 Stephanie T. Le1
Stephanie T. Le1 Alexander A. Merleev1
Alexander A. Merleev1 Guillaume Luxardi1
Guillaume Luxardi1 Michiko Shimoda1
Michiko Shimoda1 Iannis E. Adamopoulos2
Iannis E. Adamopoulos2 Lam C. Tsoi3,4
Lam C. Tsoi3,4 Jenny Z. Wang1,5Claire Alexanian1,6Siba P. Raychaudhuri2,7Samuel T. Hwang1
Jenny Z. Wang1,5Claire Alexanian1,6Siba P. Raychaudhuri2,7Samuel T. Hwang1 Johann Gudjonsson3Alina I. Marusina1
Johann Gudjonsson3Alina I. Marusina1 Emanual Maverakis1*
Emanual Maverakis1*- 1Department of Dermatology, University of California, Davis, Sacramento, CA, United States
- 2Division of Rheumatology, Allergy and Clinical Immunology, Department of Internal Medicine, University of California, Davis, Sacramento, CA, United States
- 3Department of Dermatology, University of Michigan, Ann Arbor, MI, United States
- 4Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, MI, United States
- 5Albert Einstein College of Medicine, Bronx, NY, United States
- 6Georgetown University School of Medicine, Washington, DC, United States
- 7Department of Veterans Affairs, VA Sacramento Medical Center, Northern California Health Care System, Mather, CA, United States
The present paradigm of psoriasis pathogenesis revolves around the IL-23/IL-17A axis. Dual-secreting Th17 T cells presumably are the predominant sources of the psoriasis phenotype-driving cytokines, IL-17A and IL-22. We thus conducted a meta-analysis of independently acquired RNA-seq psoriasis datasets to explore the relationship between the expression of IL17A and IL22. This analysis failed to support the existence of dual secreting IL-17A/IL-22 Th17 cells as a major source of these cytokines. However, variable relationships amongst the expression of psoriasis susceptibility genes and of IL17A, IL22, and IL23A were identified. Additionally, to shed light on gene expression relationships in psoriasis, we applied a machine learning nonlinear dimensionality reduction strategy (t-SNE) to display the entire psoriasis transcriptome as a 2-dimensonal image. This analysis revealed a variety of gene clusters, relevant to psoriasis pathophysiology but failed to support a relationship between IL17A and IL22. These results support existing theories on alternative sources of IL-17A and IL-22 in psoriasis such as a Th22 cells and non-T cell populations.
Introduction
Psoriasis is a chronic inflammatory skin condition with nail and systemic manifestations that affects ~3% of the general United States population. It is commonly associated with psoriatic arthritis and is likely linked to other comorbidities, such as cardiovascular disease and metabolic syndrome (1–4).
Of the many clinical variants, plaque psoriasis (psoriasis vulgaris) is the most common, accounting for ~80–90% of cases (1, 5). It is also the most well-characterized histologically and genetically. Plaque psoriasis was initially proposed to be driven by hyperproliferative keratinocytes. However, in 1890, neutrophil involvement was suggested after histologic evaluation revealed early neutrophil accumulation within the dermis and epidermis (i.e., microabscesses of Munro and pustules of Kogoj, respectively) (6).
Despite the clear existence of neutrophils in lesional skin, the role of the adaptive immune system in psoriasis pathophysiology became the main focus of the field after the T cell-targeting agent, cyclosporine, was shown to be an effective treatment (7–9). Thus, psoriasis researchers became very quickly focused on characterizing CD4+ and CD8+ T cell responses in normal and diseased human skin (10–12). Subsequently, experiments performed in animal models were also developed that supported the T cell-centric view of psoriasis. For example, it was demonstrated that a psoriasis-like phenotype could be induced following adoptive transfer of dysregulated CD4+ T cells (13). With this knowledge came the development of the next generation of T cell-targeting therapeutics (alefacept, efalizumab) (14–17), which further corroborated the essential role of T cells in psoriasis pathophysiology.
At the time T cells became the focus of psoriasis, adaptive immune responses were typically divided into two types, T helper type 1 (Th1) and T helper type 2 (Th2) responses. In psoriasis, the absence of Th2-defining cytokines [interleukin (IL)-4, IL-5, and IL-10] (18) and the increased presence of Th1 cytokines (interferon gamma (IFN-γ), tumor necrosis factor (TNF) and IL-12) prompted researchers to classify psoriasis as a Th1-mediated disease (18). Soon thereafter, however, it became increasingly apparent that IL-17-secreting T cells (Th17 cells) played a major role in disease pathogenesis, not only in psoriasis, but also across a wide spectrum of animal models of autoimmunity (19–22).
Psoriasis is now thought to be a predominantly Th17-driven disease (23, 24) that is maintained by the key Th-17-supporting cytokine, IL-23 (25, 26). The dominant role of the IL-23/IL-17A axis in psoriasis is also evident by the overwhelming clinical success of newly developed IL-23/IL-17A axis-targeting biologics, which could induce near complete resolution of psoriasis, even in the most severely affected individuals (27–29). IL-22 is also a highly investigated cytokine involved in psoriasis pathophysiology. It is thought to be the primary promoter of keratinocyte hyperproliferation (30, 31). The predominant view is that this cytokine is secreted by IL-17A/IL-22 dual-secreting Th17 cells (32).
However, the observed pathogenicity of IL-17A/IL-22 dual-secreting Th17 cells has never been formally demonstrated in vivo. In fact, the vast majority of evidence in support of these cells have come from animal studies and in vitro analysis of human T cells cultured under extreme polarizing conditions (32–35). Even when studied directly ex vivo, the dual secretion is usually seen only after non-physiologic T cell stimulation (36, 37). Since naturally processed autoimmune epitopes are difficult to identify (38), it is challenging to study cytokine secretion using more physiologic stimuli.
Thus, we sought evidence for the existence of dual secreting IL-17A/IL-22 Th17 cells within the psoriasis transcriptome. Weighted gene co-expression networks analysis (WGCNA) (39) have previously been used to analyze gene-gene correlations within RNA-Seq datasets. While this strategy has certain advantages, it is not ideally suited to explore gene relationships across multiple RNA-Seq datasets. Herein, we conduct meta-analyses of RNA-seq datasets to directly evaluate the current hypothesis that dual-secreting IL-17A/IL-22 Th17 cells are the dominant effector population in psoriasis. We also used this strategy to correlate the expression of IL17A, IL22, and IL23A with genes linked to psoriasis susceptibility identified through genome-wide association studies (GWAS). Finally, to explore the gene expression profile of IL17A, IL22, and IL23A in relation to other genes expressed in psoriatic plaques, we utilized a machine learning nonlinear dimensionality reduction strategy to visualize the entire psoriasis transcriptome as a 2-dimensional (2D) image. This allowed us to clearly visualize the relationship between IL17A, IL22, and IL23A and all other genes that are expressed in psoriatic skin.
Materials and Methods
Human RNA-Seq
RNA-Seq FASTQ files of human normal and psoriasis lesional skin were downloaded from the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra). Four total datasets were used: Three datasets (Accession numbers: SRP165679, SRP026042, SRP057087) and one dataset comprised of two combined experimental datasets published by the same research group (Accession numbers: SRP035988, SRP050971) (40–42).
Correlations
Correlation analyses of gene expressions were performed on read counts of each identified gene normalized with DESeq2 package (43). Values were subsequently log transformed and winsorized when necessary. Spearman's correlation coefficients were calculated (rs) using the cor.test function in R (44). P values of the correlations were estimated by algorithm AS 89.
2D Visualization of the Psoriasis Transcriptome
We computed the gene pairwise distance using a formula, 1-r2, where r represents Pearson's correlation. A visual representation of the gene co-expression network was created using a dimensionality reduction technique, t-Distributed Stochastic Neighbor Embedding (t-SNE), calculated with Rtsne package (45).
