 Ping An1*
Ping An1* Gregory D. Kirk2Sophie Limou1,3,4Elizabeth Binns-Roemer1Jeffrey B. Kopp5
Gregory D. Kirk2Sophie Limou1,3,4Elizabeth Binns-Roemer1Jeffrey B. Kopp5 Cheryl A. Winkler1*
Cheryl A. Winkler1*- 1Molecular Genetic Epidemiology Section, Basic Science Program, Basic Research Laboratory, Frederick National Laboratory for Cancer Research, National Cancer Institute, Frederick, MD, United States
- 2Departments of Epidemiology and Medicine, Johns Hopkins University, Baltimore, MD, United States
- 3CRTI UMR1064, Inserm, Université de Nantes & ITUN, CHU Nantes, Nantes, France
- 4Ecole Centrale de Nantes, Nantes, France
- 5Kidney Disease Section, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, MD, United States
Apolipoprotein L1 (APOL1) has broad innate immune functions and has been shown to restrict HIV replication in vitro by multiple mechanisms. Coding variants in APOL1 are strongly associated with HIV-associated nephropathy (HIVAN) in persons with untreated HIV infection; however, the mechanism by which APOL1 variant protein potentiates renal injury in the presence of high viral load is not resolved. Little is known about the association of APOL1 genotypes with HIV viral load, HIV acquisition, or progression to AIDS. We assessed the role of APOL1 coding variants on HIV-1 acquisition using the conditional logistic regression test, on viral load using the t-test or ANOVA, and on progression to AIDS using Cox proportional hazards models among African Americans enrolled in the ALIVE HIV natural history cohort (n = 775). APOL1 variants were not associated with susceptibility to HIV-1 acquisition by comparing genotype frequencies between HIV-1 positive and exposed or at-risk HIV-1 uninfected groups (recessive model, 12.8 vs. 12.5%, respectively; OR 1.02, 95% CI 0.62–1.70). Similar null results were observed for dominant and additive models. APOL1 variants were not associated with HIV-1 viral load or with risk of progression to AIDS [Relative hazards (RH) 1.33, 95% CI 0.30–5.89 and 0.96, 95% CI 0.49–1.88, for recessive and additive models, respectively]. In summary, we found no evidence that APOL1 variants are associated with host susceptibility to HIV-1 acquisition, set-point HIV-1 viral load or time to incident AIDS. These results suggest that APOL1 variants are unlikely to influence HIV infection or progression among individuals of African ancestry.
Introduction
Apolipoprotein L1 (APOL1) is a human innate immune factor against African trypanosomes responsible for human African trypanosomiasis (or sleeping sickness) (1). Two common APOL1 variants, G1 (rs73885319, p.S342G) and G2 (a 6-bp in-frame deletion removing two amino acids, rs71785313, p.N388_Y389del), extend APOL1 restriction to T.b.rhodesiense, the cause of acute human African trypanosomiasis. These variants are found only in individuals with recent African ancestry. The 12–14% of African Americans carrying two APOL1 renal risk alleles in the compound heterozygous or homozygous state (referred to as APOL1 high risk [HR] genotypes) have a 3-, 7-, and 17-fold increased risk for developing hypertension-attributed nephropathy, non-diabetic end-stage kidney disease, and focal segmental glomerulosclerosis, respectively, (2–4). APOL1 is most strongly associated with HIV-associated nephropathy (HIVAN), with odds ratio (OR) 29 in African Americans and OR 89 in South Africans (3, 5), suggesting a strong interaction between APOL1 and the HIV-1 virus. The lifetime risk of HIVAN, a form of collapsing focal segmental glomerulosclerosis associated with rapid progression to end-stage renal disease, is ~10% in African Americans with untreated HIV infection (6, 7). The pathogenesis of HIVAN is likely due to direct HIV infection of kidney epithelial cells, which leads to podocyte proliferation and APOL1-mediated podocyte injury and loss (8–11). APOL1 transcription is up-regulated by interferons and other pro-inflammatory cytokines (12).
