 Maria Chiriaco1*
Maria Chiriaco1* Gigliola Di Matteo2
Gigliola Di Matteo2 Francesca Conti1
Francesca Conti1 Davide Petricone2
Davide Petricone2 Maia De Luca1
Maia De Luca1 Silvia Di Cesare2
Silvia Di Cesare2 Cristina Cifaldi1Rita De Vito3Matteo Zoccolillo2,4Jessica Serafinelli1
Cristina Cifaldi1Rita De Vito3Matteo Zoccolillo2,4Jessica Serafinelli1 Noemi Poerio5
Noemi Poerio5 Maurizio Fraziano5
Maurizio Fraziano5 Immacolata Brigida4Fabio Cardinale6
Immacolata Brigida4Fabio Cardinale6 Paolo Rossi1,2Alessandro Aiuti4,7,8Caterina Cancrini1,2†
Paolo Rossi1,2Alessandro Aiuti4,7,8Caterina Cancrini1,2† Andrea Finocchi1,2*†
Andrea Finocchi1,2*†- 1University Department of Pediatrics, Unit of Immune and Infectious Diseases, Childrens' Hospital Bambino Gesù, Rome, Italy
- 2Department of Systems Medicine, University of Rome Tor Vergata, Rome, Italy
- 3Histopathology Unit, Bambino Gesù Children's Hospital-Research Institute, Rome, Italy
- 4San Raffaele Telethon Institute for Gene Therapy (SR-TIGET), IRCCS San Raffaele Scientific Institute, Milan, Italy
- 5Department of Biology, University of Rome Tor Vergata Rome, Rome, Italy
- 6Allergy, Immunology and Pediatric Pulmunology Unit, Policlinico di Bari Ospedale Giovanni XXIII, Bari, Italy
- 7Pediatric Immunohematology, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 8Vita Salute San Raffaele University, Milan, Italy
We described for the first time a female patient with the simultaneous presence of two homozygous mutations in MYD88 and CARD9 genes presenting with pyogenic bacterial infections, elevated IgE, and persistent EBV viremia. In addition to defective TLR/IL1R-signaling, we described novel functional alterations into the myeloid compartment. In particular, we demonstrated a defective production of reactive oxygen species exclusively in monocytes upon E. coli stimulation, the inability of immature mono-derived DCs (iDCs) to differentiate into mature DCs (mDCs) and the incapacity of mono-derived macrophages (MDMs) to resolve BCG infection in vitro. Our data do not provide any evidence for digenic inheritance in our patient, but rather for the association of two monogenic disorders. This case illustrates the importance of using next generation sequencing (NGS) to determine the most accurate and early diagnosis in atypical clinical and immunological phenotypes, and with particular concern in consanguineous families. Indeed, besides the increased susceptibility to recurrent invasive pyogenic bacterial infections due to MYD88 deficiency, the identification of CARD9 mutations underline the risk of developing invasive fungal infections emphasizing the careful monitoring for the occurrence of fungal infection and the opportunity of long-term antifungal prophylaxis.
Highlights
- Patient with atypical primary immunodeficiency
- Clinical manifestations: pyogenic bacterial infections, high IgE level, and persistent EBV viremia
- Next-generation sequencing reveals two homozygous mutations in MYD88 and CARD9 genes leading to complete absence of proteins
- Monocytes/macrophages function and DC differentiation were severely compromised
- NGS has a key role to determine the correct diagnosis in atypical primary immunodeficiency leading to reconsider the individualized treatment.
Introduction
Primary immunodeficiency diseases (PIDs) are a heterogeneous group of disorders characterized by poor or absent function in one or more components of the immune system which predisposes affected individuals to recurrent and severe infections, autoimmunity, aberrant inflammation, atopy, lymphoproliferation, and malignancy. To date more than 350 different disorders have been genetically characterized and new disorders continually being recognized (1). PIDs are caused by defects in the adaptive immunity, T-cell, B-cell, or combined B and T cell immunodeficiencies, or innate immunity, phagocyte, NK cell, several classes of pattern recognition receptors (PRRs), and complement disorder (2).
