 Silvia Sánchez-Ramón1*
Silvia Sánchez-Ramón1* Arancha Bermúdez2
Arancha Bermúdez2 Luis Ignacio González-Granado3
Luis Ignacio González-Granado3 Carlos Rodríguez-Gallego4Ana Sastre5
Carlos Rodríguez-Gallego4Ana Sastre5 Pere Soler-Palacín6 and the ID-Signal Onco-Haematology Group
Pere Soler-Palacín6 and the ID-Signal Onco-Haematology Group- 1Hospital Clínico San Carlos, Madrid, Spain
- 2Hospital U. Marqués de Valdecilla, Santander, Spain
- 3Hospital U. 12 Octubre, Madrid, Spain
- 4Hospital Universitario de Gran Canaria Dr. Negrín, Las Palmas de Gran Canaria, Canary Islands, Spain
- 5Hospital U. La Paz, Madrid, Spain
- 6Hospital U. Vall d'Hebron, Barcelona, Spain
Background: Immunodeficiencies (ID), in particular primary immunodeficiencies (PID), are often associated with haematological manifestations, such as peripheral cytopenias or lymphoproliferative syndromes. Early diagnosis and management have significant prognostic implications. Secondary immunodeficiencies (SID) may also be induced by oncohaematological diseases and their treatments. Haematologists and oncologists must therefore be aware of the association between blood disorders and cancer and ID, and be prepared to offer their patients appropriate treatment without delay. Our aim was to define the warning signs of primary and secondary IDs in paediatric and adult patients with oncohaematological manifestations.
Methods: A multidisciplinary group of six experts (2 haematologists, 2 immunologists, and 2 paediatricians specializing in ID) conducted a literature review and prepared a document based on agreements reached an in-person meeting. An external group of 44 IDs specialists from all over Spain assessed the document and were consulted regarding their level of agreement.
Results: This document identifies the haematological and extra-haematological diseases that should prompt a suspicion of PIDs in adults and children, in both primary care and haematology and oncology departments. Cytopenia and certain lymphoproliferative disorders are key diagnostic pointers. The diagnosis must be based on a detailed clinical history, physical exploration, complete blood count and standard laboratory tests. The immunological and haematological tests included in the diagnostic process will depend on the care level. Patients who are candidates for immunoglobulin replacement therapy must be carefully selected, and treatment should be offered as soon as possible to avoid the development of complications. Finally, this document recommends procedures for monitoring these patients.
Conclusions: This document combines scientific evidence with the opinion of a broad panel of experts, and emphasizes the importance of an early diagnosis and treatment to avoid complications. The resulting document is a useful tool for primary care physicians and specialists who see both adult and paediatric patients with oncohaematological diseases.
Introduction
Immunodeficiencies (IDs) are a group of diseases caused by quantitative and/or functional changes in the different mechanisms involved in both the innate and the adaptive immune response (1, 2). They are classified as primary immunodeficiency diseases (PIDs) if their origin is genetic, and secondary (SIDs) if their origin is acquired. Both types of IDs are associated with or predispose towards complications, such as infections, autoimmune disorders, immune dysregulation with lymphoproliferation, inflammatory disorders, lymphomas, and other types of cancer, many of which are diagnosed and treated in haematology and oncology departments. PIDs comprise a heterogeneous group of around 400 diseases (3). The Primary Immunodeficiencies Classification Committee of the International Union of Immunology Societies (IUIS) identified 8 large groups of PIDs (9 if phenocopies are included), depending on the underlying immune disorder or the predominant symptom, the most frequent being antibody deficiencies, well-defined syndromes and phagocyte function defects (3). SID, in contrast, is the result of systemic disorders [including haematological disorders, such as chronic lymphocytic leukaemia [CLL]], drugs (e.g., chemotherapeutic agents) and long-term critical or severe diseases (4), that often occur concomitantly in a single patient.
The main haematological manifestations associated with PIDs and SIDs are peripheral cytopenias and immunological dysregulation syndromes (5, 6). These disorders are of particular relevance since they are common in clinical practice, but in many cases they are not listed in the accepted compendia of warning signs for the diagnosis of IDs in children and adults, increasing the risk of failing to diagnose the underlying immunological defect.
Healthcare professionals who see patients with IDs must be aware of these manifestations, so that early diagnosis can be made and treatment can be started promptly. Improving awareness is a crucial step in preventing underdiagnosis and delayed diagnosis, and can help avoid complications (7, 8), improve patient prognosis, lessen the impact on the family, and reduce the social and economic burden of the disease (9). To achieve these aims, specific consensus documents directed at healthcare professionals who see patients with IDs are needed. This document reports the main conclusions of a wide range of specialists on the diagnosis and management of PIDs with haematological manifestations and SIDs associated with oncohaematological disease, and the treatment of these disorders. Our objective was to foster early diagnosis and appropriate treatment of this population. This study is part of the ID-Signal Project, which aims to produce a series of documents describing the clinical manifestations of IDs as they affect the different body systems. The first of this series is a recently published paper on IDs associated with respiratory diseases (10), and other forthcoming documents will focus on rheumatology and neurology.
