 Roland W. Herzog
Roland W. Herzog Veronica Kuteyeva2
Veronica Kuteyeva2 Cox Terhorst
Cox Terhorst Moanaro Biswas
Moanaro Biswas- 1Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN, United States
- 2Department of Pediatrics, College of Medicine, University of Florida, Gainesville, FL, United States
- 3Division of Immunology, Beth Israel Deaconess Medical Center (BIDMC), Harvard Medical School, Boston, MA, United States
Coagulation Factor VIII (FVIII) replacement therapy in hemophilia A patients is complicated by the development of inhibitory antibodies, which often render the treatment ineffective. Previous studies demonstrated a strong correlation between induction of regulatory T cells (Treg) and tolerance to the therapeutic protein. We, therefore, set out to evaluate whether the adoptive transfer of FVIII-specific CD4+ Treg cells prevents inhibitor response to FVIII protein therapy. To this end, we first retrovirally transduced FoxP3+ into FVIII-specific CD4+ cells, which resulted in cells that stably express FoxP3, are phenotypically similar to peripherally induced Tregs and are antigen specific suppressors, as judged by in vitro assays. Upon transfer of the FVIII-specific CD4+ FoxP3+ cells into hemophilia A mice, development of inhibitory antibodies in response to administering FVIII protein was completely suppressed. Suppression was extended for 2 months, even after transferred cells were no longer detectable in the secondary lymphoid organs of recipient animals. Upon co-transfer of FoxP3+-transduced cells with the B cell depleting anti-CD20 into mice with pre-existing inhibitory antibodies to FVIII, the escalation of inhibitory antibody titers in response to subsequent FVIII protein therapy was dramatically reduced. We conclude that reprogramed FoxP3 expressing cells are capable of inducing the in vivo conversion of endogenous FVIII peripheral Tregs, which results in sustained suppression of FVIII inhibitors caused by replacement therapy in recipient hemophilia A animals.
Introduction
Hemophilia A is one of the most common X-linked recessive disorders, affecting 1 in 5,000 male births worldwide. This blood clotting disorder is caused by mutations in the factor VIII (FVIII) gene, leading to a deficiency in FVIII production. Depending on the causative mutation, FVIII production can be either completely absent or may vary up to 5–40% of normal factor activity in blood, thus manifesting as severe, moderate, or mild forms of the disorder (1). Replacement therapy with plasma derived or recombinant FVIII infusions are the standard of care for managing hemophilia A patients, and exciting strides have been made in developing gene therapy for hemophilia A and B that have the potential to provide a lifelong cure (2–5).
The most challenging issue with conventional factor replacement therapy in the treatment of hemophilia A is the development of antibodies against infused FVIII, which occur in 25–30% of severe hemophilia A patients. Inhibitor prevalence is influenced by severity of the disorder and is often associated with large deletions/inversions in the F8 gene, which results in the lack of FVIII formation (6). Inhibitors render factor replacement therapy ineffective and can present a high risk of morbidity and mortality (7). Immune tolerance induction (ITI) for the eradication of inhibitors via frequent and high dose exposure to FVIII concentrates for a prolonged period is expensive and not always successful, especially in severe hemophilic patients (8). Mechanisms for tolerance induction by ITI are not clearly known but may include T effector cell (Teff) exhaustion/anergy, inhibition of FVIII-specific memory B-cell differentiation, or induction of regulatory T cells (Tregs) (9, 10). Conversely, there is also very little information on the immune interactions that lead to the development of inhibitors, although it has been described to be a T helper dependent process involving antigen uptake and presentation that requires the co-operation of multiple macrophage, dendritic cell or B cell subsets of antigen presenting cells (APC) (11–15).
Multiple studies have demonstrated that tolerance to replacement FVIII protein is strongly modulated by Tregs (16, 17). Co-administration of FVIII with drugs such as sirolimus (rapamycin), alone or in combination with cytokines such as IL-10 or Flt3L have been shown to induce and/or expand CD4+CD25+FoxP3+ Tregs, either through specific deletion of CD4+ Teff cells which are more sensitive to mTOR inhibition, or selective expansion of plasmacytoid dendritic cells (pDCs) (18–20). Similar results have been obtained by treatment with IL-2/anti-IL-2 complexes or oral anti-CD3 treatment (21–24).
Tregs can be naturally occurring (central or thymic), with specificity mainly toward endogenous “self” antigens, or peripherally derived (extra-thymically induced), with specificity to exogenously introduced antigens (25). The lack of endogenous FVIII protein expression in severe hemophilia A patients with large mutations in the F8 gene results in ineffective FVIII Treg induction and Teff escape during thymic selection, reflected in the higher rate of inhibitor development for these patients. Therefore, there is great interest in re-establishing tolerance to FVIII in these cases.
Cellular therapy with Tregs, either freshly isolated or ex vivo expanded, is a promising approach for tolerance induction, as has been demonstrated in several clinical trials for autoimmune disorders and in transplant studies (26–29). While autologous Tregs of a polyclonal specificity are effective, as observed in a study in hemophilia A mice (30), it is anticipated that antigen-specific Tregs would be more effective at much lower frequencies, with a significantly reduced risk for off-target suppression (31). In this study, we hypothesized that forced FoxP3 expression in conventional/effector CD4+ T cells (Tconv/Teff) from hemophilia A mice that were immunized with FVIII would yield an enriched pool of FVIII specific suppressor Treg-like cells. We examined the phenotype of these cells, and stability of FoxP3 expression over time, and were able to suggest a potential role for lasting suppression by a mechanism of conversion of Teff cells into antigen-specific endogenous Tregs. Adoptively transferred FoxP3 expressing cells from FVIII immunized mice (FoxP3FVIII) were able to successfully prevent inhibitor formation in previously untreated hemophilia A mice and, when applied as combination therapy with a B-cell depleting antibody (anti-mCD20), were able to reverse established inhibitors to FVIII. This study therefore underlines the potential of gene-engineered cells with Treg function to provide specific and lasting suppression. This cell-based tolerance approach can potentially act as stand-alone therapy or can complement conventional ITI to re-establish tolerance to FVIII replacement therapy.
Methods
Mice
All wt animals used in the experiments were 8–10-week-old male mice of the BALB/c [H-2d] background, which were purchased from Jackson Laboratories (Bar Harbor, ME). DO11.10-tg Rag2−/− mice with a transgenic T cell receptor specific for the amino acid sequence 323–339 of chicken ovalbumin (OVA), presented by MHCII I-Ad, were originally obtained from Taconic (Hudson, NY). Hemophilia A mice with a deletion in exon 16 of the F8 gene (BALB/c F8e16−/−) were originally provided by Dr. David Lillicrap (Queens, Ontario, Canada).
Plasmids and Transduction
MIGR-mFoxP3 was a gift from Dan Littman (Addgene plasmid # 24067). MIGR1 (IRES-GFP) was a gift from Warren Pear (Addgene plasmid # 27490). Both plasmids are based on the murine stem cell virus (MSCV) expression system. Transfer plasmids were introduced into the PlatE ecotropic retroviral packaging cell line (Cell Biolabs Inc, San Diego, CA) using the Viafect transfection reagent (Promega, Madison, WI) and supernatants were collected after 48 h. CD4+CD25− Tconv cells were magnetically isolated (Miltenyi Biotec) and pre-activated for 48 h with a 2:1 ratio of CD3/28 mouse activator beads (Dynabeads, Invitrogen) to cells. Cells were cultured in RPMI-1640 media (Life Technologies) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 10,000 IU/ml penicillin, 10 mg/ml streptomycin, 1X GlutaMAX-1, 1 mmol/l sodium pyruvate, 10 mmol/l HEPES, 1X non-essential amino acids and 10 μmol/l 2-mercaptoethanol. Recombinant hIL-2 (Proleukin/aldesleukin, Prometheus Therapeutics and Diagnostics, San Diego, CA) was added at a concentration of 200 IU/ml. Activated Teff cells were retrovirally transduced by spinoculation at 1,200 g for 90 min into retronectin (Takara) coated plates. GFP+ cells, representing FoxP3 (MIGR-mFoxP3 IRES-GFP) or GFP only (MIGR IRES-GFP) transduced populations of CD4+ T cells, were purified using the FACSAria II cell sorter (BD Biosciences).