The innate immune response represents the first line of defense against pathogens. Appropriate recognition of threats and induction of the downstream signaling cascades are essential steps in the removal of these organisms from the system. The failure of the innate system to identify pathogens, delays the induction of the immune response worsening outcomes of infection. Over the last two decades, several inborn errors affecting primarily components of innate immunity, conferring vulnerability to a narrow range of microorganisms, such as Mendelian susceptibility to mycobacterial disease (MSMD), herpes simplex 1 encephalitis (HSE), and monogenic susceptibility to invasive pneumococcal disease (IPD) have been identified (3).
Myeloid differentiation primary response protein 88 (MYD88) is a cytosolic adaptor protein recruited from most toll-like receptors (TLRs) and IL-1 receptors (IL-1Rs) to trigger the canonical pathway leading to NF-κB activation and inflammatory cytokine gene transcription. Patients with mutations in MYD88 present a Mendelian predisposition to invasive (meningitis and septicaemia) and non-invasive bacterial infections caused principally by S. pneumoniae, S. aureus and, less frequently, by few Gram-negative bacteria. Typically, clinical and laboratory signs of inflammation (e.g., fever, elevated C-reactive protein level) in MYD88 patients are weak or delayed even in case of severe infection (4–6). Moreover, an interesting feature is that contrary to the most of PID, in which patient's condition undergo a gradual deterioration over time, clinical status, and outcome in MYD88 deficient patients improve with age even in the absence of preventive measures, suggesting that the MyD88/TIR pathway becomes redundant once acquired immunity, is fully functional and can ensure protection.
Caspase-associated recruitment domain-containing protein 9 (CARD9) is a central regulator of innate immune response, acting as downstream regulator of Dectin-1, Dectin-2, Dectin-3, Mincle, and others involved in the recognition of fungal pathogene (7). Its deficiency causes a rare PID characterized by superficial and deep fungal infection in otherwise healthy individuals (HD). CARD9 deficiency cause defective cytokine production in response to fungal ligand, impaired T cell- dependent IL17 production and impaired neutrophil recruitment to infection sites and/or killing. Age at onset is heterogeneous and, although the most of reported patients showed an adult onset, fungal infections can occur at any age (8–11).
Here we report for the first time a female patient with inherited MYD88 and CARD9 deficiencies presenting with pyogenic bacterial infections, high level of IgE and persistent EBV viremia.
Case Report
A female infant patient was born from consanguineous parents with a family history suggestive for PID. Her sisters deceased, respectively, for meningitis and sepsis of unknown origin (Figure 1A). The umbilical cord fell at 30 days and she was fully immunized including measles, mumps, and rubella, without any complications. During the first year of life she suffered from recurrent respiratory infections and one episode of urinary tract infection due to P. aeruginosa. At the age of 2 years she was hospitalized for an intramuscular and deep abdominal abscess caused by S. aureus, without any signs of inflammation, which was successfully treated with a combined therapy of intravenous antibiotics and surgical drainage. Extensive immunological work up revealed a normal complete blood count (CBC), normal number of T-, B-, and NK-cells (including naïve and memory subsets, and CD4+CD31+), normal nitroblue tetrazolium (NBT) test, normal serum immunoglobulin levels and normal specific antibody response to T. toxoid, HBV, and HIB. We detected high levels of IgE (4,550 KU/L), mild eosinophilia (900/mcL), and non-protective response to pneumococcal vaccines. In spite of elevated circulating IgE levels, patient did not develop allergic symptoms and allergen-specific IgE was not detectable. During follow-up, despite co-trimoxazole prophylaxis, she developed urinary tract infection caused by E. coli and a relapsing necrotizing granulomatous lymphadenitis of left cervical lymph nodes, not responding to antibiotic therapy, and successfully treated with surgical removal. Moreover, the patient showed persistent EBV viremia in the blood with no evidences of organs damage. Further analysis revealed a normal frequency of Tregs and Tfh cells, and a normal level of INFγ produced by CD4+ and CD8+ T cells after PMA stimulation (data not shown). Patient showed lower frequency (3.4-fold decreased) of circulating Th17 with respect to HD (Pt 0.36 ± 0.14 vs. HDs 1.23 ± 0.19%) (Figure 1B). Moreover, a mild decreased level of memory B cells with impaired B cells responsiveness to CpG-TLR9 stimulation was observed (data not shown).
