIA inflammatory sites show unstable FOXP3 and CD25 (13), altered homing markers (9), cytokine production (6, 9), deficiency in specific chemokine production (14), and low responsiveness to IL-2 (13)—indicating impaired Treg function in JIA. Nevertheless, many reports found that JIA SF and PB Tregs are fully demethylated (8, 13), thus committed to the Treg-lineage, and suppressive in vitro (6, 8, 9, 13, 15). Hence, JIA Tregs are likely functioning inappropriately or insufficiently in the context of the inflammatory microenvironment. Interestingly, adding SF to in vitro cultures can both increase/stabilize Treg FOXP3 expression (11, 16) and in situ induce effector T cells to be resistant to Treg-mediated suppression ex vivo (17, 18). Thus, more research is needed to decipher the effects of the inflammatory microenvironment on Treg function.
In comparison, we know little about the contribution of Tregs to JDM pathogenesis. Similar to JIA, the Treg repertoire is restricted with a lack of diversity (12). FOXP3+ Tregs were found to be enriched in JDM muscle compared to muscle tissue from patients with Duchenne muscular dystrophy (19). Since the latter is already enriched in Tregs compared to normal muscle (20), this suggests a hyper-enrichment in JDM in response to autoimmune inflammation. PB Tregs of active JDM also appear less suppressive in vitro with decreased expression of CTLA4 (19). Adult DM/ polymyositis muscle biopsies are also enriched with Tregs (21). Interestingly, both Treg and effector T cell numbers decreased post immunosuppressive therapy in adult myositis, suggesting that Treg enrichment is a response to inflammation. However, juvenile and adult DM have different clinical presentation (22) and JDM PB express more Th17-type and FOXP3 transcripts (23). JDM and other myopathies are characterized by a type 1 IFN signature (24–26) and interferons may be a potential therapeutic target (27), but their effects on Tregs remain to be investigated.
Tregs are crucial in resolving muscle injury in animal studies (28) and Treg-deficient mice develop more severe myopathies in response to antigen, while adoptive Treg transfer prevents inflammation (29, 30). Thorough immune-profiling recently revealed pan-tissue and tissue-specific signatures and enhancers of murine Tregs (31). The muscle Treg signature was highly enriched in cell cycle genes, showed a dynamic response to injury and was more similar to circulating Treg signatures than to other tissue Tregs (31), indicating that muscle Tregs might acutely infiltrate muscle and are not necessarily long-term resident cells. While myopathy is a defining characteristic of JDM, skin inflammation and rash are other symptoms (5). Skin-resident Tregs are crucial for immune homeostasis (3) and have been characterized in health and various disease settings (32). However, studies on JDM-affected skin are lacking, and more work is needed to characterize JDM skin-resident Tregs.
Tregs as a Biomarker?
JIA and JDM can exhibit an unpredictable disease course. While mounting evidence indicates that an early aggressive treatment is best for severe disease (4, 27, 33, 34), the disease course is unpredictable at presentation. Additionally, due to potential short- and long-term side effects children should not be exposed to unnecessary medication. Unfortunately, once a patient appears to be in clinical remission (on or off medications), disease may flare without any notice or obvious trigger (Figure 1A). Indeed, among JIA patients who are in clinical remission, 30–50% experience flares (35, 36).
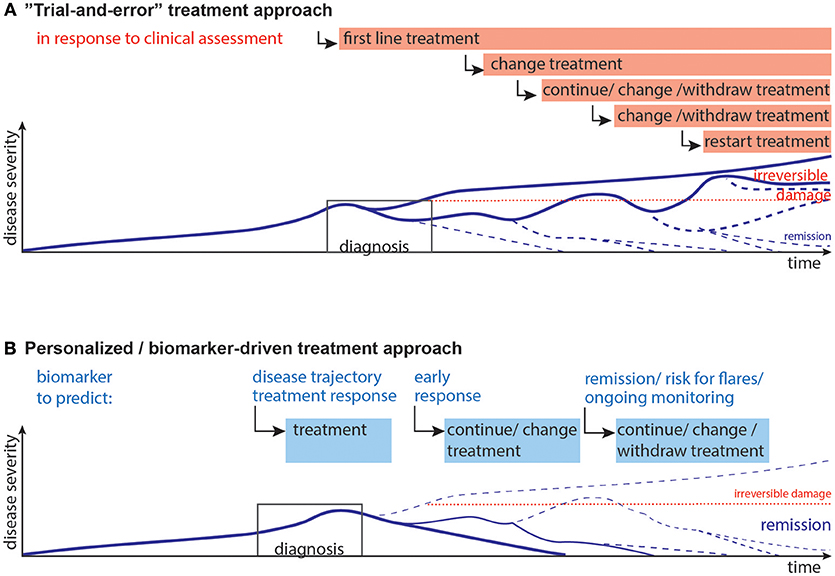
Figure 1. Current and desired disease progression models. (A) Current “Trial-and-error” model: Upon diagnosis the first line of treatment is started, which may lead to remission, or partial remission. Often, a second, third or fourth treatment strategy needs to be implemented when the previous treatments are not effective. Choice of treatments is guided by previous experience, e.g., upon presentation non-steroidal anti-inflammatory drugs with glucocorticoids are used, after a couple of month many patients are switched to methotrexate as first disease modifying therapy, often followed by anti-TNF-α agents with/without methotrexate (4). This disease progression and subsequent staggering of therapy can result in irreversible damage and long-term therapy. (B) Desired personalized/biomarker-driven model: Biomarkers could be used for prediction and aid decisions at the following stages: disease trajectory, selection of treatment, early response to therapy, remission/minimal disease activity and the risk of flare upon withdrawal of therapy, ongoing monitoring of immune activity and risk of flares. Biomarkers might thus contribute to more efficient therapy, pre-empt flares, and minimize short- and long-term effects of flares and reduce long-term damage. Lines represent models of disease progression, with line thickness representing frequency estimates.
Hence, reliable biomarkers need to predict (i) the future disease course, (ii) treatment response, and (iii) the safety for medication withdrawal during clinical remission (Figure 1B).
