 Kurt R. Lehner1
Kurt R. Lehner1 Harold A. Silverman1,2Meghan E. Addorisio2Ashbeel Roy3,4Mohammed A. Al-Onaizi3,5
Harold A. Silverman1,2Meghan E. Addorisio2Ashbeel Roy3,4Mohammed A. Al-Onaizi3,5 Yaakov Levine6
Yaakov Levine6 Peder S. Olofsson2,7
Peder S. Olofsson2,7 Sangeeta S. Chavan1,2
Sangeeta S. Chavan1,2 Robert Gros3,4,8
Robert Gros3,4,8 Neil M. Nathanson9
Neil M. Nathanson9 Yousef Al-Abed2,10
Yousef Al-Abed2,10 Christine N. Metz1,2
Christine N. Metz1,2 Vania F. Prado3,4,11,12
Vania F. Prado3,4,11,12 Marco A. M. Prado3,4,11,12Kevin J. Tracey1,2†
Marco A. M. Prado3,4,11,12Kevin J. Tracey1,2† Valentin A. Pavlov1,2*†
Valentin A. Pavlov1,2*†- 1Zucker School of Medicine at Hofstra/Northwell, Hempstead, NY, United States
- 2Center for Biomedical Science and Bioelectronic Medicine, The Feinstein Institute for Medical Research, Northwell Health, Manhasset, NY, United States
- 3Schulich School of Medicine and Dentistry, Robarts Research Institute, University of Western Ontario, London, ON, Canada
- 4Department of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada
- 5Department of Anatomy, Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
- 6SetPoint Medical Corporation, Valencia, CA, United States
- 7Department of Medicine, Center for Bioelectronic Medicine, Center for Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
- 8Department of Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada
- 9Department of Pharmacology, University of Washington, Seattle, WA, United States
- 10Department of Medicinal Chemistry, Center for Molecular Innovation, The Feinstein Institute for Medical Research, Northwell Health, Manhasset, NY, United States
- 11Department of Anatomy and Cell Biology, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada
- 12Graduate Program in Neuroscience, Schulich School of Medicine and Dentistry, University of Western Ontario, London, ON, Canada
The brain regulates physiological functions integral to survival. However, the insight into brain neuronal regulation of peripheral immune function and the neuromediator systems and pathways involved remains limited. Here, utilizing selective genetic and pharmacological approaches, we studied the role of forebrain cholinergic signaling in the regulation of peripheral immune function and inflammation. Forebrain-selective genetic ablation of acetylcholine release and vagotomy abolished the suppression of serum TNF by the centrally-acting cholinergic drug galantamine in murine endotoxemia. Selective stimulation of acetylcholine action on the M1 muscarinic acetylcholine receptor (M1 mAChR) by central administration of the positive allosteric modulator benzyl quinolone carboxylic acid (BQCA) suppressed serum TNF (TNFα) levels in murine endotoxemia. This effect was recapitulated by peripheral administration of the compound. BQCA also improved survival in murine endotoxemia and these effects were abolished in M1 mAChR knockout (KO) mice. Selective optogenetic stimulation of basal forebrain cholinergic neurons innervating brain regions with abundant M1 mAChR localization reduced serum TNF in endotoxemic mice. These findings reveal that forebrain cholinergic neurons regulate innate immune responses and inflammation, suggesting the possibility that in diseases associated with cholinergic dysfunction, including Alzheimer's disease this anti-inflammatory regulation can be impaired. These results also suggest novel anti-inflammatory approaches based on targeting forebrain cholinergic signaling in sepsis and other disorders characterized by immune dysregulation.
Introduction
The nervous system regulates and coordinates physiological functions and defense mechanisms. A major defense mechanism against pathogen invasion and tissue injury is provided by the innate immune system through inflammation (1). However, dysregulated immune responses and aberrant inflammation are implicated in the etiology of sepsis, inflammatory bowel disease, and many other life-threatening and debilitating disorders (1–6). While accumulating evidence reveals that the nervous system and specifically the vagus nerve regulate immune function and inflammation, the role of brain pathways in this context remains poorly understood (2, 7, 8). The brain regulation of peripheral inflammation and the mediating role of the vagus nerve have been indicated in studies with the experimental anti-inflammatory compound CNI-1493 (Semapimod). Administration of this molecule in the brain suppresses serum TNF (TNF-α) in murine endotoxemia and this effect is abrogated by surgical transection of the vagus nerve (vagotomy) (9). CNI-1493 binds to muscarinic acetylcholine receptors (mAChRs) (10) and administration of compounds that mimic the action of acetylcholine on mAChRs in the brain also suppresses circulating TNF and other pro-inflammatory cytokines (10–12). In addition, galantamine, an acetylcholinesterase inhibitor, which increases acetylcholine levels suppresses peripheral pro-inflammatory cytokine levels acting through a brain mAChR-mediated mechanism (13–15). These cholinergic effects in the brain are linked with activation of the vagus nerve-based inflammatory reflex (11, 13, 14, 16, 17), a physiological immunoregulatory circuit (18, 19) with recently demonstrated utility in treating human inflammatory diseases (20, 21). These studies have implicated brain cholinergic signaling in the regulation of pro-inflammatory cytokine release and inflammation. The cholinergic system in the brain has a diverse topographic neuronal organization and projection patterns (22, 23), and specific insight into the role of cholinergic pathways and receptors in the brain in peripheral immunoregulation is presently lacking.
A major collection of cholinergic neurons is localized in the basal forebrain (22, 24). These neurons project to forebrain regions with abundant expression of M1 mAChRs, including neocortical areas and the hippocampus (23), and regulate neuroplasticity, cognition, and other processes (22, 23, 25). Here, utilizing mice with selective genetic forebrain ablation of acetylcholine release, positive allosteric M1 mAChR modulation, and optogenetic stimulation of basal forebrain cholinergic neurons, we indicate a role for forebrain cholinergic signaling via M1 mAChRs in the physiological regulation of inflammation.
Results
Acetylcholine in Forebrain Mediates Cholinergic Suppression of Peripheral Pro-inflammatory Cytokine Release via a Vagus Nerve-Dependent Signaling
To investigate the role of forebrain neuronal acetylcholine in regulating peripheral inflammation, we utilized mice with selective forebrain deprivation of acetylcholine release and galantamine, a centrally-acting cholinergic drug (an acetylcholinesterase inhibitor) with anti-inflammatory properties (13, 14). A major molecular determinant of acetylcholine release is the vesicular acetylcholine transporter (VAChT), which loads acetylcholine in vesicles prior to its release in the synaptic cleft (26, 27). We used mice with Cre-loxP-based forebrain VAChT ablation, a genetic manipulation that provides a non-invasive means of selective elimination of forebrain acetylcholine release and cholinergic activity without loss of neurons (27). This is important as cholinergic neurons can also secrete GABA with ACh and targeting VAChT allows for selective manipulation of ACh release (28–31). As shown in Figure 1A, no VAChT immunoreactivity in the forebrain (hippocampus, CA3 area shown) was detected in these VAChTNkx2.1−Cre−flox/flox (VAChT−/−) mice as compared with VAChTflox/flox (VAChT+/+) control mice. This observation was consistent with the previously reported lack of VAChT immunoreactivity in several forebrain areas of VAChT−/− mice and no significant alterations in VAChT protein expression in brainstem regions as a result of genetic deletion (27). The selective forebrain VAChT ablation was not associated with differences in VAChT protein levels (immunofluorescence) in the spinal cord and peripheral neuronal varicosities in the heart (Figures 1B,C). These observations indicated that VAChT ablation was limited to the basal forebrain cholinergic system. Furthermore, no differences were observed between VAChT−/− and VAChT+/+ mice on 24 h heart rate recording (Figure 1D). There were no differences between the two groups of mice in heart rate responses to atropine (mAChR blocker) and propranolol (beta adrenergic receptor blocker) i.p. administrations (Figures 1E–G). In addition, sensitivity to post-handling stress was similar between genotypes, because heart rate responses to saline administration did not differ between the two groups of mice (Figure S1).