Material and Methods
A multidisciplinary group of experts formed of 2 haematologists, 2 immunologists, and 2 paediatricians specializing in IDs identified the issues to be addressed. A review of the literature was then performed, and this review acted as the basis for an in-person discussion of the items to be included in the document. The main conclusions and recommendations were then forwarded to an external panel of experts for their individual evaluation, depending on their speciality. The external panel consisted of 44 experts from all over Spain, with experience in the management of ID, working in Spanish National Health System centres: 14 paediatricians specializing in immunodeficiencies, 17 immunologists who see adult patients and/or perform immunological laboratory assessments of children and adults with suspected ID, 6 paediatricians specializing in haematology and 7 haematologists who see adult patients. This panel indicated their level of agreement on a scale of 1–4, with 1 reflecting “strongly disagree” and 4 “strongly agree.” The results were pooled and the percentages of votes in the categories 1 and 2 (disagreement) and in the categories 3 and 4 (agreement) were calculated. Unanimity was defined as 100% of the experts agreeing with the recommendation/conclusion; consensus, when at least 80% of the experts agreed without unanimity; majority, when >65% and <80% of the experts agreed with the recommendation/conclusion; and disagreement, when the percentage of agreement was 65% or less.
Results and Discussion
Haematological Signs That Should Prompt a Suspicion of PID
To date, the causative genetic defect has been identified in 344 of the different PIDs (3). The IUIS classification divides these PIDs into 9 categories (3). At present, autoimmunity and dysregulation of the immune system are thought to be the underlying causes of a growing number of PIDs that typically manifest initially as cytopenias. In PIDs specifically, cytopenia may be caused by cellular or humoral autoimmunity, immune dysfunction in the form of hemophagocytosis or lymphoproliferation with or without splenic sequestration, bone marrow failure with myelodysplasia or secondary myelosuppression (11). Table 1 shows the haematological signs or manifestations that, according to the panel of experts, should prompt suspicion of PIDs in primary care and haematology consultations, differentiating between paediatric and adult patients. The panel also agreed on the common haematological manifestations that might appear in each of the PIDs categories (Table 2). Some extra-haematological diseases, such as skin manifestations, gastrointestinal disorders, recurrent pneumonia, or growth retardation, among others, should also be taken into consideration.
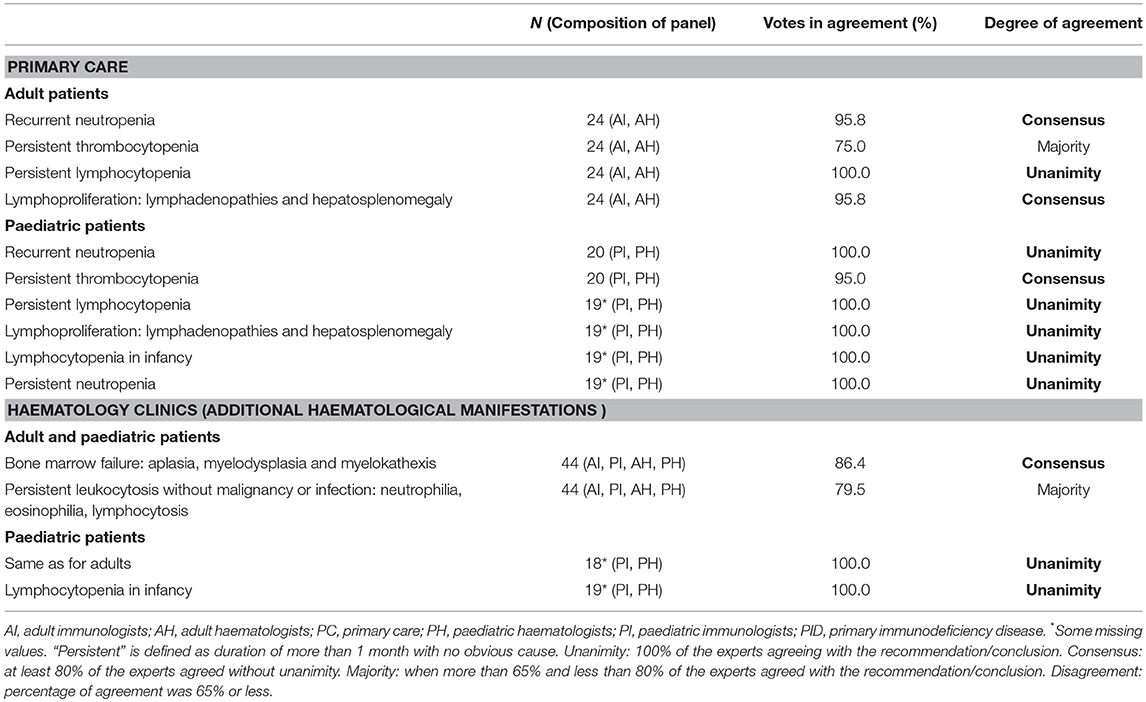
Table 1. Haematological manifestations that should prompt a suspicion of PID in primary care and haematology consultations.
