 Delphine Giusti1,2*†
Delphine Giusti1,2*† Estela Bini1†
Estela Bini1† Christine Terryn3
Christine Terryn3 Kevin Didier1
Kevin Didier1 Sébastien Le Jan1
Sébastien Le Jan1 Grégory Gatouillat1,2
Grégory Gatouillat1,2 Anne Durlach4Stéphane Nesmond1Celine Muller1
Anne Durlach4Stéphane Nesmond1Celine Muller1 Philippe Bernard1,5
Philippe Bernard1,5 Frank Antonicelli1,6‡
Frank Antonicelli1,6‡ Bach Nga Pham1,2‡
Bach Nga Pham1,2‡- 1Laboratory of Dermatology, Faculty of Medicine of Reims, University of Champagne-Ardenne, Reims, France
- 2Laboratory of Immunology, Reims University Hospital, University of Champagne-Ardenne, Reims, France
- 3PICT Platform, University of Reims Champagne-Ardenne, Reims, France
- 4Laboratory of Pathology, Reims University Hospital, Reims, France
- 5Department of Dermatology, Reims University Hospital, University of Champagne-Ardenne, Reims, France
- 6Department of Biological Sciences, Immunology, UFR Odontology, University of Reims Champagne-Ardenne, Reims, France
Background: DNA extracellular traps (ETs), released by neutrophils (NETs), or eosinophils (EETs), play a pathogenic role in several autoimmune disorders. However, to date, NETs have never been investigated in bullous pemphigoid (BP) with respect to clinical and immunological activities, both at baseline and at time of relapse which have been characterized with specific IL-17 and IL-23 patterns.
Objective: We sought to assess whether ETs were associated with BP as well as the relative contribution of IL-17 axis cytokines to NET induction.
Methods: Skin biopsy specimens were obtained from 11 patients with BP. Immuno-detection of neutrophils and eosinophils combined to DNA staining allowed us to investigate the in-situ presence of NETs and EETs using confocal scanning microscopy. NETs release was evaluated ex vivo by stimulating polymorphonuclear cells from BP patients with BP biological fluids in presence of IL-17A and IL-23 or of glucocorticoids.
Results: At baseline, ETs were observed in BP lesions at the site of dermal-epidermal cleavage. Despite an important infiltrate of eosinophils, ETs were essentially associated with neutrophils in situ and were not related to BP clinical activity at diagnosis. In situ observation of NETs was associated in 6 among 8 patients with serum capacity of NET induction. Notably both blister fluid and sera from BP patients at diagnosis and at time of relapse could induce NET formation ex vivo. In contrast, a longitudinal investigation showed a decrease of NET formation with time of treatment in patients undergoing remission. Mimicking relapse, complementation of sera from BP patients with ongoing remission with either IL-17A or IL-23 increased NET formation. Conversely, IL-17A inhibited NET formation induced by serum from BP patients with relapse supplemented or not with IL-23. Finally, glucocorticoids also inhibited NET formation ex vivo in BP.
Conclusion: NET formation is an associated phenomenon with BP. Furthermore, we showed that IL-23 favored NET formation, whereas the effects of IL-17A are environment dependent. Indeed, IL-17A displayed a protective effect on NET formation when associated with IL-23, showing for the first-time differential effects of these two cytokines in BP.
Introduction
Bullous pemphigoid (BP) is an invalidating autoimmune sub-epidermal blistering disease affecting preferentially the elderly. Eosinophils and neutrophils are the most represented cells in the skin inflammatory infiltrate of BP patients (1). Both cells have the capacity to form DNA traps (2–5) which play a pathogenic role in several autoimmune diseases (6, 7). The participation of eosinophil DNA extracellular traps (EET)s to blister formation has recently been reported in BP (7, 8). In contrast, neutrophil extracellular trap (NET) presence has not been demonstrated yet. Additionally, DNA trap formation has never been investigated in BP with respect to the clinical features of the disease i.e., activity and outcome. Also, the relationship between NET formation and the autoimmune or inflammatory responses, which characterize BP development, needs to be clarified.
Neutrophils and eosinophils are both involved in BP pathological process (8–13). Neutrophils play a crucial role in most BP experimental models to induce dermal-epidermal separation (10, 14–16). The cooperation with monocytic cell-line and the stimulation by CXCL-10, IL-17, or IL-23, lead neutrophils to release metalloproteinase MMP-9 and neutrophil elastase, which are responsible for dermal-epidermal junction (DEJ) disruption and BP180 cleavage (17–22). Besides, eosinophils are the predominant cells in the human BP inflammatory infiltrate (1). The activated eosinophils release a potent army of cytokines (9, 11) and granule proteins such as Eosinophil Cationic Protein (ECP), which serum concentrations parallel BP activity (23). The actions of both granulocytes converge to MMP-9 production, which suppresses the neutrophil elastase natural inhibitor, and thus contributes to dermal-epidermal splitting (24).
Stimuli inducing NETs release are numerous and highly variable. For instances auto-antibodies (AAb)s, as well as cytokines, appear to be able to induce this phenomenon (5, 6, 25). Indeed, AAbs from patients with rheumatoid arthritis and IL-17 were recently shown to induce NET formation in an ex vivo model of rheumatoid arthritis (25). In BP, the pathogenic role of anti-BP180 antibodies has been illustrated by both in vitro and in vivo studies, and their serum concentrations at diagnosis have been correlated with disease activity (14, 26–30). Cytokines also play a key role in BP pathogeny (17–19, 31–34). In previous studies, we showed that IL-17 levels were elevated in blister fluids, linked to a local production by neutrophils and mastocytes (17), and a relationship between IL-17 axis cytokines and BP outcome (18). More precisely, we evidenced an increased serum level of IL-23 or a high sustained serum level of IL-17 despite treatment in BP patient who later relapsed (18). Moreover, these inflammatory mediators are involved in BP pathophysiological process, as they enhance MMP-9 production by innate immune cells from patients (17, 18).
In the present study, we investigated DNA extracellular traps in BP with respect to clinical and immunological characteristics of the disease. Therefore, the aim of this study was to determine whether NETs or EETs or even both were associated with BP at tissue level, and to investigate IL-17 and IL-23 influence on NET formation ex vivo.
Materials and Methods
Patients
This prospective, observational, and translational study was conducted in our tertiary Referral Center for Autoimmune Bullous Diseases at the Reims University Hospital. Consecutive patients with newly diagnosed BP were included in this prospective study. Diagnosis of BP was made when the following criteria were met: presence of at least 3 out of 4 established clinical criteria by Vaillant et al in combination with positive direct immunofluorescence findings (30). Routine skin biopsy specimens of 11 BP patients were provided by the Pathology Department of the Reims University Hospital to perform in situ analysis. Ex-vivo assays were performed with sera from these 11 patients collected at time of diagnosis (at the same time as the biopsy) and with biological samples [sera and polymorphonuclear cells (PMN)] collected at time of diagnosis and around 150 and 360 days after, from 17 other consecutive BP patients. Sera were also collected at time of relapse in patients who underwent relapse despite treatment. PMNs used for ex vivo experiments were freshly isolated PMNs from patients with BP collected at any time point (between D1 and D360 after diagnosis) throughout the course of the study. Seven sera and PMN from healthy controls were provided by French Blood Agency and volunteers (mean age 66.4 years).