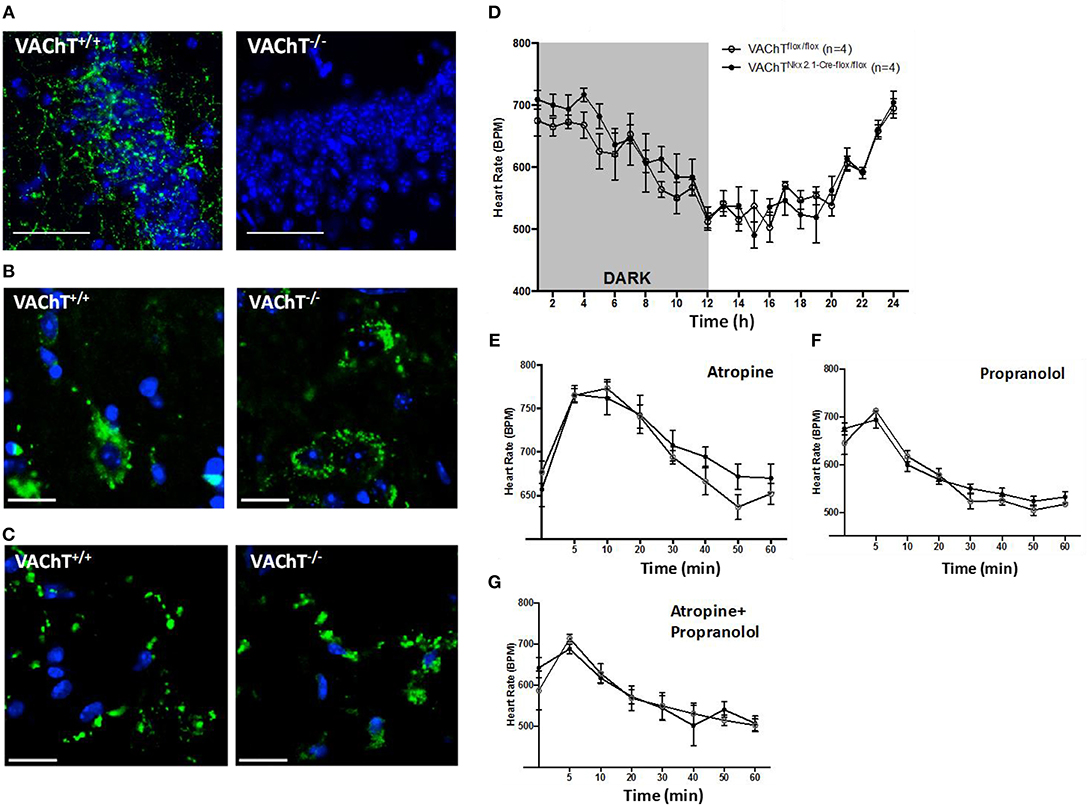
Figure 1. Morphological and functional evaluation of forebrain VAChT KO mice. (A) VAChT protein immunostaining (in green) shown in a hippocampal region of VAChT+/+ mice is not detected in VAChT−/− mice (scale bar = 100 μm). (B) VAChT protein immunostaining (in green) in spinal cord of VAChT+/+ and VAChT−/− mice (scale bar = 20 μm). (C) VAChT protein immunostaining (in green) in the heart right atrium of VAChT+/+ and VAChT−/− mice (scale bar = 20 μm). (D) Freely moving VAChTflox/flox (VAChT+/+) mice and VAChT Nkx2.1−Cre−flox/flox (VAChT−/−) mice exhibit no differences in heart rate during 24 h recording using radio-frequency telemeters. Freely moving VAChT+/+ (open circles) and VAChT−/− mice (black circles) exhibit no difference in heart rate responses to atropine (E), propranolol (F), or atropine and propranolol (G) i.p. administration (n = 4 mice/genotype).
The effects of acetylcholine in the brain can be modulated (enhanced) through inhibiting its degradation using centrally-acting acetylcholinesterase inhibitors, including galantamine. Administration (i.p.) of galantamine in VAChT+/+ mice prior to endotoxin significantly reduced serum TNF levels as compared to vehicle administration (P = 0.017) (Figure 2A). However, galantamine failed to significantly alter serum TNF levels in VAChT−/− mice (Figure 2A). In addition, serum TNF levels in VAChT−/− mice were significantly higher than in VAChT+/+ mice during endotoxemia (P = 0.009) (Figure 2A), suggesting that physiological cholinergic transmission in the forebrain regulates peripheral innate immune responses. Galantamine was previously shown to stimulate vagus nerve activity (17, 32). We next examined the role of the vagus nerve in mediating galantamine forebrain-triggered anti-inflammatory effect in endotoxemic C57BL/6 mice. Galantamine administration significantly decreased serum TNF levels as compared with vehicle during endotoxemia in sham-operated (control) mice (P = 0.019) (Figure 2B). This effect was markedly diminished in mice with cervical unilateral vagotomy (Figure 2B), thus indicating a role of the efferent vagus nerve. Collectively, these data indicate that acetylcholine derived from cholinergic neurons in forebrain plays a role in mediating the suppressive effect of galantamine on peripheral TNF levels in a vagus nerve-dependent manner during murine endotoxemia.
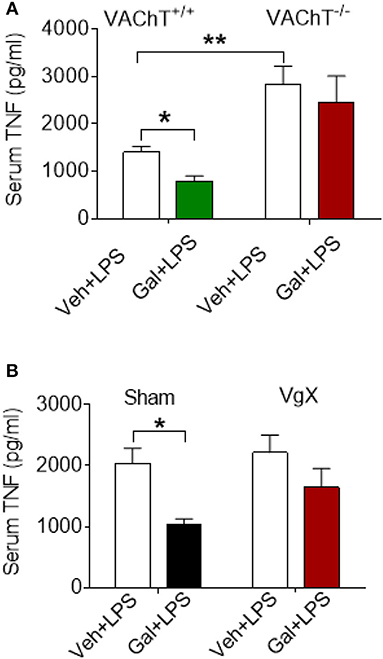
Figure 2. Selective forebrain cholinergic deficit and vagotomy alter cholinergic suppression of peripheral pro-inflammatory cytokine levels. (A) Galantamine (Gal, i.p.) as compared to vehicle (Veh), suppresses serum TNF in VAChT+/+ control mice, but not in VAChT−/− mice during endotoxemia, and serum TNF in vehicle injected VAChT−/− mice are higher as compared to VAChT+/+ control mice (*P = 0.017, **P = 0.007, two-way ANOVA, Sidak's multiple comparisons test), n = 6–7 per group. (B) Vagotomy (VgX) abolishes the suppression of serum TNF in endotoxemic mice (*P = 0.019, two-way ANOVA, Sidak's multiple comparisons test), n = 6–8 per group. See Methods for details.