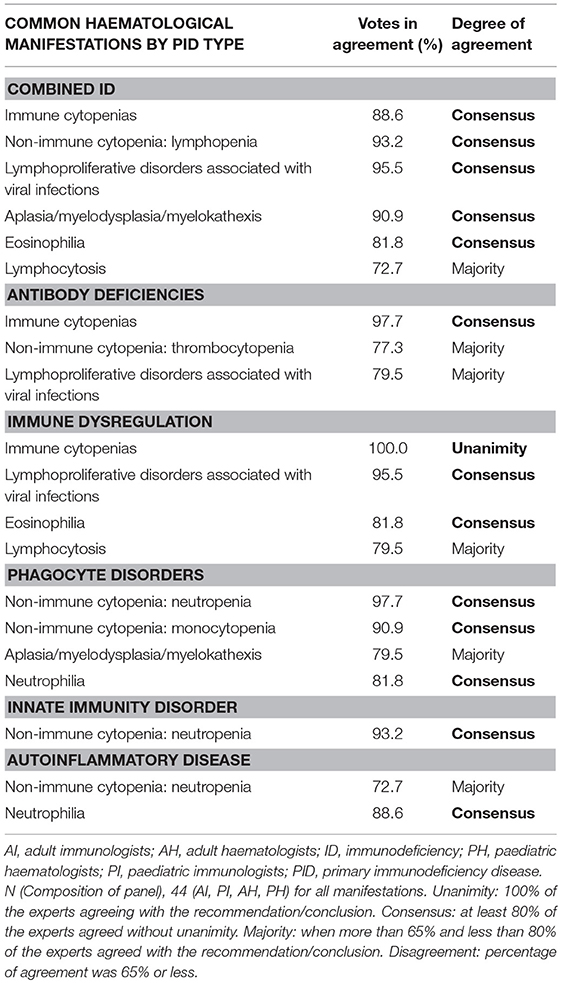
Table 2. Common haematological manifestations by PID type.
Diagnostic Approach When PID Is Suspected
Most PIDs are diagnosed with a complete, targeted clinical history, detailed physical examination, complete blood count, and standard laboratory tests, including the determination of serum immunoglobulin (Ig) levels. The clinical history should include family history of PID, consanguinity or family history of sudden death at an early age. Physical examination should include an assessment of nutritional status, sequelae from previous infections, lymph nodes, tonsils, hepatosplenomegaly, etc. A complete blood count and blood smear will rule out cytopenia or cell abnormalities (12). These basic laboratory tests should also include liver and kidney function, total protein and albumin. Determination of serum Ig (IgG, IgM, IgA, and IgE) is the first step in the evaluation of humoral immunity, and will help diagnose quantitative Ig deficiencies, such as congenital agammaglobulinemia, common variable ID, or IgA deficiency, and other antibody abnormalities associated with defects such as hyper-IgE or hyper-IgM syndrome (12).
It is important to assess the results according to the reference values for each age, since there are significant differences which, if not taken into account, can lead to the PID being overlooked or also giving a wrong diagnosis of PID (13, 14). When diagnosis remains uncertain after all these tests and suspicion is high, additional tests, such as functional or molecular studies, must be performed in reference centres. Table 3 specifies the additional immunological and haematological tests, according to care level, and in the case of immunological tests, according to the type of PID suspected. Although all the immunological tests reached consensus (Table 3), some important proposals were made that the expert panel agreed to include separately from the table. In terms of haematological assessment, cytological studies of bone marrow aspirate and biopsy specimens are strongly recommended (11). Bone marrow and/or tissue immunophenotyping is recommended, depending on the disease under consideration, for example, in the case of lymphoproliferative disease. Finally, cytogenetic studies are recommended in patients with bone marrow failure.