Inflammation markers in the serum can indicate disease activity and potentially treatment response in JIA [reviewed in (36)]. In JDM, histology of biopsies and myositis-specific auto-antibodies can indicate future disease severity or complications [reviewed in (27)].
Only a few putative biomarkers probe the immunoregulatory balance in autoimmune arthritis and myopathies. The frequency of inflammation-associated Tregs (HLA-DR+) in PB was proposed as a biomarker for disease activity in arthritis (8). TCR sequence overlap of these PB HLA-DR+ Tregs with SF Tregs in JIA, and an increase of HLA-DR+ Tregs in active rheumatoid arthritis (RA) were found. Low expression of the immunoregulatory receptor CD39 has been suggested as an indicator of methotrexate resistance in RA (37). Also, response to the TNF-α blocker adalimumab could be predicted by a Treg increase in PBMCs from RA patients cultured with adalimumab prior to treatment (38). Finally, the soluble form of the high affinity IL-2 receptor α chain (CD25), crucial for Treg phenotype and function, might be a biomarker for adult myositis disease activity (39).
In the recent past, gene signatures have been defined as multi-parameter biomarkers. Thus, far, efforts to define JIA immune-based gene biomarkers have focused on whole genome expression profiling (40–42) and epigenomic signatures (43, 44). JIA displays an altered immune signature, which changes during remission, but does not return to a state comparable to healthy controls (41, 42). Myositis is characterized by type 1 IFN signatures (27). While interesting and highlighting potential disease mechanisms, whole-genome/exome expression profiling is not feasible for routine clinical practice due to cost, logistics and data interpretation. We have recently developed a Treg gene signature associated with Treg competency using the clinically-applicable multiplex platform nanoString (45). NanoString is fast and fewer than 10,000 lysed cells are sufficient without the need to purify RNA. Although the proportion of Tregs that express FOXP3 was similar between type 1 diabetes (T1D) and controls, there was a significant change in their Treg signature (45). Future work will elucidate whether the Treg gene signature may also be used as a biomarker in JIA and other autoimmune conditions.
In summary, some progress has been made, but more biomarkers are needed for biological disease activity, prognosis, treatment success, and risk of flares. Further, a consensus of criteria to describe active/inactive disease is needed to better estimate the currently widely variable incidence of clinically inactive JIA disease (46). For JDM, a comprehensive set of criteria to assess disease activity and damage has been proposed (47).
Tregs as Therapeutic Target/Tool?
Convincing evidence demonstrates that functioning Tregs are crucial to prevent autoimmunity and our understanding of how different immunotherapies affect Tregs has improved.
(Unforeseen) Treg Effects of Immuno-Therapy
High levels of TNF-α in the inflamed JIA joint (32, 48) offer a clear rationale for anti-TNF therapy in JIA with marked success (4, 49). Anti-TNF therapy has also been used in refractory JDM (50), but with mixed evidence for its effectiveness (27, 51–53). Blocking TNF-α can, however, also elicit further autoimmune responses, particular in the skin and muscle (54–57). TNF-α itself can have both positive and negative effects on Tregs (32, 58, 59). Interestingly, the negative effects are found especially in inflamed joints (32, 58, 60), whereas positive effects of TNF-α on Treg function were reported using healthy human cells or in mice (58, 61–64). TNF-α has two receptors CD120a (TNFR1) and CD120b (TNFR2) (58). CD120b may mediate the pro-Treg functions of TNF-α, including Treg proliferation, stabilizing Tregs, and preventing disease in mouse models (58, 62–64). Little is known about the effects of ligation of CD120a in Tregs, but some research suggests targeting CD120a while sparing CD120b-TNF-interaction can alleviate collagen-induced arthritis (65). In RA, adalimumab has been shown to enhance Treg frequency and potency via CD120b (38, 66, 67). Etanercept, a soluble CD120b as TNF-α blocker, instead might predominantly affect effector T cells, by reversing their resistance to suppression in JIA (68). Unfortunately, a considerable group of JIA/JDM patients do not respond to anti-TNF therapy (27, 49, 53) and anti-TNF agents are immunogenic (69), with 50% of patients developing anti-drug antibodies leading to resistance to therapy and disease progression.
Ustekinumab is another potentially attractive anti-cytokine therapy which targets the p40 subunit of IL-12 and IL-23, key cytokines driving Th1, Th17, and Th17.1, (ex-)Th17 cells with a Th1-like phenotype, function (70–74). Ustekinumab is well-tolerated in adult and pediatric patients for treating psoriasis, psoriatic arthritis, systemic lupus erythematosus (SLE), and Crohn's disease (70, 71, 75–77), and has shown lower immunogenicity compared to most anti-TNF agents (69). Th17.1 are enriched in JIA (72, 73), and ustekinumab therapy had some success in enthesitis-related JIA (78), psoriatic arthritis (69, 79) and is in trial for various rheumatological diseases (80). While no imbalance in IL-17 has been established in JDM (19), Th17.1 have not been investigated. Ustekinumab has been suggested as a potential therapy for JDM, and a case of JDM with psoriasis was treated successfully with ustekinumab (81). Due to the reciprocal relationship between Th17 and Tregs (82), Tregs might also be affected by ustekinumab therapy, and this was indeed suggested in a case report of giant cell arteritis (83) and in T1D treated with ustekinumab (NCT02117765; Pesenacker et al.).
IL-6 also drives inflammatory environments, including skewing the Treg/Th17 balance toward Th17 (71). Anti-IL-6 receptor therapy (tocilizumab) increases Treg frequency and numbers in RA (71). IL-6 has also been implicated in JIA and JDM (11, 17, 84, 85) and is used in polyarticular, extended oligoarticular, systemic JIA (49), and refractory JDM (50), but mechanistic studies in pediatric disease are lacking.
Whether drugs such as ustekinumab and tocilizumab act on Tregs directly or through changing the microenvironment is unclear. Human Tregs can express the receptors for IL-6 (86), IL-12 (87), and IL-23 (88), but evidence for direct drug action on Tregs is lacking.