Reagents
Purified CD16/32 (Fc Block), CD3 (PerCP-Cy5.5), GITR (V500) antibodies were from BD Biosciences (San Jose, CA); CD39 (eFluor450), CTLA-4 (PE), FoxP3 (eFluor660) antibodies were purchased from eBioscience (San Diego, CA); CD25 (BV605), TNFR2 (PE), CD127 (PerCP-Cy5.5), CD62L (APC/Cy7), anti-GFP (A488), CD4 (BV421), CD4 (A700) antibodies were from Biolegend (San Diego, CA). Mouse neuropilin-1 antibody (PE) was from R&D Systems (Minneapolis, MN). Mouse CD4+ T cell isolation kit, CD4+CD25+ regulatory T cell isolation kits were from Miltenyi Biotec (Auburn, CA). OVA peptide (323–339) was synthesized by Anaspec (Fremont, CA). Recombinant human B domain deleted (BDD) FVIII (Xyntha) was from Pfizer (New York, NY). FVIII deficient plasma was from Haematologic Technologies (Essex Junction, VT). Anti-mCD20 IgG2a subtype (clone 2B8) was a kind gift from Biogen (Cambridge, MA).
Analysis of Plasma Samples
Plasma samples were collected by retro-orbital eye bleed into 0.38% sodium citrate buffer and analyzed using a modified activated partial thromboplastin time assay (aPTT). Inhibitory antibodies to FVIII were measured by Bethesda assay as described (32). One Bethesda unit (BU) is defined as the reciprocal of the dilution of test plasma at which 50% of FVIII or FIX activity is inhibited. Measurements were carried out in a Diagnostica Stago STart Hemostasis Analyzer (Parsippany, NJ). Enzyme-linked immunosorbent assay (ELISA)-based measurements of IgG1 antibodies to FVIII were carried out as described (32).
Adoptive Transfer Studies, Inhibitor Establishment, or Reversal
To generate a source of CD4+ T cells that were enriched for specificity toward FVIII, BALB/c F8e16−/− hemophilia A (HA) mice were subcutaneously immunized with 1IU FVIII/mouse delivered in adjuvant (Sigma Adjuvant System oil, Sigma-Aldrich, St. Louis, MO). A booster immunization was administered 2 weeks later. Immunized mice developed high titer antibodies (~40 BU/ml) to FVIII. 2 × 106 FoxP3 transduced cells from either FVIII immunized mice (FoxP3FVIII) or naïve animals (FoxP3naive) were adoptively transferred into recipient HA mice (n = 5–7). For prevention studies, recipient BALB/c F8e16−/− HA mice received either FoxP3FVIII, FoxP3naive, GFPFVIII, or nothing. Mice then received 1.5 IU of BDD-FVIII by weekly tail-vein injections for 2 months. Plasma samples were analyzed at 1 and 2 months for inhibitor development. For reversal studies, inhibitors were established by 1.5 IU BDD-FVIII injections for 1 month. Mice then received either FoxP3FVIII cells, mCD20 IgG2a antibody (IV, 1, and 3 weeks after inhibitor establishment) or a combination of FoxP3FVIII cells and mCD20 IgG2a antibody, following which, weekly BDD-FVIII injections were continued. Plasma samples were analyzed at 1, 2, 3- and 4-months post-inhibitor establishment.
Phenotypic Characterization of FoxP3 Transduced Cells
FoxP3 or GFP transduced cells were evaluated for expression of Treg associated markers. Since retroviral transduction requires pre-activation of CD4+ T cells, which can upregulate several phenotypic markers in a non-specific manner, we attempted to analyze cells under physiological conditions. For this, 2 × 106 FoxP3 or GFP transduced cells were adoptively transferred into wt BALB/c mice. Spleens were harvested after 48 h and GFP+ cells were identified and phenotyped. Endogenous FoxP3+ Tregs from recipient wt BALB/c mice were used as a standard to compare the difference in expression of various markers. For intracellular staining, cells were pre-fixed in 2% paraformaldehyde to retain GFP expression and intracellular staining was performed using the FoxP3 staining kit (eBioscience, San Diego, CA). Briefly, ~1 × 106 cells in a volume of 100 μl were blocked with CD16/32 for 15 min and surface labeled with antibodies at recommended concentrations. Fixation-permeabilization carried out as required and intracellular antibodies were added. Samples were acquired on the Fortessa flow cytometer (BD Biosciences) and analyzed using FCS express 6 (DeNovo Software, Los Angeles, CA).
In vitro Suppression Assay
To assay for in vitro suppression of polyclonally activated cells, CD4+CD25− responder cells from spleens of DO11.10-tg Rag2−/− mice were isolated, labeled with 3–5 μmol/l CellTrace Violet (Invitrogen, Carlsbad, CA) and cultured with CD4− total splenocytes. DO11.10-tg Rag2−/− GFP+ FoxP3 transduced CD4+ T cells were added at various Treg: T responder ratios and cells were cultured for 72 h at 37°C. Dilution of the CellTrace Violet label in GFP− proliferating responder cells (Teff) was quantified. Proliferation was determined by quantifying CellTrace Violet fluorescence intensity relative to a parent population of unstimulated responder cells (0% proliferation) and stimulated cells incubated without Treg (100% proliferation). Percentage of CD4+ responder T cell proliferation was also determined using ModFit LT analysis on FCS Express 6.
In vivo Conversion Assay
FACS-purified DO11.10-tg Rag2−/− GFP+ FoxP3 transduced CD4+ T cells were labeled with 3–5 μmol/l CellTrace Violet (Invitrogen, Carlsbad, CA) and adoptively transferred into cohorts of recipient DO11.10-tg Rag2−/− mice (n = 4) that naturally lack endogenous Tregs. Recipient mice subsequently received 50 μg OVA323−339 peptide, 3x/week for 2 weeks, delivered via i.p. injection. Control mice received no treatment. One group of mice received only FoxP3OVA cells, while another received only OVA323−339 peptide and no cells. The 4th group received both FoxP3OVA cells, as well as OVA323−339 peptide injections. In vivo conversion of endogenous OVA specific CD4+ T effectors into FoxP3+ Tregs was assessed at the end of the 2-week period. Induced endogenous Tregs were distinguished from adoptively transferred donor cells by the lack of GFP and CellTrace Violet label.
Statistical Analysis
Statistical significance was determined using either student's T-test, 1-way or 2-way ANOVA with GraphPad Prism 7 software (La Jolla, CA). Values at P < 0.05 were deemed significant and indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001. For some samples, normality was assessed with the Shapiro-Wilk normality test. Non-parametric analyses were carried out using the Kruskal Wallis test. Difference in proportions of mice that developed inhibitors was determined using Fisher's exact test.
Results
Stably Transduced FoxP3+ CD4+ T Cells Acquire in vitro Suppressor Functions
Flow cytometric analysis of retrovirally transduced murine CD4+CD25− Tconv cells revealed high expression of FoxP3, which directly correlated with GFP+ cells (Figure 1A). Transduction efficacies ranged from 25–60%. CD4+CD25− Tconv cells transduced with the control vector only expressed GFP, without concurrent FoxP3 expression (Figure 1A). FoxP3 transduced CD4+ T cells were shown to undergo antigen specific proliferation without loss of FoxP3 expression in vivo. For this, we used DO11.10 Rag2−/− mice, which lack mature B or T lymphocytes or endogenous Tregs but are transgenic for CD4+ T cells with OVA323−339 specificity. Donor FoxP3 transduced CD4+CD25− cells from DO11.10 Rag2−/− mice (FoxP3OVA), when adoptively transferred into recipient wt BALB/c mice and challenged with OVA323−339 peptide, underwent robust proliferation, as observed by dilution of the cell dye label, without compromise in FoxP3 expression (Figure 1B). This established that under non-inflammatory conditions, FoxP3 transduced cells can proliferate in an antigen dependent manner while stably retaining FoxP3 expression.
