 Daniel Ajona
Daniel Ajona Sergio Ortiz-Espinosa1,4
Sergio Ortiz-Espinosa1,4 Ruben Pio
Ruben Pio- 1Program in Solid Tumors, Center for Applied Medical Research (CIMA), University of Navarra, Pamplona, Spain
- 2Navarra Institute for Health Research (IdISNA), Pamplona, Spain
- 3Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), Madrid, Spain
- 4Department of Biochemistry and Genetics, School of Sciences, University of Navarra, Pamplona, Spain
- 5Department of Pathology, Anatomy and Physiology, School of Medicine, University of Navarra, Pamplona, Spain
The complement system represents a pillar of the innate immune response. This system, critical for host defense against pathogens, encompasses more than 50 soluble, and membrane-bound proteins. Emerging evidence underscores its clinical relevance in tumor progression and its role in metastasis, one of the hallmarks of cancer. The multistep process of metastasis entails the acquisition of advantageous functions required for the formation of secondary tumors. Thus, targeting components of the complement system could impact not only on tumor initiation but also on several crucial steps along tumor dissemination. This novel vulnerability could be concomitantly exploited with current strategies overcoming tumor-mediated immunosuppression to provide a substantial clinical benefit in the treatment of metastatic disease. In this review, we offer a tour d'horizon on recent advances in this area and their prospective potential for cancer treatment.
Introduction
The complement system represents a master component effector of innate immunity. Complement activation and regulation encompasses more than 50 soluble and membrane-bound proteins.
The function of complement, which entails the recognition and removal of pathogens and harmful entities, is accomplished by a multistep and sequential serine proteases-mediated cascade. The release of proteolytic fragments mediates key homeostatic and effector functions including: opsonization, inflammation, adaptive immune regulation, coagulation, tissue repair, neural development, bone homeostasis, angiogenesis, and host–microbiota symbiosis (1). Owing to the potentially deleterious effects of the complement system, its activity is tightly regulated at different levels by a number of soluble and membrane-bound proteins (2). Inappropriate complement activation underlies a variety of physiopathological conditions including inflammatory diseases and cancer (3).
Because many of the complement functions modulate tumor progression, their preeminent roles in promoting tumor cell dissemination are not surprising. This review focuses on recent findings on the major role of the complement system in tumor progression and highlights its key contribution to the different steps of the metastatic cascade.
Complement Activation
Complement is mainly activated via three different recognition pathways: the classical, the lectin, and the alternative pathways. These three modes of complement activation converge into the generation of C3 convertases, which cleave C3 into C3a and C3b. C3a is an anaphylatoxin displaying an inflammatory regulation role. C3b can act in the opsonization process and as a component of the C5 convertase (4).
The classical pathway is triggered by the binding of C1q to antigen-antibody complexes, dying cells, extracellular matrix proteins, pentraxins, amyloid deposits, prions, or DNA (5).
The lectin pathway starts through binding of proteins homologous to C1q (mannose-binding lectin and H-, L-, or M-ficolins) to carbohydrate structures on pathogens (6). Both the classical and the lectin pathways then sequentially cleave C4 and C2 for the generation of the classical/lectin C3 convertase (C4bC2b) (4).
Finally, the alternative pathway is initiated by the spontaneous hydrolysis of C3, also known as the “tickover” of C3, which after the formation of C3(H2O) can bind to factor B. Cleavage of factor B by factor D forms the initial alternative pathway C3 convertase, C3(H2O)Bb (7).
Although these three routes of activation differ in their mechanisms of target recognition and initiation, they converge at C3 cleavage, yielding the active fragments C3a and C3b. C3b binding to C3 convertases assembles the C5 convertase that cleaves C5 into the anaphylatoxin C5a, and C5b. The latter fragment is indispensable to assemble the membrane attack complex which mediates targeted lysis (8).
Additional pathways of complement activation include C3 and C5 extrinsic protease cleavage (9–11), the C2-bypass pathway (12), and the properdin-mediated direct convertase formation on microbial surfaces (13).
Among the complement-derived downstream effectors, C3a and C5a play diverse roles in both homeostasis and disease. These molecules bind to their cognate seven-transmembrane domain receptors C5a receptor 1 (C5aR1; CD88) and C3a receptor (C3aR), respectively. C5a can also bind to C5aR2 (14). The role of C5aR2 remains poorly understood. Recently, it has been reported that the binding of C5a to C5aR2 in carcinoma-associated fibroblasts promotes tumor formation and chemoresistance by providing a survival niche for cancer stem cells (15).
Recent discoveries have also revealed that complement activation is not only restricted to the extracellular space, as originally thought, but also occurs in the cytoplasm. The intracellular components of complement (the so-called complosome) modulate metabolic processes during T cell effector differentiation (16, 17) but so far, their intracellular functions remain largely unexplored.
Complement in Cancer Patients
Neoplastic transformation involves complex genomic and epigenomic alterations perturbing normal cell homeostasis. Local or distant dissemination of tumor cells, one of the hallmarks of cancer, represents a multistep process that entails the gain of novel cellular functions which include invasion, increased cell locomotion, intravasation, survival in the circulation, overcoming immune attack, and colonization in foreign cellular niches to form secondary tumors (18).
Overcoming immune attack is a key step in tumor progression. Altered immune recognition is achieved by a variety of mechanisms (19), including the modulation of the complement system. Complement activation has been described in cancer patients with hematological malignancies such as lymphomas (20), and in a plethora of solid tumors (21–23). Furthermore, intact complement proteins were found increased in blood of patients with lung cancer (24, 25), neuroblastoma (26), and digestive tract tumors (27). However, complement-mediated cytotoxicity is circumvented by different mechanisms, most of which include the upregulation of complement regulatory proteins (28–30). These regulators normally protect tumor cells from complement-mediated destruction, and can be grouped into two categories: membrane-bound complement regulatory proteins (mCRPs) and soluble regulators. High expression of the mCRPs membrane cofactor protein (CD46), decay-accelerating factor (CD55), and CD59 (protectin) on tumor cells is associated with increased metastatic potential, and poor prognosis in a range of tumors (31–34). Similarly, the soluble regulators factor H and FHL-1 have been found elevated in biological fluids from ovarian (35), bladder (36) and lung cancer patients (37), and are also associated with poor prognosis (38). Other soluble regulators as clusterin (39), C1 inhibitor (40), factor I and C4b-binding protein (C4BP) (41) are secreted by tumor cells into the tumor milieu and could also be detected in the circulation.