Alternatively, co-receptors can be targeted to manipulate the immunoregulatory balance. Initially established for cancer therapy (checkpoint blockade), mimicking checkpoints such as CTLA4 (CTLA4-Ig, abatacept, belatacept) is used as treatment for autoimmunity. Abatacept has been shown to be safe and effective in oligo- and polyarticular JIA (49, 89–92), adult DM/polymyositis (34, 53), a case report of steroid-sparing abatacept in complex JDM (93) and a trial in JDM is underway (27). A reduction of the T cell activation state is the main reported effect of abatacept (90, 94–96). Surprisingly, the majority of studies found abatacept decreases the frequency of Tregs (90, 95, 97–99), with some studies showing an increase in function (99). On the other hand, increased Treg frequency, but decreased activity after abatacept therapy in RA was demonstrated (100). In muscle tissue of adult DM/polymyositis, more Tregs were found following abatacept (34), suggesting that abatacept treatment could change Treg localization. Other co-receptor targeting therapies are in use/development for malignancies (e.g., anti-PD1, anti-TIM3, anti-TIGIT, etc.) and these pathways might be useful targets in autoimmunity.
Treg (-Targeted) Therapy
Adoptive transfer of Tregs has been shown to be safe and possibly effective at reducing inflammation, inducing transplant tolerance, preventing graft-vs.-host disease (GVHD) and treating autoimmunity [reviewed in (101)].
Important considerations for Treg-therapy currently under investigation are the source of therapeutic cells, antigen-specificity and possibly tailoring homing characteristics for improved activity. Isolating and expanding sufficient numbers of Tregs from patients awaiting transplantation, under immunosuppression or with autoimmune disease is feasible and can restore their function (101–103), although achieving clinically relevant Treg numbers from pediatric JIA and JDM patients might prove challenging. Third-party Tregs from umbilical cord blood have been found safe and possibly effective as GVHD prophylaxis in adults (104, 105) and pediatric thymus—routinely removed during pediatric cardiac surgery—might be a plentiful source for highly functional therapeutic Tregs (106, 107). Antigen-specific Tregs are more effective than polyclonal Tregs for therapy and with recent successes of chimeric antigen receptor (CAR) T effector therapies for cancer, there has been a surge to adapt this technology to generate CAR-Tregs [reviewed in (108)]. While generation of antigen-specific Tregs recognizing allogeneic HLA-molecules is relatively straightforward in transplantation, generation of CAR-Tregs for autoimmunity without known antigen (i.e., JIA) might be difficult. Still, CAR-Tregs reacting with antigen found at the site of inflammation (i.e., JIA joints or JDM muscle) could activate Tregs locally. Alternatively, Tregs could be conditioned in vitro to home to specific sites (107) or Tregs could be injected locally, as shown with intra-dermal injection of Tregs to inhibit murine allograft skin inflammation (109).
Since Treg cell therapies are challenging and expensive, targeting Treg expansion in vivo might be more feasible for conditions such as JIA and JDM. The most promising advances of non-cellular therapies targeting Tregs have been low-dose IL-2, IL-2 complexes, or IL-2 bio-similars (110–112). While high doses of IL-2 stimulate mainly effector cells, low-dose IL-2 [0.3–3 × 106 units/day (112)] skews the response toward Tregs. Low-dose IL-2 increases the frequency of activated, functional and fully demethylated CD25+FOXP3+ Tregs (113–115) and induces STAT5 phosphorylation in vivo (114, 115). Low-dose IL-2 therapy has been deemed safe and successful in the treatment of T1D (112, 114, 115), GVHD (116, 117), and SLE (113). Indeed, low-dose IL-2 therapy rescued Tregs with low levels of CD25 in SLE (113), indicating that it might also rescue JIA Tregs with low CD25 expression (13). To further fine-tune specificity or increase the half-life of IL-2, IL-2 complexes, and bio-similars are in development (110, 111, 118); these expand Tregs and induce phosphorylated STAT5 in vitro, in vivo, and prevent disease in animal models (118–121), including resolution of muscular dystrophy (20). Covalently linking IL-2 to anti-IL-2 (122), to non-FcRγ-binding human IgG1 (123) or CD25 (124) may enhance potential clinical application by mitigating the risk of in vivo dissociation of complexes.
However, increasing Treg numbers alone might not be sufficient to overcome the highly inflammatory environment and effector cell resistance. Thus, to achieve sustained remission combination-therapy might be necessary to reduce the inflammatory milieu paralleled with boosting Tregs to maintain a renewed tolerance.
Concluding Remarks
Taken together, it is clear that Tregs present challenges and opportunities in JIA and JDM research and clinical management (Figure 2). Their phenotype and function are clearly altered in JIA and JDM, targeting them might improve disease outcome and Tregs could be used as biomarkers to gage the state and progress of disease.
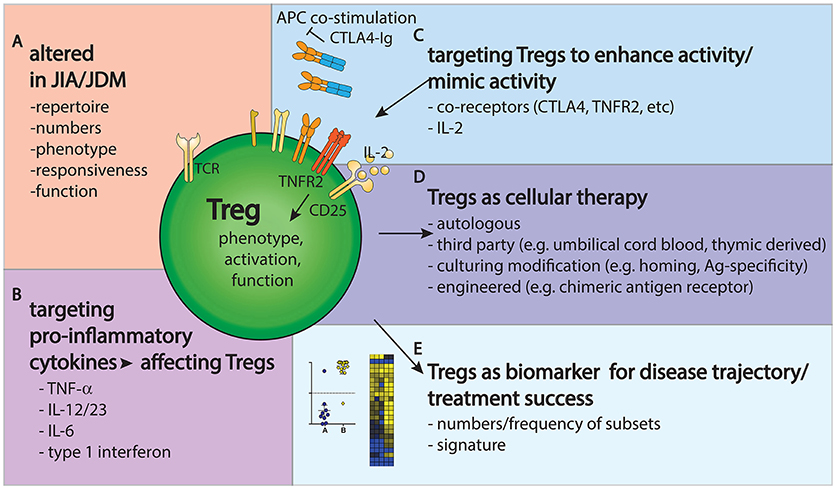
Figure 2. Tregs provide challenges and opportunities in JIA/JDM pathogenesis, treatment and monitoring. (A) Tregs are altered in repertoire, phenotype and frequency in JIA/JDM, particularly at the site of inflammation. (B–D) Treatment options to restore the immunoregulatory balance include targeting pro-inflammatory cytokines (B), targeting Tregs to enhance their activity (C) or using Tregs as a cellular therapy (D). (E) Further, changes in Treg gene signatures could aid as biomarkers to measure or predict disease/treatment response.