Recently, Taylor et al. reported that APOL1 restricts HIV-1 replication in macrophages and differentiated monocytes (12). APOL1 was shown to target HIV-1 Gag for degradation by the endolysosomal pathway and to deplete HIV-1 Vif, which counteracts the APOBEC3G host restriction factor in lysosomes (12). However, it remains unknown if variant APOL1 affects HIV acquisition, viral replication, or HIV disease progression.
APOL1 renal risk variants are most common in West Africa, where the prevalence of APOL1 HR genotypes approaches 25% but are also found throughout sub-Saharan Africa (4, 13) where HIV-1 infection is notably prevalent. Although APOL1 renal risk variants are a risk factor for kidney disease in HIV-1 infected persons, it is unknown if APOL1 renal risk variants are associated with other HIV-1 phenotypes. In the present study, we evaluate the genetic associations between APOL1 variants and HIV-1 acquisition, set-point viral load, and rate of progression to AIDS among African Americans enrolled in the ALIVE HIV-1 cohort.
Materials and Methods
Ethics Statement
Ethical approval for the study was obtained from the National Institute of Health Office of Human Subjects Research Protections (OHSRP #3314). Review Boards of participating institutions approved the study protocols, and written informed consent was obtained from all study participants.
Study Participants
Since APOL1 G1-G2 alleles are found only on African-origin chromosomes, we studied only African American participants enrolled in the ALIVE (AIDS Link to the Intravenous Experience) cohort.
The ALIVE Cohort
The epidemiological and clinical characteristics of the ALIVE cohort have been previously described (14). ALIVE is a prospective longitudinal natural cohort originally designed to characterize the incidence and natural history of HIV infection among injection drug users (IDUs) in Baltimore, MD, initiated in 1988 (14). At study entry, 88% of participants were African Americans. The participants were followed up semi-annually with blood draws for viral load and CD4+ T cell measurements and physical exam at each visit.
The study group comprises 227 African American incident HIV-1 seroconverters, 213 HIV-1 seroprevalent participants, and 335 uninfected, IDU participants. Seroconversion date was estimated as the midpoint between the last seronegative and the first seropositive HIV-1 antibody test date (mean interval 0.66 years, range 0.11–3.4 years) (15).
Genotyping of APOL1 G1-G2 Risk Variants
APOL1 renal risk variants G1 (rs73885319, p.S342G) and G2 (rs71785313, p.N388_Y389del) were genotyped by TaqMan genotyping assays (Applied Biosystems, Foster City, CA). The TaqMan allele discrimination assays were carried out on an ABI 7900HT sequencer detector system (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's protocol as previously described (3). For quality control, water controls were included on each plate and 10% of samples were duplicated. No water contamination or genotype mismatches between duplicates was observed. The genotype results were also further validated by the Sanger sequencing, following the protocol previously described (16).
Defining APOL1 Risk Haplotypes
The G1 risk allele is defined by the presence of the G allele at rs73885319 (342G) and the G2 (6-del) risk allele by the 6 base pair deletion at rs71785313 (-/TTATAA), which leads to the loss of two amino acids (388–389NYK>K) (2, 3). The G1 and G2 risk alleles are in absolute negative disequilibrium and never occur together on the same chromosome (17)). APOL1 follows a recessive inheritance model for HIVAN and other kidney diseases: APOL1 HR status for kidney disease is defined by carriage of 2 risk alleles (G1/G1, G1/G2, and G2/G2) and low-risk (LR) status is defined by carriage of 1 or 0 renal risk alleles (18).
Statistical Analysis
We assessed the potential effects of APOL1 risk genotypes using additive (2 vs. 1 vs. 0 risk alleles), dominant (2 or 1 vs. 0), and recessive (2 vs. 1 or 0 risk alleles) models. All analyses were performed using SAS version 9.12 (SAS Institute, Cary, NC).