In situ Analysis of NETs/EETs
Immunofluorescence and confocal analysis of NETs and EETs were performed on paraformaldehyde-fixed and paraffin-embedded skin biopsy specimens from 11 BP patients. DNA staining along with neutrophils and eosinophils immunostaining was performed as follow on tissues. Four consecutive deparaffanized sections were used by patient, using a distinct eosinophil marker on each. After 15 min heat retrieval in sodium citrate buffer pH6, the sections were then blocked with PBS-BSA 3% for 30 min at room temperature. Then, simultaneous staining was performed for 30 min at room temperature with mouse anti-human myeloperoxidase (1:150; R&D Systems, Minneapolis, USA) for neutrophil staining and another primary antibody (Ab) for eosinophil staining: either rabbit anti-human IL-5Rα (1:150; Proteintech, Chicago, USA), or rabbit anti-human MBP, EDN, or ECP (1:150; Novus Biologicals, Littleton, USA). This was followed by 30 min of incubation at room temperature with matched secondary Abs: chicken anti-rabbit IgG Alexa Fluor 488 and chicken anti-mouse IgG Alexa Fluor 594 (Invitrogen). Sytox Orange Nucleic Acid Stain (5 μM) (Molecular Probes, Invitrogen, Carlsbad, USA) was finally used to stain DNA prior to mounting slides with Dako® fluorescent mounting medium. The presence of NETs in situ was confirmed by an immunostaining performed with a rabbit polyclonal Ab to Histone H3 (1:100 overnight; Abcam, Cambridge, UK) revealed by a secondary chicken anti-rabbit Ab Alexa Fluor 594 (1:200. Invitrogen, Carlsbad, USA) and a mouse monoclonal Ab to human myeloperoxydase coupled to a FITC fluorochrome (1:100; Abcam, Cambridge, UK).
Extracellular DNA filaments associated to both eosinophils and neutrophils were investigated by 2 independent investigators using 3-dimensional imaging by confocal scanning microscope LSM 710 NLO Zeiss (Zeiss SAS, Germany). Image analysis was performed by using ZEISS ZEN software (Zeiss SAS, Germany) and IMARIS Software (Bitplane, Switzerland). ETs were identified as shapes of DNA extruding from cell and stained by nucleic acid probe SYTOX orange. NETs were defined as extracellular DNA filaments stained by Sytox Orange extruding from cells labeled by MPO Ab, while cell MBP, EDN, ECP or IL-5R labeling characterized EETs.
To qualify eosinophil and neutrophil cell infiltrate, hematoxylin-eosin coloration of serial sections was performed for these 11 biopsy specimens. A rating from 0 to + + + was assigned by two operators to eosinophil and neutrophil infiltrates in the papillary dermis and in the blister cavity to allow a comparative semi-quantification of these cells.
Ex vivo Analysis of NET Formation
Peripheral PMNs cells were obtained by density-gradient centrifugation from 25 mL heparinized-treated whole blood (Granulosep, Eurobio-Abcys). NET generation was performed following the protocol proposed by Brinkmann et al. (35) with the difference that SYTOX Orange was used to stain extracellular DNA. 2.105 PMNs were incubated with either 5% blister fluids or 5% sera from BP patients, following preliminary optimization assays (Supplementary Figure S1). To investigate their potential influence on NET formation, recombinant IL-17A and IL-23 were added to cell culture medium at concentrations close to those determined in the sera of BP patients (1.2 ng/mL) (18). To assess the effects of treatment on NET induction by BP sera, methylprednisolone MP (10 μM), and Compound A (10 μM) were added to the cell culture medium in presence of BP sera. Each condition was tested with biological fluids from at least 3 different subjects (patients or controls).
NETs were visualized using an inverted epifluorescence microscope (AxioObserver Z1: Zeiss, Germany; camera Cool Snap HQ2: Roper Scientific, France). For each slide, a mosaic of 4 × 4 consecutive images was taken (scale image: 0.625 μm/pixel, driven by Metamorph Software (Roper scientific, Evry, France), which represents an area of around 2 mm2 per coverslips. Fluorescence signal was collected with bandpass 545/25 excitation filter and bandpass 605/70 emission filter. For each acquisition, fluorescence, and transmitted images were taken.
To assess NETs formation, the area occupied by the DNA filaments was determined using a dedicated home-made macro based on “skeletonize function” and the “analyse particle” tool running under IMAGE J® software (ImageJ, U. S. National Institutes of Health, Bethesda, USA.). Briefly, we skeletonized the fluorescent images, setting 40 pixels as the minimal resolution, and transformed these images into drawings of DNA filaments, which were measured and processed by the “Analyze particles” tool (Supplementary Figure S1). Area of each filament was measured, and the mean was calculated, making the conversion from pixels to μm (1pixel = 0.625 μm). The total number of cells was counted using the counter cell tool of Image J® software on the transmitted image acquired simultaneously with fluorescent image. Thus, this method allowed measuring the total number of PMNs, and the total area of NETs. Results were expressed in mean area of NETs per PMN (as proposed by Rebernick et al. (36). As variations were observed when using PMNs from different subjects, results were also expressed in ratio between mean area of NETs per cell for condition X and mean area of NETs per cell for the reference condition with the same PMNs.
Statistical Analysis
As the distribution of the variables could not be assumed to be normal, comparisons between two groups were performed using the Wilcoxon matched pairs signed rank test for paired data and the Mann-Whitney test for unpaired data. When more of two matched groups were compared, Friedman test was performed. Ratio paired t-test was used to compare NETs induction by sera with and without treatment (by cytokines, methylprednisolone or compound A). Statistical difference was considered significant if p-value was 0.05 or less.
Results
DNA Extracellular Traps Mainly Originated From Neutrophils in situ
To investigate the presence of either NETs or EETs or even both in BP at site of lesion, immune-detection of neutrophils and eosinophils along with DNA staining was performed on paraffin-embedded skin biopsy specimens from 11 BP patients. The mean age of the 11 newly diagnosed BP patients (6 female (54.5%) and 5 males (45.5%), sex ratio F/M: 1.2) was of 78.9 years. At baseline, the clinical BP activity was assessed by the Bullous Pemphigoid Disease Assessment Index (BPDAI) score and by the number of new daily blisters. The BPDAI median value was of 53 [23–90], and 9 (81.8%) BP patients presented with a severe disease characterized by more than 10 blisters a day. Finally, 5 (45.5%) BP patients experienced a relapse despite treatment during the first year of follow up (Table 1).
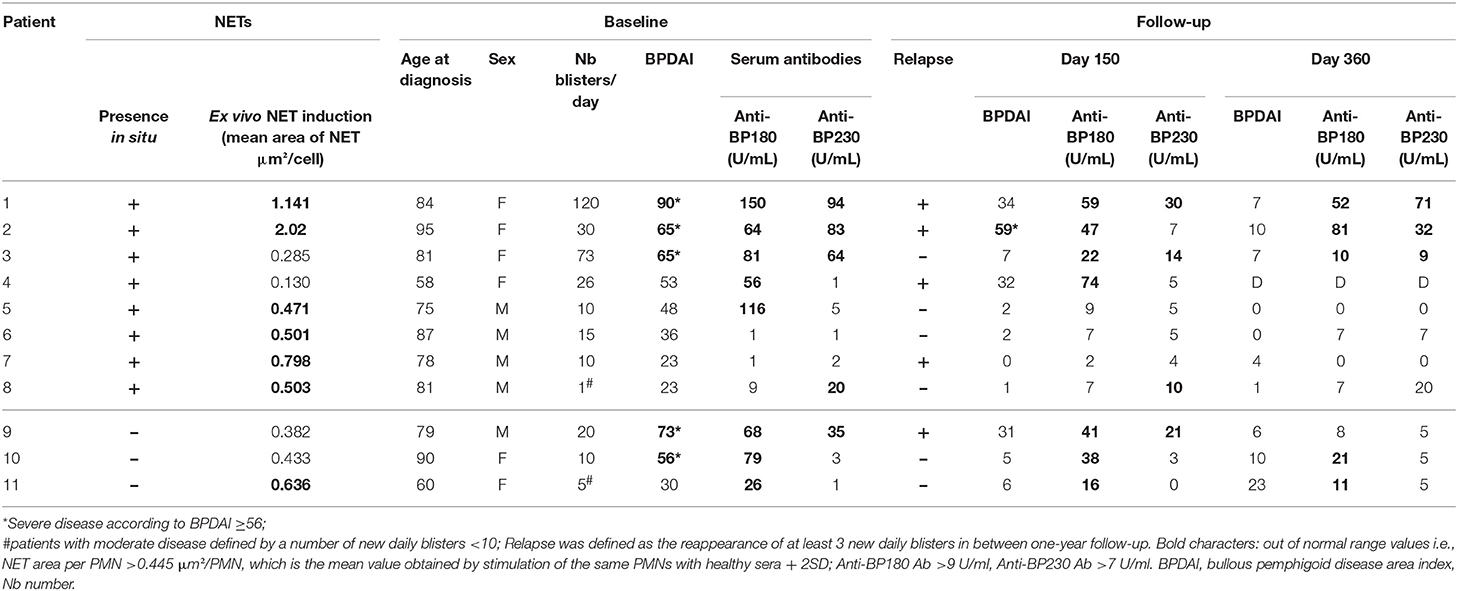
Table 1. Individual characteristics of BP patients.