The role of the microenvironment on Treg function and phenotype in JIA- and JDM-affected tissues remains to be explored further. Researchers should take advantage of biopsies taken for clinical diagnosis (JDM) and SF aspirated during therapeutic joint injections (JIA). Novel techniques, such as single cell sequencing, multidimensional mass/flow cytometry and microscopy, will aid using clinical samples to their full potential (125–127). Additionally, co-culture with SF or muscle-derived cells could highlight how the microenvironment affects Tregs. Since JDM in particular is a rare disease, collaborations between groups are crucial to increase sample size for fundamental research, biomarker-finding and -validation studies and controlled treatment trials. This could be achieved by consortiums similar to juvenile diabetes research foundation (JDRF) biomarker working group for T1D (114) and the immune tolerance network trials (128).
While there is progress toward unified measures of disease activity (46, 47), these will need to be tested and verified, followed by development of feasible, reliable and cost-effective biomarkers to predict disease activity, risk of flare and ideal treatment strategies. The ultimate goal, aided by biomarkers, is to go from a trial-and-error treatment approach toward a more efficient and personalized medicine approach with more patients achieving drug-free remission without major long-term disabilities (Figure 1).
Success of various agents affecting the immunoregulatory balance in other diseases point to potential uses in JIA and JDM. Any (new) therapy will need to be considered in regards to both effector cells AND Tregs, since some therapies might have unexpected effects on Tregs. Thus, it is important to continuously build our understanding of how various agents affect the immunoregulatory balance.
In conclusion, important recent advances might lead to valid future contributions to the widened arsenal of treatment options available to restore the immunoregulatory balance in a heterogeneous disease spectrum.
Author Contributions
AP conceived the review. RH and AP reviewed the literature and co-wrote the manuscript.
Funding
AP is funded by a Versus Arthritis career development fellowship (ARUK CDF21738).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
2. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. (2016) 1417:5–22. doi: 10.1111/nyas.13011
3. Li J, Tan J, Martino MM, Lui KO. Regulatory T-Cells: potential regulator of tissue repair and regeneration. Front Immunol. (2018) 9:585. doi: 10.3389/fimmu.2018.00585
4. Crayne CB, Beukelman T. Juvenile idiopathic arthritis: oligoarthritis and polyarthritis. Pediatr Clin North Am. (2018) 65:657–74. doi: 10.1016/j.pcl.2018.03.005
5. Rider LG, Nistala K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med. (2016) 280:24–38. doi: 10.1111/joim.12444
6. Pesenacker AM, Wedderburn LR. T regulatory cells in childhood arthritis–novel insights. Expert Rev Mol Med. (2013) 15:e13. doi: 10.1017/erm.2013.14
7. Copland A, Bending D. Foxp3 molecular dynamics in treg in juvenile idiopathic arthritis. Front Immunol. (2018) 9:2273. doi: 10.3389/fimmu.2018.02273
8. Rossetti M, Spreafico R, Consolaro A, Leong JY, Chua C, Massa M, et al. TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann Rheum Dis. (2017) 76:435–41. doi: 10.1136/annrheumdis-2015-208992
9. Duurland CL, Brown CC, O'Shaughnessy RF, Wedderburn LR. CD161(+) Tconv and CD161(+) Treg share a transcriptional and functional phenotype despite limited overlap in TCRbeta repertoire. Front Immunol. (2017) 8:103. doi: 10.3389/fimmu.2017.00103
10. Henderson LA, Volpi S, Frugoni F, Janssen E, Kim S, Sundel RP, et al. Next-generation sequencing reveals restriction and clonotypic expansion of treg cells in juvenile idiopathic arthritis. Arthritis Rheumatol. (2016) 68:1758–68. doi: 10.1002/art.39606
11. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial regulatory T cells occupy a discrete TCR niche in human arthritis and require local signals to stabilize FOXP3 protein expression. J Immunol. (2015) 195:5616–24. doi: 10.4049/jimmunol.1500391
12. Delemarre EM, van den Broek T, Mijnheer G, Meerding J, Wehrens EJ, Olek S, et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood (2016) 127:91–101. doi: 10.1182/blood-2015-06-649145
13. Bending D, Pesenacker AM, Ursu S, Wu Q, Lom H, Thirugnanabalan B, et al. Hypomethylation at the regulatory T cell-specific demethylated region in CD25hi T cells is decoupled from FOXP3 expression at the inflamed site in childhood arthritis. J Immunol. (2014) 193:2699–708. doi: 10.4049/jimmunol.1400599
14. Patterson SJ, Pesenacker AM, Wang AY, Gillies J, Mojibian M, Morishita K, et al. T regulatory cell chemokine production mediates pathogenic T cell attraction and suppression. J Clin Invest. (2016) 126:1039–51. doi: 10.1172/JCI83987
15. Bulatovic Calasan M, Vastert SJ, Scholman RC, Verweij F, Klein M, Wulffraat NM, et al. Methotrexate treatment affects effector but not regulatory T cells in juvenile idiopathic arthritis. Rheumatology (2015) 54:1724–34. doi: 10.1093/rheumatology/kev101
16. Ohl K, Nickel H, Moncrieffe H, Klemm P, Scheufen A, Foll D, et al. The transcription factor CREM drives an inflammatory phenotype of T cells in oligoarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. (2018) 16:39. doi: 10.1186/s12969-018-0253-x
17. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood (2011) 118:3538–48. doi: 10.1182/blood-2010-12-328187