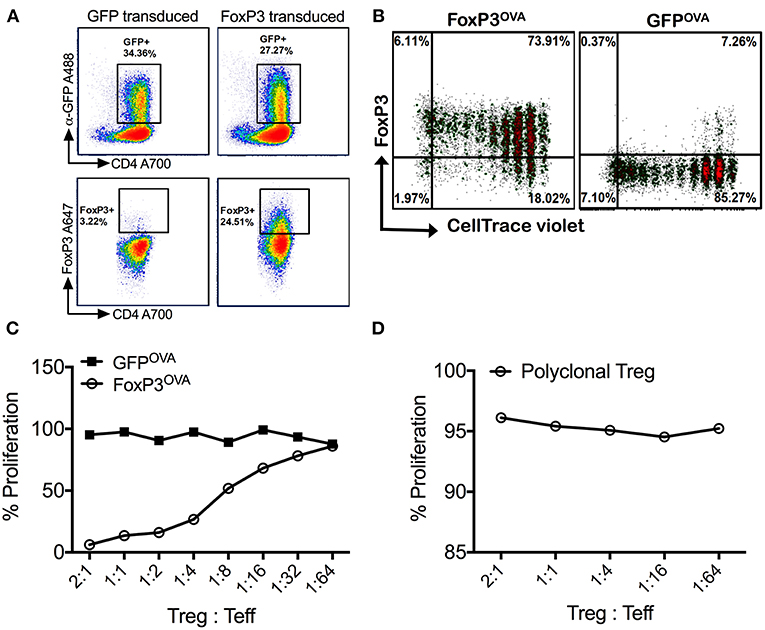
Figure 1. Retrovirally transduced cells stably express FoxP3 and are suppressive in vitro. (A) Isolated splenic murine CD4+CD25− cells transduced with the MIGR-mFoxP3 vector strongly expressed GFP (upper panel), which correlated with intracellular FoxP3 expression (lower panel). Cells transduced with the control vector MIGR1 expressed only GFP. (B) FoxP3 transduced cells from DO11.10 Rag2−/− mice were labeled with CellTrace Violet, adoptively transferred into wt BALB/c mice and recipients were injected with 100μg OVA323−339 peptide by the i.p. route. Spleens were recovered after 3 days. Robust proliferation of donor transgenic cells (FoxP3OVA) without compromise in FoxP3 expression was observed. Control DO11.10+ cells (GFPOVA) proliferated in response to OVA323−339 peptide administration but did not express FoxP3. (C) FoxP3OVA cells strongly suppressed the proliferation of CellTrace Violet labeled DO11.10+ CD4+CD25− responder cells cultured with CD4− total splenocytes and stimulated with OVA323−339 peptide. Control GFP transduced cells (GFPOVA) or (D) freshly isolated tTregs of a polyclonal specificity did not suppress the proliferation of OVA323−339 stimulated responder T cells. Data is a single representative of at least 2 independent experiments.
FoxP3 transduced DO11.10 CD4+ T cells were also shown to be functionally suppressive in an antigen specific manner in vitro. OVA323−339 peptide specific proliferation of responder DO11.10 CD4+CD25− T cells was potently suppressed on addition of FoxP3 transduced cells of the same specificity (FoxP3OVA) (Figure 1C). Suppression was still strongly evident at a Treg:Teff (responder) cell ratio of 1:16, indicating that FoxP3 transduction alone is capable of producing cells with Treg function. Control GFP transduced OVA specific cells (GFPOVA) did not suppress at any ratio (Figure 1C). To test if the observed suppression was antigen specific, the same assay was carried out with freshly isolated thymic Tregs (tTregs, Figure 1D). Because non-specific polyclonal tTregs failed to suppress the proliferation of OVA323−339 stimulated Teff cells, we conclude that suppression by FoxP3 transduced cells in response to antigen is TCR specific, and not a result of a bystander effect.
In vivo Treg Phenotype of Re-programmed FoxP3+ CD4+ T Cells
To characterize the phenotype of the reprogrammed cells under physiological conditions, 2 × 106 FoxP3 or GFP transduced cells were adoptively transferred into wt BALB/c mice and re-isolated from spleens after 48 h. FoxP3 transduced CD4+ T cells acquired a Treg-like phenotype, with high expression of CD25 and GITR, low expression of IL-7Ra chain (CD127), and FoxP3 expression levels that were higher than endogenous tTregs (Figures 2A,B). Expression of CD39 and CTLA-4 was higher than in CD4+ T cells transduced with GFP only, although expression levels did not reach those observed on endogenous tTregs. It has previously been observed that CTLA-4 expression is independent of FoxP3 (33, 34), which might explain the lack of correlation with FoxP3 overexpression. Furthermore, we had earlier shown that CTLA-4 is highly expressed in ex vivo expanded tTregs, which is a function of prolonged CD3/CD28 activation of these cells (30). FoxP3 transduced CD4+ T cells also lacked the Neuropilin-1 marker, which is generally associated with centrally derived tTregs in mice, (35, 36), thus confirming their induced nature (Figures 2A,B). Thus, reprogrammed FoxP3+ CD4+ T cells maintain a Treg phenotype in vivo and in vitro.
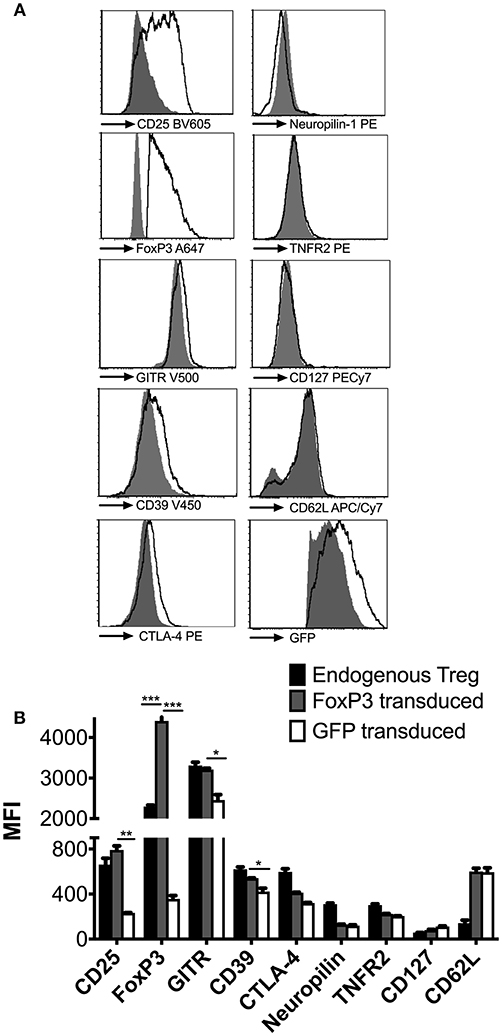
Figure 2. Phenotypic analysis of reprogrammed FoxP3 expressing Treg like cells. (A) FoxP3 transduced cells were adoptively transferred into wt BALB/c mice and re-isolated 2 days later to characterize them under in vivo conditions (empty histograms). Control GFP vector transduced cells (filled histograms) were also phenotyped for comparison. Expression levels of CD25, FoxP3, GITR, CD39, CTLA-4, NRP-1, TNFR2, CD127, CD62L, and GFP were quantified. (B) Median fluorescent intensity (MFI) of the above markers were compared between endogenous tTreg (black bars), FoxP3 transduced cells (gray bars) and GFP transduced cells (white bars). Data is representative of 2 samples and at least 2 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
FVIII-Specific Reprogrammed FoxP3+ Treg Cells Prevent Inhibitor Formation in Hemophilia A Mice
To determine whether engineered FoxP3+ CD4+ T cells could suppress the development of inhibitors to FVIII, BALB/c F8e16−/− mice with no intrinsic FVIII expression were adoptively transferred with 2 × 106 FoxP3 transduced cells. In order to simulate protein replacement therapy, mice were treated with 8 weekly i.v. injections with 1.5 IU BDD-FVIII protein (Figure 3A). Donor FoxP3+ cells were derived from mice immunized with BDD-FVIII in adjuvant, in order to enrich for FVIII specific Teff cells that could be reprogrammed into engineered Tregs (FoxP3FVIII). FoxP3 transduction resulted in 50–60% GFP+ cells, which were further purified by cell sorting (Supplementary Figure 1). As shown in Figure 3B, FoxP3 transduced cells completely prevented the formation of functional inhibitory antibodies in response to repeated administrations of BDD-FVIII. Tolerance was sustained for 2 months of BDD-FVIII exposure (Month 1: 0.04 ± 0.02, Month 2: 0.95 ± 0.49 BU/ml). Control mice, which did not receive FoxP3 transduced cells, developed high inhibitor titers (Month 1: 24.45 ± 7.8, Month 2: 160.59 ± 4.1 BU/ml). Similarly, CD4+ from FVIII immunized mice that were transduced with control GFP vector (GFPFVIII) were unable to mediate tolerance (Month 1: 3.0 ± 1.9, Month 2: 112.34 ± 43.1 BU/ml).