Stimulation of Acetylcholine Action on the M1 mAChR by Allosteric Modulation Suppresses Lethal Peripheral Inflammation
Basal forebrain cholinergic neurons project to regions with high expression of M1 mAChR, including several neocortical areas and the hippocampus. Acetylcholine released from these neurons stimulates the postsynaptically located M1 mAChR that processes cholinergic transmission (33). Positive allosteric modulation of the M1 mAChR is a selective approach of increasing endogenous acetylcholine activity at the receptor (34, 35). To study the effect of acetylcholine on brain M1 mAChR in the regulation of peripheral inflammation, we used benzyl quinolone carboxylic acid (BQCA), a positive allosteric modulator of M1 mAChR (34, 35). BQCA has previously been shown to selectively increase (up to 129-fold) the functional affinity of endogenous acetylcholine for M1 mAChR (35). Intracerebroventricular (i.c.v.) injection of BQCA (5 μg/kg) resulted in significant suppression of serum TNF as compared with vehicle administration following endotoxin challenge (P = 0.018) (Figure 3A). This observation indicated that selective activation of acetylcholine action on the M1 mAChR in the brain suppresses peripheral inflammation in murine endotoxemia. To facilitate subsequent studies with BQCA, we examined whether peripheral administration of BQCA, which is known to cross the blood brain barrier (34, 35), recapitulates anti-inflammatory effects. Intraperitoneal (i.p.) treatment of mice with BQCA (1–20 mg/kg) resulted in a dose-dependent decrease in serum TNF following endotoxin as compared to vehicle injection (P < 0.001, when 20 mg/kg BQCA was used) (Figure 3B). I.p. administration of BQCA also dose-dependently improved the survival rate in this lethal murine endotoxemia model as compared to vehicle administration (P = 0.035 when 20 mg/kg BQCA was used) (Figure 3C).
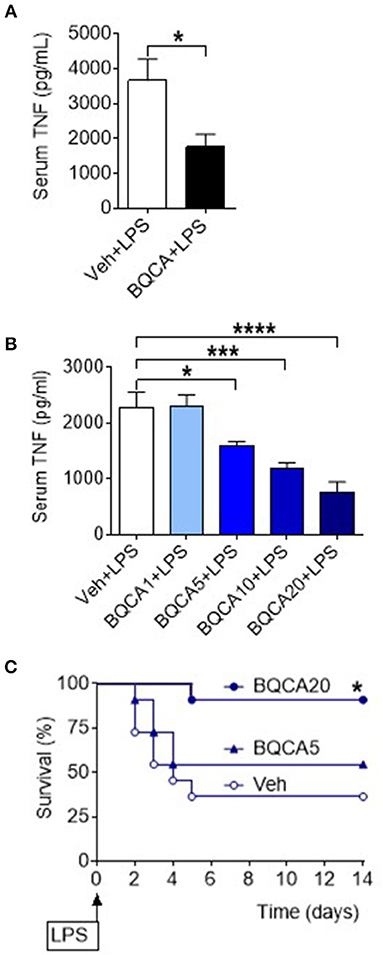
Figure 3. Activation of acetylcholine action on the M1 mAChR using a positive allosteric modulator (BQCA) suppresses serum TNF levels and improves survival in endotoxemia. (A) BQCA (5 μ/kg, i.c.v.) suppresses serum TNF (*P = 0.018, unpaired two-tailed Student's t test), n = 6–7 per group. (B) Peripheral i.p. administration of BQCA 1–20 (1–20 mg/kg) in endotoxemic mice suppresses serum TNF levels (*P = 0.027, ***P = 0.0005, ****P < 0.0001, one-way ANOVA, Dunnett's multiple comparisons test), n = 7–8 per group. (C) Peripheral i.p. administration of BQCA 20 (20 mg/kg) in endotoxemic mice improves survival in endotoxemia (*P = 0.035, Log-rank test), n = 11–12 per group.
BQCA treatment (20 mg/kg, i.p.) did not significantly alter serum TNF in M1 mAChR KO mice (as compared to vehicle treatment), while in wild type (WT) mice this drug effect was significant (P = 0.028) (Figure 4A). Previous studies have shown that activation of brain mAChR-mediated cholinergic signaling results in suppression of TNF in spleen, a major source of pro-inflammatory cytokines and an organ target of the vagus-nerve-based inflammatory reflex (36–38). BQCA (i.p.) administration 1 h prior to endotoxin significantly suppressed splenic TNF in WT mice (P = 0.047) and did not alter splenic TNF in M1 mAChR KO mice (Figure 4B). BQCA also failed to significantly alter splenic TNF in endotoxemic WT mice with unilateral cervical vagotomy (Figure 4C). In addition, BQCA (20 mg/kg, i.p.) injected 1 h prior to endotoxin was sufficient to significantly improve survival in WT mice (P = 0.028), but failed to significantly alter survival in endotoxemic M1 mAChR KO mice (Figure 4D). Together these results show that enhancement of acetylcholine activity on the M1 mAChR is sufficient to suppress inflammation in murine endotoxemia and that the efferent vagus nerve is necessary for this anti-inflammatory effect.
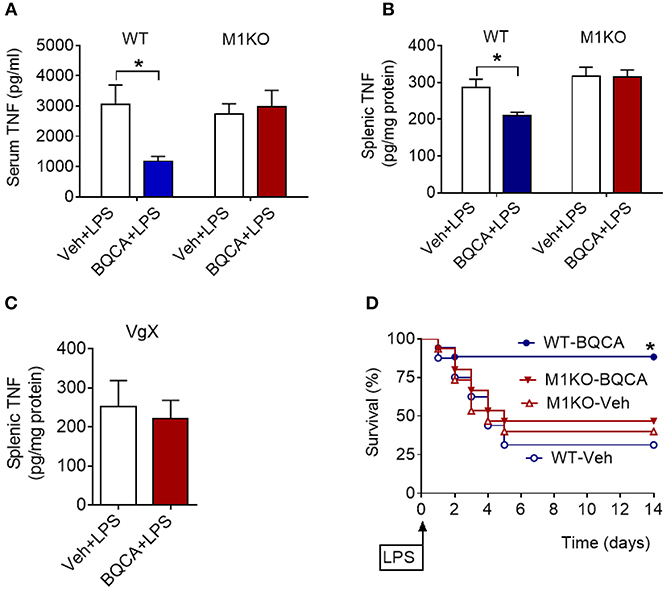
Figure 4. Anti-inflammatory effects of BQCA in endotoxemia are mediated by M1 mAChRs. (A) Peripheral (i.p.) administration of BQCA (20 mg/kg, i.p.) suppresses serum TNF in wild type (WT) mice and does not alter serum TNF in M1 mAChR KO mice during endotoxemia (*P = 0.01, two-way ANOVA, Sidak's multiple comparisons test), n = 8–10. (B) Peripheral (i.p.) administration of BQCA (20 mg/kg, i.p.) suppresses splenic TNF in WT mice and does not alter splenic TNF in M1 mAChR KO mice (*P = 0.017, two-way ANOVA, Sidak's multiple comparisons test), n = 7–10. (C) Vagotomy (VgX) abolishes the effect of BQCA on splenic TNF during endotoxemia (unpaired two-tailed Student's t test), n = 7, 8. (D) Peripheral (i.p.) administration of BQCA (20 mg/kg, i.p.) improves survival in endotoxemic WT mice and does not alter the survival rate in M1 mAChR KO mice during endotoxemia (*P = 0.028, Log-rank test), n = 15–18 per group. See Methods for details.