Gene Selection
A Pubmed search was performed to identify genes linked to psoriasis through GWAS.
Genes selected for mapping included: BTK, CD3E, CD4, CD8a, CD8b, CD19, CTSG, CXCL1, CXCL2, CXCL5, ELANE, ICAM1, IGH, IGK, IGL, IL1B, IL8, IL17A, IL22, IL23A, IL36A, IL36B, IL36G, IFNG, ITK, MPO, MS4A1 (CD20), TNF, TRA (TCRα), TRB (TCRβ), TRD (TCRδ) TRG (TCRγ).
Genes selected for meta-analysis included: B3GNT2, CARD14, CARM1, CDKAL1, CTSG, CXCL1, CXCL5, CXCR2, DDX58, DEFB4A, ELANE, FBXL19, GJB2, HLAC, IFIH1, IL12B, IL17A, IL22, IL23A, IL36RN, IL4R, KLF4, KRT1, KRT5, KRT6A, KRT6B, KRT6C, KRT10, KRT14, KRT16, KRT17, KRT37, LCE3A, LCE3B, LCE3D, MPO, NFKBIA, NOS2, NOS3, PTPN22, RELB, RUNX3, SOCS1, STAT3, STAT5A, TNFAIP3, TNFRSF9, TNIP1, TRAF3IP2, TYK2, UBE2L3, VDR, VEGFA, VEGFB.
Meta-Analysis
Meta-analysis was completed using the R package “metafor” (46). A weighted random-effects model was used to estimate a summary effect size. To estimate between-study variance, a restricted maximum-likelihood estimator was applied. A weighted estimation with inverse-variance weights was used to fit the model.
Results
The Expression of IL17A and IL22 Do Not Strongly Correlate With One Another in Psoriatic Plaques
We hypothesized that if a significant amount of IL-17A and IL-22 is produced by IL-17A/IL-22 dual secreting Th17 cells in psoriasis, then the gene expression of these two cytokines should correlate with one another. In theory, their expression would be directly linked to the number of dual-secreting Th17 cells in a psoriasis plaque. Their gene expression should also correlate with IL23A, which activates and maintains Th17 cells.
To test this hypothesis, gene expression of IL17A vs. IL22 was graphed and the Spearman's correlation coefficient (rs) was calculated (Figure 1A). These correlative studies demonstrated that the expression of IL22 does not strongly correlate with the expression of IL17A (rs = 0.04, p = 0.67). To obtain a weighted average across all four independently acquired psoriasis datasets, a meta-analysis was performed and the resulting Forest plot (Figure 1B) demonstrated again that IL17A and IL22 do not strongly correlate with one another [rs = 0.18, with a confidence interval that crosses 0 (−0.05, 0.41)] (Supplemental Figure 1).
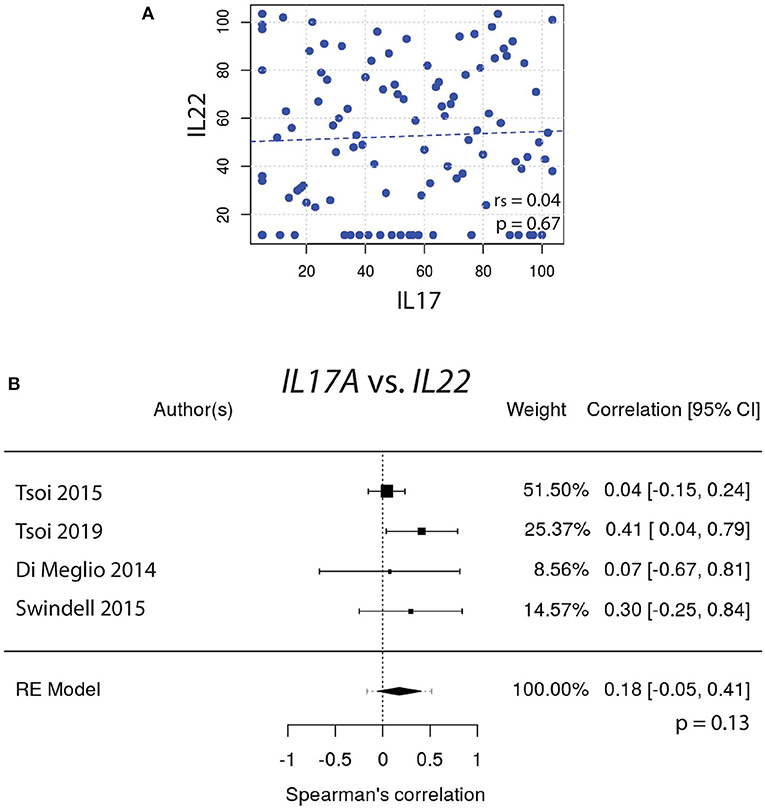
Figure 1. (A) The expression of IL17A does not strongly correlate with the expression of IL22 (rs = 0.04). (B) Meta-analysis of four datasets further supports that IL17A and IL22 expression do not strongly correlate [rs = 0.18, with a confidence interval that crosses 0 (−0.05, 0.41)].
Similarly, IL22 gene expression did not correlate well with IL23A (Figures 2A,B). In contrast, the expression of IL17A did correlate very well with IL23A (Figure 3A), a result that was consistent amongst a majority of datasets. The weighted average of this correlation across all psoriasis datasets was highly significant, [rs = 0.31 (0.12, 0.51); p = 0.0014], with no evidence (p = 0.33) of any substantial residual heterogeneity (i.e., there was no remaining variability in effect sizes that was unexplained) (Figure 3B). Our data confirms the well-characterized dependency of IL-17A on IL-23A. However, IL-22 was not found to have a similar dependency on IL-23A, casting doubt on the theory that IL-17A and IL-22 are secreted mainly by the same dual-secreting cell.
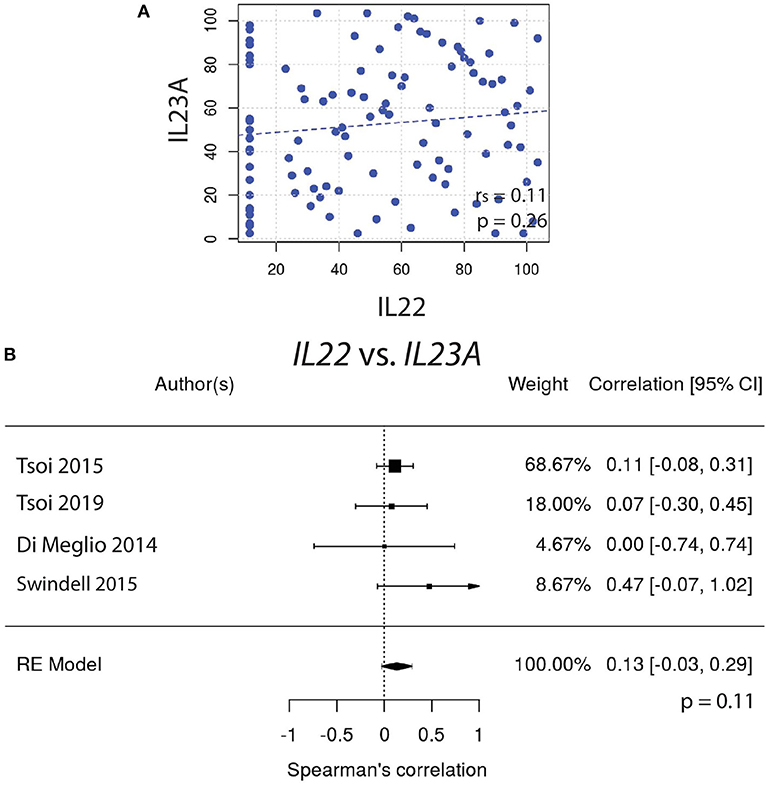
Figure 2. (A) IL22 gene expression does not correlate with IL23A (rs = 0.11). (B) Meta-analysis confirms that IL22 and IL23A do not strongly correlate [rs = 0.13, with a confidence interval that crosses 0 (−0.03, 0.29)].