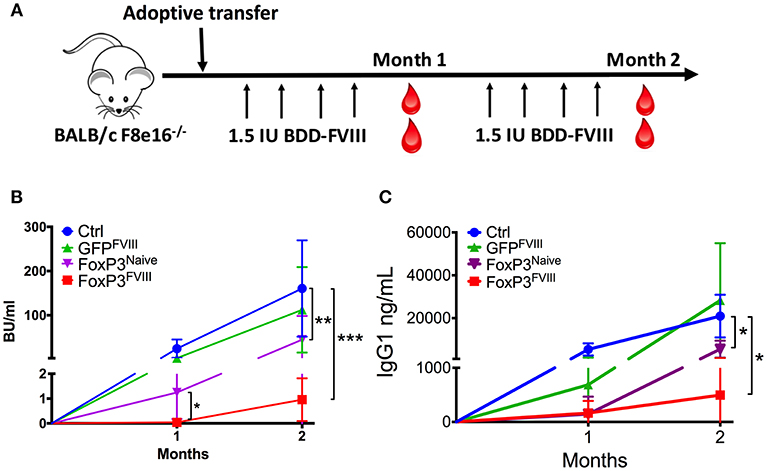
Figure 3. Prevention of inhibitor formation by FoxP3FVIII cells. (A) Experimental timeline. BALB/c F8e16−/− mice were transplanted with 2 × 106 FoxP3 transduced cells from FVIII immunized mice (FoxP3FVIII). This was immediately followed by weekly intravenous injections with BDD-FVIII. Blood was collected at Months 1 and 2 for inhibitor assessment. (B) Inhibitor titers (BU/ml) were assessed at specific time points by the Bethesda assay. Experimental groups tested were control mice that received no treatment, FoxP3 transduced cells from naïve mice (FoxP3Naïve), GFP transduced cells from FVIII immunized mice (GFPFVIII), and FoxP3 transduced cells from FVIII immunized mice (FoxP3FVIII). (C) Anti-FVIII IgG1 titers (ng/ml) at Months 1 and 2 were tested by ELISA. Data are average ±SD. Statistically significant differences are indicated for each time point. *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that enrichment of antigen specific cells in the FoxP3FVIII population contributed favorably to suppression, one group of mice received FoxP3 transduced cells from naïve mice that had not been pre-exposed to FVIII. These FoxP3Naive cells were suppressive at the 1-month time point (1.28±0.02 BU/ml), probably due to a non-specific regulatory effect at the time of BDD-FVIII introduction. However, this suppressive effect by FoxP3Naive cells was significantly lower at the 1-month time point as compared to FoxP3FVIII recipient mice (p = 0.018). The proportion of animals that developed inhibitors was also significantly greater at the 1-month time point in mice that received FoxP3Naive cells (60% vs. 0%, p = 0.045). Furthermore, the observed tolerogenic effect conferred by the FoxP3Naive cell population was not long-lasting, and high-titer inhibitors (>5BU/ml) developed in 80% of the mice by Month 2 (44.8 ± 23 BU/ml), which was in contrast to the sustained tolerance exerted by FoxP3FVIII cells (Month 2: 0.95 ± 0.49 BU/ml). Therefore, using FVIII experienced T cells for FoxP3 gene transfer resulted in more consistent and durable suppression of inhibitors upon transplant. Similar findings were obtained for FVIII-specific IgG1 titers (Figure 3C).
Taken together, we conclude that upon the adoptive transfer of re-programmed FoxP3FVIII Treg cells, anti-FVIII inhibitor formation is stably suppressed.
Combination Therapy With Reprogrammed FoxP3FVIII Cells and Anti-mCD20 Reduce Pre-existing Inhibitors in Hemophilia A Mice
To further evaluate the pre-clinical capability of engineered FoxP3FVIII cells we transferred these cells in combination with B-cell-depleting anti-mCD20 antibodies in Hemophilia A mice which contained previously induced FVIII inhibitors. We previously showed that a short course treatment with anti-mCD20 resulted in a rapid decline of inhibitors, although this effect was transient, and inhibitors rapidly rebounded to their initial titers (37). We therefore tested the ability of FoxP3FVIII cells to tolerize mice with existing inhibitor titers, alone or in combination with anti-mCD20 B cell targeting therapy. As shown in Figure 4A, BALB/c F8e16−/− mice (n = 5–7) received 4 weekly i.v. injections of BDD-FVIII to allow inhibitor development. Mice developed average inhibitor titers of 5 BU/ml, which are considered high titer. This was followed by treatment with either FoxP3FVIII cells, anti-mCD20 (2 i.v. injections spaced at a 3-week interval), or a combination treatment with anti-mCD20, followed immediately by FoxP3FVIII adoptive transfer. Mice continued to receive BDD-FVIII injections following treatment.