Activation of the complement system by tumor cells was long believed to only benefit the patient. Preclinical data suggest that complement can evoke potent complement-dependent cytotoxicity against tumor cells, and a range of therapeutic strategies have been designed to potentiate complement activation and overcome the protection mediated by complement inhibitors. This approach has been specially tailored to enhance the therapeutic efficacy of monoclonal antibodies (42). However, recent findings have challenged this view, providing evidence of the cancer-promoting potential of complement activation and the utility of complement inhibition as an anticancer therapy (43). Complement components coopted by tumor cells can lead to the acquisition of self-advantageous functions tilting the balance toward tumor progression. For instance, lung cancer cells are recognized by the complement system more efficiently than their normal counterparts. This effect is mediated by the direct binding of C1q and leads to the subsequent activation of the classical complement pathway (44). This activation is compensated by the expression of factor H/FHL-1 and CD59 (45, 46). This equilibrium in complement activity would explain the elevated levels of complement fragments found in biological fluids from these patients. Thus, C4d, a split product of the classical complement pathway, is increased in biological fluids of lung cancer patients. Detection of C4d is associated with poor prognosis, and has been proposed as a potential biomarker of clinical value in the management of lung cancer patients (44, 47, 48). Similar results were obtained in oropharyngeal tumors by detecting C4d in saliva (49). Moreover, other complement factors have been associated with cancer. Anaphylatoxin C5a is increased in plasma from lung cancer patients (50, 51), and is associated with metastatic potential in lung and gastric cancer patients (52, 53). Similarly, C1QB is one of the top-scoring genes associated with lung metastases in osteosarcoma patients (54).
Taken together, these studies indicate an association between complement activation and malignant progression.
Complement in the Tumor Microenvironment
Of all complement proteolytic fragments derived from complement activation, anaphylatoxins are by far, the best described in cancer. Anaphylatoxins C5a and C3a trigger spurious tumor intracellular signaling pathways by binding to their cognate receptors expressed in tumor and immune cells. These signaling events deeply perturb the tumor milieu by inducing the recruitment and/or tumor-promoting abilities of myeloid-derived suppressor cells (MDSC), macrophages, neutrophils, and mast cells, preventing efficient T cell-mediated responses (55).
Elevation of C5a or C5aR1 levels has been observed in solid tumors including lung (50, 53), gastric (56), ovarian (57), breast (58), urothelial (59), and clear cell renal cancers (60).
C5a induces the recruitment of MDSCs into the tumor microenvironment, and markedly dampens anti-tumor T-cell responses. C5aR1 mediates these effects on two subpopulations of MDSCs. On one side, C5a is a potent chemoattractant for granulocytic MDSCs (a neutrophil-like subpopulation) and on the other, C5a stimulates the monocytic MDSC subpopulation with the concomitant production of reactive oxygen and nitrogen species (61).
C5aR1 expressed on MDSCs is also able to bind ribosomal protein S19 (RPS19), which is released from apoptotic tumor cells into the tumor microenvironment, leading to a shift toward Th2 cell responses with increased levels of immunosuppressive TGF-β (62). Accordingly, pharmacological blockade of C5aR1 in a syngeneic model of lung cancer impaired tumor growth, decreased the percentage of splenic MDSCs, and downregulated immunosuppression-related genes including ARG1, IL6, IL10, CTLA4, LAG3, and PDL1 within the tumor milieu (50).
Besides MDSCs, C5a affects the biology of other leukocytes present in the tumor microenvironment. C5a elicits a strong pro-inflammatory infiltration with secretion of MCP-1, responsible for the recruitment of immunosuppressive macrophages, and increase of arginase-1 and IL-10 (63). Similarly, fibrinolytic enzyme-mediated generation of C5a regulates the protumorogenic properties of C5aR1+ mast cells and macrophages, leading to hampered antitumor CD8 T-cell responses in a model of squamous carcinogenesis. Interestingly, the combined treatment based on cytotoxic chemotherapy and the blockade of C5aR1 synergistically increased the recruitment and the cytotoxic properties of CXCR3+ effector memory CD8 T cells by IFNγ-dependent mechanisms (64). Ablation of PTX3, an important negative regulator of inflammation and complement activation, resulted in amplification of complement activation, MCP-1 production, and tumor-promoting macrophage recruitment. Consistently, pharmacological blockade of C5aR1 reversed these pro-tumorogenic effects (65).
Although far less studied than C5a, the anaphylatoxin C3a also preconditions a tumor-promoting microenvironment. Signaling mediated by C3a binding to C3aR contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4 T-cell responses (66). Autocrine complement C3 inhibits IL-10-mediated cytotoxic properties of tumor-infiltrating CD8 T lymphocytes through complement receptors C3aR and C5aR1, and enhances melanoma and breast cancer growth (67).
Moreover, complement activation may underlie the ability of tumors to evolve and adapt to different cues of the microenvironment increasing tumor progression. Thus, under hypoxic conditions, lung cancer cells downregulate complement inhibitors, factor H and factor I, to increase their susceptibility to complement activation (68). This phenomenon may fuel the generation of C5a which in turns may contribute to hypoxic stress in the tumor milieu to promote tumor progression through the inhibition of cell-mediated immunity. Indeed, in a syngeneic lymphoma model the impact of C5a in tumor microenvironment is dose-dependent (69).