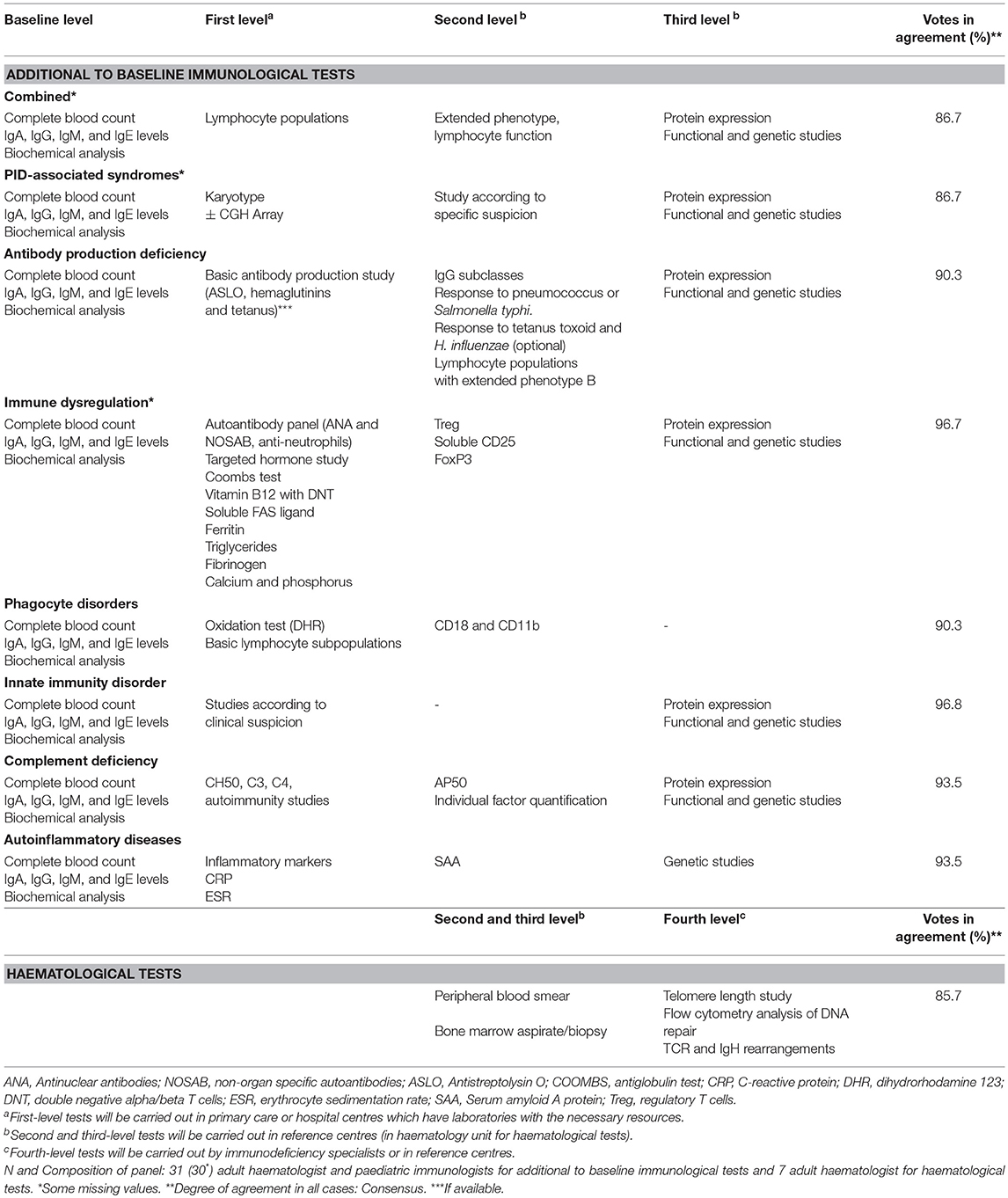
Table 3. Additional to baseline immunological and haematological tests in patients with suspected PID.
In summary, the entire diagnostic process must combine immunological and haematological approaches, especially when cytopenia is the initial manifestation of PID, in order to guide and accelerate the differential diagnosis of a significant number of autoimmune diseases, lymphoproliferative disorders and others (11). This approach will help minimize the damage that might occur if the PID diagnosis is delayed (15).
Secondary Immunodeficiencies in Oncohaematology
SID is much more common than PID in oncohaematology and in general. The causes of SID in oncohaematology are summarized as follows: i) Systemic disorders: aplastic anaemia, haematological malignancies, such as CLL, multiple myeloma (MM), Hodgkin's disease, non-Hodgkin lymphoma (NHL), graft vs. host disease, and sickle cell disease; ii) Iatrogenic disorders caused by certain drugs (chemotherapy, immunosuppressants, corticosteroids, monoclonal antibodies, such as anti-CD20 agents, and B cell differentiation and maturation inhibitors), radiation therapy, splenectomy and bone marrow ablation before transplantation; iii) Prolonged severe disease, particularly in critically ill, elderly, and hospitalized patients.
These IDs appear clinically as more frequent infections or unusual infective complications and, occasionally, as opportunistic infections (4). Immune system abnormalities induced by SIDs affect both innate and adaptive immunity; they can be subtle and clinical manifestations are often heterogeneous.
In the case of haematological malignancies, such as MM, CLL, and lymphomas, both the underlying disease and the immunosuppressive treatment can contribute in different ways to the emergence of SID.
The scientific consensus committee agreed that the more common haematological conditions and situations that may predispose to SIDs are the following:
• Post-transplantation with partial immune recovery
• CLL
• MM
• NHL
• Treatment with rituximab and new generations of anti-CD20 monoclonal antibodies, alemtuzumab, or combination protocols that include the administration of fludarabine.
• New therapeutic strategies targeting the B cell receptor signalling pathway (e.g., Syk and PI3K enzymes or mTOR transcription factor), the recruitment of cytotoxic B cells [e.g., T cell therapy with chimeric antigen receptor [CAR]], and B cell apoptosis inducers (e.g., navitoclax).
When SID is suspected, basic screening procedures, similar to those performed to rule out PID, should be conducted. This involves a complete blood count, quantification of serum immunoglobulins, and biochemistry panel (basic liver and kidney function, total protein and albumin). Antibody production assessment is also recommended when possible. Additional haematological tests recommended by the panel of experts are listed in Table 4.