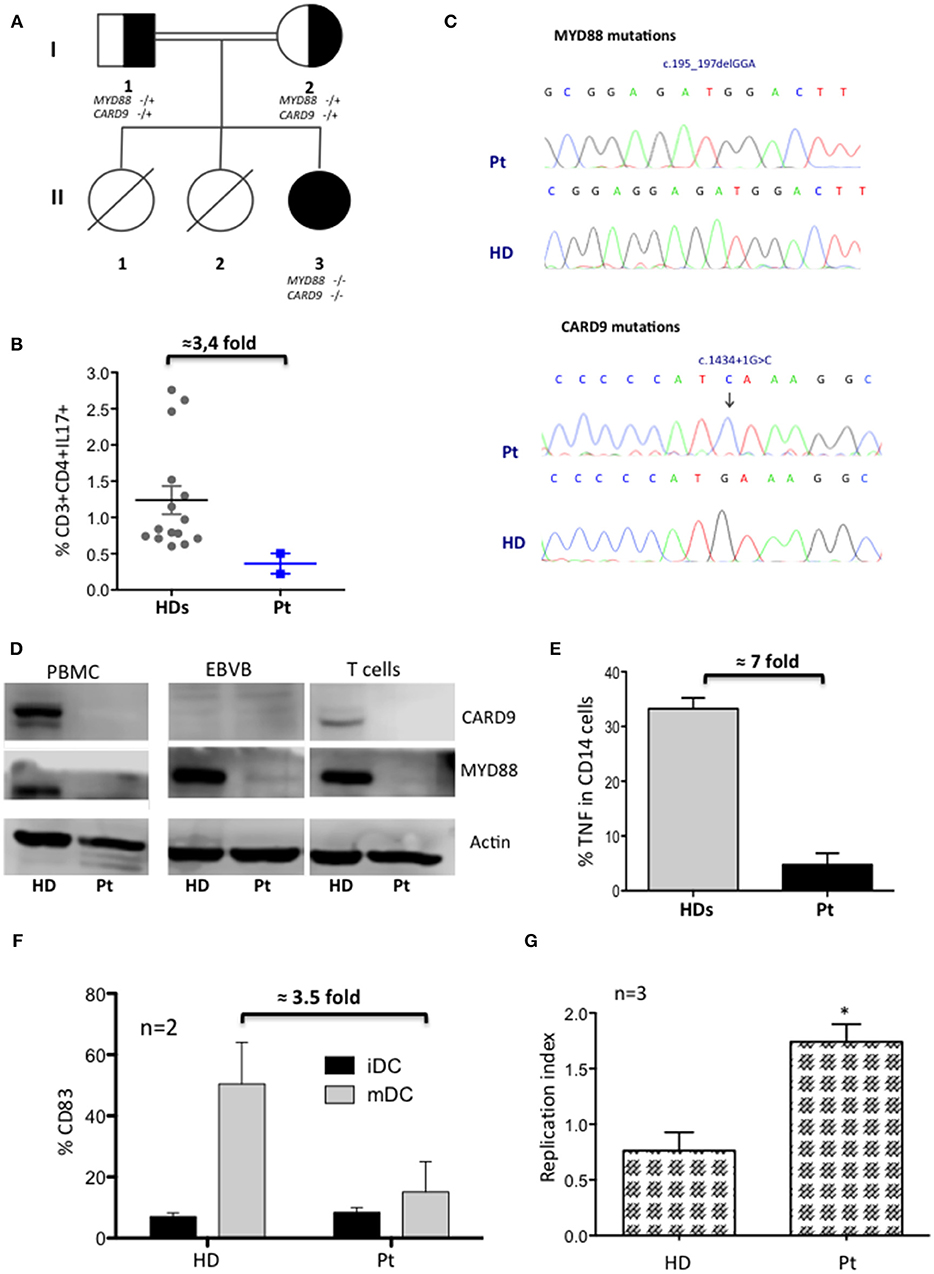
Figure 1. Characterization of MYD88-CARD9 deficiency patient: part 1. (A) Family pedigree showing proband (II3), carrier parents (I1, I2), and sisters (II1, II2). Genotype + and – indicate in the figure wild type and mutated, respectively. (B) Percentage of CD3+CD4+IL17+ cells after PMA stimulation (mean ± SEM of n = 2). (C) Sanger sequencing confirmed a homozygous in-frame deletion (c.195_197delGGA) in MYD88 gene and a homozygous splice-donor mutation (c.1434+1G>C) in CARD9 gene. (D) Western Blot of CARD9 and MYD88 proteins performed on PBMC, EBVB, and PHA derived T cell lines. (E) TNFα production by monocytes after LPS stimulation (mean ± SEM of n = 2). (F) Phenotypic analysis of iDC and mDC differentiated in vitro. Results indicate the mean percentage ± SD of CD83 maturation/activation markers in gated CD14-CD1a+HLA-DR+ subsets of iDC and mDC. (G) Capacity of MDM to kill (right panel) BCG (*P < 0.05).