Complement effectors can also affect tumor progression independently of complement activation. Factor B and factor I promote squamous cell tumor growth upon the activation of ERK1/2 (70, 71). C1q promote angiogenesis and lung metastasis in a syngeneic model of murine melanoma (72). In malignant pleural mesothelioma, C1q binds to hyaluronic acid in the tumor microenvironment and enhances tumor proliferation (73). C1q secreted by mesenchymal stromal cells mediates the activation of β-catenin in chronic lymphocytic leukemia and enhances malignant progression (74). On the other hand, properdin, a positive regulator of complement activity, induces endoplasmic reticulum-stress response and exerts a tumor suppressive role in breast cancer (75).
In summary, tumors are able to perturb complement-related immune effectors favoring tumor progression. Distorted complement homeostasis remodels the tumor microenvironment by inhibiting the anti-tumor immune responses and contributes to the metastatic dissemination of cancer cells (Figure 1).
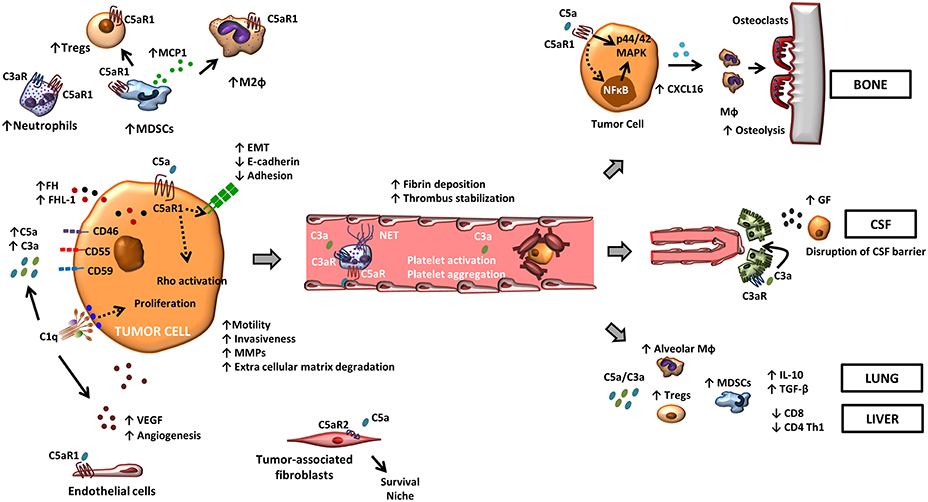
Figure 1. The role of complement in metastasis. Tumor-associated complement activation generates anaphylatoxins C5a and C3a in the tumor microenvironment. Binding of these molecules to their cognate receptors promote a range of tumor-promoting functions. C5a, through its receptor C5aR1, facilitates the recruitment and the activity of suppressive leukocyte subsets such as MDSCs, neutrophils, and Tregs in the tumor microenvironment. C3a also contributes to a suppressive tumor microenvironment by recruiting neutrophils. C5aR1 signaling affects endothelial function and tumor-associated angiogenesis, and the binding of C5a to C5aR2 in carcinoma-associated fibroblasts promotes tumor formation by providing a survival niche for cancer stem cells. In tumor cells, C5a/C5aR1 axis modulates tumor-induced MMP expression, increases tumor cell migration and invasiveness, enhances the release of pro-angiogenic factors, and induces EMT. Binding of C1q to tumor cells enhances tumor cell proliferation and favors angiogenesis in a complement activation-independent manner. Complement anaphylatoxins also facilitate tumor dissemination by stimulating a hyper-coagulation state and NETs, and adapt specific organ environments to the metastatic spread. This includes the disruption of the CSF barrier, the induction of CXCL16-mediated osteoclastogenesis, and the generation of an immunosuppressive microenvironment.
Early Metastasis Steps: Complement Effects on Tumor Cells
Epithelial–mesenchymal transition (EMT), loss of cell-cell adhesion, and increase of motility, invasiveness, and intravasation of tumor cells are at the core of the early metastatic events (18). Tumor-associated complement-activation modifies the tumor cell behavior endowing early metastatic traits. Perturbed complement activation leads to the generation of growth factors, proangiogenic factors, and other mediators that promote tumor growth and dissemination. These acquired pro-metastatic functions are mediated by the C3a and C5a stimulation of C3aR and C5aR1 in tumor cells, respectively, which triggers spurious intracellular signaling pathways. For instance, C5aR1 in lung tumor cells activates the p44/42 MAPK and NF-κB signaling pathways leading to the secretion of IL-8, VEGF, and MCP-1 to the tumor milieu (53). Complement components facilitate tumor dissemination by inducing an EMT in tumor cells which leads to the acquisition of a motile and less adherent phenotype. C5a/C5aR1 axis mediates the upregulation of transcription factor Snail and a concomitant decrease in E-cadherin and claudin-1 gene expression levels with increased invasiveness in hepatocellular carcinoma (76). In ovarian cells, TWIST1 enhances C3 expression and mediates EMT (77). According to these findings C5aR1-tumor expression was associated with tumor invasiveness, vascular and lymphatic invasion, liver metastasis, and poor outcome in patients with gastric tumors (78). Furthermore, C5aR1 inhibition hampers lung cancer cell migration, and up-regulates the expression of E-cadherin, suppressing EMT and invasiveness. Consistently, a negative correlation between the expression of C5aR1 and E-cadherin was found in lung primary tumors (79).
Initial steps for the acquisition of a metastatic phenotype also involves the secretion of stromelysins and other matrix metalloproteinases (MMPs) able to degrade different extracellular matrix components, especially the basal membrane, allowing for tumor cell intravasation and dissemination to local or distant sites (33). C5a markedly enhances cancer-mediated MMP activities and migratory and invasive tumor cell activities (80). C5a stimulation also decreases tumor adhesion to extracellular matrix proteins including collagens I and IV (53). Aberrantly expressed C5aR1 increases cell locomotion, cytoskeletal rearrangements with the formation of lamellipodia and membrane ruffling in liver bile duct malignant cells (80). C5aR1 signaling promotes motility and invasiveness through the activation of RhoA, and leads to enhanced invasion and vascular invasion in gastric cancer cells (56). ERK and PI3K, downstream C5aR1 activation, mediate an increase in cell invasiveness in renal cancer cells (81).