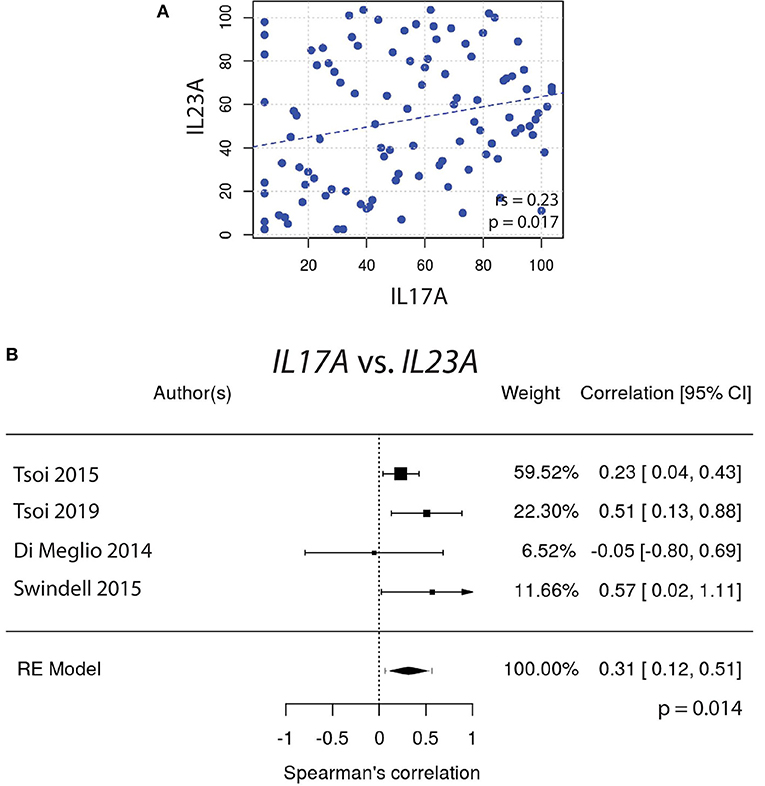
Figure 3. (A) A significant Spearman correlation (rs = 0.23, p = 0.017) is seen between IL17A and IL23A gene expression. (B) Meta-analysis supports a strong correlation between IL17A and IL23A [rs = 0.31, (0.12, 0.51); p = 0.0014].
2D Visualization of the Psoriasis Transcriptome Reveals T Cell, B Cell, Inflammatory Cytokines, Neutrophil-Recruiting, and Neutrophil Gene Clusters
To determine how genes expressed in psoriatic plaques are related to one another, correlation coefficients were calculated for all pairwise comparisons. The distances between each gene pair was calculated as described in the methods. The resulting distance matrix was then used to construct a 2D image using t-SNE.
In the 2D plot (Figure 4), genes that highly correlate with one another tend to be located in the same region, known as a cluster. Genes that do not cluster near each other do not correlate well. Figure 4 clearly demonstrates that genes associated with B cells [BTK, CD19, IGH, IGK, IGL, MS4A1 (CD20)], T cells (CD3E, CD4, CD8a, CD8b, ICAM1, ITK, TRA TRB, TRD, TRG), neutrophils (CTSG, ELANE, MPO), neutrophil-recruiting (CXCL1, CXCL2, CXCL5, IL8), and psoriasis-associated inflammatory cytokines (IL1B, IL17A, IL23A, IL36A, IL36B, IL36G) cluster well together in distinct groups, which supports this method as a means to visualize the entire psoriasis transcriptome.
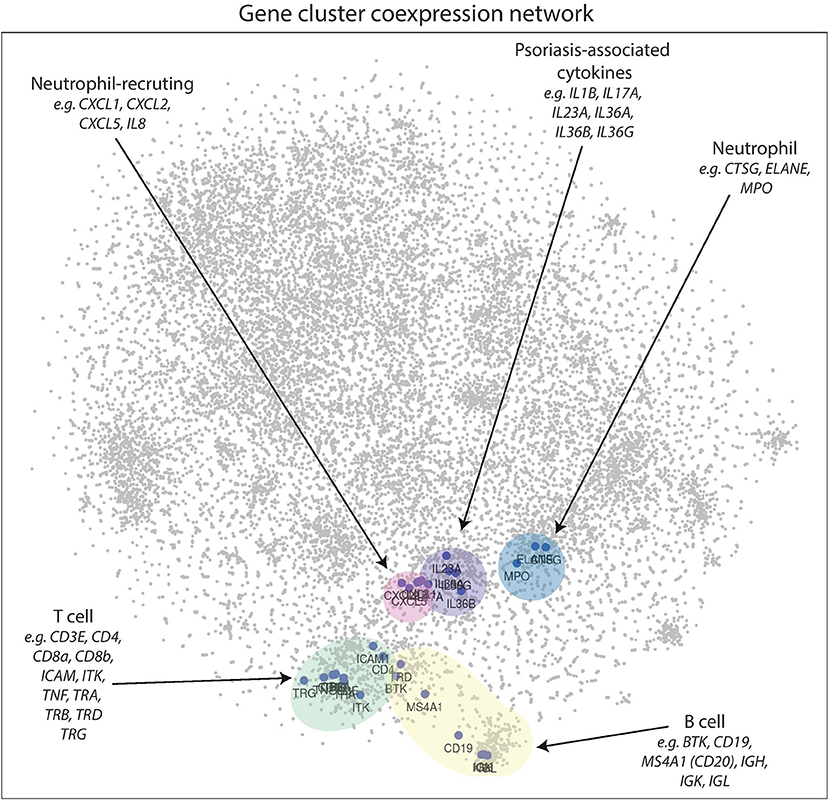
Figure 4. 2D gene coexpression network illustrates gene expression relationships of the psoriasis plaque. Points closer together represent genes that have higher correlation coefficients, while genes at further distances generally do not correlate well. Gene clusters appear to correspond to neutrophil-recruiting cytokines, neutrophil effector molecules, T cell, B-cell, and psoriasis-associated genes in lesional psoriatic skin.
IL22 Does Not Cluster With Other Inflammatory Cytokines Involved in Psoriasis, Including IL17A
With respect to other cytokines and chemokines involved in psoriasis pathophysiology, IL22 is located peripherally at a relatively great distance away on the 2D plot of the psoriasis transcriptome (Figure 5). This supports our results from the meta-analyses and suggests that IL22 does not correlate well with IL23A. Interestingly, IL22 does not cluster well with any of the most commonly implicated cytokines in psoriasis pathophysiology. In contrast, IL17A clusters together with IL23A and the other cytokines thought to be involved in psoriasis pathophysiology.