Although the clinical phenotype was not typical of the hyper IgE syndromes (HIES), family history, elevated levels of IgE, eosinophilia, and decreased Th17 cells led us to exclude by Sanger sequencing genetic defects associated with HIES (STAT3, DOCK8, and TYK2). Moreover, IRAK4 PID variants were excluded considering the absence of fever and of a significant increase of inflammatory markers during severe infections. HaloPlex targeted sequencing panel, including 630 causative and candidate PID genes (Cifaldi et al., manuscript submitted) identified two homozygous mutations in two different genes (Figures 1C,D and Supplementary Table 1). A known pathogenic in-frame deletion c.195_197delGGA (AF of 8.13180e-06_1000 Genomes) in exon1 of the MYD88 gene leading to absent protein expression resulting in a loss-of-function mutation (4), and a homozygous splice-donor variant c.1434+1G>C (AF of G = 0.0008/4_1000 Genomes) in the CARD9 gene, with absent protein, previously described as heterozygous variant (11). Sanger sequencing confirmed the mutations and both parents resulted heterozygous (data not shown).
Since both mutated proteins caused impaired innate immunity we deeply investigated the myeloid compartment. Patient's monocytes showed a marked reduction of TNFα-production (7-fold decreased) after LPS-stimulation (Figure 1E), although PB-monocytes (classical, intermediate, and non-classical), and major subtypes of dendritic cells (mDC1, mDC2, and pDC) were normally represented (data not shown). Moreover, immature monocytes-derived DC had a defective ability to become mature/activated-DC (mDC) (3.5-fold decrease) (Figure 1F), while maintaining their phagocytic capacity (data not shown). Additionally, patient's monocytes-derived macrophages (MDMs) failed to kill Bacillus Calmette–Guérin (BCG) in vitro (Figure 1G) after engulfment (12–14).
Next, we investigated the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity by dihydrorhodamine (DHR) flow cytometric assay using PMA and opsonized E. coli to stimulate neutrophils and monocytes. Neutrophils from our patient performed a normal respiratory burst (Figure 2A), in agreement with what previously observed in MYD88 and CARD9 deficient patients. Conversely, we found an impaired capacity to produce ROS in patient's monocytes selectively stimulated with E. coli (7-fold decreased respect to HD) (Figure 2B). Interestingly, these results showed the impaired contact signal/trigger to external stimuli by monocytes, which could in part explain, the susceptibility to urinary tract infection. To confirm this inability, we tested the capacity of monocytes to stimulate T cells via LPS to produce INFγ and found a defective LPS/monocytes-triggered INFγ production by patient's CD3 cells (7-fold decreased) as compared to HD (Figure 2C).