Analysis of Risk to HIV-1 Acquisition
We assessed the impact of APOL1 G1-G2 variants on HIV-1 infection susceptibility by comparing frequencies between the HIV-1 infected group comprising HIV-1 seroincident and seroprevalent subjects and the HIV-1 at-risk, uninfected group. Odds ratios (OR) and P-values were obtained by using a conditional logistic regression test. Statistical power was calculated with GAS-power-calculator available at http://csg.sph.umich.edu/abecasis/gas_power_calculator/.
Analysis of Viral Load
For the seroincident participants, HIV-1 viral load set-point was defined as the mean log10-transformed HIV-1 RNA plasma copies measured between 6 and 33 months after seroconversion (2–5 measurements). Viral load measurements exceeding 3-fold (0.5 log10) from the average of all remaining points were excluded, as previously suggested (19). We ran t-tests to estimate the difference of viral load means between APOL1 HR and LR subgroups. We used the one-way analysis of variance (ANOVA) to determine whether there were any statistically significant differences among the means for carriage of 2, 1, or 0 APOL1 risk alleles.
Analysis of Disease Progression to AIDS
In the ALIVE cohort, we tested the association of APOL1 risk alleles on disease progression to AIDS using Cox proportional hazards model (Cox model) and Kaplan-Meier survival curve analyses for incident HIV seroconverters. The disease progression endpoints were: CD4 T-cell <200 cells/mm3, or clinical AIDS diagnosis (20). The median time from seroconversion to AIDS was 7 years. To avoid the confounding effect of anti-retroviral therapy (ART) on disease progression, we censored the data after July 31, 1997 as few ALIVE participants received ART prior to this date (21). We included known genetic factors modifying AIDS progression as covariates in the adjusted Cox model analysis: HLA-B*57 and HLA Class I homozygosity (22). The analyses were stratified by sex and by age at seroconversion: 0–20, 20–40, and >40 years. Two-tailed P-values were computed using Wald tests.
Results
Association of APOL1 Risk Alleles on the Risk of HIV-1 Acquisition
To determine whether APOL1 G1 or G2 variants affect host susceptibility to HIV-1 acquisition, we compared the distribution of G1 and G2 variants in HIV-1 seroincident subjects (n = 227) with at risk, seronegative individuals (n = 335) (Table 1). No associations with HIV-1 acquisition were observed for the additive (P = 0.61), dominant (P = 0.56) or recessive genetic models (P = 0.87) (Table 1). To increase power, we combined seroconverters and seroprevalents) but results remained non-significant (Table 1). Adjusting for sex and age did not affect the results (Table 1). These results suggest that APOL1 risk variants have no impact on host susceptibility to HIV-1 acquisition.