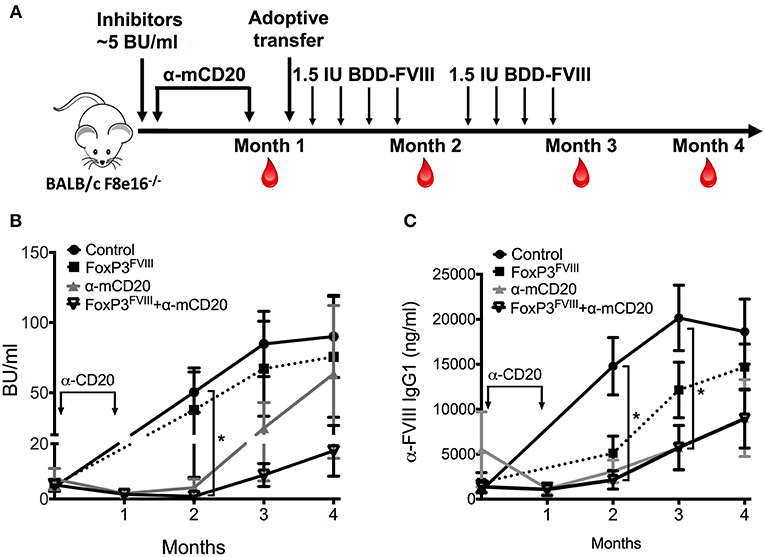
Figure 4. Reduction of pre-existing inhibitors by combination therapy with FoxP3FVIII cells and anti-mCD20. (A) Experimental timeline. Inhibitors were established in BALB/c F8e16−/− mice by 4 weekly injections of BDD-FVIII. Mice received 2 IV injections of anti-mCD20 spaced 3 weeks apart (α-mCD20 group) following which, inhibitor titers were quantified (Month 1). Another group received 2 × 106 FoxP3FVIII cells 1 week after the last anti-mCD20 injection (FoxP3 FVIII + α-mCD20 group). Control groups received only FoxP3 FVIII cells or no treatment (Control). BDD-FVIII administrations were continued for another 2 months following treatment. Blood was collected on Months 2, 3 and 4, respectively. (B) Inhibitor titers (BU/ml) measured over time from control and treated mice that received α-mCD20 only, FoxP3FVIII cells only, or a combination of α-mCD20 + FoxP3 FVIII cells. (C) Anti-FVIII IgG1 titers (ng/ml) over time were tested by ELISA. Data are average ±SD. Statistically significant differences are indicated for each time point. *P < 0.05, **P < 0.01, ***P < 0.001.
Mice that received FoxP3FVIII cells developed high titer inhibitors that were comparable to control mice that received only BDD-FVIII injections. Immediately following anti-mCD20 treatment (Month 1), both experimental groups (anti-mCD20 and anti-mCD20+FoxP3FVIII group) saw a decline in inhibitor titers (1.67–1.98 BU/ml), which was sustained for another month after resuming BDD-FVIII injections (Month 2, anti-mCD20 group: 4.09±3.12 BU/ml, anti-mCD20+FoxP3FVIII group: 0.948±0.42 BU/ml) (Figure 4B). However, tolerance was short-lived, and titers escalated when BDD-FVIII injections were continued at the Month 3 timepoint (anti-mCD20 group: 24.77±18.24 BU/ml, anti-mCD20+FoxP3FVIII group: 8.65±4.15 BU/ml). As observed, mice that received the treatment combination of anti-mCD20+FoxP3FVIII had lower inhibitor titers as compared to mice that received anti-mCD20 only. This difference was more obvious at the Month 4 timepoint (anti-mCD20 group: 116.447±104.784 BU/ml, anti-mCD20+FoxP3FVIII group: 22.27 ± 8.015 BU/ml). It therefore appears that whilst anti-mCD20 treatment is effective at short term inhibitor reduction, the re-establishment of inhibitors in response to continued BDD-FVIII injections is more effectively controlled by anti-mCD20+FoxP3FVIII treatment, hinting at a more sustained tolerance mediated by the transplanted engineered Tregs. IgG1 ELISA titers (Figure 4C) were unable to completely recapitulate this difference in functional antibody titers, but this could be attributed to the development of non-neutralizing IgG1 antibodies, which do not interfere with functional activity. Quantification of total B cell, memory B cell, and plasma cell (Supplementary Figure 2) populations from spleen and bone marrow of anti-mCD20 treated mice after the Month 4 time point confirmed that B cell depletion was transient and the decrease in inhibitor titers at latter time points was not due to generalized immunosuppression. We have earlier shown that immunosuppression mediated by anti-mCD20 is transient and B cell populations in various immune compartments completely recover within 2 months after depletion, while frequencies of T cells remain unaffected by anti-mCD20 treatment [(37); Supplementary Figure 1].
Conversion of Teff Into Tregs by Reprogrammed FoxP3 Cells as a Putative Mechanism of Lasting Tolerance
Previous observations by us and others indicated that Tregs that are adoptively transferred into immuno-competent recipients are only transiently detectable for a period of 2–3 weeks (21, 30, 38). Not surprisingly, when we tested a subset of the experimental animals in Figure 3 one-month post-transfer for the presence of donor-derived FoxP3 Tregs, using GFP as an identifier, we were unable to detect any donor cells (data not shown). We therefore looked at the possibility of in vivo conversion of host endogenous antigen specific Teff cells into Tregs, which would account for a more long-lasting suppression of FVIII inhibitors. For this, we again used the surrogate CD4+ T cell epitope system recognizing OVA323−339. We showed that adoptive transfer of CellTrace Violet+FoxP3-GFP+ DO11.10+ CD4+ T cells into recipient animals of the same strain was capable of inducing antigen specific Tregs in the recipient mice, which was dependent on the presence of antigen (50 μg OVA323−339 peptide injected 3 × /week × 2 weeks) (Figure 5).
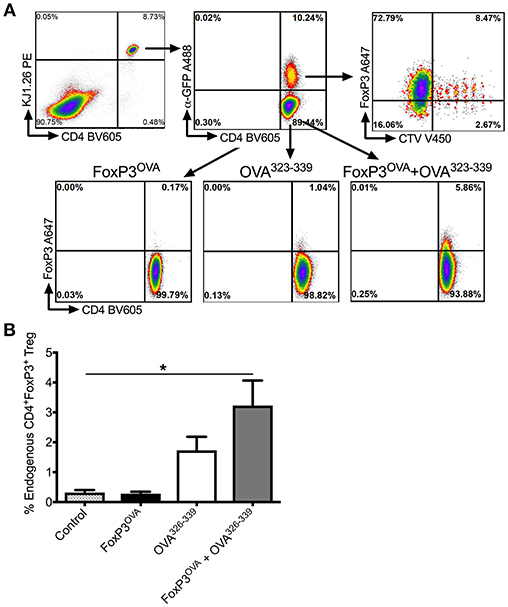
Figure 5. In vivo conversion into antigen specific Tregs by transplanted FoxP3 expressing cells. (A) Representative dot plots depicting gating scheme. Transplanted FoxP3OVA cells are KJ1.26+CD4+FoxP3+GFP+CTV+ and thus distinguishable from endogenous de novo induced Tregs that are KJ1.26+CD4+FoxP3+GFP−CTV−. OVA specific FoxP3+ Treg induction in DO11.10 Rag2−/− mice is compared between either control group that received no treatment, mice that received FoxP3OVA transplanted cells, administration of OVA323−339 peptide only, and mice that received FoxP3OVA cells and OVA323−339 peptide, delivered i.p. for 2 weeks. (B) Bar graphs of induced DO11.10+FoxP3+ Tregs in treated DO11.10 Rag2−/− mice, indicated as a percentage of total CD4+ T cells. Mice received no treatment, FoxP3OVA cells, OVA323−339 peptide, or FoxP3OVA cells + OVA323−339 peptide. Data are average ±SD of at least 4 animals per group and are a single representative of 2 independent experiments. Statistically significant differences are indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
As DO11.10 Rag−/− mice lack endogenous FoxP3+ CD4+ Tregs, were identified with the OVA specific TCR DO11.10 antibody (KJ1.26) and were distinguished from donor DO11.10+ FoxP3 transduced Tregs, which were dually positive for GFP and the CellTrace Violet label (Figure 5A). As shown in Figure 5B, transplanting only donor DO11.10+ FoxP3 transduced Tregs (FoxP3OVA) did not induce conversion of endogenous CD4+ Teffs into OVA specific Tregs in recipient mice spleens (% DO11.10+ Tregs: 0.28 ± 0.06 of total CD4+ T cells). OVA323−339 administration without FoxP3OVA transfer yielded a small percentage of OVA specific Tregs (% DO11.10+ Tregs: 1.73 ± 0.45). These numbers were however, significantly higher in spleens of mice that received FoxP3OVA transfer combined with successive OVA323−339 injections (3.228±0.83). Therefore, administration of donor antigen specific FoxP3+ cells induced the emergence of endogenous Tregs of the same specificity in recipients, ensuring the persistence of tolerance.