18. Haufe S, Haug M, Schepp C, Kuemmerle-Deschner J, Hansmann S, Rieber N, et al. Impaired suppression of synovial fluid CD4+CD25- T cells from patients with juvenile idiopathic arthritis by CD4+CD25+ Treg cells. Arthritis Rheum. (2011) 63:3153–62. doi: 10.1002/art.30503
19. Vercoulen Y, Bellutti Enders F, Meerding J, Plantinga M, Elst EF, Varsani H, et al. Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood. PLoS ONE (2014) 9:e105353. doi: 10.1371/journal.pone.0105353
20. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med. (2014) 6:258ra142. doi: 10.1126/scitranslmed.3009925
21. Pandya JM, Loell I, Hossain MS, Zong M, Alexanderson H, Raghavan S, et al. Effects of conventional immunosuppressive treatment on CD244+ (CD28null) and FOXP3+ T cells in the inflamed muscle of patients with polymyositis and dermatomyositis. Arthritis Res Ther. (2016) 18:80. doi: 10.1186/s13075-016-0974-5
22. Tansley SL, McHugh NJ, Wedderburn LR. Adult and juvenile dermatomyositis: are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms? Arthritis Res Ther. (2013) 15:211. doi: 10.1186/ar4198
23. Lopez De Padilla CM, Crowson CS, Hein MS, Pendegraft RS, Strausbauch MA, Niewold TB, et al. Gene expression profiling in blood and affected muscle tissues reveals differential activation pathways in patients with new-onset juvenile and adult dermatomyositis. J Rheumatol. (2017) 44:117–24. doi: 10.3899/jrheum.160293
24. Gitiaux C, Latroche C, Weiss-Gayet M, Rodero MP, Duffy D, Bader-Meunier B, et al. Myogenic progenitor cells exhibit type I interferon-driven proangiogenic properties and molecular signature during juvenile dermatomyositis. Arthritis Rheumatol. (2018) 70:134–45. doi: 10.1002/art.40328
25. Greenberg SA. A gene expression approach to study perturbed pathways in myositis. Curr Opin Rheumatol. (2007) 19:536–41. doi: 10.1097/BOR.0b013e3282efe261
26. Zhu W, Streicher K, Shen N, Higgs BW, Morehouse C, Greenlees L, et al. Genomic signatures characterize leukocyte infiltration in myositis muscles. BMC Med Genomics (2012) 5:53. doi: 10.1186/1755-8794-5-53
27. Wu Q, Wedderburn LR, McCann LJ. Juvenile dermatomyositis: latest advances. Best Pract Res Clin Rheumatol. (2017) 31:535–57. doi: 10.1016/j.berh.2017.12.003
28. Deyhle MR, Hyldahl RD. The role of T lymphocytes in skeletal muscle repair from traumatic and contraction-induced injury. Front Physiol. (2018) 9:768. doi: 10.3389/fphys.2018.00768
29. Young NA, Sharma R, Friedman AK, Kaffenberger BH, Bolon B, Jarjour WN. Aberrant muscle antigen exposure in mice is sufficient to cause myositis in a Treg cell-deficient milieu. Arthritis Rheum. (2013) 65:3259–70. doi: 10.1002/art.38184
30. Allenbach Y, Solly S, Gregoire S, Dubourg O, Salomon B, Butler-Browne G, et al. Role of regulatory T cells in a new mouse model of experimental autoimmune myositis. Am J Pathol. (2009) 174:989–98. doi: 10.2353/ajpath.2009.080422
31. DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM, et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol. (2018) 3:eaat5861. doi: 10.1126/sciimmunol.aat5861
32. Pesenacker AM, Broady R, Levings MK. Control of tissue-localized immune responses by human regulatory T cells. Eur J Immunol. (2015) 45:333–43. doi: 10.1002/eji.201344205
33. Wallace CA, Ringold S, Bohnsack J, Spalding SJ, Brunner HI, Milojevic D, et al. Extension study of participants from the trial of early aggressive therapy in juvenile idiopathic arthritis. J Rheumatol. (2014) 41:2459–65. doi: 10.3899/jrheum.140347
34. Tjarnlund A, Tang Q, Wick C, Dastmalchi M, Mann H, Tomasova Studynkova J, et al. Abatacept in the treatment of adult dermatomyositis and polymyositis: a randomised, phase IIb treatment delayed-start trial. Ann Rheum Dis. (2018) 77:55–62. doi: 10.1136/annrheumdis-2017-211751
35. Wallace CA, Huang B, Bandeira M, Ravelli A, Giannini EH. Patterns of clinical remission in select categories of juvenile idiopathic arthritis. Arthritis Rheum. (2005) 52:3554–62. doi: 10.1002/art.21389
36. Duurland CL, Wedderburn LR. Current developments in the use of biomarkers for juvenile idiopathic arthritis. Curr Rheumatol Rep. (2014) 16:406. doi: 10.1007/s11926-013-0406-3
37. Peres RS, Liew FY, Talbot J, Carregaro V, Oliveira RD, Almeida SL, et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci USA. (2015) 112:2509–14. doi: 10.1073/pnas.1424792112
38. Nguyen DX, Cotton A, Attipoe L, Ciurtin C, Doré CJ, Ehrenstein MR. Regulatory T cells as a biomarker for response to adalimumab in rheumatoid arthritis. J Allergy Clin Immunol. (2018) 142:978–80.e979. doi: 10.1016/j.jaci.2018.04.026
39. Tournadre A, Dubost JJ, Soubrier M, Ruivard M, Souteyrand P, Schmidt J, et al. Soluble IL-2 receptor: a biomarker for assessing myositis activity. Dis Markers (2014) 2014:472624. doi: 10.1155/2014/472624
40. Cui A, Quon G, Rosenberg AM, Yeung RS, Morris Q, Consortium BS. Gene expression deconvolution for uncovering molecular signatures in response to therapy in juvenile idiopathic arthritis. PLoS ONE (2016) 11:e0156055. doi: 10.1371/journal.pone.0156055
41. Jiang K, Frank M, Chen Y, Osban J, Jarvis JN. Genomic characterization of remission in juvenile idiopathic arthritis. Arthritis Res Ther. (2013) 15:R100. doi: 10.1186/ar4280
42. Moncrieffe H, Bennett MF, Tsoras M, Luyrink LK, Johnson AL, Xu H, et al. Transcriptional profiles of JIA patient blood with subsequent poor response to methotrexate. Rheumatology (2017) 56:1542–51. doi: 10.1093/rheumatology/kex206
43. Peeters JG, Vervoort SJ, Tan SC, Mijnheer G, de Roock S, Vastert SJ, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived T Cells reduces disease-associated gene expression. Cell Rep. (2015) 12:1986–96. doi: 10.1016/j.celrep.2015.08.046