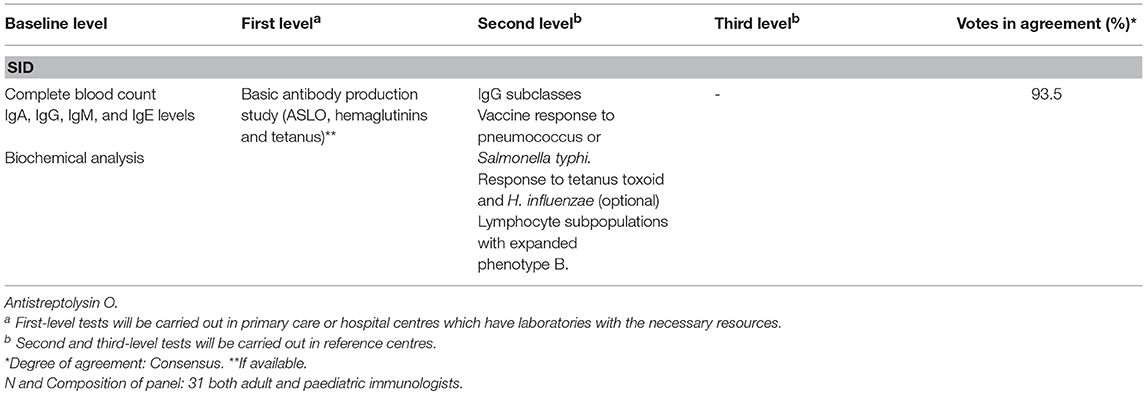
Table 4. Haematological tests to be performed in patients with suspected SID.
Intravenous and Subcutaneous Immunoglobulin Replacement Therapy for PID and SID With Haematological Manifestations
Immunoglobulin replacement therapy (IGRT) is essential in patients with PIDs and SIDs that directly affect B cell function and antibody production and, in the case of SID, that present with severe or recurrent infections (16). The indications for IGRT continue to expand, as the characteristics, effects, and clinical presentations of IDs become better known.
Treatment of Primary Immunodeficiencies
In currently characterized PIDs phenotypes, intravenous, or subcutaneous IGRT is indicated in agammaglobulinemia due to the absence of B cells, and in hypogammaglobulinemia with deficient antibody production. However, the use of IGRT must be individually evaluated in patients with normal IgG and defective antibody function, hypogammaglobulinemia, and normal antibody function, isolated deficiency of IgG subclasses with recurrent infections, and recurrent infections due to complex, uncharacterized immunological mechanisms (17, 18).
Treatment of Secondary Immunodeficiencies
The use of subcutaneous or intravenous IGRT for SIDs is less well defined than for PIDs and is mainly based on experience in the treatment of the latter (18, 19). IGRT may be considered in highly selected patients with CLL who also have hypogammaglobulinemia with recurrent bacterial infections and deficient production of specific antibodies, since infections are currently the main cause of morbidity and mortality in these patients. Specific antibodies tests have been assigned to reference centres (second level) because with the exception of anti-tetanus IgG and Antistreptolysin O, other specific antibodies as pneumococcus or Salmonella typhi, are usually not available in primary care and even some regional hospitals. Moreover, it seems reasonable, at least in those cases without a clear evidence of primary antibody deficiencies to initially test for IgG levels and measure specific antibodies in a second level. IGRT is also an option in patients with MM and recurrent serious bacterial infections, in patients who have received haematopoietic stem cell transplantation, and in patients with many other diseases (cancer and inflammatory or autoimmune diseases) who have received treatments that deplete B cell levels (16, 20–22). The immunological impact on SIDs of new therapies that target key molecules in the differentiation, maturation or apoptosis of B cells has not yet been explored (23). Experience acquired with the first generation of anti-CD20 monoclonal antibodies has shown that low baseline levels of IgM and IgG in serum are associated with an increased risk of infection and a cumulative effect after repeated cycles has been observed (24). Secondary antibody responses do not seem to be affected following therapy with anti-CD20 monoclonal antibodies, but several immunization studies have shown that primary responses are markedly impaired (25). Recent data suggest that CD19-targeted CAR therapy might preserve patients' humoral immunity (26).
With regard to the choice between subcutaneous or intravenous administration, both routes have shown equivalence in terms of efficacy and safety (17, 27, 28), although systemic adverse effects are more common with the intravenous route, and milder local effects are observed with the subcutaneous route (28–30). Despite the need to train the patient and the requirement for simple devices for proper administration, the subcutaneous route has a series of significant advantages: it is associated with improved quality of life for both patients and their caregivers (28, 31–33); it has been shown to be more cost-effective, mainly due to fewer days missed from work and school (28); and it is a better solution for patients with problems for venous access and mobility issues. Finally, patient preferences are an essential part of the decision-making process (34).
Monitoring IDs Patients With Haematological Involvement
The number of infections and subsequent hospitalizations has decreased in patients with certain IDs in recent years, thanks to improvements in diagnosis and treatment. However, these patients still require regular clinical monitoring (12). Personalized decisions on the periodicity of monitoring can be made on the basis of the patients' age, clinical status and type of ID. Patients should be assessed by an immunologist at least once every 6–12 months, and a full clinical history, physical examination, complete blood count and biochemistry with LDH and immunoglobulins must be obtained (12). During dose adjustment, IgG values should be determined more frequently. Table 5 summarizes the agreements reached by the expert group.