Discussion
Regulatory T cells are mainly specific for autologous antigens, thus maintaining self-tolerance (39). During autoimmune or inflammatory conditions, the ratio of Tregs to immune effector T cells is perturbed, and an immune response is mounted against self (40). Ex vivo expansion of thymic Tregs can increase the population of self-antigen specific Tregs. However, for genetic disorders like severe hemophilia A where significant mutations do not allow expression of the protein of interest, central tolerance often cannot be established resulting in very few antigen specific Tregs.
While the formation of inhibitors during clotting factor replacement therapy has been shown to be dependent on T helper cells, T cell help can in turn be suppressed by Tregs (41), which are instrumental in peripheral tolerance induction. Tregs can also directly interact with and regulate B cells and plasma cells in various niches (42, 43), although this has not yet been demonstrated for hemophilia. Extra thymically induced peripheral or induced Tregs can be FoxP3+, LAP+ (Th3), or LAG3+ (TR1). Of these subsets, FoxP3+ Tregs have been the most extensively studied in hemophilia (44, 45). However, this does not discount the role of other Treg subtypes, as LAP+ Tregs have been shown to be involved in mediating tolerance to FVIII by oral delivery of antigen (46).
For this study, we reprogrammed antigen enriched Teff cells by ectopic FoxP3 expression, to generate a large pool of engineered cells with Treg function. FoxP3 expression alone has been shown to strongly regulate Treg development and function (47–49) by controlling a transcriptional network of target genes that are involved in promoting the suppressive phenotype among other immunological and non-immunological functions (50–53), as we observed in our studies. FoxP3 transduced cells expressed many Treg associated markers and were strongly suppressive both in vitro and in vivo. There is valid concern that reprogrammed FoxP3 expressing cells may revert into Th2 cells, or pathogenic Th17 cells in vivo (54–56). While this possibility exists, given the plastic nature of T-lineage cells (57, 58), it may depend on attenuation of FoxP3 expression, or may require an inflammatory trigger, which is more common with autoimmune disease. FoxP3 tTregs have also been shown to be remarkably stable in vivo and it is possible that this stability extends to peripherally induced Tregs (59). We were able to confirm stable FoxP3 expression in proliferating engineered Tregs in vivo after adoptive transfer.
Some pre-clinical studies have been carried out using adoptive therapy with FoxP3 transduced cells, particularly in autoimmune disease and in transplant rejection (33, 60–62). In a few of these studies, antigen specificity has been re-directed by the addition of either a TCR of a single specificity or a chimeric antigen receptor (CAR) molecule upstream of the FoxP3 gene construct (63–65). All of these studies showed efficacy. Using transgenic mice, Jaeckel et al. were able to report that FoxP3 transduction of naïve polyclonal CD4+ T cells was not completely effective in suppressing established type I diabetes in mice. Instead, antigen specific FoxP3 transduced cells were highly effective at reversing recent onset diabetes (66). This complements other studies that suggest that antigen specific Treg are superior to their polyclonal counterparts (67–69). Indeed, our data implies that FoxP3 transduction of a population of Teffs that are antigen experienced for FVIII and thus enriched for antigen specificity leads to sustained tolerance. The lack of an MHCII tetramer system to identify and isolate FVIII specific Th2 cells in BALB/c mice limits the scope of this study to ascertain the effective minimum dose of antigen specific FoxP3+ cells required to suppress an inhibitor response in the hemophilia A experimental model.
For clinical application, the ratios of transduced FVIII specific Tregs and untransduced Teffs may vary between patients. However, it is expected that patients with a stronger Teff response would in turn generate more FVIII-specific Tregs. FoxP3 transduction of a mixed population of FVIII specific and non-specific cells may also result in the generation of unwanted Tregs against non-specific antigens. This can be resolved by identifying antigen specific Teffs upon short term FVIII specific stimulation in vitro. Rapid cell-surface upregulation of activation markers such as CD154 (CD40L) and CD137 (41BB) on antigen-specific triggering (70, 71) allows for detection and isolation of activated cells by cell-sorting before FoxP3 transduction. Ex vivo expansion can increase the pool of reprogramed antigen-specific Tregs before infusion.
Among other limitations, the use of the retroviral LTR promoter system for FoxP3 expression has been shown to lead to fluctuations in FoxP3 expression in other studies, depending on the activation status of the transduced cell (33). However, ours is only a proof-of concept study that can be easily adapted to the lentiviral delivery system, as has successfully been shown in PBMC from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome patients, which was able to generate a large pool of FoxP3 expressing Tregs, phenotypically and functionally identical to Tregs from healthy donors (72).
Our in vivo studies reveal that a single adoptive transfer with FoxP3FVIII Tregs is able to completely prevent the development of inhibitors, using an antigen dose of 1.5IU of FVIII administered weekly. This was not sufficient to reverse established inhibitors, however, and FoxP3FVIII treatment alone was ineffective to prevent inhibitor escalation in response to continuous FVIII administration. We have observed this in previous studies, where ex vivo expanded polyclonal Tregs were unable to completely reverse pre-existing inhibitors to FVIII, although they did halt any further increases in inhibitor titers (30). This is understandable since although it has been shown that Tregs can interact with B cells and plasma cells, it is unclear whether this interaction can lead to a suppressive or cytotoxic outcome. Previously, we have shown that targeting both the B and T cellular compartments by combination therapy with the murine equivalent of rituximab (anti-CD20) and rapamycin (sirolimus) can have a positive outcome in inhibitor reversal (37), which was corroborated in a recent study on a hemophilia B patient with inhibitors (73). On applying this combination therapy to this study, we observed that FoxP3FVIII and anti-CD20 together were superior to either treatment alone and that Treg therapy appeared to prolong the delay in inhibitor re-emergence.
Studies have shown that both murine and human Treg can initiate infectious tolerance, transmitting suppressive capacity from the Treg to the target cell (74, 75). Conversion of CD4+CD25−FoxP3− Teff cells into Tregs may occur under several conditions, where a number of triggers may induce FoxP3 expression. These may include IL-2 deprivation by Tregs, which reduces the availability of this cytokine for Teff proliferation (76), or the production of suppressive cytokines or receptors by Tregs, such as IL-10 (77), IL-35 (78), TGF-B (79, 80), or CTLA-4 (81). This is also observed in tumors, where tumor antigen specific CD4+ T cells in the tumor draining lymph node (TDLN) are activated, but diverted either into anergy or Treg formation, which is enhanced by already present Tregs in the TDLN (82). The complete mechanism is not understood, but strongly depends on Treg: DC interactions in both a contact dependent and independent manner, inducing suboptimal presentation of antigen (83–85). This is usually enhanced by immunosuppressants like rapamycin, which inhibits the mTOR pathway, by interfering with T cell co-stimulation, activation and proliferation (86). We and others have shown that infused Tregs can generate a similar suppressive microenvironment that can promote antigen presentation in a tolerogenic manner, thus inducing the conversion of endogenous Teff cells into antigen specific Tregs (30, 80, 87). In this study, we were able to extend our earlier findings to show that reprogramed FoxP3 expressing cells are capable of inducing de novo Tregs of a desired antigen specificity in host animals. We speculate that co-administration of FoxP3FVIII Tregs and FVIII antigen initially generates an immunosuppressive environment due to the high proportion of infused non-specific FoxP3 Tregs. Moreover, the small population of antigen specific Tregs can directly interact with DC to enhance sub-optimal presentation, thus diverting antigen specific Teff cells into Tregs. Tolerance to FVIII protein administration is thus due to a combination of various mechanisms: initial non-specific suppression, interaction of antigen-specific FoxP3 Tregs with DC in the context of antigen presentation, and conversion of Teff cells into endogenous antigen-specific Tregs to confer lasting suppression. Both transplanted FVIII-specific and non-specific Tregs may enhance the latter. Our results imply that a transplant that includes FVIII-specific cells directs a more consistent and durable effect.