44. Spreafico R, Rossetti M, Whitaker JW, Wang W, Lovell DJ, Albani S. Epipolymorphisms associated with the clinical outcome of autoimmune arthritis affect CD4+ T cell activation pathways. Proc Natl Acad Sci USA. (2016) 113:13845–50. doi: 10.1073/pnas.1524056113
45. Pesenacker AM, Wang AY, Singh A, Gillies J, Kim YW, Piccirillo CA, et al. A Treg gene signature is a specific and sensitive biomarker to identify children with new onset type 1 diabetes. Diabetes (2016) 65:1031–9. doi: 10.2337/db15-0572
46. Shoop-Worrall SJW, Verstappen SMM, Baildam E, Chieng A, Davidson J, Foster H, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. (2017) 76:1381–8. doi: 10.1136/annrheumdis-2016-210511
47. McCann LJ, Pilkington CA, Huber AM, Ravelli A, Appelbe D, Kirkham JJ, et al. Development of a consensus core dataset in juvenile dermatomyositis for clinical use to inform research. Ann Rheum Dis. (2018) 77:241–50. doi: 10.1136/annrheumdis-2017-212141
48. de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis. (2007) 66:589–98. doi: 10.1136/ard.2006.061853
49. Shepherd J, Cooper K, Harris P, Picot J, Rose M. The clinical effectiveness and cost-effectiveness of abatacept, adalimumab, etanercept and tocilizumab for treating juvenile idiopathic arthritis: a systematic review and economic evaluation. Health Technol Assess. (2016) 20:1–222. doi: 10.3310/hta20340
50. Spencer CH, Rouster-Stevens K, Gewanter H, Syverson G, Modica R, Schmidt K, et al. Biologic therapies for refractory juvenile dermatomyositis: five years of experience of the childhood arthritis and rheumatology research alliance in North America. Pediatr Rheumatol Online J. (2017) 15:50. doi: 10.1186/s12969-017-0174-0
51. Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res (2014) 66:783–7. doi: 10.1002/acr.22198
52. Wang CR. Successful treatment of refractory juvenile dermatomyositis with adalimumab. J Clin Rheumatol. (2017) 23:174–5. doi: 10.1097/RHU.0000000000000514
53. Sasaki H, Kohsaka H. Current diagnosis and treatment of polymyositis and dermatomyositis. Mod Rheumatol. 28:913–21. doi: 10.1080/14397595.2018.1467257
54. Omran NE, Noorwali AA. Nephritis, cerebritis, and myositis after adalimumab therapy in a patient with rheumatoid arthritis: a case report. Int J Gen Med. (2018) 11:151–4. doi: 10.2147/IJGM.S154835
55. Brunasso AM, Aberer W, Massone C. New onset of dermatomyositis/polymyositis during anti-TNF-alpha therapies: a systematic literature review. ScientificWorldJournal (2014) 2014:179180. doi: 10.1155/2014/179180
56. Liu SW, Velez NF, Lam C, Femia A, Granter SR, Townsend HB, et al. Dermatomyositis induced by anti-tumor necrosis factor in a patient with juvenile idiopathic arthritis. JAMA Dermatol. (2013) 149:1204–8. doi: 10.1001/jamadermatol.2013.5220
57. Ko JM, Gottlieb AB, Kerbleski JF. Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J Dermatolog Treat. (2009) 20:100–8. doi: 10.1080/09546630802441234
58. Goldstein JD, Perol L, Zaragoza B, Baeyens A, Marodon G, Piaggio E. Role of cytokines in thymus- versus peripherally derived-regulatory T cell differentiation and function. Front Immunol. (2013) 4:155. doi: 10.3389/fimmu.2013.00155
59. Nie H, Zheng Y, Li R, Zhang J. Reply to suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. (2016) 22:18–9. doi: 10.1038/nm.4018
60. Wang J, van Dongen H, Scherer HU, Huizinga TW, Toes RE. Suppressor activity among CD4+,CD25++ T cells is discriminated by membrane-bound tumor necrosis factor alpha. Arthritis Rheum. (2008) 58:1609–18. doi: 10.1002/art.23460
61. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. (2016) 22:16–7. doi: 10.1038/nm.4019
62. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 ligation in human T regulatory cells enhances IL2-induced cell proliferation through the non-canonical NF-kappaB pathway. Sci Rep. (2018) 8:12079. doi: 10.1038/s41598-018-30621-4
63. Fischer R, Proske M, Duffey M, Stangl H, Martinez GF, Peters N, et al. Selective activation of tumor necrosis factor receptor ii induces antiinflammatory responses and alleviates experimental arthritis. Arthritis Rheumatol. (2018) 70:722–35. doi: 10.1002/art.40413
64. Lamontain V, Schmid T, Weber-Steffens D, Zeller D, Jenei-Lanzl Z, Wajant H, et al. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell Mol Immunol. (2018) 16:76–85. doi: 10.1038/cmi.2017.138
65. McCann FE, Perocheau DP, Ruspi G, Blazek K, Davies ML, Feldmann M, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. (2014) 66:2728–38. doi: 10.1002/art.38755
66. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. (2012) 64:3129–38. doi: 10.1002/art.34565
67. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII bindin–g in rheumatoid arthritis. J Exp Med. (2016) 213:1241–53. doi: 10.1084/jem.20151255
68. Petrelli A, Wehrens EJ, Scholman RC, Prakken BJ, Vastert SJ, van Wijk F. Self-sustained resistance to suppression of CD8+ Teff cells at the site of autoimmune inflammation can be reversed by tumor necrosis factor and interferon-gamma blockade. Arthritis Rheumatol. (2016) 68:229–36. doi: 10.1002/art.39418
69. Strand V, Balsa A, Al-Saleh J, Barile-Fabris L, Horiuchi T, Takeuchi T, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs (2017) 31:299–316. doi: 10.1007/s40259-017-0231-8
70. Elliott M, Benson J, Blank M, Brodmerkel C, Baker D, Sharples KR, et al. Ustekinumab: lessons learned from targeting interleukin-12/23p40 in immune-mediated diseases. Ann N Y Acad Sci. (2009) 1182:97–110. doi: 10.1111/j.1749-6632.2009.05070.x
71. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic potential of targeting the Th17/Treg axis in autoimmune disorders. Molecules (2017) 22:E134. doi: 10.3390/molecules22010134
72. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA. (2010) 107:14751–6. doi: 10.1073/pnas.1003852107
73. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. (2011) 63:2504–15. doi: 10.1002/art.30332
74. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR, et al. T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. (2014) 66:1955–60. doi: 10.1002/art.38647
75. Papp KA, Griffiths CE, Gordon K, Lebwohl M, Szapary PO, Wasfi Y, et al. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: final results from 5 years of follow-up. Br J Dermatol. (2013) 168:844–54. doi: 10.1111/bjd.12214
76. Kellen R, Silverberg NB, Lebwohl M. Efficacy and safety of ustekinumab in adolescents. Pediatric Health Med Ther. (2016) 7:109–20. doi: 10.2147/PHMT.S75836
77. van Vollenhoven RF, Hahn BH, Tsokos GC, Wagner CL, Lipsky P, Touma Z, et al. Efficacy and safety of ustekinumab, an IL-12 and IL-23 inhibitor, in patients with active systemic lupus erythematosus: results of a multicentre, double-blind, phase 2, randomised, controlled study. Lancet (2018) 392:1330–9. doi: 10.1016/S0140-6736(18)32167-6
78. Mannion ML, McAllister L, Cron RQ, Stoll ML. Ustekinumab as a therapeutic option for children with refractory enthesitis-related arthritis. J Clin Rheumatol. (2016) 22:282–4. doi: 10.1097/RHU.0000000000000408
79. Kavanaugh A, Puig L, Gottlieb AB, Ritchlin C, Li S, Wang Y, et al. Maintenance of clinical efficacy and radiographic benefit through two years of ustekinumab therapy in patients with active psoriatic arthritis: results from a randomized, placebo-controlled phase III trial. Arthritis Care Res (Hoboken). (2015) 67:1739–49. doi: 10.1002/acr.22645
80. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
81. Montoya CL, Gonzalez ML, Ospina FE, Tobon GJ. A rare case of amyopathic juvenile dermatomyositis associated with psoriasis successfully treated with ustekinumab. J Clin Rheumatol. (2017) 23:129–30. doi: 10.1097/RHU.0000000000000430
82. Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. (2009) 9:883–9. doi: 10.1038/nri2660
83. Samson M, Ghesquiere T, Berthier S, Bonnotte B. Ustekinumab inhibits Th1 and Th17 polarisation in a patient with giant cell arteritis. Ann Rheum Dis. (2018) 77:e6. doi: 10.1136/annrheumdis-2017-211622
84. Barnes MG, Grom AA, Thompson SD, Griffin TA, Pavlidis P, Itert L, et al. Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum. (2009) 60:2102–12. doi: 10.1002/art.24601
85. Nistala K, Varsani H, Wittkowski H, Vogl T, Krol P, Shah V, et al. Myeloid related protein induces muscle derived inflammatory mediators in juvenile dermatomyositis. Arthritis Res Ther. (2013) 15:R131. doi: 10.1186/ar4311
86. Ferreira RC, Rainbow DB, Rubio Garcia A, Pekalski ML, Porter L, Oliveira JJ, et al. In-depth immunophenotyping data of IL-6R on the human peripheral regulatory T cell (Treg) compartment. Data Brief (2017) 12:676–91. doi: 10.1016/j.dib.2017.04.043
87. Bhairavabhotla R, Kim YC, Glass DD, Escobar TM, Patel MC, Zahr R, et al. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum Immunol. (2016) 77:201–13. doi: 10.1016/j.humimm.2015.12.004
88. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn LR. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood (2013) 121:2647–58. doi: 10.1182/blood-2012-08-443473
89. Brunner HI, Tzaribachev N, Vega-Cornejo G, Louw I, Berman A, Calvo Penades I, et al. Subcutaneous abatacept in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase III open-label study. Arthritis Rheumatol. (2018) 70:1144–54. doi: 10.1002/art.40466
90. Maggi L, Cimaz R, Capone M, Santarlasci V, Rossi MC, Mazzoni A, et al. Immunosuppressive activity of abatacept on circulating T helper lymphocytes from juvenile idiopathic arthritis patients. Int Arch Allergy Immunol. (2016) 171:45–53. doi: 10.1159/000450948
91. Lovell DJ, Ruperto N, Mouy R, Paz E, Rubio-Perez N, Silva CA, et al. Long-term safety, efficacy, and quality of life in patients with juvenile idiopathic arthritis treated with intravenous abatacept for up to seven years. Arthritis Rheumatol. (2015) 67:2759–70. doi: 10.1002/art.39234
92. Ruperto N, Lovell DJ, Quartier P, Paz E, Rubio-Perez N, Silva CA, et al. Abatacept in children with juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled withdrawal trial. Lancet (2008) 372:383–91. doi: 10.1016/S0140-6736(08)60998-8
93. Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr. (2012) 160:520–2. doi: 10.1016/j.jpeds.2011.11.057
94. Sumitomo S, Nagafuchi Y, Tsuchida Y, Tsuchiya H, Ota M, Ishigaki K, et al. A gene module associated with dysregulated TCR signaling pathways in CD4(+) T cell subsets in rheumatoid arthritis. J Autoimmun. (2018) 89:21–9. doi: 10.1016/j.jaut.2017.11.001
95. Orban T, Beam CA, Xu P, Moore K, Jiang Q, Deng J, et al. Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower C-peptide decline. Diabetes (2014) 63:3449–57. doi: 10.2337/db14-0047
96. Rochman Y, Yukawa M, Kartashov AV, Barski A. Functional characterization of human T cell hyporesponsiveness induced by CTLA4-Ig. PLoS ONE (2015) 10:e0122198. doi: 10.1371/journal.pone.0122198
97. Szentpetery A, Heffernan E, Gogarty M, Mellerick L, McCormack J, Haroon M, et al. Abatacept reduces synovial regulatory T-cell expression in patients with psoriatic arthritis. Arthritis Res Ther. (2017) 19:158. doi: 10.1186/s13075-017-1364-3
98. Langdon K, Haleagrahara N. Regulatory T-cell dynamics with abatacept treatment in rheumatoid arthritis. Int Rev Immunol. 37:206–14. doi: 10.1080/08830185.2018.1465943