C3a-mediated stimulation elicits an increase in p42/44, p38 MAPK, and PKB/AKT activation and downregulates inducible hemeoxygenase-1 (HO-1) in leukemic cells (82). Autocrine stimulation of C5aR1 and C3aR upon C5a and C3a binding leads to PI3K/AKT signaling and regulates the proliferation and invasiveness of ovarian tumor cells (57).
In summary, complement-mediated effects are crucial in the early stages of metastasis, involving changes in tumor cell adherence to surrounding stroma and neighboring cells, increasing local invasiveness and promoting lymphatic and hematogenous dissemination.
Complement Effects on Dissemination
The host microenvironment at local or distant sites provides signals permissive for tumor promotion. Critical pathways triggered in the surrounding stroma and/or endothelial or lymphatic cells are required for proper cell-cell and cell-matrix engagement and for the secretion of a panoply of protumorogenic factors (83, 84). In addition, vascular or lymphatic vessels provide a major route by which tumor cells exit the primary tumor site, enter the circulation and establish metastasis (85). Furthermore, tumor vascular density is a prognostic indicator of metastatic dissemination. In cancer, complement may be involved in the modulation of the angiogenic program in the tumor microenvironment, although the specific role of complement in angiogenesis is highly dependent on the tumor type. For instance, C5aR1 blockade does not affect tumor angiogenesis in murine models of lung or cervical cancer (50, 61). In contrast, genetic inhibition of C3 and C5aR1 impairs endothelial cell function in an ovarian cancer model (86). C5a also supports an angiogenic program displayed by infiltrating macrophages in squamous cell carcinoma (64). C1q deposition on melanoma cells increases tumor vascular density and facilitates tumor progression (72). The evidence that complement has a role in endothelial homeostasis might have implications also at secondary metastatic sites, a possibility which remains largely unexplored.
Once in the circulation, tumor cells have to overcome the mechanical constraints imposed by sheer-stress, anoikis induced by cell anchorage-independency, and the immune attack. A role of platelets, together with fibrin and thrombin, has been invoked for the establishment of distant metastasis by protecting circulating tumor cells from mechanical stress and facilitating engraftment at target sites (87).
Complement components contribute to a hyper-coagulation state allowing tumor cell survival in the circulation. C3a induces platelet activation and aggregation favoring a pro-thrombogenic state (88). Similarly, C5a stimulates neutrophils to release Tissue Factor, inducing a prothrombotic phenotype (89). C3aR in neutrophils stimulates neutrophils extracellular traps (NETs) (90), extracellular structures composed of chromatin and degrading enzymes (myeloperoxidase, cathepsin G, and elastase) that contribute to form a three-dimensional scaffold that supports fibrin deposition and thrombus stabilization and entraps platelets, erythrocytes and tumor cells, driving a protumorogenic state (91).
This pro-tumorogenic milieu also favors the subsequent dissemination of tumor cells to neighboring or distant sites. Homing of tumor cells to target sites could also be actively mediated by factors released by target organs that act as potent tumor cell chemoattractants (92). But tumors also precondition target organs creating a hospitable niche by the mobilization of bone marrow-derived myeloid cells, tumor secreted factors such as VEGF, TGF, TNF (93–95), and tumor released-exosomes which also modulate the tumor microenvironment (96, 97). These nanometer-sized vesicles, which contain a complex cargo of membrane receptors, nucleic acids, cytoskeletal components, and intracellular proteins, act as unique vehicles for transport to local or distant organs. Tumor derived exosomes, which are more abundantly released in inflammation, represent another mechanism of immunosuppression. As observed for tumor cells, exosomes display CD55 and CD59, conferring resistance against complement-mediated lysis (98), and potentially regulating the exosome-mediated cross-talk associated with the metastatic program.
These events largely studied in murine models collectively contribute to prepare the “fertile soil” invoked by the Paget's hypothesis (99), and crystallize the concept of “premetastatic niche” (100). The premetastatic niche consists in the accumulation of aberrant immune cells and extracellular matrix proteins in target organs (101). Emerging data demonstrate that C5a contributes to the lung premetastatic niche by regulating the expression of TGF-β and IL-10 by immature myeloid cells and the subsequent accumulation of regulatory T cells, the proliferation of resident alveolar macrophages in the premetastatic lungs, and a decrease in the number and the maturation status of lung dendritic cells. As a consequence, effector CD4 T-cell responses skew toward Th1 responses (102, 103).
Late Steps of Metastasis
A similar paradigm to which occurs in the primary tumor could also influence metastatic behavior in the target organ. Tumor cells need to overcome the constraints imposed by the “foreign soil” and require compatibility with the hosting milieu. Each organ provides unique opportunities which could be exploited in the benefit of tumor cells by propelling the growth of micro to macrometastases (104). An increasing body of evidence indicates that complement is involved in this process, resulting in tumor outgrowths at secondary sites.
Genetic abrogation of C5aR1 in the host dampens M2-polarized tumor associated macrophages, leading to a decrease of liver and lung metastases in a syngeneic colon cancer model (52). Pharmacological inhibition of C5aR1 increases the infiltration of CD8 cytotoxic T cells in metastatic nodules, and impairs lung and liver metastatic processes with no effect detected in primary tumors. Thus, genetic or pharmacological inhibition of C5aR1 results in impaired metastasis (103).