Selective Optogenetic Stimulation of Basal Forebrain Medial Septum Cholinergic Neurons Suppresses Serum TNF Levels
We next examined whether the anti-inflammatory effect of acetylcholine acting on M1 mAChR (achieved by allosteric modulation with BQCA) could be replicated by direct stimulation of basal forebrain cholinergic neurons in vivo. The medial septum (medial septal nucleus) is a major nucleus in the basal forebrain cholinergic system (22, 23). The medial septum also plays an important role as a relay of afferent vagus nerve signaling in the forebrain as recently demonstrated (39). Accordingly, we stimulated basal forebrain medial septum cholinergic neurons using a selective optogenetic approach in transgenic mice. These mice express channelrhodopsin-2 coupled to a yellow fluorescent protein (ChR2-YFP) under the control of the choline acetyltransferase (ChAT) promoter. Immunofluorescent staining of brain slices confirmed the neuronal colocalization of ChAT and ChR2-YFP in the medial septum (Figure 5A) and the abundant expression of ChR2-YFP in the medial septum and the adjacent vertical limb of the diagonal band of Broca (Figure S2). Photoactivation of medial septum neurons by laser light (473 nm) significantly suppressed serum TNF levels compared with sham stimulation (P = 0.039) (Figure 5B) in mice with confirmed (by microscopic histochemical evaluation) location of the fiber tip in the medial septum (Figure S3; Supplementary Methods). Laser light exposure of medial septum neurons in C57BL/6 mice not expressing ChR2-YFP on cholinergic neurons (non-carriers) was performed to control for possible confounding effects of heat and other non-thermal effects of light. This manipulation failed to alter serum TNF levels (Figure 5C) thus confirming the specific cholinergic nature of the mechanism. As ChAT-ChR2-YFP mice have been shown to overexpress VAChT (40, 41) and present increased cholinergic tone, LPS was administered to ChAT-ChR2-YFP and non-carrier mice not subjected to anesthesia and surgical manipulation. No differences in serum TNF levels upon LPS administration were observed between the two groups of mice (Figure 5D), indicating that the genetic modification by itself did not alter the inflammatory response. These results show that selective activation of basal forebrain medial septum cholinergic neurons decreases peripheral inflammation in endotoxemia.
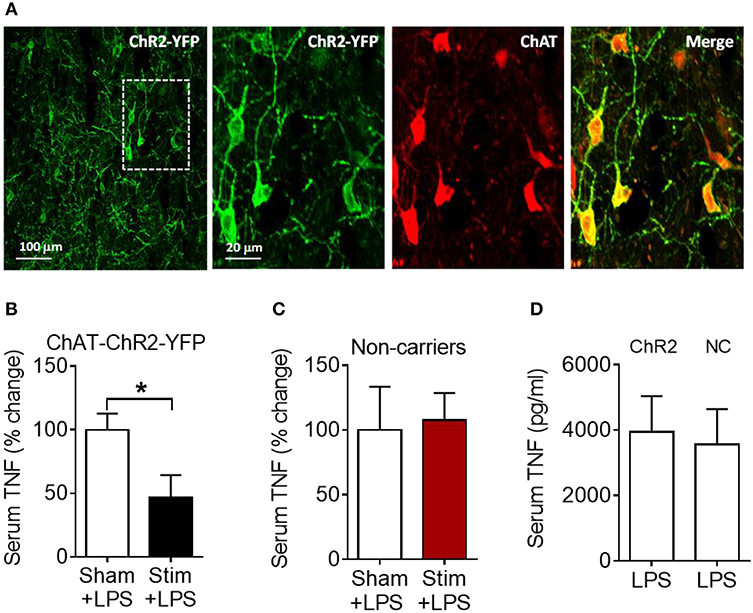
Figure 5. Optogenetic stimulation of basal forebrain cholinergic neurons in the medial septum suppresses serum TNF in endotoxemic mice. (A) Immunostaining of medial septum neurons in a brain section of a ChAT-ChR2-EYFP mouse: EYFP immunostaining (first and second panel); ChAT immunostaining of the same area (third panel); and double immunostaining (fourth panel). (B) Optogenetic stimulation suppresses serum TNF during endotoxemia (*P = 0.039, unpaired two-tailed Student's t test), n = 5–6 per group. (C) Laser light exposure of medial septum neurons in control (non-carrier) mice does not significantly alter serum TNF levels during endotoxemia (unpaired two-tailed Student's t test), n = 6–7 per group. (D) LPS (i.p.) administration to ChAT-ChR2-EYFP (ChR2) and control (non-carrier, NC) mice does not result in statistically different serum TNF levels, (unpaired two-tailed Student's t test), n = 9 per group. See Methods for details.
Discussion
Here, we show a role for forebrain cholinergic signaling and the M1 mAChR in the brain neuronal regulation of inflammation through vagus nerve-mediated signaling.
Genetic ablation of VAChT, which eliminates synaptic acetylcholine release from basal forebrain cholinergic neurons (27), abolished the anti-inflammatory effect of the centrally-acting acetylcholinesterase inhibitor galantamine. These findings point to forebrain acetylcholine as a major mediator of galantamine anti-inflammatory effects in endotoxemia. The selective forebrain cholinergic deficit (in VAChT−/− mice) also resulted in increased circulatory TNF levels in endotoxemic mice, thus suggesting a tonic anti-inflammatory role of acetylcholine in forebrain.
Forebrain areas, including the cortex (neocortex) and the hippocampus have abundant (predominantly post-synaptic) localizations of the M1 mAChR, which plays a major role in processing the effects of acetylcholine released from basal forebrain cholinergic neurons (22). Positive allosteric modulation of the M1 mAChR by BQCA is a selective approach of increasing the activity of the endogenous acetylcholine on the receptor (34, 35). The anti-inflammatory effects of the centrally-acting BQCA demonstrated here indicate a role of acetylcholine acting on the M1 mAChR in controlling peripheral inflammatory responses. Unilateral vagotomy attenuated the anti-inflammatory effects of galantamine and BQCA, thus indicating a brain-to periphery mediating role for the vagus nerve. The vagus nerve is an important neuroanatomical component of the inflammatory reflex (18) in which sensory and motor vagus nerve signaling regulates cytokine production by immune cells in the spleen, and alleviates inflammation (2, 19). Importantly, recent studies demonstrated that afferent vagus nerve signaling reaches the forebrain (hippocampus and cortex), and identified basal forebrain cholinergic nuclei, including the medial septum as major relay components (39, 42, 43). Together with these findings, our observations suggest a role for the medial septum in a forebrain regulatory hub of the inflammatory reflex. This regulation possibly involves other brain regions through multisynaptic pathways with brainstem nuclei providing peripheral vagus nerve projections. These brain networks remain to be further elucidated.