Extracellular DNA filaments were identified in situ in 8 out of the 11 (73%) BP patients (Figures 1A–C). NETs were observed on lesional skin biopsy specimen, in the papillary dermis at the edge of the dermal-epidermal separation (Figures 1A–G). A 3-dimensional reconstruction picture corresponding to Figure 1E allowed defining NETs as shapes of DNA extruding from neutrophils stained by MPO (Supplementary Video S1; Figure 1F). EETs were only observed in one patient despite a higher number of eosinophils than neutrophils in BP lesional skin, corroborated by both HES staining and immunofluorescence using specific eosinophils markers including MBP, EDN, ECP, or IL-5Rα (Figure 1H; Table 2). DNA extracellular trap was observed neither in blister cavity nor in perilesional dermis, despite the presence of numerous inflammatory cells. Besides, in those 8 BP patients, NETs were absent from skin biopsy specimens performed at distance of the lesions (data not shown). Also, no DNA traps were evidenced in 4 plastic surgical normal skin specimens (mean age of controls 66.5 years) (data not shown).
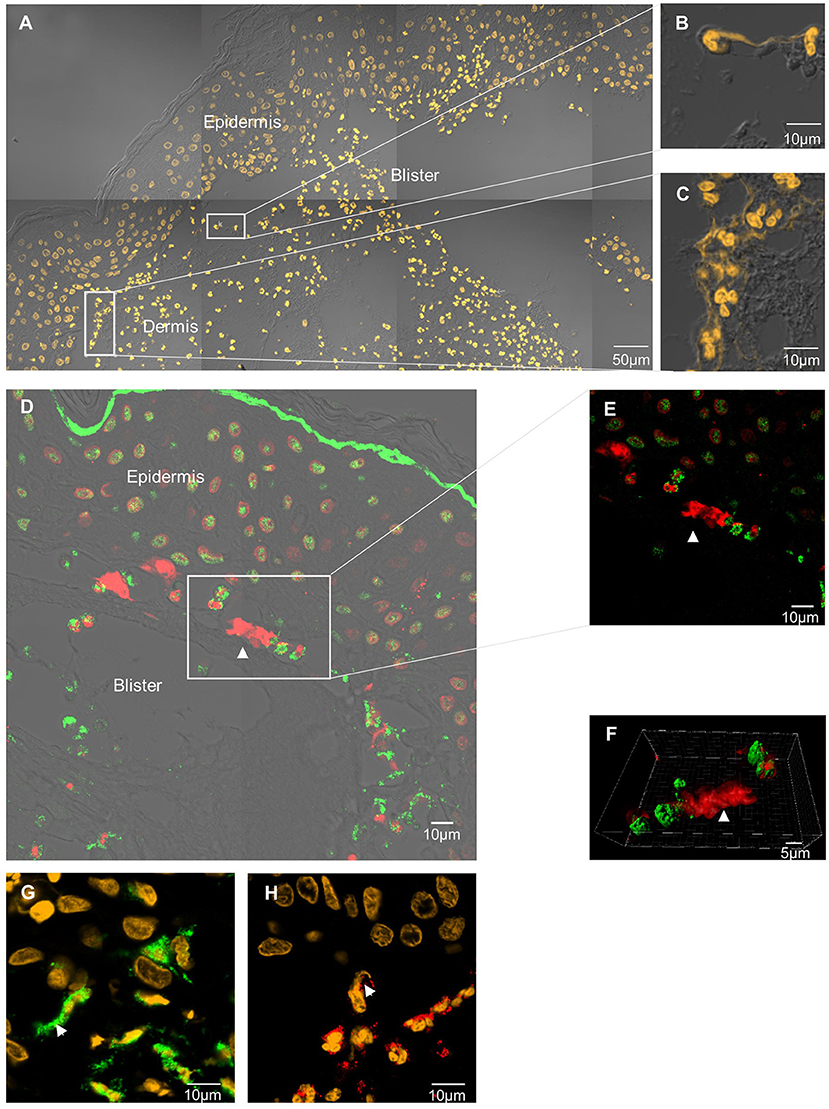
Figure 1. In situ exploration of DNA traps in BP. DNA extracellular traps are located at the edge of the DEJ splitting, in the papillary dermis of BP patient skin. Skin biopsy specimen stained by SYTOX Orange nucleic acid probe (A–C). Neutrophil extracellular trap stained by anti-histone H3 Ab (red) and anti-myeloperoxidase Ab (green) (D–F). 3-dimensional reconstruction imaging by confocal microscopy and IMARIS® software of this NET (F). Magnification 80x, numerical aperture of 1, z-stack with 0.67μm step-size. Neutrophil extracellular trap stained by Sytox orange and anti-myeloperoxidase (green) (G). Eosinophil extracellular trap stained by Sytox orange and anti-eosinophil-derived-neurotoxin Ab (red) (H).
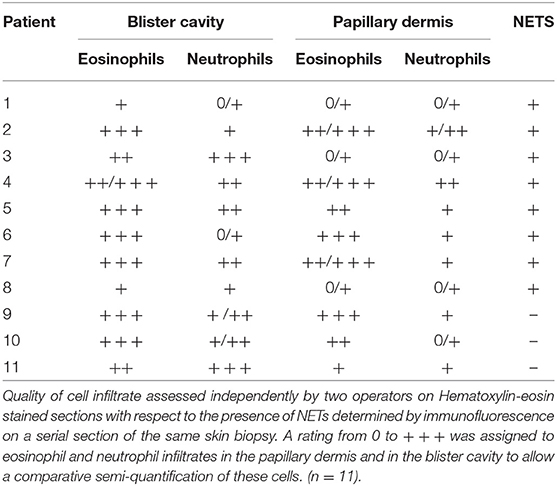
Table 2. Inflammatory cell infiltrate in BP skin according to the presence of NETs forming cells in situ.
In situ occurrence of NETs appeared to be independent of the disease activity at time of diagnosis. Indeed, patients in whom NETs were seen, showed miscellaneous BPDAI scores with values ranged from 23 to 90 and a median of 50.5 (Tables 1, 3). Furthermore, these 8 BP patients were evenly distributed according to the BPDAI score, as 3 patients displayed a BPDAI score superior to 56, 2 with a BPDAI close to mean value between 48 and 53, and 3 with a BPDAI inferior to 36. Also, NETs were observed in both patients with a multibullous BP (displaying more than 10 new blisters per day and up to 120 new daily blisters), and in those with a limited disease (down to only one blister per day). Likewise, the presence of NETs was not associated with BP relapse, as among the 8 BP patients displaying NETs at baseline, 4 later underwent a relapse and 4 remained on remission over the one-year of clinical follow-up (Table 1).