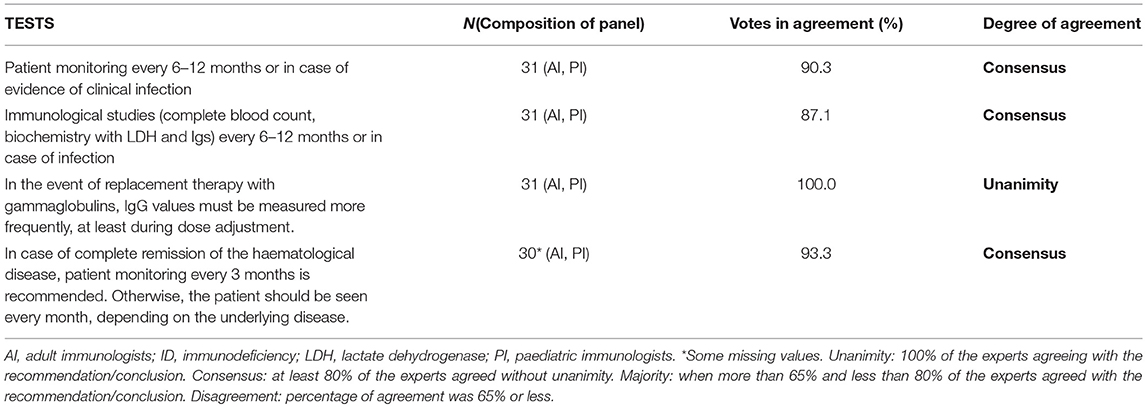
Table 5. Minimum tests to be performed during monitoring of the patient with ID and haematological symptoms.
Conclusions
Peripheral cytopenias and several lymphoproliferative disorders should prompt a suspicion of PID. The diagnosis must be based primarily on the clinical history, a detailed physical exploration, a complete blood count and the determination of serum Ig levels. Further immunological and haematological tests included in the diagnostic process will depend on the patient's clinical phenotype and health care level at which they are seen. Immunoglobulin replacement therapy, when indicated, should be offered as soon as possible to avoid complicated infections and organ damage.
This study is the first to focus on both primary and secondary IDs from an oncohaematological perspective. It addresses the diagnosis, treatment and follow-up of both adult and paediatric patients, and reflects the opinion and clinical experience of more than 40 interdisciplinary experts from different fields and specialities who regularly treat these patients. This document is intended to be a useful tool for doctors working in any field who might see patients with ID, to help them to identify the warning signs of an ID associated with haematology. Other approaches to promote the early identification of PIDs and SIDs should include the training of professionals.
Author Contributions
SS-R and PS-P made substantial contributions to the conception and design of the document, to the first draft of the document (before the evaluation of the external panel of experts), and also to the analysis and interpretation of data from the external panel of experts; they have also been involved in drafting and revising the manuscript critically for important intellectual content and have given final approval of the version to be published. AB, LG-G, CR-G, and AS made contributions to the first draft of the document (before the evaluation of the external panel of experts) and to the interpretation of data from the external panel of experts; they also have been involved in revising the manuscript and have given final approval of the version to be published. ID-Signal Onco-Haematology Group evaluated the conclusions and recommendations individually.
Funding
This article received funding fee from CSL Behring.
Conflict of Interest Statement
SS-R has served as speaker, consultant, and advisory member for or has received research funding from CSL Behring, Grifols, Shire, and Octapharma. AB reports personal fees from CSL Behring, outside the submitted work. LG-G reports grants from Fondo De Investigación Sanitaria Carlos III, from Shire, grants and personal fees from CSL Behring, during the conduct of the study. CR-G reports personal fees from CSL Behring, during the conduct of the study, and personal fees from Shire SL outside the submitted work. AS reports personal fees from CSL, during the conduct of the study. PS-P reports personal fees from CSL Behring, grants from Jeffrey Modell Foundation, during the conduct of the study; personal fees from Shire SL, personal fees from Grifols, outside the submitted work.
Acknowledgments
The authors of this work would like to thank CSL Behring for the financial support provided for the conduct of this study (grant to SS-R, PS-P, AB, LG-G, CR-G, and AS), the oncohaematology ID-Signal Group for their participation as members of the external panel, and Cristina Valiente and Verónica Albert of GOC Networking, for their methodological support throughout the course of the study. The consensus document has received the scientific endorsement of the Spanish Society of Immunology.
Consortium/Group Members
The ID-Signal Oncohaematology Group consists of the following members: Laura Alonso, Luis Allende, Laia Alsina, Ana María Bielsa, Sara Calleja-Antolín, Carmen Cámara, Javier Carbone, Carmen Carreras, Ana De Andrés Martín, Angela Deyá, Cristina Díaz de Heredia, Romina Dieli-Crimi, José Luis Díez, Nerea Domínguez-Pinilla, Luis Fernández-Pereira, José Ma García, Juana Gil-Herrera, Antonio Gutiérrez, Isidro Jarque, Manel Juan, Francisco Lendínez, Pilar Llobet, Mónica López, Ana López de la Guía, Marcos López-Hoyos, Andrea Martín-Nalda, Mónica Martínez, Josefa Melero, Ana Méndez-Echevarría, Pedro Moral, Olaf Neth, María Núñez, Gonzalo Ocejo-Vinyals, Juliana Ochoa-Grullón, Peter Olbrich, Raquel Oña, Manuel Pérez-Encinas, Jaime Pons, Carmen Rodríguez, Berta Sánchez, Juan Luis Santos-Pérez, Ma Elena Seoane, Alexandru Vlagea.