Moreover, activation of C5aR1 in tumor cells leads to an increased prometastatic activity. For instance, in a lung cancer model of bone metastasis, C5a/C5aR1 axis induced the production of pro-osteoclastogenic factors favoring skeletal metastases. Among these factors, CXCL16 released upon C5aR1 signaling led to osteoclastogenic activation and osteolytic lesions. These effects were blocked by C5a inhibition or genetic silencing of C5aR1 in tumor cells, suggesting its implication in skeletal metastases (53). Indeed, complement is involved in bone homeostasis and turnover (105). Bone-forming osteoblasts and bone-resorbing osteoclasts are tightly regulated to ensure a balanced bone mass. Receptor activator of nuclear factor k-B ligand (RANKL), which is secreted by osteoblasts, binds to its receptor on the membrane of committed monocytes to differentiate into osteoclasts (106). Complement modulates osteoclasts differentiation in vitro and in vivo through C5aR1, but no effects were exerted in osteoblast differentiation (107). However, C3aR and C5aR1 signaling by C3a and C5a in osteoblasts modulates the release of pro-inflammatory pro-osteoclastogenic cytokines IL-6 and IL-8, and C5a increases RANKL in osteoblasts, overall favoring a pro-osteoclastogenic milieu (108). Because of these bone-specific mechanisms, the complement system might be specially relevant in skeletal metastases (53). Indeed, lung primary tumors that metastasize to bone show higher C5aR1 levels than those that metastasize to other locations, suggesting its major role in the tumor-induced skeletal lesions. Nevertheless, this axis also mediates lung metastases, since lung tumor colonization was decreased when lung cancer cells were devoid of C5aR1 (53).
In brain metastases, an elegant study by Massagué et al. unveiled a different prometastatic mechanism. C3 was upregulated in four leptomeningeal metastatic models and proved necessary for tumor growth within the leptomeningeal space. C3a, generated after C3 cleavage and bound to the C3aR expressed on the choroid plexus, was able to disrupt the blood-cerebrospinal fluid barrier. This effect was critical since blockade of this step provided a survival benefit in these models. However, C3 did not mediate cancer cell entry into the cerebrospinal fluid but other determinants were required for full tumor cell colonization (109).
Inhibition of complement-related proteins, and specially anaphylatoxins (14), has been proposed as a therapeutic option for maximizing the clinical efficacy of current immunotherapies. Recent studies have provided support of this idea after combined inhibition of anaphylatoxins and PD-1 signaling for the treatment of metastatic cancer. Administration of PD-1/PD-L1 blocking antibodies resulted in intratumoral complement activation and the subsequent accumulation of C5a within the tumor milieu (110). Importantly, the combination of C5a and PD-1 blockade reversed CD8 T-cell exhaustion, and markedly reduced lung cancer metastasis in two syngeneic animal models (111).
Conclusions
The complement system represents an important player in tumorigenesis and metastasis. Its relevance stems from its ability to foster a protumorogenic milieu by modulating tumor-immune responses. It also endows tumor cells with cell functions required for metastatic dissemination. Preclinical studies support the idea that the therapeutic blockade of complement has potential in combinatorial immunotherapy to effectively eradicate primary tumors and distant metastases. A better understanding of the mechanisms of interaction of the complement system with tumor cells and their microenvironment is required for designing combined novel immunotherapeutic regimens able to effectively target established tumors.
Author Contributions
DA, RP, and FL designed the concept. DA and FL wrote the manuscript. SO-E prepared the figure. All authors read and approved the final version of the manuscript.
Funding
Authors' work is supported by FIMA (Foundation for Applied Medical Research), and CIBERONC (CB16/12/00443). DA and RP are funded by Fundación Científica de la Asociación Española Contra el Cáncer, Fundación Ramón Areces, Juan Serrano, and Fondo de Investigación Sanitaria-Fondo Europeo de Desarrollo Regional Una manera de hacer Europa (FEDER, PI17/00411). FL is funded by La Caixa Foundation, Caja Navarra Foundation and the Spanish Ministry of Economy and Competitiveness (SAF2015-71606R). SO-E was supported by a predoctoral fellowship from the Asociación de Amigos de la Universidad de Navarra and now is supported by an FPU fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. (2017) 18:1288–98. doi: 10.1038/ni.3858
2. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. (2016) 274:152–71. doi: 10.1111/imr.12475
3. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
4. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
5. Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. (2009) 30:83–90. doi: 10.1016/j.it.2008.11.003
6. Hein E, Garred P. The lectin pathway of complement and biocompatibility. Adv Exp Med Biol. (2015) 865:77–92. doi: 10.1007/978-3-319-18603-0_5
7. Rodriguez de Cordoba S, Harris CL, Morgan BP, Llorca O. Lessons from functional and structural analyses of disease-associated genetic variants in the complement alternative pathway. Biochim Biophys Acta. (2011) 1812:12–22. doi: 10.1016/j.bbadis.2010.09.002
8. Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol. (2014) 772:229–62. doi: 10.1007/978-1-4614-5915-6_11
9. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. (2006) 12:682–7. doi: 10.1038/nm1419
10. Clark A, Weymann A, Hartman E, Turmelle Y, Carroll M, Thurman JM, et al. Evidence for non-traditional activation of complement factor C3 during murine liver regeneration. Mol Immunol. (2008) 45:3125–32. doi: 10.1016/j.molimm.2008.03.008
11. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
12. Selander B, Mårtensson U, Weintraub A, Holmström E, Matsushita M, Thiel S, et al. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest. (2006) 116:1425–34. doi: 10.1172/JCI25982
13. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. (2007) 179:2600–8. doi: 10.4049/jimmunol.179.4.2600
14. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: emerging roles in cancer progression and treatment. Semin Cell Dev Biol. (2017) 85:153–63. doi: 10.1016/j.semcdb.2017.11.023
15. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56 e816. doi: 10.1016/j.cell.2018.01.009
16. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
17. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science. (2016) 352:aad1210. doi: 10.1126/science.aad1210
18. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. (2018) 18:533–48. doi: 10.1038/s41568-018-0038-z
19. Pardoll D. Does the immune system see tumors as foreign or self? Ann Rev Immunol. (2003) 21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135
20. Bu X, Zheng Z, Wang C, Yu Y. Significance of C4d deposition in the follicular lymphoma and MALT lymphoma and their relationship with follicular dendritic cells. Pathol Res Pract. (2007) 203:163–7. doi: 10.1016/j.prp.2006.11.004
21. Lucas SD, Karlsson-Parra A, Nilsson B, Grimelius L, Akerström G, Rastad J, et al. Tumor-specific deposition of immunoglobulin G and complement in papillary thyroid carcinoma. Hum Pathol. (1996) 27:1329–35.