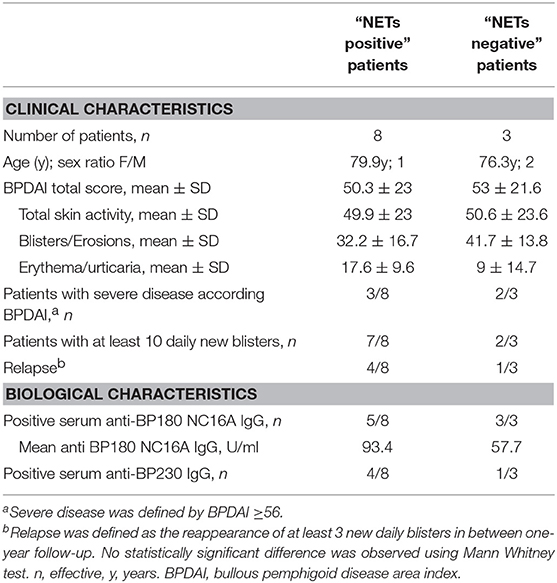
Table 3. Clinical and biological characteristics of BP patients according to the in-situ observation of NETs.
Both Neutrophils and Biological Fluids From Patients With BP Were Required for NET Formation
To further investigate the factors involved in NET formation associated with BP, we established an ex vivo NET generation model with peripheral blood PMNs from patients with BP stimulated with blister fluid collected at time of diagnosis (Figure 2A). Similar NET formation level was observed when PMNs from BP patients were stimulated with BP serum (Figures 2B–D, p = 0.25, Wilcoxon matched pairs test). Besides, immunostaining of myeloperoxydase and citrullinated histones allowed to confirm that neutrophils were the source of the observed NETs in this ex vivo model (Figure 2C). The induction of NETs by blister fluids was independent from BPDAI score at diagnosis and from BP 180 Ab levels in blister fluids (Figures 2E,F). We then investigated the capacity of the sera from the 11 BP patients for whom in situ exploration of NETs was performed, to induce NET formation. Among the 8 patients previously identified with NETs in situ, 6 showed capacity of NET induction compared with 1 patient among the 3 for whom in-situ assessment was negative (Table 1).
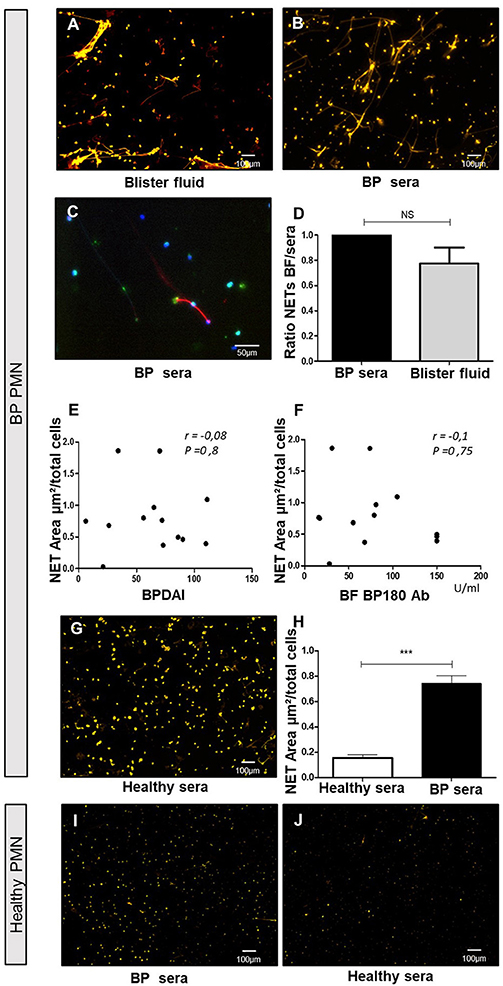
Figure 2. Blister fluids and BP sera induce NET formation by BP PMN but not by control PMN. Ex vivo stimulation of BP PMNs with BP blister fluids (BF) (A), and BP sera (B,C) stained by Sytox orange (A,B) or anti-Histone H3 and anti-MPO Ab (C). NET area induced by BP sera vs. BF from the same patients on heterologous BP PMNs (D) (n = 3); results expressed in ratio of NET area induced by BF to NET area induced by serum of the same patient, results represent mean ± SEM, NS, non-significant. Correlation of NET area induced by heterologous BF with BPDAI (E) and BF concentration of BP180 Ab (F). Stimulation of BP PMN with healthy control sera (G) and comparison with NET area induced by BP sera (H) (n = 7), ***p < 0.001, results represent mean ± SEM. Stimulation of PMNs from healthy controls by BP sera (I), or heterologous healthy sera (J). Magnification 20x. BPDAI, Bullous Pemphigoid Disease Area Index; BF BP180 Ab, concentrations of anti-BP180 antibodies in blister fluids.
In contrast, the capability of sera from healthy donors to induce NET formation by PMN from BP patients was significantly lower than sera from BP patients (0.15 vs. 0.74 μm2/cell, p = 0.0006 Mann-Whitney test) (Figures 2G,H). To investigate whether circulating PMNs from BP patients were primed to release NETs, we assessed the ability of PMNs isolated from healthy donor peripheral blood to generate NETs upon BP serum stimulation. As showed in Figure 2I, NET quantification remained low in those conditions. Also, when PMNs from healthy donors were stimulated with heterologous sera from healthy donors, NET area remained low (Figure 2J).
BP Serum Capacity to Induce NET Formation Decreased in BP Patients With Ongoing Remission
Then, we evaluated the ability of BP sera collected during patient's follow up to induce NET release. Compared with baseline values, NET formation was gradually reduced with time of treatment as illustrated by ex vivo experiments conducted with sera collected at day150 and day 360 (0.18, 0.13, and 0.06 μm2/cell, p = 0.0001Friedman test) (Figures 3A–C,E). Conversely, NET levels remained as elevated as with BP serum at diagnosis when NET generation was performed with the BP sera collected at time of relapse from the same patients (0.13 at baseline vs. 0.15 μm2/cell at time of relapse, p = 0.68 Wilcoxon matched pairs test) (Figures 3D,F).
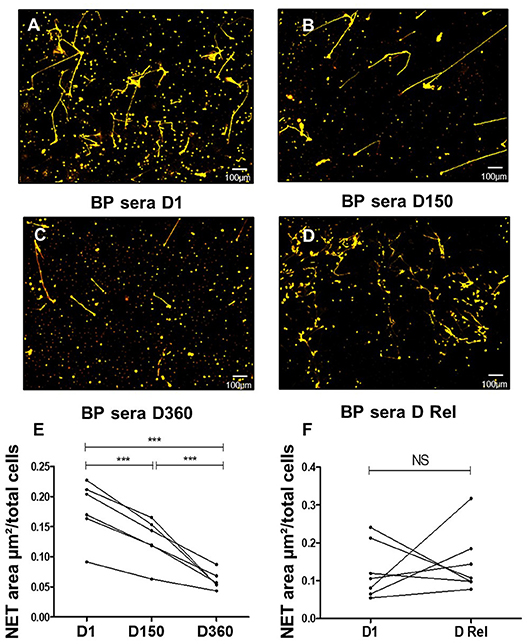
Figure 3. NETs are induced more efficiently by BP sera at diagnosis and at time of relapse. Ex vivo stimulation of BP PMNs with heterologous BP sera collected: at baseline (A), around 150 days after diagnosis in patients with controlled disease (B), around 360 days after diagnosis in patients undergoing remission (C), or at time of relapse in patients undergoing relapse (D). Magnification 20x. NET area measured for each condition: stimulation of BP PMNs with BP sera collected Baseline (D1) vs matched sera collected at Day 150 and Day 360 (E) (n = 6), ***p < 0.0001. NET area measured after stimulation of BP PMNs with BP sera collected at baseline (D1) vs. matched sera collected at time of relapse (D Rel) from the same patients (F) (n = 7), NS: non-significant.