References
1. Espanol T, Hernandez M, Giner MT, Casas C, Gurbindo D, Marco T, et al. Directory of diagnostic tests in primary immunodeficiencies. Allergol Immunopathol (Madr). (2005) 33:157–61. doi: 10.1157/13075699
2. García Martínez J, Santos-Díez L, Dopazo L. Diagnóstico de las inmunodeficiencias primarias. Protoc diagn Ter pediatr. (2013) 1:81–92. Available online at: https://www.aeped.es/sites/default/files/documentos/7-inmunodeficiencias_primarias_0.pdf
3. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
4. Chinen J, Shearer WT. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol. (2010) 125 (2 Suppl. 2):S195–203. doi: 10.1016/j.jaci.2009.08.040
5. Costa-Carvalho BT, Grumach AS, Franco JL, Espinosa-Rosales FJ, Leiva LE, King A, et al. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J Clin Immunol. (2014) 34:10–22. doi: 10.1007/s10875-013-9954-6
6. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers related to immunodeficiencies: update and perspectives. Front Immunol. (2016) 7:365. doi: 10.3389/fimmu.2016.00365
7. Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol. (2002) 104:221–30. doi: 10.1006/clim.2002.5241
8. Ballow M, Notarangelo L, Grimbacher B, Cunningham-Rundles C, Stein M, Helbert M, et al. Immunodeficiencies. Clin Exp Immunol. (2009) 158 (Suppl. 1):14–22. doi: 10.1111/j.1365-2249.2009.04023.x
9. Modell V, Gee B, Lewis DB, Orange JS, Roifman CM, Routes JM, et al. Global study of primary immunodeficiency diseases (PI)–diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol res. (2011) 51:61–70. doi: 10.1007/s12026-011-8241-y
10. Soler-Palacín P, de Gracia J, González-Granado LI, Martín C, Rodríguez-Gallego C, Sánchez-Ramón S, et al. Primary immunodeficiency diseases in lung disease: warning signs, diagnosis and management. Respir Res. (2018) 19:219. doi: 10.1186/s12931-018-0923-8
11. Seidel MG. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood. (2014) 124:2337–44. doi: 10.1182/blood-2014-06-583260
12. Roxo Junior P. Primary immunodeficiency diseases: relevant aspects for pulmonologists. J bras pneumol. (2009) 35:1008–17. doi: 10.1590/S1806-37132009001000010
13. Garcia-Prat M, Vila-Pijoan G, Martos Gutierrez S, Gala Yerga G, García Guantes E, Martínez-Gallo M, et al. Age-specific pediatric reference ranges for immunoglobulins and complement proteins on the optilite. J Clin Lab Anal. (2018) 30:e22420. doi: 10.1002/jcla.22420
14. Garcia-Prat M, Álvarez-Sierra D, Aguiló-Cucurull A, Salgado-Perandrés S, Briongos-Sebastian S, Franco-Jarava C, et al. Extended immunophenotyping reference values in a healthy pediatric population. Cytometry B Clin cytom. (2018) doi: 10.1002/cyto.b.21728 [Epubh ahead of print].