In addition to positive allosteric modulation of acetylcholine action on M1 mAChR, suppression of serum TNF levels during endotoxemia was also achieved by selective optogenetic stimulation of a subset of basal forebrain cholinergic neurons in the medial septum. Although optogenetic stimulation was performed in animals under isoflurane anesthesia and there is some evidence that relatively high doses of isoflurane can increase blood-brain barrier permeability (44), the proper sham stimulation pointed to the specificity of the effect. Medial septum cholinergic neurons innervate the hippocampus and the parahippocampal region, which are interconnected with several neocortical and subcortical areas (23, 24, 39). Multisynaptic connections between the hippocampus and the hypothalamus have also been described. Although we stereotactically targeted optogenetic stimulation of the medial septum, a spread of stimulation to adjacent cholinergic neurons in the diagonal band of Broca cannot be excluded. Cholinergic neurons in the diagonal band of Broca project to cortical areas and the amygdala (22–24). These neural networks involving cholinergic circuits suggest that multiple points of regulation can be further explored in studying the brain control of peripheral inflammation. Some of these networks have been previously associated with the regulation of autonomic responses, including modulation of vagus nerve activity (2). These previous observations are consistent with our findings that signaling through the vagus nerve mediates brain cholinergic modulation of peripheral inflammation. Our findings have clinical relevance. Galantamine is in clinical use for alleviation of cognitive deterioration in patients with Alzheimer's disease. We have also recently shown that treatment with galantamine alleviates inflammation and insulin resistance in patients with the metabolic syndrome (45). BQCA and other centrally-acting positive allosteric modulators of the M1 mAChR have also been preclinically developed in the search of efficient treatments of Alzheimer's disease and other neurological conditions (34, 35). Our results suggest considering these two types of therapeutics as anti-inflammatory agents. While optogenetics provide a very selective approach for spatiotemporal neuronal control in studying neural circuitries (46), this technology currently has limited therapeutic implications (47–49). However, medial septum deep brain (electrical) stimulation has been successfully used for improving spatial working memory and cognition in experimental settings of traumatic brain injury (50, 51). Future insights into brain networks triggered by medial septum neuronal stimulation and possibly other regions may further inform implications of deep brain stimulation and non-invasive approaches, including transcranial magnetic stimulation and transcranial direct current stimulation (52–54) in therapeutic anti-inflammatory strategies.
Basal forebrain cholinergic signaling has a documented role in attention, learning and memory, and degeneration of basal forebrain cholinergic neurons, which results in diminished release of acetylcholine in the forebrain, is one of the hallmarks of Alzheimer's disease (23). Intriguingly, increased peripheral TNF and other pro-inflammatory cytokine levels have been found in Alzheimer's disease (55), and peripheral inflammation has been linked to exacerbation of brain pathogenesis and neurodegeneration, affecting cholinergic neurons (56, 57). This may be of importance given that cholinergic neurotransmission is dysfunctional in different types of dementia (58), suggesting a potential mechanism by which patients with Alzheimer's disease and other types of dementia may be more susceptible to distinct types of infection (59).
Peripheral inflammation in sepsis, liver disease, and other inflammatory conditions also is linked with brain inflammation (2, 60) and deterioration in brain function and delirium within the scope of characteristic encephalopathies (2, 61–64). Dysregulation in cholinergic signaling has a documented role in brain derangements, including delirium and encephalopathies in sepsis and other inflammatory conditions (60, 65–67). Together with these previously published findings, our observations suggest a bidirectional relationship between brain cholinergic signaling and peripheral inflammation. In this context targeted pharmacological or device-generated modulation of forebrain cholinergic signaling may have broader therapeutic implications.
Materials and Methods
Animals
Male mice were used in all experiments. BALB/c mice (24–28 g) were purchased from Taconic. Mice with genetic deletion of VAChT in the forebrain (VAChT–/– mice) were generated as previously described (27). Briefly, VAChTflox/flox mouse line (crossed for five generations with C57BL/6J) was crossed with the Nkx2.1-Cre mouse line (C57BL/6J-Tg(Nkx2-1-cre)2Sand/J) (Jackson Laboratory, stock No: 008661). Control VAChT+/+ mice were VAChTflox/flox littermates. ChAT-ChR2-YFP BAC transgenic mice (Jackson Laboratory, stock No: 014546) (68) and non-carrier wild type mice (on the C57BL/6 background) were used in experiments with laser light exposure of the medial septum neurons. M1 mAChR (Chrm1−/−) KO mice (69) were generated on the C57Bl/6 background as previously described (70). M1 mAChR KO and wild type (WT) littermates were received from Charles River. Animals were allowed to acclimate for at least 2 weeks prior to the corresponding experiment. All animals were housed in standard conditions (a 12 h light–dark cycle) with access to regular chow and water.
Chemicals
Galantamine (Galantamine hydrobromide) (purity ≥95% by HPLC) was purchased from Calbiochem. Galantamine working solutions were prepared in sterile saline. Benzyl quinolone carboxylic acid (BQCA) (purity: ≥97% (HPLC) was purchased from Enzo Life Sciences. BQCA working solutions were prepared in betacyclodextrin and contained 5% betacyclodextrin in sterile saline (35). Atropine (purity: ≥99% (TLC) and propranolol (purity: ≥98% (TLC) were purchased from Sigma and working solutions prepared in sterile saline.
Endotoxemia and Drug Treatment
Endotoxemia in mice was induced by administering LPS (endotoxin, Sigma L4130 O111:B4 for consistency with our previous studies (13, 36, 71, 72) in doses as indicated, injected i.p.) Groups of animals were treated i.p. with galantamine (3 mg/kg) or different doses of BQCA or vehicle (sterile saline as a vehicle in galantamine treatments or 5% betacyclodextrin containing sterile saline as a vehicle in BQCA treatments) 1 h prior to endotoxin administration. Vehicle or BQCA (5 μg/kg) [based on the information available about the effect of the compound on M1 mAChR (34, 35)] was also administered i.c.v. 1 h prior to endotoxin (8 mg/kg, i.p.). In experiments with optogenetic stimulation LPS was injected i.p. 1 h after the end of stimulation or sham stimulation. Animals were euthanized by CO2 asphyxiation 1.5 h after endotoxin administration, and blood was collected via cardiac puncture (from the right ventricle) for cytokine (TNF) determination. In other sets of survival experiments, groups of mice were treated with BQCA or vehicle (i.p.) 1 h prior to endotoxin (8 mg/kg, i.p.) injection. Mice were monitored for survival twice daily for the first 5 days, and then daily for the remainder of the 14 day experiments.
Vagotomy
To avoid potential high lethality associated with bilateral cervical vagotomy, a unilateral cervical vagotomy was performed. Mice were anesthetized by isoflurane inhalation and the right cervical vagus nerve was exposed, ligated with a 4–0 silk suture, and divided. In sham-operated animals, the cervical vagus nerve was visualized, but was neither isolated from the surrounding tissues nor transected. All animals were permitted to recover for 7 days following the surgical procedure and before their inclusion in endotoxemia experiments.
Brain Surgical Manipulations and Optogenetic Stimulation
In experiments with optogenetic stimulation, mice were anesthetized with isoflurane and placed on a stereotactic frame (Kopf Instruments). Body temperature was maintained between 36.5 and 37°C using a feedback controlled rectal thermometer and heating pad. Using aseptic technique, the scalp was incised to expose the skull and the overlying connective tissue was removed. A small (~500 μm) craniotomy was performed on the desired locations for fiber insertion. Mice expressing light-activated cation ChR2 tagged with a fluorescent protein (ChR2-YFP) under the control of the choline acetyltransferase (ChAT) promoter were used in optogenetic experiments. The optic fiber was inserted slowly over 3 min, targeting the dorsal cholinergic neurons in the medial septum at a location 0.8 mm anterior to Bregma and 0.7 mm lateral to a depth of 3.5 mm at a 10 degree angle below dura, which was opened with a 27 gauge needle. Laser light was delivered via a 200 μm diameter fiber (Thorlabs) inserted into the craniotomy. For ChR2 activation, a 473 nm laser (Optoengine, LLC) was used at a power of 10 mW at the tip. The laser was controlled by a waveform generator (Agilent). Stimulation was performed by using square pulses at 20 Hz for 10 min. Sham stimulation was carried out the same way with the exception that following optic fiber insertion, no laser stimulation was performed. Following optogenetic stimulation or sham stimulation, the fiber was removed over 3 min, the craniotomy was covered using paraffin wax, and the animal recovered for 1 h on a heating pad at 37°C prior to LPS injection. Laser light exposure of medial septum neurons or sham laser light exposure was performed following the same protocol in non-carrier mice.