IL-17 and IL-23 Display Differential Effects on NET Formation
As we previously reported that IL-17 and IL-23 remained elevated or even increased in the sera of patients at risk of relapse (18), we further investigated whether these cytokines participated to NET induction in BP. In this line, we complemented BP sera collected after 150 days from patients with ongoing remission with either IL-17A or IL-23 or even both. Both IL-17A and IL-23 independently enhanced NET formation with an even greater effect under IL-23 stimulation (1.9-fold increase with IL-17, p = 0.029, and 10-fold increase with IL-23 with respect to the stimulation with D150 sera alone, p = 0.04 ratio paired t-test, Figures 4A–D,F,G). We then assessed the combined effects of both cytokines, IL-17A and IL-23 on NET generation. The high NET formation level induced by addition of IL-23 was reduced when IL-17A was added to the cell culture medium (Figures 4E,H).
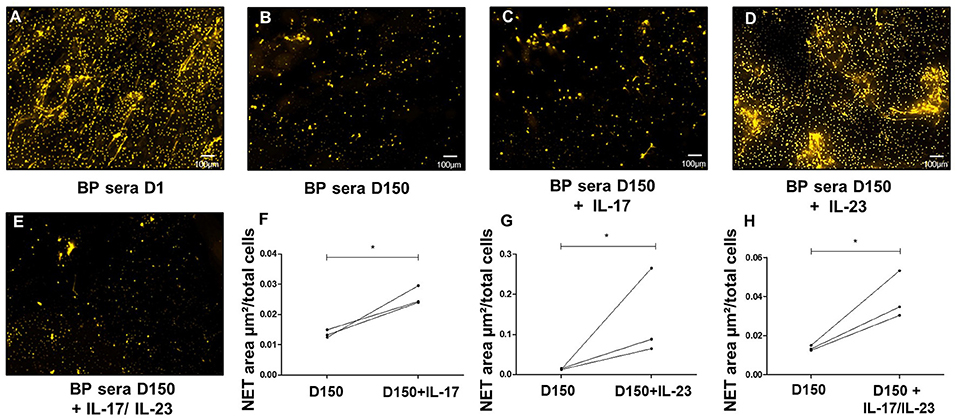
Figure 4. IL23 is critical to NETs formation. Ex-vivo stimulation of BP PMNs with BP sera collected at baseline (A), around 150 days after diagnosis in patients with controlled disease (B), and day 150 sera added with either IL-17A (C), IL-23 (D), or both IL-17A and IL-23 (E) (1.2 ng/mL for both cytokines). Comparison of NET area measured when cytokines IL-17A, IL-23, or both were added to day 150-serum with NET area induced by day 150- serum alone (F–H) (n = 3), * p < 0.05.
To further investigate the potential lowering effects of IL-17A on IL-23-induced NETs release, we added IL-17A to the potent NET inducer sera collected at the time of relapse. Supporting the above-mentioned observation, IL-17A addition (alone or combined with IL-23) significantly decreased the area occupied by NETs with respect to the area obtained with the serum of BP patients collected at time of relapse (0.09, and 0.17 vs. 0.45 μm2/cell, p = 0.05 and p = 0.02, respectively, ratio paired t-test) (Figures 5A–C,E,F). In contrast, supplementation of these sera collected at time of relapse with IL-23, did not affect their capacity to induce NET formation (0.48 vs. 0.45 μm2/cell, p = 0.89 ratio paired t-test) (Figures 5D,G).
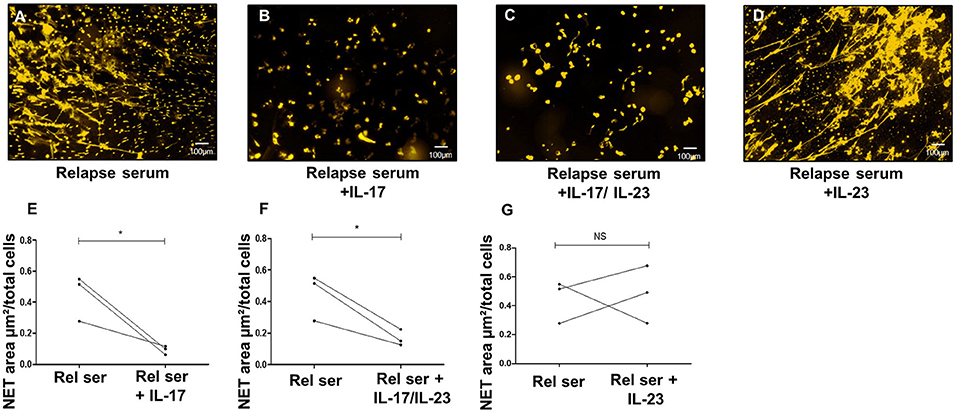
Figure 5. IL-17A inhibits NET formation by BP sera at time of relapse. Ex vivo stimulation of BP PMNs with BP sera collected at time of relapse alone (A), or in presence of either IL-17 (B), or both IL-17 and IL-23 (C), or IL-23 (D). Comparison of NET area measured when cytokines were added to serum collected at time of relapse with NET level generated in presence of this relapse serum (Rel Ser) alone (E–G) (n = 3), *p < 0.05.
NET Formation Is Inhibited by Methylprednisolone and by Compound A
Having demonstrated that the capacity of BP sera to induce NET release progressively decreased in BP patients with ongoing remission upon treatment, we then wondered whether corticosteroid therapy also reduced the capability of BP sera collected at baseline to induce NET formation. NET quantification revealed that both methylprednisolone (10 μM) and compound A (also named 2-(4-acetoxyphenyl)-2-chloro-N-ethyl ammonium chloride, a natural glucocorticoid receptor ligand (37), 10 μM) significantly decreased the capacity of BP serum to induce NET formation by BP PMNs (0.05 and 0.04 vs. 0.14 μm2/cell, p = 0.04 and p = 0.01, ratio paired t-test) (Figure 6).
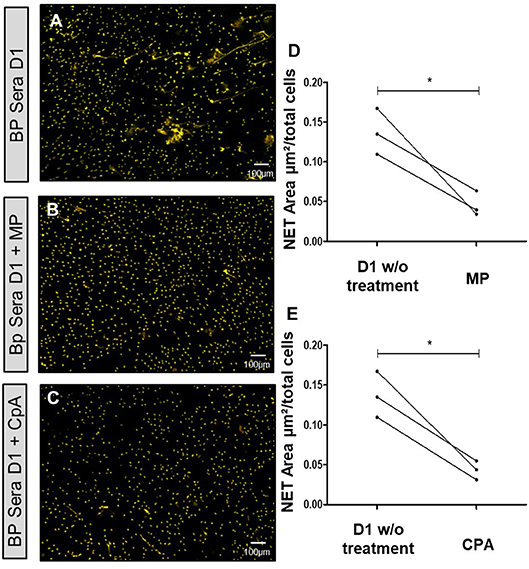
Figure 6. NET formation is inhibited by methylprednisolone or Compound A. Ex vivo stimulation of BP PMNs with BP sera collected at diagnosis (D1) alone (A), or in presence of either methylprednisolone (B), or compound A (C). Comparison of NET area measured when treatments were added to baseline serum with NET formation by this baseline serum without treatment (D,E) (n = 3). *p < 0.05, mean ± SEM are represented. MP, Methylprednisolone; CpA, Compound A; w/o, without.