22. Makela K, Helen P, Haapasalo H, Paavonen T. Complement activation in astrocytomas: deposition of C4d and patient outcome. BMC Cancer. (2012) 12:565. doi: 10.1186/1471-2407-12-565
23. Niculescu F, Rus HG, Retegan M, Vlaicu R. Persistent complement activation on tumor cells in breast cancer. Am J Pathol. (1992) 140:1039–43.
24. Narayanasamy A, Ahn JM, Sung HJ, Kong DH, Ha KS, Lee SY, et al. Fucosylated glycoproteomic approach to identify a complement component 9 associated with squamous cell lung cancer (SQLC). J Proteomics. (2011) 74:2948–2958. doi: 10.1016/j.jprot.2011.07.019
25. Gminski J, Mykala-Ciesla J, Machalski M, Drozdz M, Najda J. Immunoglobulins and complement components levels in patients with lung cancer. Roman J Intern Med. (1992) 30:39–44.
26. Kim PY, Tan O, Diakiw SM, Carter D, Sekerye EO, Wasinger VC, et al. Identification of plasma complement C3 as a potential biomarker for neuroblastoma using a quantitative proteomic approach. J Proteomics. (2014) 96:1–12. doi: 10.1016/j.jprot.2013.10.032
27. Chong PK, Lee H, Loh MC, Choong LY, Lin Q, So JB, et al. Upregulation of plasma C9 protein in gastric cancer patients. Proteomics. (2010) 10:3210–21. doi: 10.1002/pmic.201000127
28. Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. (2003) 40:109–23. doi: 10.1016/S0161-5890(03)00112-3
29. Gancz D, Fishelson Z. Cancer resistance to complement-dependent cytotoxicity (CDC): problem-oriented research and development. Mol Immunol. (2009) 46:2794–800. doi: 10.1016/j.molimm.2009.05.009
30. Yan J, Allendorf DJ, Li B, Yan R, Hansen R, Donev R. The role of membrane complement regulatory proteins in cancer immunotherapy. Adv Exp Med Biol. (2008) 632:159–74. doi: 10.1007/978-0-387-78952-1_12
31. Maciejczyk A, Szelachowska J, Szynglarewicz B, Szulc R, Szulc A, Wysocka T, et al. CD46 Expression is an unfavorable prognostic factor in breast cancer cases. Appl Immunohistochem Mol Morphol. (2011) 19:540–6. doi: 10.1097/PAI.0b013e31821a0be9
32. Meng ZW, Liu MC, Hong HJ, Du Q, Chen YL. Expression and prognostic value of soluble CD97 and its ligand CD55 in intrahepatic cholangiocarcinoma. Tumour Biol. (2017) 39:1010428317694319. doi: 10.1177/1010428317694319
33. Brown GT, Murray GI. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol. (2015) 237:273–81. doi: 10.1002/path.4586
34. Ouyang Q, Zhang L, Jiang Y, Ni X, Chen S, Ye F, et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int J Oncol. (2016) 48:2015–24. doi: 10.3892/ijo.2016.3408
35. Junnikkala S, Hakulinen J, Jarva H, Manuelian T, Bjørge L, Bützow R, et al. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br J Cancer. (2002) 87:1119–27. doi: 10.1038/sj.bjc.6600614
36. Cheng ZZ, Corey MJ, Pärepalo M, Majno S, Hellwage J, Zipfel PF, et al. Complement factor H as a marker for detection of bladder cancer. Clin Chem. (2005) 51:856–63. doi: 10.1373/clinchem.2004.042192
37. Pio R, Garcia J, Corrales L, Ajona D, Fleischhacker M, Pajares MJ, et al. Complement factor H is elevated in bronchoalveolar lavage fluid and sputum from patients with lung cancer. Cancer Epidemiol Biomarkers Prev. (2010) 19:2665–72. doi: 10.1158/1055-9965.EPI-10-0467
38. Cui T, Chen Y, Knösel T, Yang L, Zöller K, Galler K, et al. Human complement factor H is a novel diagnostic marker for lung adenocarcinoma. Int J Oncol. (2011) 39:161–8. doi: 10.3892/ijo.2011.1010
39. Koltai T. Clusterin: a key player in cancer chemoresistance and its inhibition. OncoTargets Ther. (2014) 7:447–56. doi: 10.2147/OTT.S58622
40. Gasque P, Thomas A, Fontaine M, Morgan BP. Complement activation on human neuroblastoma cell lines in vitro: route of activation and expression of functional complement regulatory proteins. J Neuroimmunol. (1996) 66:29–40.