Finally, adoptive immunotherapy with Tregs has reported safety and therapeutic efficacy in clinical trials for diseases ranging from autoimmune disorders to transplantation (88). For this treatment to reach its full potential, ex vivo expansion of Treg to sufficient numbers of a desired purity needs to be fully optimized. This is particularly challenging in cases where there are perturbations in Treg numbers, or when a desired specificity is required. Engineering conventional T cells to exhibit Treg function and/or antigen specificity therefore has the potential to enhance both Treg numbers and function.
Ethics Statement
Animals were housed under specific pathogen-free conditions at the University of Florida and treated under Institutional Animal Care and Use Committee-approved protocols.
Author Contributions
RH, CT, and MB designed experiments, interpreted data, and wrote the manuscript. VK, RS, and MB performed experiments. MB supervised the study.
Funding
This work was supported by NIH grants R01 HL131093 (to RH and CT), R01 AI51390, and U54 HL142012 (to RH). MB was supported by AWD00215 from the Bayer Hemophilia Awards Program and by grant 072395-00002B from the National Hemophilia Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00274/full#supplementary-material
References
1. Sherman A, Biswas M, Herzog RW. Innovative approaches for immune tolerance to factor VIII in the treatment of hemophilia A. Front Immunol. (2017) 8:1604. doi: 10.3389/fimmu.2017.01604
2. Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science (2018) 359:eaan4672. doi: 10.1126/science.aan4672
3. George LA, Sullivan SK, Giermasz AJ., Rasko EJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. (2017) 377:2215–27. doi: 10.1056/NEJMoa1708538
4. Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, et al. AAV5-factor VIII gene transfer in severe hemophilia A. N Engl J Med. (2017) 377:2519–30. doi: 10.1056/NEJMoa1708483
5. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. (2014) 371:1994–2004. doi: 10.1056/NEJMoa1407309
6. Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia (2002) 8:23–9. doi: 10.1046/j.1351-8216.2001.00134.x
7. Witmer C, Young G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol. (2013) 4:59–72. doi: 10.1177/2040620712464509
8. Barg AA, Livnat T, Kenet G. Inhibitors in hemophilia: treatment challenges and novel options. Semin Thromb Hemost. (2017) 44:544–50 doi: 10.1055/s-0037-1612626
9. Schep SJR., Schutgens EG, Fischer K, Boes ML. Review of immune tolerance induction in hemophilia A. Blood Rev. (2018) 32:326–38. doi: 10.1016/j.blre.2018.02.003
10. Miao CH. Immunomodulation for inhibitors in hemophilia A: the important role of Treg cells. Expert Rev Hematol. (2010) 3:469–83. doi: 10.1586/ehm.10.33
11. Scott DW. Inhibitors - cellular aspects and novel approaches for tolerance. Haemophilia (2014) 20 (Suppl 4):80–6. doi: 10.1111/hae.12407
12. Zerra PE, Cox C, Baldwin WH, Patel SR, Arthur CM, Lollar P, et al. Marginal zone B cells are critical to factor VIII inhibitor formation in mice with hemophilia A. Blood (2017) 130:2559–68. doi: 10.1182/blood-2017-05-782912
13. Georgescu MT, Lai JD, Hough C, Lillicrap D. War and peace: factor VIII and the adaptive immune response. Cell Immun. (2016) 301:2–7. doi: 10.1016/j.cellimm.2015.11.008
14. Herczenik E, van Haren SD, Wroblewska A, Kaijen P, van den Biggelaar M, Meijer AB. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J Allergy Clin Immunol. (2012) 129:501–9.e1-5. doi: 10.1016/j.jaci.2011.08.029
15. Navarrete A, Dasgupta S, Delignat S, Caligiuri G, Christophe OD, Bayry J, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. (2009) 7:1816–23. doi: 10.1111/j.1538-7836.2009.03571.x
16. Cao O, Loduca PA, Herzog RW. Role of regulatory T cells in tolerance to coagulation factors. J Thromb Haemost (2009) 7 (Suppl 1):88–91. doi: 10.1111/j.1538-7836.2009.03417.x
17. Wang X, Terhorst C, Herzog RW. In vivo induction of regulatory T cells for immune tolerance in hemophilia. Cell Immunol. (2016) 301:18–29. doi: 10.1016/j.cellimm.2015.10.001
18. Biswas M, Sarkar D, Kumar SR, Nayak S, Rogers GL, Markusic DM, et al. Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD4+CD25+FoxP3+ Treg. Blood (2015) 125:2937–47. doi: 10.1182/blood-2014-09-599266
19. Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW. Induction of tolerance to factor VIII by transient co-administration with rapamycin. J Thromb Haemost. (2011) 9:1524–33. doi: 10.1111/j.1538-7836.2011.04351.x
20. Nayak S, Sarkar D, Perrin GQ, Moghimi B, Hoffman BE, Zhou S, et al. Prevention and reversal of antibody responses against factor IX in gene therapy for hemophilia B. Front Microbiol. (2011) 2:244. doi: 10.3389/fmicb.2011.00244
21. Liu CL, Ye P, Yen BC, Miao CH. In vivo expansion of regulatory T cells with IL-2/IL-2 mAb complexes prevents anti-factor VIII immune responses in hemophilia A mice treated with factor VIII plasmid-mediated gene therapy. Mol Ther. (2011) 19:1511–20. doi: 10.1038/mt.2011.61
22. Kuhn C, Weiner HL. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy (2016) 8:889–906. doi: 10.2217/imt-2016-0049
23. Forster K, Goethel A, Chan CW, Zanello G, Streutker C, Croitoru K. An oral CD3-specific antibody suppresses T-cell-induced colitis and alters cytokine responses to T-cell activation in mice. Gastroenterology (2012) 143:1298–307. doi: 10.1053/j.gastro.2012.07.019
24. Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, et al. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood (2009) 113:193–203. doi: 10.1182/blood-2008-04-151597
25. Elkord E. Thymus-derived, peripherally derived, and in vitro-induced T regulatory cells. Front Immunol. (2014) 5:17. doi: 10.3389/fimmu.2014.00017
26. Biswas M, Kumar SRP, Terhorst C, Herzog RW. Gene therapy with regulatory T cells: a beneficial alliance. Front Immunol. (2018) 9:554. doi: 10.3389/fimmu.2018.00554
27. Mathew JM, Voss JH, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. (2018) 8:7428. doi: 10.1038/s41598-018-25574-7
28. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
29. Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e465. doi: 10.1212/NXI.0000000000000465
30. Sarkar D, Biswas M, Liao G, Seay HR, Perrin GQ, Markusic DM, et al. Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol Ther Methods Clin Dev. (2014) 1:14030 doi: 10.1038/mtm.2014.30
31. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. (2013) 19:1271–3. doi: 10.1016/j.bbmt.2013.06.004
32. Cao O, Hoffman BE, Moghimi B, Nayak S, Cooper M, Zhou S, et al. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol Ther. (2009) 17:1733–42. doi: 10.1038/mt.2009.159
33. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther. (2008) 16:194–202. doi: 10.1038/sj.mt.6300341
34. Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
35. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. (2012) 209:1713–22. doi: 10.1084/jem.20120822
36. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. (2012) 209:1723–42. doi: 10.1084/jem.20120914
37. Biswas M, Rogers GL, Sherman A, Byrne BJ, Markusic DM, Jiang H, et al. Combination therapy for inhibitor reversal in haemophilia A using monoclonal anti-CD20 and rapamycin. Thromb Haemost. (2017) 117:33–43. doi: 10.1160/TH16-05-0404
38. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
39. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. (2006) 212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x
40. Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology (2006) 117:289–300. doi: 10.1111/j.1365-2567.2005.02317.x
41. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458:351–6. doi: 10.1038/nature07674
42. Iikuni N, Lourenco EV, Hahn BH, La Cava A. Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol. (2009) 183:1518–22. doi: 10.4049/jimmunol.0901163