15. Burroughs L, Woolfrey A. Hematopoietic cell transplantation for treatment of primary immune deficiencies. Cell ther transplant. (2010) 2. doi: 10.3205/ctt-2010-en-000077.01
16. Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. (2017) 139 (3s):S1–46. doi: 10.1016/j.jaci.2016.09.023
17. Stiehm ER, Orange JS, Ballow M, Lehman H. Therapeutic use of immunoglobulins. Adv pediatr. (2010) 57:185–218. doi: 10.1016/j.yapd.2010.08.005
18. Jolles S, Chapel H, Litzman J. When to initiate immunoglobulin replacement therapy (IGRT) in antibody deficiency: a practical approach. Clin Exp Immunol. (2017) 188:333–41. doi: 10.1111/cei.12915
19. Srivastava S, Wood P. Secondary antibody deficiency - causes and approach to diagnosis. Clin Med (Lond). (2016) 16:571–6. doi: 10.7861/clinmedicine.16-6-571
20. Mikulska M, Lanini S, Gudiol C, Drgona L, Ippolito G, Fernández-Ruiz M, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) consensus document on the safety of targeted and biological therapies: an infectious diseases perspective (agents targeting lymphoid cells surface antigens [I]: CD19, CD20 and CD52). Clin microbiol infect. (2018) 24 (Suppl. 2):S71–82. doi: 10.1016/j.cmi.2018.02.003
21. Bonanni A, Calatroni M, D'Alessandro M, Signa S, Bertelli E, Cioni M, et al. Adverse events linked with the use of chimeric and humanized anti-CD20 antibodies in children with idiopathic nephrotic syndrome. Br J Clin Pharmacol. (2018) 84:1238–49. doi: 10.1111/bcp.13548
22. Franks SE, Getahun A, Hogarth PM, Cambier JC. Targeting B cells in treatment of autoimmunity. Curr Opin Immunol. (2016) 43:39–45. doi: 10.1016/j.coi.2016.09.003
23. Sanchez-Ramon S, Dhalla F, Chapel H. Challenges in the role of gammaglobulin replacement therapy and vaccination strategies for hematological malignancy. Front Immunol. (2016) 7:317. doi: 10.3389/fimmu.2016.00317
24. Roberts DM, Jones RB, Smith RM, Alberici F, Kumaratne DS, Burns S, et al. Rituximab-associated hypogammaglobulinemia: incidence, predictors and outcomes in patients with multi-system autoimmune disease. J autoimmun. (2015) 57:60–5. doi: 10.1016/j.jaut.2014.11.009
25. Pescovitz MD, Torgerson TR, Ochs HD, Ocheltree E, McGee P, Krause-Steinrauf H, et al. Effect of rituximab on human in vivo antibody immune responses. J Allergy Clin Immunol. (2011) 128:1295–302.e5. doi: 10.1016/j.jaci.2011.08.008
26. Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. (2016) 128:360–70. doi: 10.1182/blood-2016-01-694356
27. Gardulf A, Nicolay U, Asensio O, Bernatowska E, Böck A, Carvalho BC, et al. Rapid subcutaneous IgG replacement therapy is effective and safe in children and adults with primary immunodeficiencies–a prospective, multi-national study. J Clin Immunol. (2006) 26:177–85. doi: 10.1007/s10875-006-9002-x
28. Lingman-Framme J, Fasth A. Subcutaneous immunoglobulin for primary and secondary immunodeficiencies: an evidence-based review. Drugs. (2013) 73:1307–19. doi: 10.1007/s40265-013-0094-3
29. Haddad É, Barnes D, Kafal A. Home therapy with subcutaneous immunoglobulins for patients with primary immunodeficiency diseases. Transfus apher sci. (2012) 46:315–21. doi: 10.1016/j.transci.2012.03.022
30. Jiang F, Torgerson TR, Ayars AG. Health-related quality of life in patients with primary immunodeficiency disease. Allergy Asthma Clin Immunol. (2015) 11:27. doi: 10.1186/s13223-015-0092-y
31. Gardulf A, Nicolay U, Math D, Asensio O, Bernatowska E, Bock A, et al. Children and adults with primary antibody deficiencies gain quality of life by subcutaneous IgG self-infusions at home. J Allergy Clin Immunol. (2004) 114:936–42. doi: 10.1016/j.jaci.2004.06.053
32. Misbah S, Sturzenegger MH, Borte M, Shapiro RS, Wasserman RL, Berger M, et al. Subcutaneous immunoglobulin: opportunities and outlook. Clin Exp Immunol. (2009) 158(Suppl. 1):51–9. doi: 10.1111/j.1365-2249.2009.04027.x
33. Berger M, Murphy E, Riley P, Bergman GE, Investigators VT. Improved quality of life, immunoglobulin G levels, and infection rates in patients with primary immunodeficiency diseases during self-treatment with subcutaneous immunoglobulin G. South Med J. (2010) 103:856–63. doi: 10.1097/SMJ.0b013e3181eba6ea
34. Pasquet M, Pellier I, Aladjidi N, Auvrignon A, Cherin P, Clerson P, et al. A cohort of french pediatric patients with primary immunodeficiencies: are patient preferences regarding replacement immunotherapy fulfilled in real-life conditions? Patient prefer adherence. (2017) 11:1171–80. doi: 10.2147/PPA.S123363
Keywords: immunoglobulins/deficiency, antibodies/deficiency, immunoglobulins/administration and dosage, autoimmunity, hematologic neoplasms, immunologic deficiency syndromes
Citation: Sánchez-Ramón S, Bermúdez A, González-Granado LI, Rodríguez-Gallego C, Sastre A, Soler-Palacín P and the ID-Signal Onco-Haematology Group (2019) Primary and Secondary Immunodeficiency Diseases in Oncohaematology: Warning Signs, Diagnosis, and Management. Front. Immunol. 10:586. doi: 10.3389/fimmu.2019.00586
Received: 16 December 2018; Accepted: 05 March 2019;
Published: 26 March 2019.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Francisco Javier Espinosa-Rosales, Fundación Mexicana para Niñas y Niños con Inmunodeficiencias (FUMENI), MexicoRicardo U. Sorensen, LSU Health Sciences Center New Orleans, United States
Copyright © 2019 Sánchez-Ramón, Bermúdez, González-Granado, Rodríguez-Gallego, Sastre, Soler-Palacín and the ID-Signal Onco-Haematology Group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Sánchez-Ramón, c3NyYW1vbkBzYWx1ZC5tYWRyaWQub3Jn