Discussion
NET formation has recently been reported in several autoimmune diseases as a phenomenon potentially associated to tissue damage and/or loss of tolerance mechanisms (6). In this study, we brought in situ evidence of the presence of NETs in BP at the extremity of the blister where dermis separates from epidermis. Using an ex vivo model, we also showed that both BP neutrophils and biological fluids (blister fluid, serum) from BP patients were required for NET formation. Analysis of NET presence at tissue level with respect to BP clinical and to immunological data revealed that NET formation was not associated with specific characteristics of the disease at baseline. However, a longitudinal analysis of NET formation using our ex vivo model discriminated sera from BP patients with ongoing remission under treatment from those collected at time of relapse. Based on our previous studies (17, 18), we identified IL-23 as a potent NETs enhancer in BP. In contrast, we evidenced in BP a protective role of IL-17A despite the presence of potent inducers such as IL-23 or relapse sera, demonstrating for the first time in BP antagonist functions for these 2 cytokines.
To the best of our knowledge, we here evidenced for the first time the presence in situ of NETs in BP. Actually, NETs were observed in 8 out of 11 patients with BP at baseline. In contrast, EETs were present only in 1 case despite the use of 4 different markers on 4 sequential sections for eosinophil immunostaining. The absence of EETs could not be explained by the quality of the inflammatory infiltrate, as eosinophils were the most represented immune cells where NETs were detected. It is possible that EETs formation rather occurs in the skin of BP patients with tissue eosinophilia as previously described (7). So far, in our study NETs were observed in the skin biopsy from BP patients whatever disease activity, although the quantity of NETs observed remained low in all cases. Thus, our observations suggest that NET formation is a common but quantitatively rare process associated with BP.
Noticeably, NETs localized precisely at the very edge of dermal-epidermal separation. NET formation could be dictated by skin microenvironment. Furthermore, most patients for whom in situ exploration was positive, displayed at the same time capacity to induce NET formation ex vivo when BP PMNs were cultured in presence of their sera. Actually, we showed that both body fluid and PMNs from BP patients were required for NET formation. Such specific NET localization at the extremity of blister where the epidermis separates from the dermis could be related to a role of NETs in blister formation as it was demonstrated using an ex vivo model that degradation of DNA filaments inhibited DEJ splitting (8).
Although immunoglobulins have been associated with NET formation (38–42), such process could not be foreseen in BP. Indeed, no correlation could be drawn between NETs' presence in situ or NET formation ex vivo and serum or blister fluids levels of anti-BP180 AAb. Besides, the fact that NETs were observed in most skin biopsy specimen independently of disease activity, suggests that NET inducers may be present in all body fluids from BP patients at baseline. Indeed, sera from BP patients in clinical remission were less efficient to induce NET formation. Altogether, this suggests that NET induction is rather associated with inflammatory mediators than with the autoimmune response in BP. In this setting, it was shown that inflammatory molecules such as cytokines and complement components were potent inducers of DNA traps (5, 6, 43–45).
Neutrophils' priming is a required step for NET formation in BP. In BP skin environment, the presence of cytokines able to activate innate cells such as neutrophils, eosinophils and monocytes has already been largely described (17–19, 31). Such inflammatory response may be responsible for neutrophils' priming and explain why NET formation level remained low in neutrophils from healthy donors even when stimulated with BP serum. Accordingly, it has been shown that neutrophils from normal individuals released NETs upon IL-17A stimulation only after having been primed by TNF-α (25). In BP, the importance of neutrophils was also highlighted by the capacity of both classical corticoids and Compound A to inhibit NET generation induced by serum collected at baseline. However, further studies are required to determine the cytokines involved in neutrophils sensitization to NET formation in BP.
Serum from BP patients with relapse holds on high level of NET formation. This suggests that NETs' triggers remained present in the serum of those patients even under therapy. In this line, we previously reported that the concentration of IL-17 and IL-23 remained elevated or even increased in the sera of BP patients at risk of relapse (18). Noteworthy, using our ex vivo model, we demonstrated that IL-23, and to a lesser extent IL-17A, could enhance NET formation in BP neutrophils. While IL-17 is known to potently recruit neutrophils, a role and the mechanisms associated with this cytokine in the induction of NET have still not been fully elucidated. Activation of p38-Mitogen-activated protein kinase (p38 MAPK) and Nuclear Factor Kappa B (NF-κB) pathways by IL-17A (46–49) may explain this property, as both are involved in NET formation (50–52), and p38 MAPK pathway mediates IL-17 induced ROS production (46). To our knowledge, the role of IL-23 in NET generation had not been evaluated yet. Neutrophils constitutively express low amount of IL-23R, which are up-regulated upon activation (53). Three activation cascades may be activated following IL-23 transduction signal among which the above-mentioned p38 MAPK and NF-κB pathways but also the mammalian target of rapamycin (mTOR) pathway which play a pivotal role in NET formation and may explain the higher capacity of IL-23 to induce NET formation in BP (50, 51, 53, 54). In this line, no correlation could be drawn between IL-17 or IL-23 blister fluids concentrations and NET formation (r = −0.5, p = 0.14, and r = −0.71, p = 0.06 respectively), suggesting that IL-17A and IL-23 were not the only triggers of NET formation in BP. Observed effects of these cytokines may also result from synergic actions of several cytokines and further studies are needed to understand mechanisms of NET induction in BP.
This study is the first to our knowledge to demonstrate tendentious functions of IL-17A with respect to IL-23 presence. Indeed, we observed a protective effect of IL-17A supplementation on NET formation induced either by IL-23 itself or by sera from BP patients with relapse, highlighting the role of disease specific microenvironment in cytokine's function. In the same line, blocking IL-17 signaling in inflammatory bowel diseases resulted in an increased expression of pro-inflammatory chemokines and cytokines (55, 56). Some molecules have recently been described in such regulation mechanisms. Suppressor of cytokine signaling 3 (SOCS3) is therefore known to inhibit Janus kinase 2 activity, thereby decreasing IL-23 induced effects (57–59). Yet, SOCS3 expression has recently been correlated with severity of inflammation, expression of proinflammatory cytokines, and activation of p38 MAPK pathway (60). SOCS3 expression is also induced by IL-17 family cytokines (61, 62). Then, although this has to be proven, we hypothesized that in BP IL-17A attenuates IL-23 induced NET formation by inducing SOCS3 expression as previously demonstrated in the airway epithelium (62). Such effects could explain the limited quantity of NETs observed in the skin of BP patients. Furthermore, our present results demonstrate that, in the future, patients with bullous pemphigoid could be further divided into subgroups according to the biomarkers expressed to identify an adequate candidate for biotherapy maintenance in relay of corticosteroid in patients at risk of relapse.
NETs are involved in loss of tolerance mechanisms in several autoimmune diseases (6). In BP, NETs release could participate to both autoimmune and inflammatory responses. Auto-antigens may be present at the surface of NETs, which prolongs their exposition, subsequently favoring an autoimmune response (63, 64). In this line, NET formation is associated with production of reactive oxygen species, and granule enzymes cover the DNA filaments (2, 6, 65). Several studies showed the involvement of NETs in tissue damage (66, 67). Brinkmann et al. reported the association of NETs with various proteases such as neutrophil elastase (5). Yet, neutrophil elastase has largely been implicated in BP pathological processes, as well as matrix metalloproteinase MMP-9 (14, 68). Thus, although NET formation does not directly correlate with BP activity, we showed in this study that NET formation depends on the presence of inflammatory cytokines, and therefore that the inflammatory response associated with BP may participate to BP antigen immunogenicity or to the perpetuation of the disease. Subsequently, our results showing NETs at the dermal-epidermal splitting area, along with the serum capacity to induce NETs at the same time, really advocate for a role of neutrophil in extracellular DNA release, although NET implication in BP still needs to be further demonstrated. Based on the literature, we hypothesize that NET-associated mechanisms may be involved in BP180 antigen immunogenicity, and further in epitope spreading in BP.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
The investigation was conducted under the approval of the Ethic Committee of the University Hospital of Reims (CNIL authorization DR-2013-320), and all of the subjects gave their informed and written consent before participating in the study in accordance with the Helsinki Declaration.