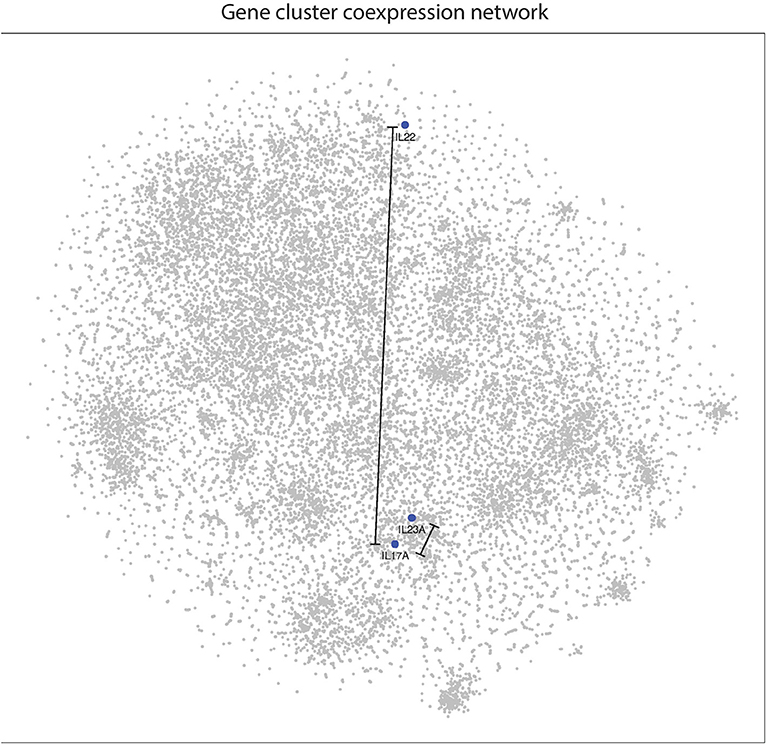
Figure 5. IL22 is located at a relatively great distance away from IL17A and IL23A in the psoriasis transcriptome, as illustrated in the 2D gene expression network map of psoriasis. These results suggest that IL22 is not highly linked to either IL17A or IL23A in psoriasis pathophysiology.
IL22 Correlates With Keratins
Several studies have demonstrated that IL-22 stimulates keratinocytes. There is a variety of evidence, including data obtained from in vitro studies with skin-like organoid cultures, that support IL-22 as the main cytokine responsible for epidermal hyperplasia, a hallmark of psoriasis (47, 48).
Because IL22 failed to strongly correlate with IL17A and IL23A (Figures 1A, 2A), the relationships between IL22 and keratin genes were explored across the four independently acquired RNA-Seq psoriasis datasets. Again, Spearman's correlation coefficients (rs) were calculated for each keratin gene's relationship with IL17A, IL22, and IL23A. These correlative studies demonstrate that the expression of IL22 did indeed strongly correlate with the expression of the different keratin genes (Figure 6), especially KRT6C (keratin 6C) (rs = 0.32, p = 0.0011). To obtain a weighted average across all four independent psoriasis datasets, a meta-analysis was performed and the resulting Forest plots (Figure 7) confirm the close relationship between IL22 expression and keratin gene expression [KRT6C: rs = 0.34, with a confidence interval that did not cross 0 (0.18–0.50)]. The weighted average of this correlation across all psoriasis datasets was highly significant (p = 0.000025), with no evidence (p = 0.56) of any substantial residual heterogeneity (i.e., there was no remaining variability in effect sizes that was unexplained). Additional genes that were found to positively correlate with IL22 expression are listed in Supplemental Figure 2.
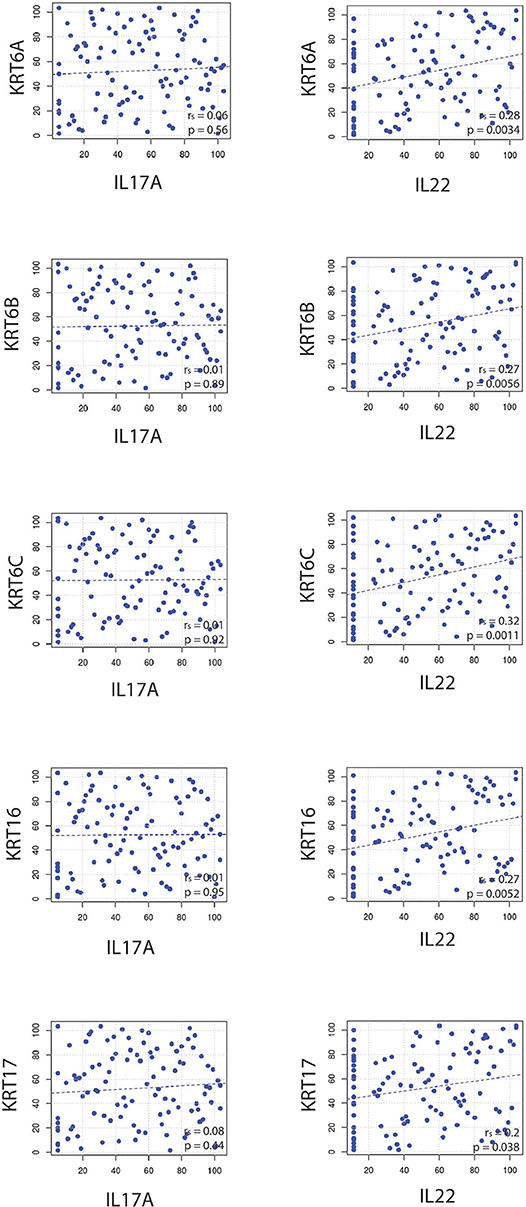
Figure 6. Correlative studies demonstrate that IL22 strongly correlates with commonly psoriasis-associated keratin genes (KRT6A, KRT6B, KRT6C, KRT16, KRT17), a finding that was not true for IL17A.
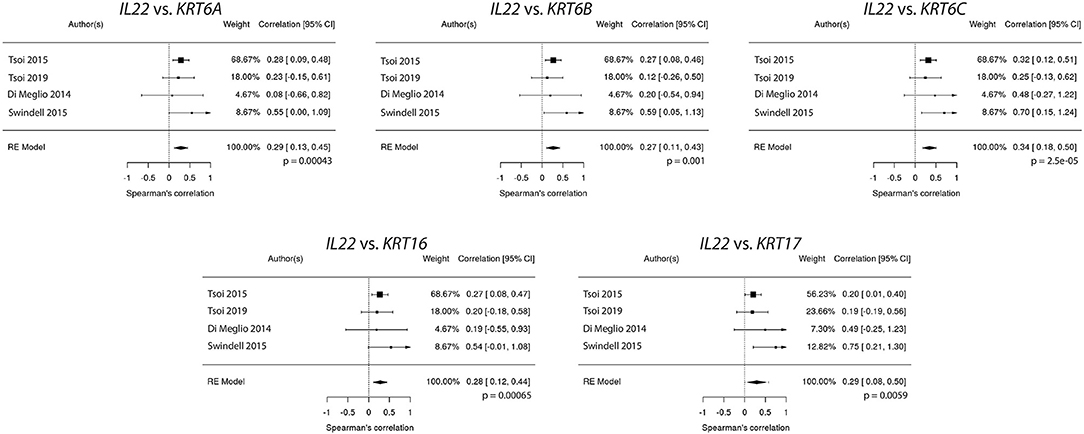
Figure 7. Meta-analysis confirms a close relationship between IL22 expression and keratin (KRT6A, KRT6B, KRT6C, KRT16, KRT17) expression.
In contrast to IL22's relationship with keratin gene expression, IL17A did not correlate well with the keratins (Figure 6), a finding that was confirmed by a meta-analysis across all four RNA-Seq datasets.
IL23A Correlates With Other Genes Besides IL17A
IL-23A is known for its ability to support Th17 T cells but it likely has a variety of functions independent of this role. To investigate this, IL23A's ability to independently correlate with other immune-relevant genes was explored. Figure 8 reveals that IL23A correlates with several genes unrelated to IL17A, a finding confirmed by meta-analyses across all psoriasis RNA-Seq datasets. Included in the analysis were genes identified by GWAS to be linked to psoriasis. Of these genes, CARM1, KRT14, KRT37, TNFAIP3, UBE2L3 are elevated in psoriasis plaques compared to control healthy skin (Table 1). Thus, IL23A appears to be linked to other genes putatively involved in the pathophysiology of psoriasis that are unrelated to IL17A.