Brain Surgical Manipulations and I.C.V. Drug Administration
Craniotomies were performed as described above at a location 0.6 mm posterior and 1.2 mm lateral to Bregma targeting the right lateral ventricle. A 10 μL Hamilton microsyringe in a microinjector pump (UMP3-1, World Precision Instruments) was lowered into the right lateral ventricle over 3 min to a depth of 2.1 mm. Following 5 min of equilibration, BQCA (5 ug/kg) or vehicle was delivered over 3 min. Following injection, the syringe remained in place for 5 min to prevent backflow. Then the needle was slowly removed over 3 min. The craniotomy was covered using paraffin wax and the animal recovered for 1 h on a heating pad at 37° C prior to LPS injection.
Immunohistochemistry and Immunofluorescent Microscopy
Expression of ChR2-EYFP in cholinergic neurons of the medial septum was confirmed with immunofluorescent staining for choline acetyltransferase (ChAT) and ChR2-YFP. Mice underwent intracardiac perfusion with 1 × PBS followed by 4% paraformaldehyde. Brains were harvested and cryoprotected with subsequent incubations of 15% and 30% sucrose. They were then stored in O.C.T. at −20°C. 20 μm sections were collected on gelatin subbed slides (SouthernBiotech) using a cryostat (Leica Microsystems). Slides were rinsed in 1 × PBS followed by washing with 0.25% Triton X-100/1 × PBS (PBT). Blocking was performed with a solution of 10% MeOH, 0.1% bovine serum albumin (BSA), 3% normal donkey serum (NDS), and 0.05% hydrogen peroxide. Slides were then washed in PBT. Cholinergic neurons were visualized using goat anti-ChAT (Millipore, AB144 1:200, dilution) as a primary antibody and specific ChR2-EYFP expression was visualized using rabbit anti-GFP Alexa Fluor 488 (ThermoFisher Scientific, A21311, 1:400 dilution) in a solution of PBT containing 1% NDS. Donkey anti-goat Alexafluor 555 (Thermo Fisher Scientific, A21432, 1:200 dilution) in a solution of 1% NDS/PBT was used to visualize Goat anti-ChAT, and slides were mounted and coverslipped with DAPI-Fluoromount G (Southern BioTech). Images were taken using an Olympus FluoView FV300 Confocal Laser Scanning Microscope.
In experiments with VAChT−/− and VAChT+/+ animals, mice were anesthetized with ketamine (100 mg/kg) and xylazine (25 mg/kg) in 0.9% sodium chloride, and then sacrificed by trans-cardial perfusion. Brains were harvested and placed in 4% paraformaldehyde in 1 × PBS overnight at 4°C. The brains were isolated and 40 μm sections of the tissue were obtained using a vibratome. Brain sections were incubated in a blocking solution of 1 × PBS/0.4% Triton X-100 containing 0.1% glycine (wt/vol), 0.1% lysine (wt/vol), 1% BSA (wt/vol), and 1% normal donkey serum (wt/vol). Sections were incubated with anti-VAChT primary antibody (catalog no. 139103; Synaptic Systems) overnight. Sections were then incubated with the Alexa Fluor 488 anti-rabbit secondary antibody (1:1,000; Life Technologies) for 1 h. The nuclei were labeled with Hoechst. Images were acquired using the Zeiss LSM 510 Meta confocal system as previously described (27).
Serum Isolation and Cytokine Determination
The blood was allowed to clot for 80 min following collection. It was then centrifuged at 5,000 rpm (1,500 × g) for 10 min, and the supernatant (serum) was collected and stored at −20°C until analysis. Serum TNF was quantified by using ELISA per the manufacturer's instructions (eBioscience).
Electrocardiography (ECG)
Electrocardiograms were recorded using radiotelemeters. The radio frequency transmitters were implanted subcutaneously under anesthesia and ECG recordings were initiated following a minimum recovery period of 7 days post-implantation. Heart rate was continuously measured in awake, freely moving mice over 24 h to obtain baseline recordings. To determine the effect of autonomic blockade, heart rate was recorded for 60 min following administration of atropine (1 mg/kg, i.p.), propranolol (1 mg/kg, i.p.), or atropine + propranolol. All data were collected using the Dataquest A.R.T. software (Transoma Medical). Experiments were performed as previously described (73, 74).
Statistical Analysis
GraphPad Prism 6.0 software was used for all statistical analysis. Values are presented as mean ± SEM. One-way or two-way ANOVA, followed by appropriate post-hoc tests for multiple comparisons, and a two-tailed two-sample equal variance Student's t-test were performed to determine statistical significance. The statistical significance of differences between groups of animals in survival experiments was analyzed by Log-rank test. P values equal to or below 0.05 were considered significant.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
All animal experiments were performed in accordance with the National Institutes of Health Guidelines under protocols approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee of the Feinstein Institute for Medical Research, Northwell Health, Manhasset, NY and the Institutional Animal Care and Use Committee at the University of Western Ontario (Protocols 2018-103 and 2018-104).
Author Contributions
KRL, MAMP, KJT, and VAP designed research. KRL, MEA, HAS, MA-O, AR, YL, PSO, SSC, RG, VFP, and VAP performed research. NMN, YA-A, RG, VFP, and MAMP contributed reagents, analytic tools and knockout and transgenic mice. KRL, HAS, RG, YA-A, CNM, VFP, MAMP, KJT, and VAP analyzed and interpreted data. KRL, KJT, and VAP wrote the manuscript. NMN, CNM, PSO, YA-A, and MAMP provided additional comments to finalize the paper.