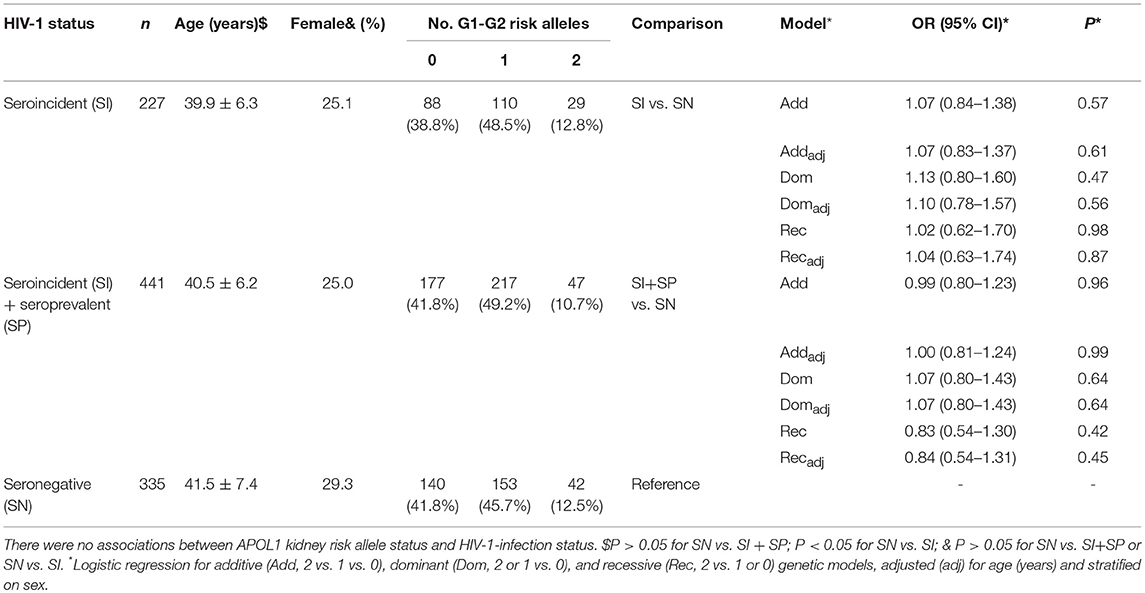
Table 1. Association of APOL1 G1-G2 variants with HIV-1 acquisition.
Association of APOL1 Risk Alleles on HIV-1 Viral Load
Among HIV-1 seroincident participants, set-point HIV-1 viral load levels were found to be similar for carriers of 2, 1, or 0 APOL1 risk alleles (P = 0.79, ANOVA). In the recessive model comparing viral load between carriers of HR genotypes (VL = 4.23 ± 0.64, N = 27) and those with LR APOL1 genotypes (VL = 4.20 ± 0.73, N = 177), we also observed no differences in viral load (P = 0.48, Table 2).
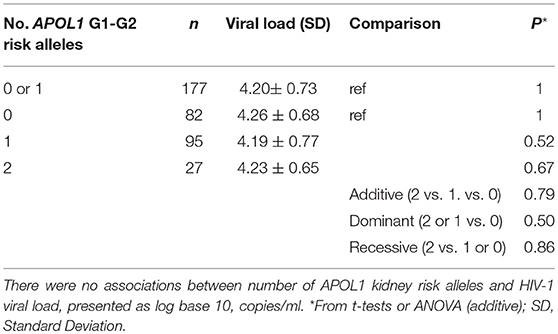
Table 2. Association between APOL1 G1-G2 variant alleles and HIV-1 viral load.
Association of APOL1 Risk Alleles on HIV-1 Disease Progression
To assess the impact of APOL1 HR on disease progression in untreated individuals from date of seroconversion to CD4 <200 cell/mm3 and to incident AIDS, we performed time-to-event analysis for 227 African American seroincident participants. APOL1 genotypes were not associated with the rate of progression to CD4 <200 cells/mm3 (Figure 1A) or AIDS in Kaplan-Meier survival analyses (Figure 1B, additive model, P > 0.12 for log-rank or Wilcoxon tests). In the crude (data not shown) and adjusted Cox models, APOL1 HR genotype (recessive model), APOL1 risk allele number (additive) or risk allele carriage (dominant model) were not associated with the rate of progression to AIDS (P > 0.70, Table 3).