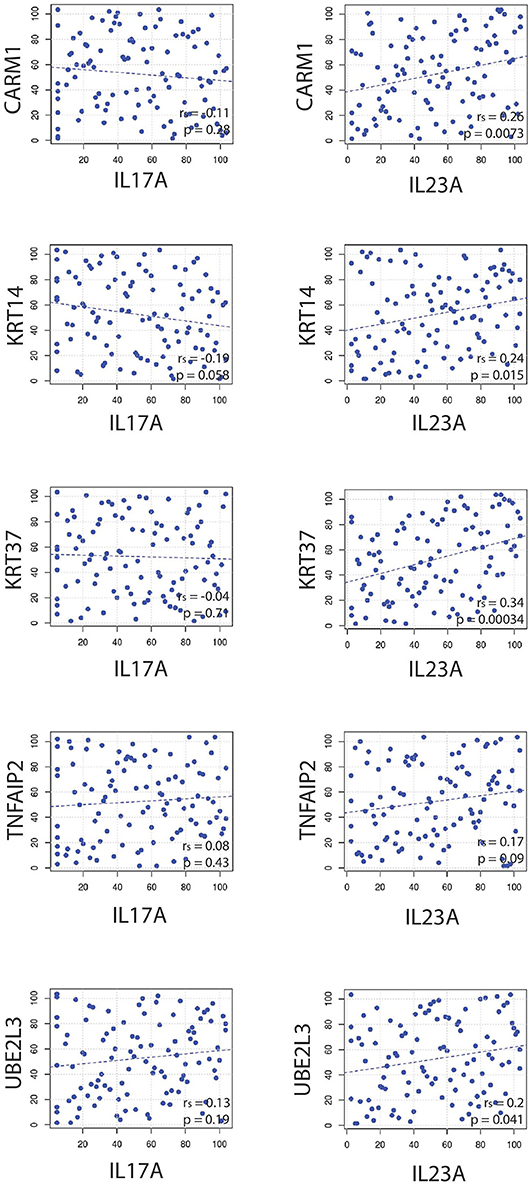
Figure 8. Spearman correlations reveal that IL23A correlates with several genes that do not seem to be strongly related to IL17A.
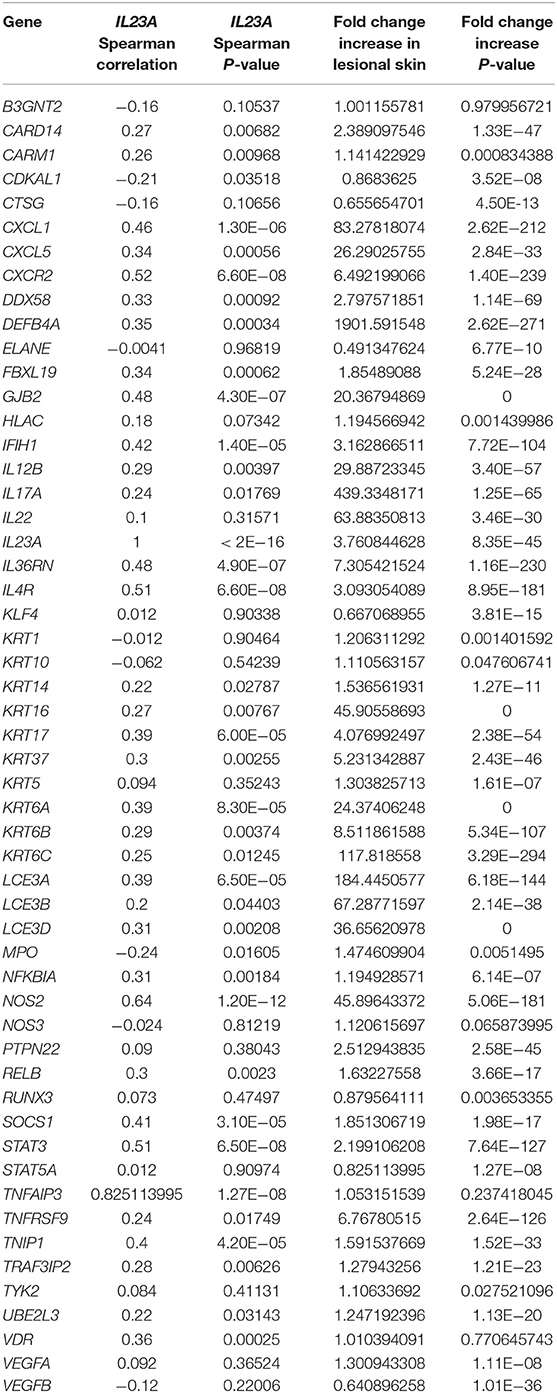
Table 1. Expression of genes linked to psoriasis identified through GWAS and associated IL23A Spearman correlation and p-value and fold change increase in lesional skin and p-value.
IL17A, IL22, and IL23A Expression Correlates With Psoriasis Susceptibility Genes
A variety of genes have been linked to psoriasis susceptibility through GWAS (49–55). Table 2 demonstrates that many of these genes are differentially regulated in the setting of psoriasis. We thus sought to determine how the expression of genes located at psoriasis susceptibility loci correlated with the expression of IL17A, IL22, and IL23A, genes known to be linked to the pathophysiology of psoriasis. For this analysis, the expression of IL17A, IL22, and IL23A was plotted against the expression of each of the genes identified through GWAS studies. Spearman's correlation coefficients (rs) were then calculated, which demonstrated a variety of significant correlations (Table 2) between GWAS-identified genes and IL17A, IL22, and IL23A. Correlation values between atopic dermatitis GWAS-identified genes and IL17A, IL22 and IL23A expression in psoriasis samples were also obtained for comparison (Table 3). To obtain a weighted average across all four independent psoriasis data sets, meta-analysis was performed. The resulting Forest plots are depicted in Figure 9, which confirm the close relationship between IL17A, IL22, and IL23A and the different genes linked to psoriasis susceptibility. These results support a direct or indirect link between IL17A, IL22, and IL23A and these genes. Of note, the genes that significantly correlated with IL17A, IL22, and IL23A varied for each cytokine. These results will hopefully help investigators better understand the pathophysiology of psoriasis.
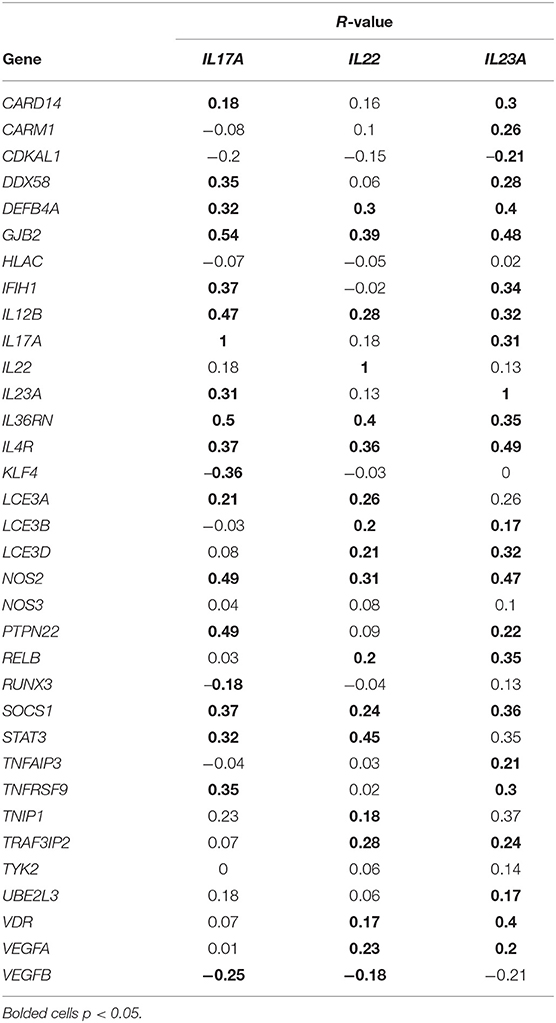
Table 2. Correlation (R-values) of IL17A, IL22, and IL23A with genes linked to psoriasis susceptibility through genome-wide association studies.