Author Contributions
PB, FA, and BP conceived the study. DG, EB, SL, GG, PB, FA, and BN contributed to the study design and data analysis. AD, CM, PB, and FA contributed to the clinical and histological metadata collection. DG, EB, CT, KD, SL, SN, CM, and BP contributed to the sample processing. CT conceived the macro allowing ex vivo NET quantification. DG, EB, and SL performed the statistical analyses. DG, PB, FA, and BP wrote the manuscript and all authors reviewed and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by research grants from the French Department of Health's Project Hospitalier de Recherche Clinique (PHRC) Interrégional 2012 and the Association pour la Recherche en Dermatologie. We also thank Christine Terryn for technical assistance during the study and the PICT platform for providing microscopy equipments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00701/full#supplementary-material
Supplementary Figure S1. Optimal culture conditions to induce NETosis with BP biological fluids. Representative microphotographs of NETs and their respective drawings generated by ex vivo stimulation of BP PMNs with either healthy sera (A, A', B, B'), BP sera (C, C', D, D') or BP blister fluids (E, E', F, F') collected at baseline. PMNs were incubated with 5% biological fluids during either 1 h (A, A', C, C', E, E') or 3 h (B, B', D, D', F, F'). NETs area generated by BP PMNs after 1 or 3 h stimulation by healthy sera, BP sera and BP BF (G). BF: blister fluids.
Supplementary Video S1. 3-dimensional-reconstruction of the NET corresponding to Figures 1E,F 3D-reconstruction of a netting neutrophil in situ by IMARIS® (Bitplane® software, Belfast, UK). Red, Histone; Green, myeloperoxidase. Magnification 80x, numerical aperture of 1, z-stack with 0.67 μm step-size.
References
1. Dubertret L, Bertaux B, Fosse M, Touraine R. Cellular events leading to blister formation in bullous pemphigoid. Br J Dermatol. (1980) 103:615–24. doi: 10.1111/j.1365-2133.1980.tb01683.x
2. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
3. Ueki S, Tokunaga T, Fujieda S, Honda K, Hirokawa M, Spencer LA, et al. Eosinophil ETosis and DNA traps: a new look at eosinophilic inflammation. Curr Allergy Asthma Rep. (2016) 16:54. doi: 10.1007/s11882-016-0634-5
4. Yousefi S, Simon D, Simon H-U. Eosinophil extracellular DNA traps: molecular mechanisms and potential roles in disease. Curr Opin Immunol. (2012) 24:736–9. doi: 10.1016/j.coi.2012.08.010
5. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
6. Lee KH, Kronbichler A, Park DD-Y, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev. (2017) 16:1160–73. doi: 10.1016/j.autrev.2017.09.012
7. Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon H-U. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol. (2011) 127:194–9. doi: 10.1016/j.jaci.2010.11.002
8. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon H-U, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy. (2017) 72:1105–13. doi: 10.1111/all.13131
9. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med. (2018) 5:201. doi: 10.3389/fmed.2018.00201
10. Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. (1997) 100:1256–63. doi: 10.1172/JCI119639
11. Simon D, Borradori L, Simon H-U. Eosinophils as putative therapeutic targets in bullous pemphigoid. Exp Dermatol. (2017) 26:1187–92. doi: 10.1111/exd.13416
12. Hellberg L, Samavedam UKSRL, Holdorf K, Hänsel M, Recke A, Beckmann T, et al. Methylprednisolone blocks autoantibody-induced tissue damage in experimental models of bullous pemphigoid and epidermolysis bullosa acquisita through inhibition of neutrophil activation. J Invest Dermatol. (2013) 133:2390–9. doi: 10.1038/jid.2013.91
13. Fang H, Zhang Y, Li N, Wang G, Liu Z. The autoimmune skin disease bullous pemphigoid: the role of mast cells in autoantibody-induced tissue injury. Front Immunol. (2018) 9:407. doi: 10.3389/fimmu.2018.00407
14. Bieber K, Koga H, Nishie W. In vitro and in vivo models to investigate the pathomechanisms and novel treatments for pemphigoid diseases. Exp Dermatol. (2017) 26:1163–70. doi: 10.1111/exd.13415
15. Leighty L, Li N, Diaz LA, Liu Z. Experimental models for the autoimmune and inflammatory blistering disease, Bullous pemphigoid. Arch Dermatol Res. (2007) 299:417–22. doi: 10.1007/s00403-007-0790-5
16. Chen R, Fairley JA, Zhao M-L, Giudice GJ, Zillikens D, Diaz LA, et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol. (2002) 169:3987–92. doi: 10.4049/jimmunol.169.7.3987
17. Le Jan S, Plée J, Vallerand D, Dupont A, Delanez E, Durlach A, et al. Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol. (2014) 134:2908–17. doi: 10.1038/jid.2014.263
18. Plée J, Le Jan S, Giustiniani J, Barbe C, Joly P, Bedane C, et al. Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci Rep. (2015) 5:18001. doi: 10.1038/srep18001
19. Riani M, Le Jan S, Plée J, Durlach A, Le Naour R, Haegeman G, et al. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. J Allergy Clin Immunol. (2016) 139:863–72.e3. doi: 10.1016/j.jaci.2016.08.012
20. de Graauw E, Sitaru C, Horn MP, Borradori L, Yousefi S, Simon D, et al. Monocytes enhance neutrophil-induced blister formation in an ex vivo model of bullous pemphigoid. Allergy. (2018) 73:1119–30. doi: 10.1111/all.13376
21. Lin L, Betsuyaku T, Heimbach L, Li N, Rubenstein D, Shapiro SD, et al. Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol. (2012) 31:38–44. doi: 10.1016/j.matbio.2011.09.003
22. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol. (2001) 117:1091–6. doi: 10.1046/j.0022-202x.2001.01521.x
23. Giusti D, Gatouillat G, Le Jan S, Plée J, Bernard P, Antonicelli F, et al. Eosinophil Cationic Protein (ECP), a predictive marker of bullous pemphigoid severity and outcome. Sci Rep. (2017) 7:4833. doi: 10.1038/s41598-017-04687-5
24. Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. (2005) 115:879–87. doi: 10.1172/JCI23977
25. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
26. Gammon WR, Merritt CC, Lewis DM, Sams WM, Carlo JR, Wheeler CE. An in vitro model of immune complex-mediated basement membrane zone separation caused by pemphigoid antibodies, leukocytes, and complement. J Invest Dermatol. (1982) 78:285–90. doi: 10.1111/1523-1747.ep12507222
27. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. (1993) 92:2480–8. doi: 10.1172/JCI116856
28. Sitaru C, Dähnrich C, Probst C, Komorowski L, Blöcker I, Schmidt E, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. (2007) 16:770–7. doi: 10.1111/j.1600-0625.2007.00592.x
29. Charneux J, Lorin J, Vitry F, Antonicelli F, Reguiai Z, Barbe C, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid: a retrospective study of 138 patients. Arch Dermatol. (2011) 147:286–91. doi: 10.1001/archdermatol.2011.23
30. Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. (2017) 18:513–28. doi: 10.1007/s40257-017-0264-2
31. Giusti D, Le Jan S, Gatouillat G, Bernard P, Pham BN, Antonicelli F. Biomarkers related to bullous pemphigoid activity and outcome. Exp Dermatol. (2017) 26:1240–7. doi: 10.1111/exd.13459
32. Chakievska L, Holtsche MM, Künstner A, Goletz S, Petersen B-S, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun. (2018) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
33. D'Auria L, Cordiali Fei P, Ameglio F. Cytokines and bullous pemphigoid. Eur Cytokine Netw. (1999) 10:123–34.
34. Ameglio F, D'auria L, Bonifati C, Ferraro C, Mastroianni A, Giacalone B. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. (1998) 138:611–4.