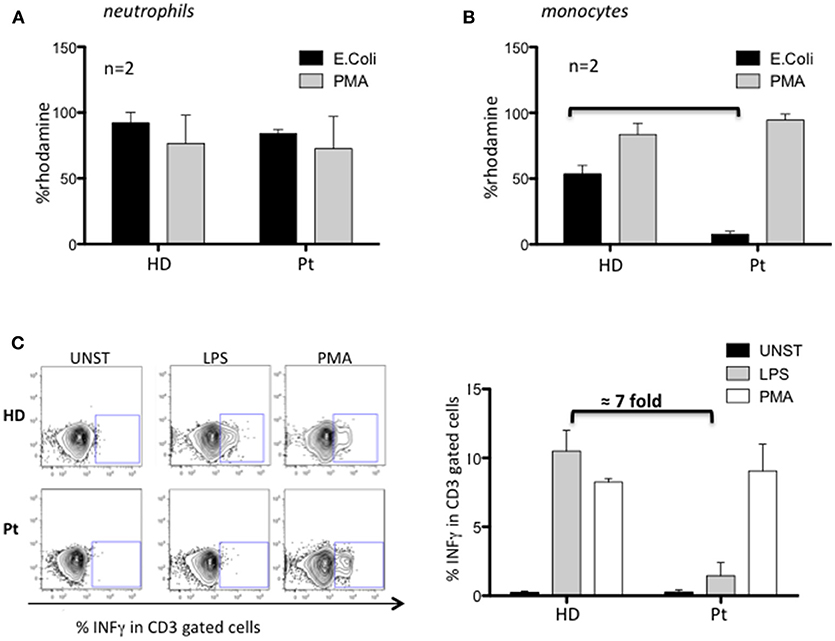
Figure 2. Characterization of MYD88-CARD9 deficiency patient: part 2. (A,B) NADPH oxidase activity evaluated by dihydrorhodamine (DHR) assay in patient's neutrophils and monocytes stimulated with PMA or E. coli. The production of ROS is determined by the oxidation of DHR into the fluorescent rhodamine. (C) INFγ production by patient's CD3 cells stimulated with PMA or LPS/monocytes. Left panel shows representative plots and right panel shows mean ± SEM of n = 2.
Discussion
Here we describe a patient with a phenotype of recurrent respiratory and deep tissue bacterial infections with granuloma inflammation in whom two pathogenic homozygous mutations in MYD88 and CARD9 genes were identified by NGS. In agreement with MYD88- and CARD9-deficiencies, we showed a defective TLR/IL1R-signaling leading to a severe decrease of TNFα and IL17 pro-inflammatory cytokines production, that compromise neutrophils recruitment to the site of acute inflammation (Supplementary Figure 1) and the capacity of the immune system to definitely resolve bacterial infections (15). The patient has a severely compromised myeloid compartment. Indeed, peripheral monocytes were unable to differentiate into mDCs, and MDMs cells do not resolve in vitro BCG infection. Although NADPH-oxidase respiratory burst is normally functioning after PMA-stimulation, monocytes are severely unable to produce ROS in response to E. coli. These data highlight the inability of patient monocytes to receive and process external signals and could explain the development of P. aeruginosa, S. aureus, and E. coli infections in our patient. Additionally, we investigated if MYD88-CARD9 mutations hinder the development of an appropriate T cell response. We found a marked defect (7-fold decrease) of INFγ production by CD3 cells after LPS-monocytes stimulation revealing a compromised cooperation between monocytes and T cells.
High level of serum IgE, family history, recurrent bacterial infections, and low level of circulating Th17 initially suggested us a diagnosis of HIES or IRAK4 deficiency shortly refuted by molecular diagnosis of CARD9 and MYD88 deficiencies. Frans et al. (16) similarly to our patient, described an adult female patient who was at first phenotypically diagnosed with HIES because of severe bacterial and fungal infections, and elevated serum IgE levels, but finally defined as suffering from IRAK-4 deficiency. These findings highlight the importance of using NGS to identify the genetic cause of the disease in a patient presenting with atypical clinical and immunological phenotype. Considering IRAK-4 deficiency as a phenocopy of MYD88 deficiency, although both CARD9 (10) and MYD88 (6) deficiency showed moderately increased level of IgE compared to the very high levels found in HIES patients (17), we believe that MYD88 plays a crucial role in determining the patient's phenotype.
We further demonstrate that MDMs were unable to resolve in vitro BCG infection. Interestingly, MYD88 deficiency mice showed an increased susceptibility and mortality to the experimental infection with three Mycobacteria species (M. avium, M. bovis, and M. tubercolosis) and Mycobacterium avium infection has been reported in one IRAK4-deficient patient (18) thus suggesting a possible role for MYD88/IRAK4 signaling axis in protective immunity to Mycobacteria.