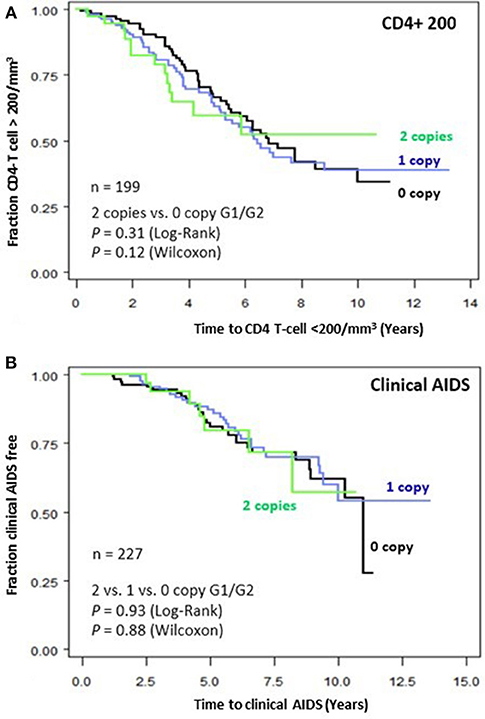
Figure 1. Genetic effects of APOL1 G1-G2 variants on progression of HIV disease. Kaplan-Meier survival curves for carriage of 0, 1, and 2 APOL1 risk allele for progression to (A) CD4+ T-cell <200/mm3 and (B) clinical AIDS. RH and adjusted P-values were estimated from Cox proportional hazards models. P-values for survival curves were obtained from a log-rank test.
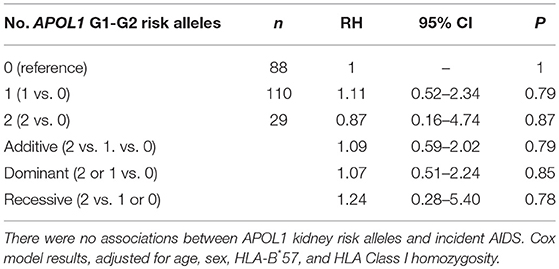
Table 3. Association between APOL1 G1-G2 variant alleles and incident clinical AIDS.
Discussion
In this genetic epidemiological study of an HIV-1 at-risk and natural progression cohort, we observed no evidence of association between APOL1 renal risk alleles and HIV-1 acquisition, HIV-1 viral load, and rate of progression to CD4 <200, AIDS, or the composite outcome. Our results indicate that APOL1 renal risk variants, which are highly prevalent among African Americans and sub-Saharan Africans, do not significantly contribute to the HIV-1 epidemic by increasing viral burden or potentiating HIV-1 transmission.
A recent in vitro study reported that APOL1 protein can inhibit HIV-1 infection of macrophage and monocytes by multiple mechanisms, including inhibition of transcription and degradation of HIV-1 Gag and Vif proteins (12). If APOL1 protein effectively inhibits HIV-1 in vitro, APOL1 coding variants might confer differential impact on HIV replication or disease progression by enhancing or attenuating the anti-HIV properties of APOL1 protein. However, our genetic association study revealed no in vivo evidence of association of APOL1 renal risk alleles with HIV-1 infection acquisition or disease progression. Our findings are supported by the observation that APOL1 gene expression is undetectable in CD4+ T cells, the primary target of HIV infection even with IFN-γ stimulation (12). In contrast, APOL1 gene expression is highly inducible by IFN-γ stimulation in monocytes and macrophages, which were used in the in vitro experiments testing for APOL1 restriction of HIV replication (12). CD4+ T lymphocytes are the principal target of HIV, while infected macrophages play a supportive role in viral pathogenesis involving HIV cell-to-cell spread, and certain tissue infections including lungs, gut and brain (23, 24). The in vivo role of APOL1 in HIV-1 pathogenesis thus warrants further investigation. An implication of this study is that development of HIVAN and eGFR decline among those with APOL1 HR status (18, 25, 26), is likely due to local podocyte injury in a setting of high viral load in patients with untreated HIV infection. A recent study demonstrated that variant APOL1 protein increases accumulation of HIV-1 in podocytes, inducing inflammatory responses via IL-1β priming (11).