Conflict of Interest Statement
VAP, SSC, and KJT are inventors on patents broadly related to the topic of this paper and have assigned their rights to the Feinstein Institute for Medical Research. YL was employed by SetPoint Medical Corporation (Valencia, CA 91355, USA).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the following grants from the National Institute of General Medical Sciences, NIH: RO1GM089807 (to VAP and KJT), and RO1GM057226 (to KJT) and CIHR (MP 93651, 12600, 89919) (to VFP and MAMP).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00585/full#supplementary-material
References
1. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428–35. doi: 10.1038/nature07201
2. Pavlov VA, Chavan SS, Tracey KJ. Molecular and functional neuroscience in immunity. Annu. Rev. Immunol. (2018) 36:783–812. doi: 10.1146/annurev-immunol-042617-053158
3. Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci USA. (2014) 111:12157–62. doi: 10.1073/pnas.1401712111
4. Xu H, Turnquist HR, Hoffman R, Billiar TR. Role of the IL-33-ST2 axis in sepsis. Mil Med Res. (2017) 4:3. doi: 10.1186/s40779-017-0115-8
5. Chen J, Kieswich JE, Chiazza F, Moyes AJ, Gobbetti T, Purvis GS, et al. IkappaB kinase inhibitor attenuates sepsis-induced cardiac dysfunction in CKD. J Am Soc Nephrol. (2017) 28:94–105. doi: 10.1681/ASN.2015060670
6. Rothbard JB, Rothbard JJ, Soares L, Fathman CG, Steinman L. Identification of a common immune regulatory pathway induced by small heat shock proteins, amyloid fibrils, and nicotine. Proc Natl. Acad Sci USA. (2018) 115:7081–6. doi: 10.1073/pnas.1804599115
7. Hoover DB. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol. Ther. (2017) 179:1–16. doi: 10.1016/j.pharmthera.2017.05.002
8. Yuan PQ, Tache Y. Abdominal surgery induced gastric ileus and activation of M1-like macrophages in the gastric myenteric plexus: prevention by central vagal activation in rats. Am J Physiol Gastrointest Liver Physiol. (2017) 313:G320–9. doi: 10.1152/ajpgi.00121.2017
9. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. (2002) 195:781–8. doi: 10.1084/jem.20011714
10. Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, et al. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci USA. (2006) 103:5219–23. doi: 10.1073/pnas.0600506103
11. Munyaka P, Rabbi MF, Pavlov VA, Tracey KJ, Khafipour E, Ghia JE. Central muscarinic cholinergic activation alters interaction between splenic dendritic cell and CD4+CD25- T cells in experimental colitis. PLoS ONE. (2014) 9:e109272. doi: 10.1371/journal.pone.0109272
12. Lee ST, Chu K, Jung KH, Kang KM, Kim JH, Bahn JJ, et al. Cholinergic anti-inflammatory pathway in intracerebral hemorrhage. Brain Res. (2010) 1309:164–71. doi: 10.1016/j.brainres.2009.10.076
13. Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, et al. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. (2009) 23:41–5. doi: 10.1016/j.bbi.2008.06.011
14. Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, Ghia JE. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. (2014) 7:335–47. doi: 10.1038/mi.2013.52
15. Yang Y, Peng Y, Yang J. Galantamine protects against hydrochloric acid aspiration-induced acute respiratory distress syndrome in rabbits. Trop J Pharm Res. (2018) 17:669–73. doi: 10.4314/tjpr.v17i4.15
16. Song JG, Li HH, Cao YF, Lv X, Zhang P, Li YS, et al. Electroacupuncture improves survival in rats with lethal endotoxemia via the autonomic nervous system. Anesthesiology. (2012) 116:406–14. doi: 10.1097/ALN.0b013e3182426ebd
17. Pham GS, Wang LA, Mathis KW. Pharmacological potentiation of the efferent vagus nerve attenuates blood pressure and renal injury in a murine model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. (2018) 315:R1261–71. doi: 10.1152/ajpregu.00362.2017
19. Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. (2011) 334:98–101. doi: 10.1126/science.1209985
20. Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci USA. (2016) 113:8284–9. doi: 10.1073/pnas.1605635113
21. Bonaz B, Sinniger V, Hoffmann D, Clarencon D, Mathieu N, Dantzer C, et al. Chronic vagus nerve stimulation in Crohn's disease: a 6-month follow-up pilot study. Neurogastroenterol Motility. (2016) 28:948–53. doi: 10.1111/nmo.12792
22. Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience. (1983) 10:1185–201. doi: 10.1016/0306-4522(83)90108-2
23. Ballinger EC, Ananth M, Talmage DA, Role LW. Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron. (2016) 91:1199–218. doi: 10.1016/j.neuron.2016.09.006
24. Gielow MR, Zaborszky L. The Input-Output Relationship of the Cholinergic Basal Forebrain. Cell Rep. (2017) 18:1817–30. doi: 10.1016/j.celrep.2017.01.060
25. Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. (2012) 76:116–29. doi: 10.1016/j.neuron.2012.08.036
26. Prado MA, Reis RA, Prado VF, de Mello MC, Gomez MV, de Mello FG. Regulation of acetylcholine synthesis and storage. Neurochem Int. (2002) 41:291–9. doi: 10.1016/S0197-0186(02)00044-X
27. Al-Onaizi MA, Parfitt GM, Kolisnyk B, Law CS, Guzman MS, Barros DM, et al. Regulation of cognitive processing by hippocampal cholinergic tone. Cereb. Cortex. (2017) 27:1615–28. doi: 10.1093/cercor/bhv349
28. Takacs VT, Cserep C, Schlingloff D, Posfai B, Szonyi A, Sos KE, et al. Co-transmission of acetylcholine and GABA regulates hippocampal states. Nat. Commun. (2018) 9:2848. doi: 10.1038/s41467-018-05136-1
29. Case DT, Burton SD, Gedeon JY, Williams SG, Urban NN, Seal RP. Layer- and cell type-selective co-transmission by a basal forebrain cholinergic projection to the olfactory bulb. Nat Commun. (2017) 8:652. doi: 10.1038/s41467-017-00765-4
30. Kljakic O, Janickova H, Prado VF, Prado MAM. Cholinergic/glutamatergic co-transmission in striatal cholinergic interneurons: new mechanisms regulating striatal computation. J Neurochem. (2017) 142(Suppl. 2):90–102. doi: 10.1111/jnc.14003
31. Saunders A, Granger AJ, Sabatini BL. Corelease of acetylcholine and GABA from cholinergic forebrain neurons. eLife. (2015) 4:e06412. doi: 10.7554/eLife.06412
32. Waldburger JM, Boyle DL, Edgar M, Sorkin LS, Levine YA, Pavlov VA, et al. Spinal p38 MAP kinase regulates peripheral cholinergic outflow. Arthritis Rheum. (2008) 58:2919–21. doi: 10.1002/art.23807
33. Volpicelli LA, Levey AI. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Progr Brain Res. (2004) 145:59–66. doi: 10.1016/S0079-6123(03)45003-6
34. Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. (2009) 29:14271–86. doi: 10.1523/JNEUROSCI.3930-09.2009
35. Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA. (2009) 106:15950–5. doi: 10.1073/pnas.0900903106
36. Rosas-Ballina M, Valdes-Ferrer SI, Dancho ME, Ochani M, Katz D, Cheng KF, et al. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav Immun. (2015) 44:19–27. doi: 10.1016/j.bbi.2014.07.010
37. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. (2006) 203:1623–8. doi: 10.1084/jem.20052362
38. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci USA. (2008) 105:11008–13. doi: 10.1073/pnas.0803237105
39. Suarez AN, Hsu TM, Liu CM, Noble EE, Cortella AM, Nakamoto EM, et al. Gut vagal sensory signaling regulates hippocampus function through multi-order pathways. Nat Commun. (2018) 9:2181. doi: 10.1038/s41467-018-04639-1
40. Kolisnyk B, Guzman MS, Raulic S, Fan J, Magalhaes AC, Feng G, et al. (2013). ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J Neurosci. 33: 10427–38. doi: 10.1523/JNEUROSCI.0395-13.2013
41. Janickova H, Rosborough K, Al-Onaizi M, Kljakic O, Guzman MS, Gros R, et al. (2017). Deletion of the vesicular acetylcholine transporter from pedunculopontine/laterodorsal tegmental neurons modifies gait. J Neurochem. 140:787–98. doi: 10.1111/jnc.13910
42. Broncel A, Bocian R, Klos-Wojtczak P, Konopacki J. Medial septal cholinergic mediation of hippocampal theta rhythm induced by vagal nerve stimulation. PLoS ONE. (2018) 13:e0206532. doi: 10.1371/journal.pone.0206532
43. Hickman JL PX, Donegan D, Welle CG. Temporally precise vagus nerve stimulation (VNS) enhances motor learning and performance of a skilled forelimb reach task. In: Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience Online, Society for Neuroscience (2018).