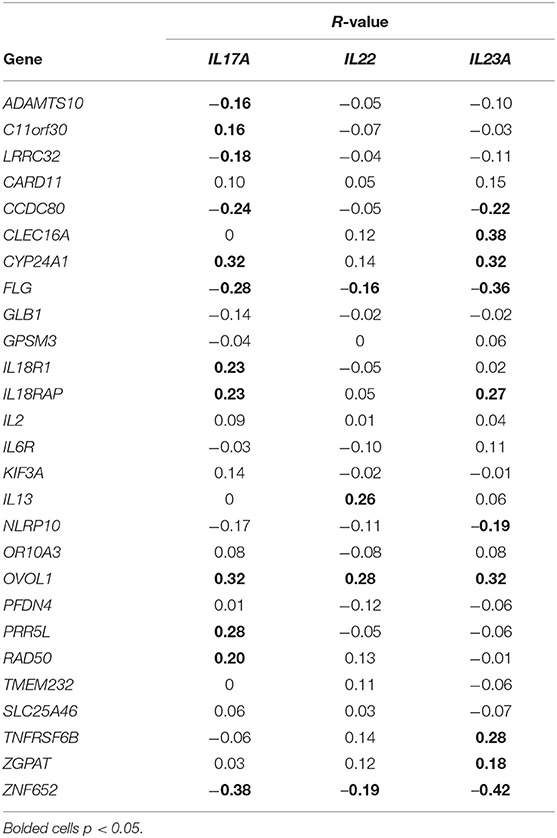
Table 3. Correlation (R-values) of IL17A, IL22 and IL23A in psoriasis with genes linked to atopic dermatitis susceptibility through genome-wide association studies (56–61).

Figure 9. Psoriasis susceptibility genes that positively correlate across IL17A, IL22, and IL23A, supporting a link between these cytokines and genes that have been linked to psoriasis through GWAS.
Discussion
Investigators have employed numerous genetic strategies to characterize the immune response in the setting of psoriasis. Microarray and RNA-Seq have provided insight into the psoriatic transcriptome, identifying thousands of differentially expressed genes (40). However, differential expression alone does not necessarily mean that the gene is involved in psoriasis pathogenesis. For example, a gene that is normally downregulated in psoriatic T cells may actually appear falsely upregulated in psoriasis simply because there are more T cells in a psoriatic plaque. With the rising popularity of single cell sequencing, investigators are now focused on re-characterizing the psoriasis transcriptome at a greater cellular resolution, not previously obtained with whole tissue transcriptomics. However, single cell sequencing is also not without its drawbacks. Purifying immune cell populations from skin biopsy specimens can alter their transcriptome, especially for cells isolated by positive selection or flow cytometry. Furthermore, immune cells within the skin will undoubtedly have different purification yields. T cells in particular are especially difficult to analyze because once purified, they require additional non-physiologic in vitro stimulation with lectins or anti-CD3/anti-CD28 antibodies to identify their cytokine secretion profiles. How closely the garnered information from these studies will relate to in vivo cellular function remains unclear. Although each technique will yield important discoveries, none can perfectly decipher the in vivo pathogenic immune response.
With these limitations in mind, we have focused on developing new methods to characterize immune responses from whole tissue RNA-Seq (12, 62). We view this strategy to be an important complement to work currently being conducted by other investigators. The main advantage being that the data is not subject to experimentally-induced changes in gene expression. Its main disadvantage, however, is that it cannot discriminate between direct or indirect correlations between genes of interest.
In our current study, we utilize a machine-learning 2D visualization strategy, t-SNE, to characterize IL17A, IL22, and IL23A gene expression in the context of the entire psoriatic transcriptome. The 2D map of the psoriatic transcriptome revealed distinct gene clusters corresponding to common immune cell types (e.g., B cells, T cells, neutrophils).
Our data did not support the existence of a dual-secreting IL-17A/IL-22 Th17 cell as the major source of these cytokines in psoriasis. In fact, in the 2D model, these genes are located far from one another. As such, IL22 correlated with several genes that did not appear to have a relationship with IL17A. In addition, a set of genes identified to be involved in psoriasis pathophysiology (CARD14, CXCL5, CXCR2, DDX58, IFIH, PTPN22, and TNFRSF9) correlated with IL17A and IL23A, but did not correlate with IL22.
Though, IL-22 is commonly considered a hallmark Th17 cytokine (63), our results are in line with studies demonstrating the existence of uniquely secreting IL-17 and IL-22 T cells or the existence of other cytokine-secreting phenotypes (48, 64–69), although these other studies usually relied upon non-physiologic ex vivo T cell stimulation. Another possibility is that other cell types, such as γδ T cells or mast cells, contribute to the IL-22 production in psoriasis (48, 65, 70). Even neutrophils have been implicated as major producers of IL-22 and IL-17A (71) and recent animal models have re-explored their role as effector cells in psoriasis pathophysiology (22, 72, 73). Indeed, there are numerous studies supporting a key function of these cells (71, 74–78). Single cell sequencing may provide information to verify the relationship between IL17A and IL22 expression. Although it is possible that dual-secreting IL-17A/IL-22 Th17 cells exist, our results suggest that they are not a major source of IL-22.
Although we did not find evidence for a strong link between IL22 with IL17A or IL23A, our results do support a strong correlation between the expression of IL22 and the keratin genes, such as KRT6A, KRT6B, KRT6C, KRT16, KRT17, a finding in accord with IL-22's ability to induce epidermal hyperproliferation (48). IL-22 is clearly a major cytokine involved in psoriasis pathophysiology. In animal models, it has been demonstrated to simulate psoriasis-like epidermal changes (47, 79) and elevated levels of IL-22 positively correlate with disease severity in humans, as measured by Psoriasis Area Severity Index (PASI) scores (80–83).
Conclusion
Although dual-secreting T cells may exist, our results demonstrate that it is unlikely that the classical Th17 cells (IL-17A/ IL-22 dual-secreting T cells) play a universal role in psoriasis pathophysiology. RNA-Seq analysis revealed that the expression of these cytokines seems to be largely unrelated to one another in the psoriasis transcriptome. However, the expression of IL17A did correlate with IL23A but, interestingly, unique relationships between IL23A and genes unrelated to IL17A were also established, supporting a broad function of IL-23.
Taken together, these results do not support the current dogma that IL-17A/IL-22 dual-secreting Th17 T cells are the major driver of psoriasis pathophysiology. In addition, our results support unique functions of IL-23 that are unrelated to its known role in supporting Th17 responses. Finally, we demonstrate that the expression of genes linked to psoriasis susceptibility also correlate with expression of either IL17A, IL22, or IL23. This supports the aforementioned cytokines' involvement in multiple avenues of psoriasis susceptibility.
2D mapping of inflammatory transcriptomes is an exciting innovative modality that may help us visualize relationships of all genes expressed in a disease process. When applied to gene expression relationships in psoriatic lesional skin, distinct clusters of cell lineage genes could be identified, supporting the presence of a complex crosstalk among separate cell lines in disease development. In the near future, single cell transcriptome analysis will provide additional insight into psoriasis pathogenesis. Identifying the cells responsible for the psoriasis phenotype will bring us one step closer to developing a cure for psoriasis.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: “http://www.ncbi.nlm.nih.gov/Traces/sra”.
Author Contributions
EM, AAM, AIM, GL, MS, SR, and SH contributed to the conception and design of the study; LT and JG organized the database; AAM performed the statistical analysis; SL, JW, and CA performed data mining, SL and EM wrote the first draft of the manuscript; SL, IA, and EM wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00589/full#supplementary-material
Supplemental Figure 1. Funnel plot representation demonstrating RNA-seq datasets analyzed in the meta-analysis of IL17A and IL22. All data points, representing individual data sets, fall within the 95% confidence interval. In our meta-analysis, the p value for residual heterogeneity did not reach significance (p = 0.34), indicating that all datasets are within the variation that is expected for this particular meta-analysis.