This study has both strengths and limitations. A strength is that the ALIVE cohort is one of few well-characterized HIV natural history cohorts enrolling African Americans prior to the ART era, and the large number of treatment-naïve seroconverters makes it a choice cohort for unbiased exploration of HIV-related outcomes. The relatively modest sample size is balanced by the combined high frequency of these variants in the African American population. We had 80% power to detect a potential association of APOL1 G1-G2 with HIV-1 infection, with an OR 1.35 for additive model and 1.93 for recessive model. We were unable to control for mortality due to APOL1-associated ESKD or to HIVAN since biopsy data were unavailable; however, only 1 death was observed among 29 APOL1 HR individuals prior to censoring on July 31, 1997, suggesting that our null results are not due to frailty bias resulting from excess HIVAN or ESKD-related deaths in the HR group.
In summary, this population genetic study found no evidence that APOL1 renal risk variants contribute to the risk of HIV-1 acquisition or progression of HIV-1 disease progression to AIDS. APOL1 variants are unlikely to contribute to the prevalence of HIV infection in subSaharan Africa or among African Americans.
Author Contributions
PA and CW conceived the study, designed the analyses, and wrote the manuscript. PA performed the analyses. EB-R performed genotyping. GK provided clinical data and DNA samples. GK, SL, and JK contributed to data interpretation and manuscript revisions. All authors reviewed the manuscript.
Funding
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of health, under contract HHSN26120080001E. This Research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and of the Kidney Disease Section, National Institute of Diabetes and Digestive and Kidney Diseases. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. ALIVE is supported by National Institute on Drug Abuse (U01-036297 and R01-12586). SL's work was realized in the context of the LabEx IGO program that is supported by the National Research Agency via the Investment Into The Future program (ANR-11-LABX-0016-01).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
HIV-1, HIV type 1; OR, Odds ratio; RH, Relative Hazards; SNP, Single Nucleotide Polymorphism.
References
1. Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Perez-Morga D. The trypanolytic factor of human serum. Nat Rev Microbiol. (2006) 4:477–86. doi: 10.1038/nrmicro1428
2. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329:841–5. doi: 10.1126/science.1193032
3. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (2011) 22:2129–37. doi: 10.1681/ASN.2011040388
4. Limou S, Dummer PD, Nelson GW, Kopp JB, Winkler CA. APOL1 toxin, innate immunity, and kidney injury. Kidney Int. (2015) 88:28–34. doi: 10.1038/ki.2015.109
5. Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol. (2015) 26:2882–90. doi: 10.1681/ASN.2014050469
6. Shahinian V, Rajaraman S, Borucki M, Grady J, Hollander WM, Ahuja TS. Prevalence of HIV-associated nephropathy in autopsies of HIV-infected patients. Am J kidney Dis. (2000) 35:884–8. doi: 10.1016/s0272-6386(00)70259-9
7. Lucas GM, Eustace JA, Sozio S, Mentari EK, Appiah KA, Moore RD. Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy: a 12-year cohort study. AIDS (2004) 18:541–6. doi: 10.1097/00002030-200402200-00022
8. Chandel N, Sharma B, Husain M, Salhan D, Singh T, Rai P, et al. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol. (2013) 304:F1347–57. doi: 10.1152/ajprenal.00717.2012
9. Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who's interfering with whom? Nat Rev Microbiol. (2015) 13:403–13. doi: 10.1038/nrmicro3449
10. Kopp JB, Heymann J, Winkler CA. APOL1 renal risk variants: fertile soil for hiv-associated nephropathy. Semin Nephrol. (2017) 37:514–9. doi: 10.1016/j.semnephrol.2017.07.004
11. Mikulak J, Oriolo F, Portale F, Tentorio P, Lan X, Saleem MA, et al. Impact of APOL1 polymorphism and IL-1beta priming in the entry and persistence of HIV-1 in human podocytes. Retrovirology (2016) 13:63. doi: 10.1186/s12977-016-0296-3
12. Taylor HE, Khatua AK, Popik W. The innate immune factor apolipoprotein L1 restricts HIV-1 infection. J Virol. (2014) 88:592–603. doi: 10.1128/JVI.02828-13
13. Limou S, Nelson GW, Kopp JB, Winkler CA. APOL1 kidney risk alleles: population genetics and disease associations. Adv Chronic Kidney Dis. (2014) 21:426–33. doi: 10.1053/j.ackd.2014.06.005
14. Vlahov D, Graham N, Hoover D, Flynn C, Bartlett JG, Margolick JB, et al. Prognostic indicators for AIDS and infectious disease death in HIV-infected injection drug users: plasma viral load and CD4+ cell count. JAMA (1998) 279:35–40.