Interestingly our patient had a persistent-asymptomatic EBV viremia commonly reported in several PID patients but not typically associated with CARD9- or MYD88-deficiency (19). The impaired TLR2/MYD88 axis observed in our patients by the absence of Myd88 protein could induce a defective cytokines production that allows EBV to establish persistent infections evading immune surveillance (20, 21). The role of Card9 protein during EBV infection still remains unknown. This condition could be depend by the simultaneous presence of both defects that may create the proper context to allow the EBV to replicate, or by the presence of any other gene unknown variants that could increase the susceptibility to EBV. Despite the persistent asymptomatic EBV viremia represents the only new feature as compared to MYD88- and CARD9- deficiency phenotype, we do not feel to define our patient as affected by digenic PID (22, 23), but rather with two monogenic disorders. Accordingly to the patient's age the predominant clinical phenotype is the one associated with MYD88-deficiency (Figure 3). The age at onset of fungal infection, characteristic for CARD9-deficiency, is highly heterogeneous and could arise from childhood to adulthood (10), with incomplete clinical penetrance in younger individuals (24).
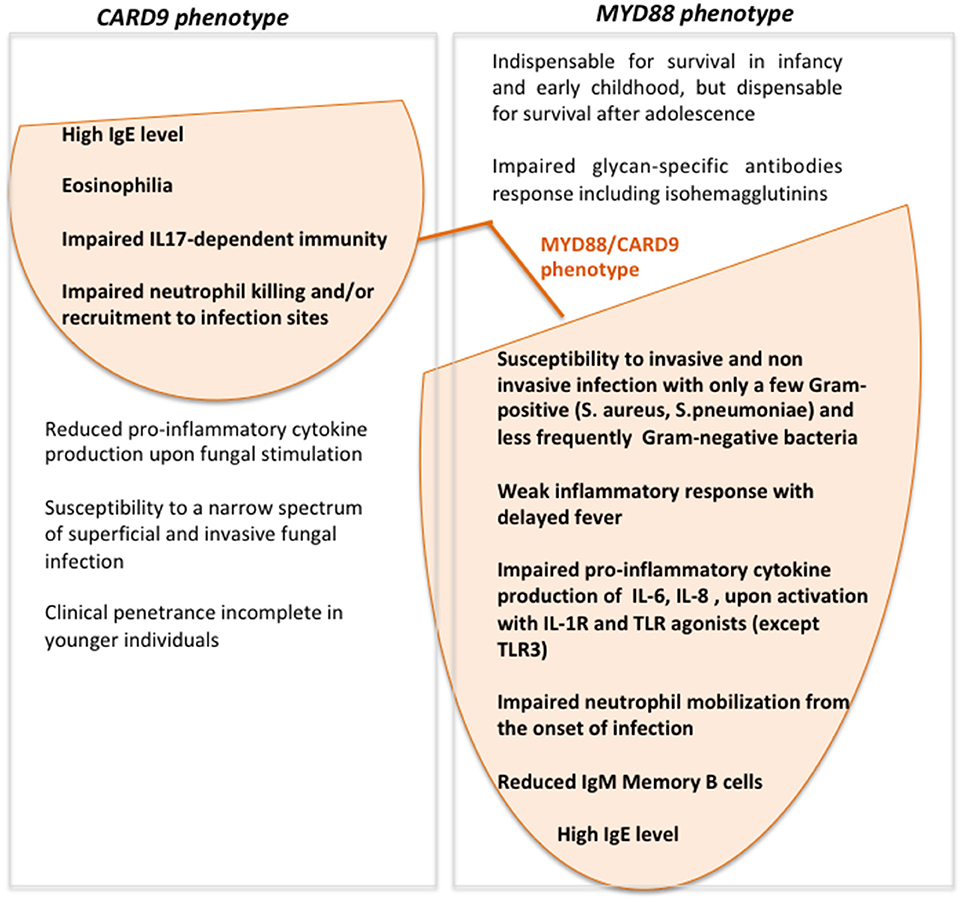
Figure 3. Venn diagram showing MYD88- and CARD9-deficiency features and clinical/laboratory data of our patient with the simultaneous presence of MYD88 and CARD9 mutations.
Concluding Remarks
We described for the first time a patient with the simultaneous presence of two homozygous variant mutations in MYD88 and CARD9 genes presenting with pyogenic bacterial infections, elevated IgE and persistent EBV viremia. In addition to the defective TLR/IL1R-signaling, we demonstrated a selective impaired monocytes/macrophages function and DC differentiation. This study shows the difficult path to perform a definitive diagnosis if patient's phenotype is atypical. Although NGS technologies have a key role to discover mutated genes, a continuous critical comparison with the patient clinical manifestations is needed to improve clinical management. Indeed, besides the increased susceptibility to recurrent invasive pyogenic bacterial infections due to MYD88-deficiency, the identification of CARD9 mutations underline the risk of developing invasive fungal infections emphasizing the importance of careful monitoring for the occurrence of fungal infection and the opportunity of long-term antifungal prophylaxis.