Supplemental Figure 2. Additional genes that positively correlate with IL22 expression.
References
1. Johnson MA, Armstrong AW. Clinical and histologic diagnostic guidelines for psoriasis: a critical review. Clin Rev Allergy Immunol. (2013) 44:166–72. doi: 10.1007/s12016-012-8305-3
2. Raychaudhuri SP, Wilken R, Sukhov AC, Raychaudhuri SK, Maverakis E. Management of psoriatic arthritis: early diagnosis, monitoring of disease severity and cutting edge therapies. J Autoimmun. (2017) 76:21–37. doi: 10.1016/j.jaut.2016.10.009
3. Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA. (2006) 296:1735–41. doi: 10.1001/jama.296.14.1735
4. Patrick MT, Stuart PE, Raja K, Gudjonsson JE, Tejasvi T, Yang J, et al. Genetic signature to provide robust risk assessment of psoriatic arthritis development in psoriasis patients. Nat Commun. (2018) 9:4178. doi: 10.1038/s41467-018-06672-6
5. Raychaudhuri SK, Maverakis E, Raychaudhuri SP. Diagnosis and classification of psoriasis. Autoimmun Rev. (2014) 13:490–5. doi: 10.1016/j.autrev.2014.01.008
7. Mueller W, Herrmann B. Cyclosporin A for psoriasis. N Engl J Med. (1979) 301:555. doi: 10.1056/NEJM197909063011015
8. Ellis CN, Gorsulowsky DC, Hamilton TA, Billings JK, Brown MD, Headington JT, et al. Cyclosporine improves psoriasis in a double-blind study. JAMA. (1986) 256:3110–6. doi: 10.1001/jama.1986.03380220076026
9. Griffiths CE, Powles AV, Leonard JN, Fry L, Baker BS, Valdimarsson H. Clearance of psoriasis with low dose cyclosporin. Br Med J. (1986) 293:731–2. doi: 10.1136/bmj.293.6549.731
10. Bos JD, Hagenaars C, Das PK, Krieg SR, Voorn WJ, Kapsenberg ML. Predominance of “memory” T cells (CD4+, CDw29+) over “naive” T cells (CD4+, CD45R+) in both normal and diseased human skin. Arch Dermatol Res. (1989) 281:24–30. doi: 10.1007/BF00424268
11. Chang JC, Smith LR, Froning KJ, Schwabe BJ, Laxer JA, Caralli LL, et al. CD8+ T cells in psoriatic lesions preferentially use T-cell receptor V beta 3 and/or V beta 13.1 genes. Proc Natl Acad Sci USA. (1994) 91:9282–6. doi: 10.1073/pnas.91.20.9282
12. Merleev AA, Marusina AI, Ma C, Elder JT, Tsoi LC, Raychaudhuri SP, et al. Meta-analysis of RNA sequencing datasets reveals an association between TRAJ23, psoriasis, and IL-17A. JCI Insight. (2018) 3:120682. doi: 10.1172/jci.insight.120682
13. Schon MP, Detmar M, Parker CM. Murine psoriasis-like disorder induced by naive CD4+ T cells. Nature Med. (1997) 3:183–8. doi: 10.1038/nm0297-183
14. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. (1999) 103:1243–52. doi: 10.1172/JCI5857
15. Krueger GG. Clinical response to alefacept: results of a phase 3 study of intravenous administration of alefacept in patients with chronic plaque psoriasis. J Eur Acad Dermatol Venereol. (2003) 17(Suppl. 2):17–24. doi: 10.1046/j.1468-3083.17.s2.4.x
16. Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. (2003) 139:719–27. doi: 10.1001/archderm.139.6.719
17. Sivamani RK, Goodarzi H, Garcia MS, Raychaudhuri SP, Wehrli LN, Ono Y, et al. Biologic therapies in the treatment of psoriasis: a comprehensive evidence-based basic science and clinical review and a practical guide to tuberculosis monitoring. Clin Rev Allergy Immunol. (2013) 44:121–40. doi: 10.1007/s12016-012-8301-7
18. Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. (2004) 25:295–305. doi: 10.1016/j.it.2004.03.006
19. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
20. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
21. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
22. Adamopoulos IE, Suzuki E, Chao CC, Gorman D, Adda S, Maverakis E, et al. IL-17A gene transfer induces bone loss and epidermal hyperplasia associated with psoriatic arthritis. Ann Rheum Dis. (2015) 74:1284–92. doi: 10.1136/annrheumdis-2013-204782
23. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. (2009) 129:2175–83. doi: 10.1038/jid.2009.65
24. Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. (2008) 128:1207–11. doi: 10.1038/sj.jid.5701213
25. Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. (2006) 176:1908–15. doi: 10.4049/jimmunol.176.3.1908
26. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
27. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CEM, Papp K, et al. Secukinumab in plaque psoriasis — results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
28. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. (2015) 373:1318–28. doi: 10.1056/NEJMoa1503824
29. Wang EA, Suzuki E, Maverakis E, Adamopoulos IE. Targeting IL-17 in psoriatic arthritis. Eur J Rheumatol. (2017) 4:272–7. doi: 10.5152/eurjrheum.2017.17037
30. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
31. Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. (2007) 178:2229–40. doi: 10.4049/jimmunol.178.4.2229
32. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
33. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. (2009) 206:35–41. doi: 10.1084/jem.20072713
34. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
35. Chung Y, Yang X, Chang SH, Ma L, Tian Q, Dong C. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. (2006) 16:902–7. doi: 10.1038/sj.cr.7310106
36. Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. (2008) 118:597–607. doi: 10.1172/JCI33263
37. Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R. Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J Immunol. (2012) 188:5438–47. doi: 10.4049/jimmunol.1103801
38. Sercarz EE, Maverakis E. Mhc-guided processing: binding of large antigen fragments. Nat Rev Immunol. (2003) 3:621–9. doi: 10.1038/nri1149
39. Ahn R, Yan D, Chang HW, Lee K, Bhattarai S, Huang ZM, et al. RNA-seq and flow-cytometry of conventional, scalp, and palmoplantar psoriasis reveal shared and distinct molecular pathways. Sci Rep. (2018) 8:11368. doi: 10.1038/s41598-018-29472-w
40. Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, et al. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol. (2014) 134:1828–38. doi: 10.1038/jid.2014.28
41. Tsoi LC, Iyer MK, Stuart PE, Swindell WR, Gudjonsson JE, Tejasvi T, et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. (2015) 16:24. doi: 10.1186/s13059-014-0570-4
42. Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic dermatitis is an IL-13 dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol. (2019) S0022-202X(19)30007-7. doi: 10.1016/j.jid.2018.12.018. [Epub ahead of print].
43. Love M, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
44. Team RC. R: A Language and Environment for Statistical Computing. Available online at: http://www.R-project.org/.
45. Krijthe JH. Rtsne: T-Distributed Stochastic Neighbor Embedding Using a Barnes-Hut Implementation. Available online at: https://github.com/jkrijthe/Rtsne.
46. Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. (2010) 36:1–48. doi: 10.18637/jss.v036.i03
47. Hao JQ. Targeting interleukin-22 in psoriasis. Inflammation. (2014) 37:94–9. doi: 10.1007/s10753-013-9715-y
48. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. (2013) 252:116–32. doi: 10.1111/imr.12027
49. Mahil SK, Capon F, Barker JN. Genetics of psoriasis. Dermatol Clin. (2015) 33:1–11. doi: 10.1016/j.det.2014.09.001
50. Nedoszytko B, Sokolowska-Wojdylo M, Ruckemann-Dziurdzinska K, Roszkiewicz J, Nowicki RJ. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol Alergol. (2014) 31:84–91. doi: 10.5114/pdia.2014.40920
51. Singh S, Pradhan D, Puri P, Ramesh V, Aggarwal S, Nayek A, et al. Genomic alterations driving psoriasis pathogenesis. Gene. (2019) 683:61–71. doi: 10.1016/j.gene.2018.09.042
52. Osmola-Mankowska A, Teresiak-Mikolajczak E, Skrzypczak-Zielinska M, Adamski Z. Genetic polymorphism in psoriasis and its meaning for the treatment efficacy in the future. Postepy Dermatol Alergol. (2018) 35:331–7. doi: 10.5114/ada.2018.77661
53. Xu X, Zhang HY. The immunogenetics of psoriasis and implications for drug repositioning. Int J Mol Sci. (2017) 18:E2650. doi: 10.3390/ijms18122650
54. Harden JL, Krueger JG, Bowcock AM. The immunogenetics of Psoriasis: a comprehensive review. J Autoimmun. (2015) 64:66–73. doi: 10.1016/j.jaut.2015.07.008
55. Puig L, Julia A, Marsal S. The pathogenesis and genetics of psoriasis. Actas Dermosifiliogr. (2014) 105:535–45. doi: 10.1016/j.adengl.2014.05.013
56. Bin L, Leung DY. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin Immunol. (2016) 12:52.
57. Hirota T, Nakayama T, Sato S, Yanagida N, Matsui T, Sugiura S, et al. Association study of childhood food allergy with genome-wide association studies-discovered loci of atopic dermatitis and eosinophilic esophagitis. J Allergy Clin Immunol. (2017) 140:1713–6. doi: 10.1016/j.jaci.2017.05.034
58. Liang Y, Chang C, Lu Q. The genetics and epigenetics of atopic dermatitis-filaggrin and other polymorphisms. Clin Rev Allergy Immunol. (2016) 51:315–28. doi: 10.1007/s12016-015-8508-5
59. Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. (2015) 47:1449–56. doi: 10.1038/ng.3424
60. Schaarschmidt H, Ellinghaus D, Rodriguez E, Kretschmer A, Baurecht H, Lipinski S, et al. A genome-wide association study reveals 2 new susceptibility loci for atopic dermatitis. J Allergy Clin Immunol. (2015) 136:802–6. doi: 10.1016/j.jaci.2015.01.047
61. Stemmler S, Hoffjan S. Trying to understand the genetics of atopic dermatitis. Mol Cell Probes. (2016) 30:374–85. doi: 10.1016/j.mcp.2016.10.004
62. Dillen CA, Pinsker BL, Marusina AI, Merleev AA, Farber ON, Liu H, et al. Clonally expanded gammadelta T cells protect against Staphylococcus aureus skin reinfection. J Clin Invest. (2018) 128:1026–42. doi: 10.1172/JCI96481
63. Zhao H, Li Y, Wang S, Yang Y, Wang J, Ruan X, et al. Whole transcriptome RNA-seq analysis: tumorigenesis and metastasis of melanoma. Gene. (2014) 548:234–43. doi: 10.1016/j.gene.2014.07.038
64. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. (2008) 159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
65. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
66. Jia L, Wu C. The biology and functions of Th22 cells. Adv Exp Med Biol. (2014) 841:209–30. doi: 10.1007/978-94-017-9487-9_8
67. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63. doi: 10.1038/ni.1767
68. Mirshafiey A, Simhag A, El Rouby NM, Azizi G. T-helper 22 cells as a new player in chronic inflammatory skin disorders. Int J Dermatol. (2015) 54:880–8. doi: 10.1111/ijd.12883
69. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. (2009) 10:864–71. doi: 10.1038/ni.1770
70. Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol. (2015) 136:351–9 e1. doi: 10.1016/j.jaci.2015.01.033
71. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
72. Schon M, Denzer D, Kubitza RC, Ruzicka T, Schon MP. Critical role of neutrophils for the generation of psoriasiform skin lesions in flaky skin mice. J Invest Dermatol. (2000) 114:976–83. doi: 10.1046/j.1523-1747.2000.00953.x
73. Suzuki E, Maverakis E, Sarin R, Bouchareychas L, Kuchroo VK, Nestle FO, et al. T Cell-independent mechanisms associated with neutrophil extracellular trap formation and selective autophagy in IL-17A-mediated epidermal hyperplasia. J Immunol. (2016) 197:4403–12. doi: 10.4049/jimmunol.1600383
74. Reich K, Papp KA, Matheson RT, Tu JH, Bissonnette R, Bourcier M, et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp Dermatol. (2015) 24:529–35. doi: 10.1111/exd.12710
75. Keijsers R, Hendriks AGM, van Erp PEJ, van Cranenbroek B, van de Kerkhof PCM, Koenen H, et al. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap-forming neutrophils expressing RORgammat and IL-17. J Invest Dermatol. (2014) 134:1276–84. doi: 10.1038/jid.2013.526
76. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. (2016) 14:708–22. doi: 10.1016/j.celrep.2015.12.072
77. Terui T, Ozawa M, Tagami H. Role of neutrophils in induction of acute inflammation in T-cell-mediated immune dermatosis, psoriasis: a neutrophil-associated inflammation-boosting loop. Exp Dermatol. (2000) 9:1–10. doi: 10.1034/j.1600-0625.2000.009001001.x
78. Sanda GE, Belur AD, Teague HL, Mehta NN. Emerging associations between neutrophils, atherosclerosis, and psoriasis. Curr Atheroscler Rep. (2017) 19:53. doi: 10.1007/s11883-017-0692-8
79. Sukhov A, Burrall B, Maverakis E. The history of open access medical publishing: a comprehensive review. Dermatol Online J. (2016) 22:13030/qt6578w9f8.
80. Boniface K, Guignouard E, Pedretti N, Garcia M, Delwail A, Bernard FX, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. (2007) 150:407–15. doi: 10.1111/j.1365-2249.2007.03511.x
81. Lo YH, Torii K, Saito C, Furuhashi T, Maeda A, Morita A. Serum IL-22 correlates with psoriatic severity and serum IL-6 correlates with susceptibility to phototherapy. J Dermatol Sci. (2010) 58:225–7. doi: 10.1016/j.jdermsci.2010.03.018
82. Shimauchi T, Hirakawa S, Suzuki T, Yasuma A, Majima Y, Tatsuno K, et al. Serum interleukin-22 and vascular endothelial growth factor serve as sensitive biomarkers but not as predictors of therapeutic response to biologics in patients with psoriasis. J Dermatol. (2013) 40:805–12. doi: 10.1111/1346-8138.12248
Keywords: IL17, IL22, machine learning, neutrophil, psoriasis, RNA-seq, T cell, transcriptome
Citation: Le ST, Merleev AA, Luxardi G, Shimoda M, Adamopoulos IE, Tsoi LC, Wang JZ, Alexanian C, Raychaudhuri SP, Hwang ST, Gudjonsson J, Marusina AI and Maverakis E (2019) 2D Visualization of the Psoriasis Transcriptome Fails to Support the Existence of Dual-Secreting IL-17A/IL-22 Th17 T Cells. Front. Immunol. 10:589. doi: 10.3389/fimmu.2019.00589
Received: 16 December 2018; Accepted: 05 March 2019;
Published: 04 April 2019.
Edited by:
Angelo Valerio Marzano, University of Milan, ItalyReviewed by:
Howard A. Young, National Cancer Institute at Frederick, United StatesWilson Liao, University of California, San Francisco, United States
Copyright © 2019 Le, Merleev, Luxardi, Shimoda, Adamopoulos, Tsoi, Wang, Alexanian, Raychaudhuri, Hwang, Gudjonsson, Marusina and Maverakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanual Maverakis, ZW1hdmVyYWtpc0B1Y2RhdmlzLmVkdQ==