15. An P, Duggal P, Wang LH, O'Brien SJ, Donfield S, Goedert JJ, et al. Polymorphisms of CUL5 are associated with CD4+ T cell loss in HIV-1 infected individuals. PLoS Genet. (2007) 3:e19. doi: 10.1371/journal.pgen.0030019
16. Ko WY, Rajan P, Gomez F, Scheinfeldt L, An P, Winkler CA, et al. Identifying Darwinian selection acting on different human APOL1 variants among diverse African populations. Am J Hum Genet. (2013) 93:54–66. doi: 10.1016/j.ajhg.2013.05.014
17. Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, et al. APOL1-mediated cell injury involves disruption of conserved trafficking processes. J Am Soc Nephrol. (2017) 28:1117–30. doi: 10.1681/ASN.2016050546
18. Estrella MM, Li M, Tin A, Abraham AG, Shlipak MG, Penugonda S, et al. The association between APOL1 risk alleles and longitudinal kidney function differs by HIV viral suppression status. Clin Infect Dis. (2015) 60:646–52. doi: 10.1093/cid/ciu765
19. Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. (2009) 5:e1000791. doi: 10.1371/journal.pgen.1000791
20. CDC. Revision of the CDC surveillance case definition for acquired immunodeficiency syndrome. MMWR (1987) 36(Suppl. 1):1S−15S.
21. Celentano DD, Galai N, Sethi AK, Shah NG, Strathdee SA, Vlahov D, et al. Time to initiating highly active antiretroviral therapy among HIV-infected injection drug users. AIDS (2001) 15:1707–15. doi: 10.1097/00002030-200109070-00015
22. O'Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet. (2004) 36:565–74. doi: 10.1038/ng1369
23. Sattentau QJ, Stevenson M. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe (2016) 19:304–10. doi: 10.1016/j.chom.2016.02.013
24. Koppensteiner H, Brack-Werner R, Schindler M. Macrophages and their relevance in Human Immunodeficiency Virus Type I infection. Retrovirology (2012) 9:82. doi: 10.1186/1742-4690-9-82
25. Jotwani V, Shlipak MG, Scherzer R, Parekh RS, Kao WH, Bennett M, et al. APOL1 genotype and glomerular and tubular kidney injury in women with HIV. Am J Kid Dis. (2015) 65:889–98. doi: 10.1053/j.ajkd.2015.02.329
Keywords: HIV-1, AIDS, APOL1, host susceptibility, genetic epidemiology
Citation: An P, Kirk GD, Limou S, Binns-Roemer E, Kopp JB and Winkler CA (2019) Impact of APOL1 Genetic Variants on HIV-1 Infection and Disease Progression. Front. Immunol. 10:53. doi: 10.3389/fimmu.2019.00053
Received: 26 October 2018; Accepted: 09 January 2019;
Published: 24 January 2019.
Edited by:
Guido Poli, Vita-Salute San Raffaele University, ItalyReviewed by:
Jacques Fellay, École Polytechnique Fédérale de Lausanne, SwitzerlandJoanna Mikulak, Humanitas Research Hospital, Italy
Copyright © 2019 An, Kirk, Limou, Binns-Roemer, Kopp and Winkler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping An, cGluZy5hbkBuaWguZ292
Cheryl A. Winkler, d2lua2xlcmNAbWFpbC5uaWguZ292