35. Brinkmann V, Laube B, Abed UA, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp. (2010) 24:1724. doi: 10.3791/1724
36. Rebernick R, Fahmy L, Glover C, Bawadekar M, Shim D, Holmes CL, et al. DNA area and NETosis Analysis (DANA): a high-throughput method to quantify neutrophil extracellular traps in fluorescent microscope images. Biol Proced Online. (2018) 20:7. doi: 10.1186/s12575-018-0072-y
37. Lesovaya E, Yemelyanov A, Swart AC, Swart P, Haegeman G, Budunova I. Discovery of Compound A – a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget. (2015) 6:30730–44. doi: 10.18632/oncotarget.5078
38. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential use of human neutrophil Fcγ receptors for inducing neutrophil extracellular trap formation. J Immunol Res. (2016) 2016:2908034. doi: 10.1155/2016/2908034
39. Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J Immunol Baltim Md. (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
40. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Transforming growth factor-β-activated kinase 1 is required for human FcγRIIIb-induced neutrophil extracellular trap formation. Front Immunol. (2016) 7:277. doi: 10.3389/fimmu.2016.00277
41. Rosales C. Fcγ receptor heterogeneity in leukocyte functional responses. Front Immunol. (2017) 8:280. doi: 10.3389/fimmu.2017.00280
42. Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M, et al. Regulatory T cells suppress inflammation and blistering in pemphigoid diseases. Front Immunol. (2017) 8:1628. doi: 10.3389/fimmu.2017.01628
43. Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodontal Res. (2016) 51:70–6. doi: 10.1111/jre.12284
44. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
45. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
46. Pietrowski E, Bender B, Huppert J, White R, Luhmann HJ, Kuhlmann CRW. Pro-inflammatory effects of interleukin-17A on vascular smooth muscle cells involve NAD(P)H- oxidase derived reactive oxygen species. J Vasc Res. (2011) 48:52–8. doi: 10.1159/000317400
47. Tokuda H, Kanno Y, Ishisaki A, Takenaka M, Harada A, Kozawa O. Interleukin (IL)-17 enhances tumor necrosis factor-alpha-stimulated IL-6 synthesis via p38 mitogen-activated protein kinase in osteoblasts. J Cell Biochem. (2004) 91:1053–61. doi: 10.1002/jcb.20004
48. Laan M, Lötvall J, Chung KF, Lindén A. IL-17-induced cytokine release in human bronchial epithelial cells in vitro: role of mitogen-activated protein (MAP) kinases. Br J Pharmacol. (2001) 133:200–6. doi: 10.1038/sj.bjp.0704063
49. Gaffen SL. Structure and signalling in the IL-17 receptor superfamily. Nat Rev Immunol. (2009) 9:556. doi: 10.1038/nri2586
50. Tatsiy O, McDonald PP. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. (2018) 9:2036. doi: 10.3389/fimmu.2018.02036
51. Lapponi MJ, Carestia A, Landoni VI, Rivadeneyra L, Etulain J, Negrotto S, et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Ther. (2013) 345:430–7. doi: 10.1124/jpet.112.202879
52. Nel JG, Theron AJ, Pool R, Durandt C, Tintinger GR, Anderson R. Neutrophil extracellular traps and their role in health and disease. South Afr J Sci. (2016) 112:w14352. doi: 10.4414/smw.2016.14352
53. Chen F, Cao A, Yao S, Evans-Marin HL, Liu H, Wu W, et al. mTOR Mediates IL-23 induction of neutrophil IL-17 and IL-22 production. J Immunol. (2016) 196:4390–9. doi: 10.4049/jimmunol.1501541
54. Itakura A, McCarty OJT. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol - Cell Physiol. (2013) 305:C348–54. doi: 10.1152/ajpcell.00108.2013
55. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. (2015) 43:739–50. doi: 10.1016/j.immuni.2015.08.019
56. Song X, Dai D, He X, Zhu S, Yao Y, Gao H, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. (2015) 43:488–501. doi: 10.1016/j.immuni.2015.06.024
57. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. (2012) 36:239–50. doi: 10.1016/j.immuni.2011.12.015
58. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA. (2006) 103:8137–42. doi: 10.1073/pnas.0600666103
59. Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. (2014) 5:58. doi: 10.3389/fimmu.2014.00058
60. Chaves de Souza JA, Nogueira AVB, Chaves de Souza PP, Kim YJ, Silva Lobo C, Pimentel Lopes de Oliveira GJ, et al. SOCS3 expression correlates with severity of inflammation, expression of proinflammatory cytokines, and activation of STAT3 and p38 MAPK in LPS-induced inflammation in vivo. Mediators Inflamm. (2013) 2013:650812. doi: 10.1155/2013/650812
61. Caruso R, Stolfi C, Sarra M, Rizzo A, Fantini MC, Pallone F, et al. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase-dependent induction of Socs-3. Blood. (2009) 113:3512–9. doi: 10.1182/blood-2008-08-172767
62. Niwa M, Fujisawa T, Mori K, Yamanaka K, Yasui H, Suzuki Y, et al. IL-17A Attenuates IFN-λ expression by inducing suppressor of cytokine signaling expression in airway epithelium. J Immunol. (2018) 201:2392–402. doi: 10.4049/jimmunol.1800147
63. Bruschi M, Petretto A, Bertelli R, Galetti M, Bonanni A, Pratesi F, et al. Post-translational modified proteins are biomarkers of autoimmune-processes: NETosis and the inflammatory–autoimmunity connection. Clin Chim Acta. (2017) 464:12–16. doi: 10.1016/j.cca.2016.11.006
64. Bruschi M, Petretto A, Vaglio A, Santucci L, Candiano G, Ghiggeri GM. Annexin A1 and autoimmunity: from basic science to clinical applications. Int J Mol Sci. (2018) 19:E1348. doi: 10.3390/ijms19051348
65. Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol. (2016) 28:159–73. doi: 10.1016/j.smim.2016.03.004
66. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. (2015) 6:6673. doi: 10.1038/ncomms7673
67. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
Keywords: neutrophil extracellular traps, eosinophil extracellular traps, cytokine, autoimmunity, inflammation, bullous pemphigoid
Citation: Giusti D, Bini E, Terryn C, Didier K, Le Jan S, Gatouillat G, Durlach A, Nesmond S, Muller C, Bernard P, Antonicelli F and Pham BN (2019) NET Formation in Bullous Pemphigoid Patients With Relapse Is Modulated by IL-17 and IL-23 Interplay. Front. Immunol. 10:701. doi: 10.3389/fimmu.2019.00701
Received: 04 January 2019; Accepted: 14 March 2019;
Published: 04 April 2019.
Edited by:
Takashi Hashimoto, Osaka University, JapanReviewed by:
Katja Bieber, Universität zu Lübeck, GermanyMingyue Wang, Peking University First Hospital, China
Copyright © 2019 Giusti, Bini, Terryn, Didier, Le Jan, Gatouillat, Durlach, Nesmond, Muller, Bernard, Antonicelli and Pham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Delphine Giusti, ZGdpdXN0aUBjaHUtcmVpbXMuZnI=
†These authors have contributed equally to this work
‡These authors have contributed equally to this work