41. Okroj M, Hsu YF, Ajona D, Pio R, Blom AM. Non-small cell lung cancer cells produce a functional set of complement factor I and its soluble cofactors. Mol Immunol. (2008) 45:169–79. doi: 10.1016/j.molimm.2007.04.025
42. Kolev M, Towner L, Donev R. Complement in cancer and cancer immunotherapy. Arch Immunol Ther Exp. (2011) 59:407–19. doi: 10.1007/s00005-011-0146-x
43. Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. (2009) 30:286–92. doi: 10.1016/j.it.2009.04.002
44. Ajona D, Pajares MJ, Corrales L, Perez-Gracia JL, Agorreta J, Lozano MD, et al. Investigation of complement activation product c4d as a diagnostic and prognostic biomarker for lung cancer. J Natl Cancer Institute. (2013) 105:1385–93. doi: 10.1093/jnci/djt205
45. Ajona D, Castaño Z, Garayoa M, Zudaire E, Pajares MJ, Martinez A, et al. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. (2004) 64:6310–8. doi: 10.1158/0008-5472.CAN-03-2328
46. Ajona D, Hsu YF, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. (2007) 178:5991–8. doi: 10.4049/jimmunol.178.9.5991
47. Ajona D, Okrój M, Pajares MJ, Agorreta J, Lozano MD, Zulueta JJ, et al. Complement C4d-specific antibodies for the diagnosis of lung cancer. Oncotarget. (2018) 9:6346–55. doi: 10.18632/oncotarget.23690
48. Ajona D, Razquin C, Pastor MD, Pajares MJ, Garcia J, Cardenal F, et al. Elevated levels of the complement activation product C4d in bronchial fluids for the diagnosis of lung cancer. PLoS ONE. (2015) 10:e0119878. doi: 10.1371/journal.pone.0119878
49. Ajona D, Pajares MJ, Chiara MD, Rodrigo JP, Jantus-Lewintre E, Camps C, et al. Complement activation product C4d in oral and oropharyngeal squamous cell carcinoma. Oral Dis. (2015) 21:899–904. doi: 10.1111/odi.12363
50. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
51. Zhao C, Li Y, Qiu W, He F, Zhang W, Zhao D, et al. C5a induces A549 cell proliferation of non-small cell lung cancer via GDF15 gene activation mediated by GCN5-dependent KLF5 acetylation. Oncogene. (2018) 37:4821–37. doi: 10.1038/s41388-018-0298-9
52. Piao C, Zhang WM, Li TT, Zhang CC, Qiu S, Liu Y, et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res. (2018) 366:127–38. doi: 10.1016/j.yexcr.2018.03.009
53. Ajona D, Zandueta C, Corrales L, Moreno H, Pajares MJ, Ortiz-Espinosa S, et al. Blockade of the complement C5a/C5aR1 axis impairs lung cancer bone metastasis by CXCL16-mediated effects. Am J Respir Critic Care Med. (2018) 197:1164–76. doi: 10.1164/rccm.201703-0660OC
54. Zandueta C, Ormazábal C, Perurena N, Martínez-Canarias S, Zalacaín M, Julián MS, et al. Matrix-Gla protein promotes osteosarcoma lung metastasis and associates with poor prognosis. J Pathol. (2016) 239:438–49. doi: 10.1002/path.4740
55. Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. (2017) 127:780–9. doi: 10.1172/JCI90962
56. Kaida T, Nitta H, Kitano Y, Yamamura K, Arima K, Izumi D, et al. C5a receptor (CD88) promotes motility and invasiveness of gastric cancer by activating RhoA. Oncotarget. (2016) 7:84798–809. doi: 10.18632/oncotarget.12656
57. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. (2014) 6:1085–95. doi: 10.1016/j.celrep.2014.02.014
58. Imamura T, Yamamoto-Ibusuki M, Sueta A, Kubo T, Irie A, Kikuchi K, et al. Influence of the C5a-C5a receptor system on breast cancer progression and patient prognosis. Breast Cancer. (2016) 23:876–85. doi: 10.1007/s12282-015-0654-3
59. Wada Y, Maeda Y, Kubo T, Kikuchi K, Eto M, Imamura T. C5a receptor expression is associated with poor prognosis in urothelial cell carcinoma patients treated with radical cystectomy or nephroureterectomy. Oncol Lett. (2016) 12:3995–4000. doi: 10.3892/ol.2016.5137
60. Xi W, Liu L, Wang J, Xia Y, Bai Q, Long Q, et al. High Level of Anaphylatoxin C5a Predicts Poor Clinical Outcome in Patients with Clear Cell Renal Cell Carcinoma. Sci Rep. (2016) 6:29177. doi: 10.1038/srep29177
61. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
62. Markiewski MM, Vadrevu SK, Sharma SK, Chintala NK, Ghouse S, Cho JH, et al. The ribosomal protein S19 suppresses antitumor immune responses via the complement C5a receptor 1. J Immunol. (2017) 198:2989–99. doi: 10.4049/jimmunol.1602057
63. Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J Biol Chem. (2015) 290:10667–76. doi: 10.1074/jbc.M114.612622
64. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell. (2018) 34:561–578 e566. doi: 10.1016/j.ccell.2018.09.003
65. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. (2015) 160:700–14. doi: 10.1016/j.cell.2015.01.004
66. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. (2016) 196:4783–92. doi: 10.4049/jimmunol.1600210
67. Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. (2016) 6:1022–35. doi: 10.1158/2159-8290.CD-15-1412
68. Okroj M, Corrales L, Stokowska A, Pio R, Blom AM. Hypoxia increases susceptibility of non-small cell lung cancer cells to complement attack. Cancer Immunol Immunother. (2009) 58:1771–80. doi: 10.1007/s00262-009-0685-8
69. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. (2012) 189:2985–94. doi: 10.4049/jimmunol.1200846
70. Riihila P, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, Meri S, et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am J Pathol. (2017) 187:1186–97. doi: 10.1016/j.ajpath.2017.01.006
71. Riihila P, Nissinen L, Farshchian M, Kivisaari A, Ala-Aho R, Kallajoki M, et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J Invest Dermatol. (2015) 135:579–88. doi: 10.1038/jid.2014.376
72. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. (2016) 7:10346. doi: 10.1038/ncomms10346
73. Agostinis C, Vidergar R, Belmonte B, Mangogna A, Amadio L, Geri P, et al. Complement protein C1q binds to hyaluronic acid in the malignant pleural mesothelioma microenvironment and promotes tumor growth. Front Immunol. (2017) 8:1559. doi: 10.3389/fimmu.2017.01559
74. Mangolini M, Götte F, Moore A, Ammon T, Oelsner M, Lutzny-Geier G, et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat Commun. (2018) 9:3839. doi: 10.1038/s41467-018-06069-5