99. Alvarez-Quiroga C, Abud-Mendoza C, Doniz-Padilla L, Juarez-Reyes A, Monsivais-Urenda A, Baranda L, et al. CTLA-4-Ig therapy diminishes the frequency but enhances the function of Treg cells in patients with rheumatoid arthritis. J Clin Immunol. (2011) 31:588–95. doi: 10.1007/s10875-011-9527-5
100. Bonelli M, Goschl L, Bluml S, Karonitsch T, Hirahara K, Ferner E, et al. Abatacept (CTLA-4Ig) treatment reduces T cell apoptosis and regulatory T cell suppression in patients with rheumatoid arthritis. Rheumatology (Oxford) (2016) 55:710–20. doi: 10.1093/rheumatology/kev403
101. Duggleby R, Danby RD, Madrigal JA, Saudemont A. Clinical grade regulatory CD4(+) T cells (Tregs): moving toward cellular-based immunomodulatory therapies. Front Immunol. (2018) 9:252. doi: 10.3389/fimmu.2018.00252
102. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
103. Tang Q, Vincenti F. Transplant trials with Tregs: perils and promises. J Clin Invest. (2017) 127:2505–12. doi: 10.1172/JCI90598
104. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
105. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
106. Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant. (2015) 16:58–71. doi: 10.1111/ajt.13456
107. Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplant. (2018) 19:62–76. doi: 10.1111/ajt.14936
108. Sicard A, Boardman DA, Levings MK. Taking regulatory T-cell therapy one step further. Curr Opin Organ Transplant. (2018) 23:509–15. doi: 10.1097/MOT.0000000000000566
109. Landman S, de Oliveira VL, van Erp PEJ, Fasse E, Bauland SCG, Joosten I, et al. Intradermal injection of low dose human regulatory T cells inhibits skin inflammation in a humanized mouse model. Sci Rep. (2018) 8:10044. doi: 10.1038/s41598-018-28346-5
110. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. (2018) 18:648–59. doi: 10.1038/s41577-018-0046-y
111. Leon K, Garcia-Martinez K, Carmenate T, Rojas G. Combining computational and experimental biology to develop therapeutically valuable IL2 muteins. Semin Oncol. (2018) 45:95–104. doi: 10.1053/j.seminoncol.2018.04.001
112. Dwyer CJ, Ward NC, Pugliese A, Malek TR. Promoting immune regulation in type 1 diabetes using low-dose interleukin-2. Curr Diab Rep. (2016) 16:46. doi: 10.1007/s11892-016-0739-1
113. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis. (2015) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
114. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med. (2016) 13:e1002139. doi: 10.1371/journal.pmed.1002139
115. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
116. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. (2013) 5:179ra143. doi: 10.1126/scitranslmed.3005265
117. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood (2016) 128:130–7. doi: 10.1182/blood-2016-02-702852
118. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med. (2018) 24:1005–14. doi: 10.1038/s41591-018-0070-2
119. Lee SY, Cho ML, Oh HJ, Ryu JG, Park MJ, Jhun JY, et al. Interleukin-2/anti-interleukin-2 monoclonal antibody immune complex suppresses collagen-induced arthritis in mice by fortifying interleukin-2/STAT5 signalling pathways. Immunology (2012) 137:305–16. doi: 10.1111/imm.12008
120. Yokoyama Y, Iwasaki T, Kitano S, Satake A, Nomura S, Furukawa T, et al. IL-2-Anti-IL-2 monoclonal antibody immune complexes inhibit collagen-induced arthritis by augmenting regulatory T cell functions. J Immunol. (2018) 201:1899–906. doi: 10.4049/jimmunol.1701502
121. Zeng Z, Yu K, Chen L, Li W, Xiao H, Huang Z. Interleukin-2/Anti-Interleukin-2 immune complex attenuates cardiac remodeling after myocardial infarction through expansion of regulatory T cells. J Immunol Res. (2016) 2016:8493767. doi: 10.1155/2016/8493767
122. Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS, et al. Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol. (2018) 201:2094–106. doi: 10.4049/jimmunol.1800578
123. Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML, et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun. (2015) 56:66–80. doi: 10.1016/j.jaut.2014.10.002
124. Ward NC, Yu A, Moro A, Ban Y, Chen X, Hsiung S, et al. IL-2/CD25: a long-acting fusion protein that promotes immune tolerance by selectively targeting the IL-2 receptor on regulatory T cells. J Immunol. (2018) 201:2579–92. doi: 10.4049/jimmunol.1800907
125. Kunz DJ, Gomes T, James KR. Immune cell dynamics unfolded by single-cell technologies. Front Immunol. (2018) 9:1435. doi: 10.3389/fimmu.2018.01435
126. Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell (2018) 174, 968–81 e915. doi: 10.1016/j.cell.2018.07.010
127. Dixon AR, Bathany C, Tsuei M, White J, Barald KF, Takayama S. Recent developments in multiplexing techniques for immunohistochemistry. Expert Rev Mol Diagn. (2015) 15:1171–86. doi: 10.1586/14737159.2015.1069182
Keywords: regulatory T cells, juvenile idiopathic arthritis, juvenile dermatomyositis, biomarker, therapy
Citation: Hoeppli RE and Pesenacker AM (2019) Targeting Tregs in Juvenile Idiopathic Arthritis and Juvenile Dermatomyositis—Insights From Other Diseases. Front. Immunol. 10:46. doi: 10.3389/fimmu.2019.00046
Received: 31 October 2018; Accepted: 09 January 2019;
Published: 25 January 2019.
Edited by:
David Bending, University of Birmingham, United KingdomReviewed by:
Jorg Van Loosdregt, University Medical Center Utrecht, NetherlandsKlaus Tenbrock, RWTH Aachen Universität, Germany
Copyright © 2019 Hoeppli and Pesenacker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne M. Pesenacker, YS5wZXNlbmFja2VyQHVjbC5hYy51aw==