Ethics Statement
All procedures performed in the study were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration. Written informed consent, following standard ethical procedures with approval of the Children's Hospital Bambino Gesù Ethical Committee, was obtained from parents of patient.
Author Contributions
MC designed and performed experiments, analyzed the data, and wrote the paper. GD performed NGS and contributed to revision the manuscript. FrC and FaC contributed to therapeutic approach and wrote the paper. DP and MZ performed the bioinformatics analyses. SD, CrC, NP, MF, IB, and RD performed laboratory studies and contributed to data interpretation. MD, JS, and PR have been important part in complicated diagnostic and therapeutic course. AA supervise the project and contributed to interpretation and revision of the manuscript. CaC and AF supervised the project and wrote the manuscript.
Funding
This work was supported by the European Commission—Advanced Cell-based Therapies for the treatment of Primary Immunodeficiency (HEALTH-F5-2010-261387, CELL-PID), the ERARE-3-JTC 2015 EUROCID to AA, the Italian TELETHON foundation (grant number GGP15109) to AF, Italian Ministero della Salute (NET-2011-02350069) to AA, CaC, TELETHON foundation (TIGET Core grant C6) to AA and Ospedale Pediatrico Bambino Gesù Ricerca Corrente (201702P003966) to CaC. MZ conducted this study as part of his Ph.D. in Immunologia Medicina Molecolare e Biotecnologie Applicate, Tor Vergata University, Rome, Italy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patient, her parents and the Ospedale Pediatrico Bambino Gesù staff for their participation in this study. IB received a fellowship from LaSpes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00130/full#supplementary-material
Abbreviations
AF, allelic frequency; BCG, bacillus calmette-guerin; CARD9, caspase-associated recruitment domain-containing protein 9; CBC, complete blood count; EBV, epstein-barr virus; IL-1Rs, IL1 receptors; IPD, invasive pneumococcal disease; HBV, hepatitis B virus; HD, healthy donor; HIB, Haemophilus Influenzae B; HIES, hyper IgE syndrome; HSE, herpes simplex virus 1 encephalitis; MDCs, mono-derived dendritic cells; MDMs, mono-derived macrophages; MSMD, mendelian susceptibility to mycobacterial disease; MYD88, myeloid differentiation primary response protein 88; NADPH, nicotinamide adenine dinucleotide phosphate; NGS, next-generation sequencing; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PB, peripheral blood; PCR, polymerase chain reaction; PID, primary Immunedeficiency; PRRs, pattern recognition receptors; ROS, reactive oxygen species; Tfh, T follicular helper cells; TLRs, Toll-like receptors; Tregs, T regulatory cells.
References
1. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. Primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
2. McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. (2018) 14 (Suppl. 2):61. doi: 10.1186/s13223-018-0290-5
3. Bucciol G, Moens L, Bosch B, Bossuyt X, Casanova JL, Puel A, et al. Lessons learned from the study of human inborn errors of innate immunity. J Allergy Clin Immunol. (2018). doi: 10.1016/j.jaci.2018.07.013. [Epub ahead of print].