75. Block I, Müller C, Sdogati D, Pedersen H, List M, Jaskot AM, et al. CFP suppresses breast cancer cell growth by TES-mediated upregulation of the transcription factor DDIT3. Oncogene. (2019) 2019:739. doi: 10.1038/s41388-019-0739-0
76. Hu WH, Hu Z, Shen X, Dong LY, Zhou WZ, Yu XX. C5a receptor enhances hepatocellular carcinoma cell invasiveness via activating ERK1/2-mediated epithelial-mesenchymal transition. Exp Mol Pathol. (2016) 100:101–8. doi: 10.1016/j.yexmp.2015.10.001
77. Cho MS, Rupaimoole R, Choi HJ, Noh K, Chen J, Hu Q, et al. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J Immunol. (2016) 196:1412–8. doi: 10.4049/jimmunol.1501886
78. Nitta H, Shimose T, Emi Y, Imamura T, Ohnishi K, Kusumoto T, et al. Expression of the anaphylatoxin C5a receptor in gastric cancer: implications for vascular invasion and patient outcomes. Med Oncol. (2016) 33:118. doi: 10.1007/s12032-016-0834-9
79. Gu J, Ding JY, Lu CL, Lin ZW, Chu YW, Zhao GY, et al. Overexpression of CD88 predicts poor prognosis in non-small-cell lung cancer. Lung Cancer. (2013) 81:259–65. doi: 10.1016/j.lungcan.2013.04.020
80. Nitta H, Wada Y, Kawano Y, Murakami Y, Irie A, Taniguchi K, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin Cancer Res. (2013) 19:2004–13. doi: 10.1158/1078-0432.CCR-12-1204
81. Maeda Y, Kawano Y, Wada Y, Yatsuda J, Motoshima T, Murakami Y, et al. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. (2015) 33:1844–50. doi: 10.3892/or.2015.3800
82. Abdelbaset-Ismail A, Borkowska-Rzeszotek S, Kubis E, Bujko K, Brzezniakiewicz-Janus K, Bolkun L, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. Leukemia. (2017) 31:446–58. doi: 10.1038/leu.2016.198
83. Catena R, Luis-Ravelo D, Antón I, Zandueta C, Salazar-Colocho P, Larzábal L, et al. PDGFR signaling blockade in marrow stroma impairs lung cancer bone metastasis. Cancer Res. (2011) 71:164–74. doi: 10.1158/0008-5472.CAN-10-1708
84. Hernandez I, Moreno JL, Zandueta C, Montuenga L, Lecanda F. Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer. Oncogene. (2010) 29:3758–69. doi: 10.1038/onc.2010.130
85. Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J. (2015) 21:267–73. doi: 10.1097/PPO.0000000000000138
86. Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. (2012) 14:994–1004. doi: 10.1593/neo.121262
87. Konstantopoulos K, McIntire LV. Effects of fluid dynamic forces on vascular cell adhesion. J Clin Invest. (1996) 98:2661–5. doi: 10.1172/JCI119088
88. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med. (1983) 158:603–15.
89. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
90. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. (2016) 7:11037. doi: 10.1038/ncomms11037
91. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
92. Lecanda F. Tumor-stroma: tumor microenvironment as a target to combat metastasis. Dis Models Mech. (2011) 8:87–93. doi: 10.1016/j.ddmod.2011.07.010
93. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. (2009) 9:285–93. doi: 10.1038/nrc2621
94. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. (2002) 2:289–300. doi: 10.1016/S1535-6108(02)00153-8
95. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. (2005) 438:820–7. doi: 10.1038/nature04186
96. Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. (2012) 18:883–91. doi: 10.1038/nm.2753
97. Valencia K, Luis-Ravelo D, Bovy N, Antón I, Martínez-Canarias S, Zandueta C, et al. miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol Oncol. (2014) 8:689–703. doi: 10.1016/j.molonc.2014.01.012
98. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol. (2003) 33:522–31. doi: 10.1002/immu.200310028
99. Vicent S, Perurena N, Govindan R, Lecanda F. Bone metastases in lung cancer. Potential novel approaches to therapy. Am J Respir Critic Care Med. (2015) 192:799–809. doi: 10.1164/rccm.201503-0440SO
100. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
101. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
102. Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol. (2015) 194:5529–38. doi: 10.4049/jimmunol.1403215
103. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. (2014) 74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157
104. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. (2016) 529:298–306. doi: 10.1038/nature17038
105. Schoengraf P, Lambris JD, Recknagel S, Kreja L, Liedert A, Brenner RE, et al. Does complement play a role in bone development and regeneration? Immunobiology. (2013) 218:1–9. doi: 10.1016/j.imbio.2012.01.020
106. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. (2011) 11:411–25. doi: 10.1038/nrc3055
107. Kovtun A, Bergdolt S, Hägele Y, Matthes R, Lambris JD, Huber-Lang M, et al. Complement receptors C5aR1 and C5aR2 act differentially during the early immune response after bone fracture but are similarly involved in bone repair. Sci Rep. (2017) 7:14061. doi: 10.1038/s41598-017-14444-3
108. Ignatius A, Schoengraf P, Kreja L, Liedert A, Recknagel S, Kandert S, et al. Complement C3a and C5a modulate osteoclast formation and inflammatory response of osteoblasts in synergism with IL-1beta. J Cell Biochem. (2011) 112:2594–605. doi: 10.1002/jcb.23186
109. Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massagué J, et al. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell. (2017) 168:1101–13 e1113. doi: 10.1016/j.cell.2017.02.025
110. Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, et al. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Oncoimmunology. (2017) 6:e1349587. doi: 10.1080/2162402X.2017.1349587
Keywords: cancer, metastasis, complement, tumor microenvironment, anaphylatoxin, bone colonization
Citation: Ajona D, Ortiz-Espinosa S, Pio R and Lecanda F (2019) Complement in Metastasis: A Comp in the Camp. Front. Immunol. 10:669. doi: 10.3389/fimmu.2019.00669
Received: 10 January 2019; Accepted: 12 March 2019;
Published: 03 April 2019.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Uday Kishore, Brunel University London, United KingdomChiara Agostinis, IRCCS Materno Infantile Burlo Garofolo, Italy
Michael Kirschfink, Universität Heidelberg, Germany
Copyright © 2019 Ajona, Ortiz-Espinosa, Pio and Lecanda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruben Pio, cnBpb0B1bmF2LmVz