43. Glatman Zaretsky A, Konradt C, Depis F, Wing JB, Goenka R, Atria DG, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. (2017) 18:1906–16. doi: 10.1016/j.celrep.2017.01.067
44. Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood (2007) 110:1132–40. doi: 10.1182/blood-2007-02-073304
45. Miao CH, Harmeling BR, Ziegler SF, Yen BC, Torgerson T, Chen L, et al. CD4+FOXP3+ regulatory T cells confer long-term regulation of factor VIII-specific immune responses in plasmid-mediated gene therapy-treated hemophilia mice. Blood (2009) 114:4034–44. doi: 10.1182/blood-2009-06-228155
46. Wang X, Su J, Sherman A, Rogers GL, Liao G, Hoffman BE, et al. Plant-based oral tolerance to hemophilia therapy employs a complex immune regulatory response including LAP+CD4+ T cells. Blood (2015) 125:2418–27. doi: 10.1182/blood-2014-08-597070
47. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
48. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. (2017) 17:703–17. doi: 10.1038/nri.2017.75
49. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
50. Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol. (2010) 185:1071–81. doi: 10.4049/jimmunol.1000082
51. Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci USA. (2010) 107:5919–24. doi: 10.1073/pnas.1002006107
52. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature (2007) 445:936–40. doi: 10.1038/nature05563
53. Birzele F, Fauti T, Stahl H, Lenter MC, Simon E, Knebel D, et al. Next-generation insights into regulatory T cells: expression profiling and FoxP3 occupancy in Human. Nucleic Acids Res. (2011) 39:7946–60. doi: 10.1093/nar/gkr444
54. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood (2008) 112:2340–52. doi: 10.1182/blood-2008-01-133967
55. Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci USA. (2009) 106:8635–40. doi: 10.1073/pnas.0900621106
56. Wang Y, Souabni A, Flavell RA, Wan YY. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J Immunol. (2010) 185:5983–92. doi: 10.4049/jimmunol.1001255
57. Overacre AE, Vignali DA. T(reg) stability: to be or not to be. Curr Opin Immunol. (2016) 39:39–43. doi: 10.1016/j.coi.2015.12.009
58. Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. (2014) 259:173–91. doi: 10.1111/imr.12173
59. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329:1667–71. doi: 10.1126/science.1191996
60. Wright GP, Notley CA, Xue SA, Bendle GM, Holler A, Schumacher TN, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci USA. (2009) 106:19078–83. doi: 10.1073/pnas.0907396106
61. Beavis PA, Gregory B, Green P, Cribbs AP, Kennedy A, Amjadi P, et al. Resistance to regulatory T cell-mediated suppression in rheumatoid arthritis can be bypassed by ectopic foxp3 expression in pathogenic synovial T cells. Proc Natl Acad Sci USA. (2011) 108:16717–22. doi: 10.1073/pnas.1112722108
62. Loser K, Hansen W, Apelt J, Balkow S, Buer J, Beissert S. In vitro-generated regulatory T cells induced by Foxp3-retrovirus infection control murine contact allergy and systemic autoimmunity. Gene Ther. (2005) 12:1294–304. doi: 10.1038/sj.gt.3302567
63. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi: 10.1186/1742-2094-9-112
64. Haque M, Song J, Fino K, Sandhu P, Song X, Lei F, et al. Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci Rep. (2016) 6:20588. doi: 10.1038/srep20588
65. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125:1107–15. doi: 10.1182/blood-2014-04-566786
66. Jaeckel E, von Boehmer H, Manns MP. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes (2005) 54:306–10. doi: 10.2337/diabetes.54.2.306
67. van Herwijnen MJ, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci USA. (2012) 109:14134–9. doi: 10.1073/pnas.1206803109
68. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene therapy-induced antigen-specific tregs inhibit neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol Ther. (2018) 26:173–83. doi: 10.1016/j.ymthe.2017.09.001
69. Mathew JM, Voss JH, McEwen ST, Konieczna I, Chakraborty A, Huang X, et al. Generation and characterization of alloantigen-specific regulatory T cells for clinical transplant tolerance. Sci Rep. (2018) 8:1136. doi: 10.1038/s41598-018-19621-6
70. Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, et al. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med. (2005) 11:1118–24. doi: 10.1038/nm1292
71. Stuehler C, Nowakowska J, Bernardini C, Topp MS, Battegay M, Passweg J, et al. Multispecific aspergillus T cells selected by CD137 or CD154 induce protective immune responses against the most relevant mold infections. J Infect Dis. (2015) 211:1251–61. doi: 10.1093/infdis/jiu607
72. Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4(+) T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. (2013) 5:215ra174. doi: 10.1126/scitranslmed.3007320
73. Malec L, Abshire T, Jobe S, White G. rFIXFc for immune tolerance induction in a severe hemophilia B patient with an inhibitor and prior history of ITI related nephrotic syndrome. Haemophilia (2018) 24: e294–6 doi: 10.1111/hae.13583
74. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med. (2002) 196:255–60. doi: 10.1084/jem.20020394
75. Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev. (2006) 212:301–13. doi: 10.1111/j.0105-2896.2006.00406.x
76. Letourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. (2009) 123:758–62. doi: 10.1016/j.jaci.2009.02.011
77. Dieckmann D, Bruett CH, Ploettner H, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory, contact-dependent T cells induce interleukin 10-producing, contact-independent type 1-like regulatory T cells [corrected]. J Exp Med. (2002) 196:247–53. doi: 10.1084/jem.20020642
78. Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol. (2011) 186:6661–6. doi: 10.4049/jimmunol.1100315
79. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
80. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. (2008) 205:1975–81. doi: 10.1084/jem.20080308
81. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
82. Alonso R, Flament H, Lemoine S, Sedlik C, Bottasso E, Peguillet I, et al. Induction of anergic or regulatory tumor-specific CD4(+) T cells in the tumor-draining lymph node. Nat Commun. (2018) 9:2113. doi: 10.1038/s41467-018-04524-x
83. Mavin E, Nicholson L, Rafez Ahmed S, Gao F, Dickinson A, Wang XN. Human regulatory T cells mediate transcriptional modulation of dendritic cell function. J Immunol. (2017) 198:138–46. doi: 10.4049/jimmunol.1502487
84. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA. (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
85. Kushwah R, Hu J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. (2011) 1:20. doi: 10.1186/2045-3701-1-20
86. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood (2008) 111:453–62. doi: 10.1182/blood-2007-06-094482
87. Pasquet L, Douet JY, Sparwasser T, Romagnoli P, van Meerwijk JP. Long-term prevention of chronic allograft rejection by regulatory T-cell immunotherapy involves host Foxp3-expressing T cells. Blood (2013) 121:4303–10. doi: 10.1182/blood-2012-08-452037
Keywords: hemophilia A, tolerance, FoxP3, treg, cell therapy, immunotherapy
Citation: Herzog RW, Kuteyeva V, Saboungi R, Terhorst C and Biswas M (2019) Reprogrammed CD4+ T Cells That Express FoxP3+ Control Inhibitory Antibody Formation in Hemophilia A Mice. Front. Immunol. 10:274. doi: 10.3389/fimmu.2019.00274
Received: 22 August 2018; Accepted: 31 January 2019;
Published: 20 February 2019.
Edited by:
Herman Waldmann, University of Oxford, United KingdomReviewed by:
Carol H. Miao, University of Washington, United StatesDavid William Scott, Uniformed Services University of the Health Sciences, United States
Copyright © 2019 Herzog, Kuteyeva, Saboungi, Terhorst and Biswas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moanaro Biswas, bmJpc3dhc0BpdS5lZHU=