4. Alsina L, Israelsson E, Altman MC, Dang KK, Ghandil P, Israel L, et al. A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MYD88 or IRAK4. Nat Immunol. (2014) 15:1134–42. doi: 10.1038/ni.3028
5. Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev. (2011) 24:490–7. doi: 10.1128/CMR.00001-11
6. Von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi: 10.1126/science.1158298
7. Drummond RA, Franco LM, Lionakis MS. Human CARD9: a critical molecule of fungal immune surveillance. Front Immunol. (2018) 9:1836. doi: 10.3389/fimmu.2018.01836
8. Glocker EO, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. (2009) 361: 1727–35. doi: 10.1056/NEJMoa0810719
9. Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J Allergy Clin Immunol. (2015) 135:1558–68.e2. doi: 10.1016/j.jaci.2014.12.1930
10. Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl Med. (2013) 369:1704–14. doi: 10.1056/NEJMoa1208487
11. Drummond RA, Lionakis MS. Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front Cell Infect Microbiol. (2016) 6:39. doi: 10.3389/fcimb.2016.00039
12. Szymanski EP, Leung JM, Fowler CJ, Haney C, Hsu AP, Chen F, et al. Pulmonary nontuberculous mycobacterial infection. a multisystem, multigenic disease. Am J Respir Crit Care Med. (2015) 192:618–28. doi: 10.1164/rccm.201502-0387OC
13. Berod L, Stüve P, Swallow M, Arnold-Schrauf C, Kruse F, Gentilini MV, et al. MyD88 signalling in myeloid cells is sufficient to prevent chronic mycobacterial infection. Eur J Immunol. (2014) 44:1399–409. doi: 10.1002/eji.201344039
14. Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. (2010) 207:777–92. doi: 10.1084/jem.20090067
15. Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight (2016) 1:e89890. doi: 10.1172/jci.insight.89890
16. Frans G, Moens L, Schrijvers R, Wuyts G, Bouckaert B, Schaballie H, et al. PID in disguise: molecular diagnosis of IRAK-4 deficiency in an adult previously misdiagnosed with autosomal dominant hyper IgE syndrome. Clin Immunol. (2015) 35:739–44. doi: 10.1007/s10875-015-0205-x
17. Al-Shaikhly T, Ochs HD. Hyper IgE syndromes: clinical and molecular characteristics. Immunol Cell Biol. (2018). doi: 10.1111/imcb.12209. [Epub ahead of print].
18. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (2010) 89:403–25. doi: 10.1097/MD.0b013e3181fd8ec3
19. Cohen JI. Primary immunodeficiencies associated with EBV disease. Curr Top Microbiol Immunol. (2015) 390 (Pt 1):241–65. doi: 10.1007/978-3-319-22822-8_10
20. Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J Immunol. (2009) 182:851–9. doi: 10.4049/jimmunol.182.2.851
21. Chijioke O, Azzi T, Nadal D, Münz C. Innate immune responses against Epstein Barr virus infection. J Leukoc Biol. (2013) 94:1185–90. doi: 10.1189/jlb.0313173
22. Ameratunga R, Woon ST, Bryant VL, Steele R, Slade C, Leung EY, et al. Clinical implications of digenic inheritance and epistasis in primary immunodeficiency disorders. Front Immunol. (2018) 8:1965. doi: 10.3389/fimmu.2017.01965
23. Ehlayel M, de Beaucoudrey L, Fike F, Nahas SA, Feinberg J, Casanova JL, et al. Simultaneous presentation of 2 rare hereditary immunodeficiencies: IL-12 receptor beta1 deficiency and ataxia-telangiectasia. J Allergy Clin Immunol. (2008) 122:1217–9. doi: 10.1016/j.jaci.2008.07.005
Keywords: MYD88, CARD9, NGS, primary immune deficiency (PID), pyogenic infections
Citation: Chiriaco M, Di Matteo G, Conti F, Petricone D, De Luca M, Di Cesare S, Cifaldi C, De Vito R, Zoccolillo M, Serafinelli J, Poerio N, Fraziano M, Brigida I, Cardinale F, Rossi P, Aiuti A, Cancrini C and Finocchi A (2019) First Case of Patient With Two Homozygous Mutations in MYD88 and CARD9 Genes Presenting With Pyogenic Bacterial Infections, Elevated IgE, and Persistent EBV Viremia. Front. Immunol. 10:130. doi: 10.3389/fimmu.2019.00130
Received: 31 October 2018; Accepted: 16 January 2019;
Published: 14 February 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Saul Oswaldo Lugo Reyes, National Institute of Pediatrics, MexicoRuben Martinez-Barricarte, Rockefeller University, United States
Copyright © 2019 Chiriaco, Di Matteo, Conti, Petricone, De Luca, Di Cesare, Cifaldi, De Vito, Zoccolillo, Serafinelli, Poerio, Fraziano, Brigida, Cardinale, Rossi, Aiuti, Cancrini and Finocchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Chiriaco, bWFyeS5jaGlyaWFjb0BnbWFpbC5jb20=
Andrea Finocchi, YW5kcmVhLmZpbm9jY2hpQHVuaXJvbWEyLml0
†These authors share last authorship