44. Tetrault S, Chever O, Sik A, Amzica F. Opening of the blood-brain barrier during isoflurane anaesthesia. Eur J Neurosci. (2008) 28:1330–41. doi: 10.1111/j.1460-9568.2008.06443.x
45. Consolim-Colombo FM, Sangaleti CT, Costa FO, Morais TL, Lopes HF, Motta JM, et al. Galantamine alleviates inflammation and insulin resistance in patients with metabolic syndrome in a randomized trial. JCI Insight. (2017) 2:93340. doi: 10.1172/jci.insight.93340
46. Ye H, Fussenegger M. Optogenetic medicine: synthetic therapeutic solutions precision-guided by light. Cold Spring Harb Perspect Med. (2018). doi: 10.1101/cshperspect.a034371
47. Boyden ES. Optogenetics and the future of neuroscience. Nat Neurosci. (2015) 18:1200–1. doi: 10.1038/nn.4094
48. Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci. (2015) 18:1213–25. doi: 10.1038/nn.4091
49. Montgomery KL, Iyer SM, Christensen AJ, Deisseroth K, Delp SL. Beyond the brain: Optogenetic control in the spinal cord and peripheral nervous system. Sci Transl Med. (2016) 8:337rv335. doi: 10.1126/scitranslmed.aad7577
50. Lee DJ, Gurkoff GG, Izadi A, Berman RF, Ekstrom AD, Muizelaar JP, et al. Medial septal nucleus theta frequency deep brain stimulation improves spatial working memory after traumatic brain injury. J Neurotrauma. (2013) 30:131–9. doi: 10.1089/neu.2012.2646
51. Lee DJ, Gurkoff GG, Izadi A, Seidl SE, Echeverri A, Melnik M, et al. Septohippocampal neuromodulation improves cognition after traumatic brain injury. J Neurotrauma. (2015) 32:1822–32. doi: 10.1089/neu.2014.3744
52. Zimerman M, Hummel FC. Non-invasive brain stimulation: enhancing motor and cognitive functions in healthy old subjects. Front Aging Neurosci. (2010) 2:149. doi: 10.3389/fnagi.2010.00149
53. Cabrera LY, Evans EL, Hamilton RH. Ethics of the electrified mind: defining issues and perspectives on the principled use of brain stimulation in medical research and clinical care. Brain Topogr. (2014) 27:33–45. doi: 10.1007/s10548-013-0296-8
54. Fregni F, Pascual-Leone A. Technology insight: noninvasive brain stimulation in neurology-perspectives on the therapeutic potential of rTMS and tDCS. Nat Clin Pract Neurol. (2007) 3:383–93. doi: 10.1038/ncpneuro0530
55. Kokras N, Stamouli E, Sotiropoulos I, Katirtzoglou EA, Siarkos KT, Dalagiorgou G, et al. Acetyl cholinesterase inhibitors and cell-derived peripheral inflammatory cytokines in early stages of alzheimer's disease. J Clin Psychopharmacol. (2018) 38:138–43. doi: 10.1097/JCP.0000000000000840
56. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. (2007) 7:161–7. doi: 10.1038/nri2015
57. Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. (2009) 73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95
58. Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. (2018) 141:1917–33. doi: 10.1093/brain/awy132
59. McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. (2017) 9:14. doi: 10.1186/s13195-017-0241-2
60. Zaghloul N, Addorisio ME, Silverman HA, Patel HL, Valdes-Ferrer SI, Ayasolla KR, et al. Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front Immunol. (2017) 8:1673. doi: 10.3389/fimmu.2017.01673
61. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. (2008) 9:46–56. doi: 10.1038/nrn2297
62. Coltart I, Tranah TH, Shawcross DL. Inflammation and hepatic encephalopathy. Arch Biochem Biophys. (2013) 536:189–96. doi: 10.1016/j.abb.2013.03.016
63. Felipo V. Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci. (2013) 14:851–8. doi: 10.1038/nrn3587
64. Metz CN, Pavlov VA. Vagus nerve cholinergic circuitry to the liver and the gastrointestinal tract in the neuroimmune communicatome. Am J Physiol Gastrointest Liver Physiol. (2018) 315:G651–8. doi: 10.1152/ajpgi.00195.2018
65. Garcia-Ayllon MS, Cauli O, Silveyra MX, Rodrigo R, Candela A, Compan A, et al. Brain cholinergic impairment in liver failure. Brain. (2008) 131:2946–56. doi: 10.1093/brain/awn209
66. Maldonado JR. Neuropathogenesis of delirium: review of current etiologic theories and common pathways. Am J Geriatr Psychiatry. (2013) 21:1190–222. doi: 10.1016/j.jagp.2013.09.005
67. Hshieh TT, Fong TG, Marcantonio ER, Inouye SK. Cholinergic deficiency hypothesis in delirium: a synthesis of current evidence. J Gerontol Ser A Biol Sci Med Sci. (2008) 63:764–72. doi: 10.1093/gerona/63.7.764
68. Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. (2011) 8:745–52. doi: 10.1038/nmeth.1668
69. Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, et al. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci USA. (1997) 94:13311–6. doi: 10.1073/pnas.94.24.13311
70. Kow RL, Cheng EM, Jiang K, Le JH, Stella N, Nathanson NM. (2015). Muscarinic M1 receptor and cannabinoid CB1 receptor do not modulate paraoxon-induced seizures. Pharmacol Res Perspect. 3:e00100. doi: 10.1002/prp2.100
71. Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, et al. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. (2008) 14:567–74. doi: 10.2119/2008-00079.Parrish
72. Tarnawski L, Reardon C, Caravaca AS, Rosas-Ballina M, Tusche MW, Drake AR, et al. Adenylyl cyclase 6 mediates inhibition of TNF in the inflammatory reflex. Front Immunol. (2018) 9:2648. doi: 10.3389/fimmu.2018.02648
73. Roy A, Dakroub M, Tezini GC, Liu Y, Guatimosim S, Feng Q, et al. Cardiac acetylcholine inhibits ventricular remodeling and dysfunction under pathologic conditions. FASEB J. (2016) 30:688–701. doi: 10.1096/fj.15-277046
Keywords: forebrain cholinergic, cytokines, inflammation, vagus nerve, endotoxemia, sepsis, neural regulation
Citation: Lehner KR, Silverman HA, Addorisio ME, Roy A, Al-Onaizi MA, Levine Y, Olofsson PS, Chavan SS, Gros R, Nathanson NM, Al-Abed Y, Metz CN, Prado VF, Prado MAM, Tracey KJ and Pavlov VA (2019) Forebrain Cholinergic Signaling Regulates Innate Immune Responses and Inflammation. Front. Immunol. 10:585. doi: 10.3389/fimmu.2019.00585
Received: 14 December 2018; Accepted: 05 March 2019;
Published: 02 April 2019.
Edited by:
Pietro Ghezzi, Brighton and Sussex Medical School, United KingdomReviewed by:
Bruno Bonaz, Centre Hospitalier Universitaire de Grenoble, FranceEgle Solito, Queen Mary University of London, United Kingdom
Copyright © 2019 Lehner, Silverman, Addorisio, Roy, Al-Onaizi, Levine, Olofsson, Chavan, Gros, Nathanson, Al-Abed, Metz, Prado, Prado, Tracey and Pavlov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentin A. Pavlov, dnBhdmxvdkBub3J0aHdlbGwuZWR1
†These authors have contributed equally to this work