 Klara Kostovcikova1,2
Klara Kostovcikova1,2 Stepan Coufal1Natalie Galanova1
Stepan Coufal1Natalie Galanova1 Alena Fajstova1Tomas Hudcovic3Martin Kostovcik4
Alena Fajstova1Tomas Hudcovic3Martin Kostovcik4 Petra Prochazkova1
Petra Prochazkova1 Zuzana Jiraskova Zakostelska1
Zuzana Jiraskova Zakostelska1 Martina Cermakova5Blanka Sediva5,6
Martina Cermakova5Blanka Sediva5,6 Marek Kuzma5
Marek Kuzma5 Helena Tlaskalova-Hogenova1
Helena Tlaskalova-Hogenova1 Miloslav Kverka1,7*
Miloslav Kverka1,7*- 1Laboratory of Cellular and Molecular Immunology, Institute of Microbiology of the CAS, v.v.i., Prague, Czechia
- 2Laboratory of Cell and Developmental Biology, Institute of Molecular Genetics of the CAS, v.v.i., Prague, Czechia
- 3Laboratory of Gnotobiology, Institute of Microbiology of the CAS, v.v.i., Nový Hrádek, Czechia
- 4Laboratory of Fungal Genetics and Metabolism, Institute of Microbiology of the CAS, v.v.i., Prague, Czechia
- 5Laboratory of Molecular Structure Characterization, Institute of Microbiology of the CAS, v.v.i., Prague, Czechia
- 6Faculty of Applied Sciences, University of West Bohemia, Pilsen, Czechia
- 7Department of Pharmacology, Institute of Experimental Medicine of the CAS, v.v.i., Prague, Czechia
Diet is a major factor determining gut microbiota composition and perturbances in this complex ecosystem are associated with the inflammatory bowel disease (IBD). Here, we used gnotobiotic approach to analyze, how interaction between diet rich in proteins and gut microbiota influences the sensitivity to intestinal inflammation in murine model of ulcerative colitis. We found that diet rich in animal protein (aHPD) exacerbates acute dextran sulfate sodium (DSS)-induced colitis while diet rich in plant protein (pHPD) does not. The deleterious effect of aHPD was also apparent in chronic DSS colitis and was associated with distinct changes in gut bacteria and fungi. Therefore, we induced acute DSS-colitis in germ-free mice and transferred gut microbiota from aCD or aHPD fed mice to find that this effect requires presence of microbes and aHPD at the same time. The aHPD did not change the number of regulatory T cells or Th17 cells and still worsened the colitis in immuno-deficient RAG2 knock-out mice suggesting that this effect was not dependent on adaptive immunity. The pro-inflammatory effect of aHPD was, however, abrogated when splenic macrophages were depleted with clodronate liposomes. This treatment prevented aHPD induced increase in colonic Ly-6Chigh pro-inflammatory monocytes, but the ratio of resident Ly-6C−/low macrophages was not changed. These data show that the interactions between dietary protein of animal origin and gut microbiota increase sensitivity to intestinal inflammation by promoting pro-inflammatory response of monocytes.
Introduction
Commensal gut microbiota significantly influences the host's metabolism. Its catabolic and anabolic pathways allow it to use a broad spectrum of substrates including ingested items, substances secreted into the gut lumen or those directly produced by (other) microbes (1). Out of these sources, diet represents the major bulk, and since it is the easiest one to influence, dietary intervention of microbiota and health brings most attention. Changes in diet drive microbes to adapt to a new substrate, thus induce profound changes in microbiota that could improve the host's ability to adapt to the environment. The changes induced by dietary extremes may persist for a long time and thus reflect the host's diet (2). These adaptive changes are similar across different mammalian lineages and have important implications for host's health (3). Dietary interventions can also induce rapid and reproducible changes in microbiota and enrich the community with transient food-borne microbes that are not capable of long-time colonization (4). Indeed, ~15% of microbial operational taxonomic units (OTUs) show strong diurnal fluctuation due to the timing of food intake thus synchronize circadian clock down to the level of metabolic processes. Interestingly, long-term violation of this fine-tuned system in shift workers and frequent flyers could cause temporal dysbiosis with serious metabolic consequences (5).
Disruption of gut microbiota, i.e., dysbiosis, has been recently linked to pathogenesis of inflammatory bowel disease (IBD) (6), metabolic syndrome (7, 8), cardiovascular diseases (9), neurological disorders (10), and even cancer (11). There is an increasing body of evidence that dietary macronutrients may both promote or counteract dysbiosis and its consequences (12). Both short- and long-term dietary interventions lead to substantial changes in gut microbiota and can influence pro-inflammatory status of gut mucosa (13, 14). There is a well-recognized link between diet and IBD. Retrospective studies found that high intake of sucrose, red meat, or margarine increases the relative risk for IBD, consumption of cereal, fruit and vegetable or high-fiber food in general decreases it (15–18).
Increased dietary intake of animal proteins has been proposed as a factor contributing to the Crohn's disease development already several decades ago (19, 20). Metabolism of proteins mainly takes place in the distal colon where carbohydrate-based sources are reduced and bacteria can switch their metabolism to asaccharolytic one. Depending on the source of the protein, animal, and diary proteins are almost completely degraded during passage whereas plant proteins are less degraded and reach large intestine in bigger amounts (21). Degradation of proteins, peptides and amino acids leads to production of various biologically active metabolites, such as branched-chain fatty acids, ammonium, hydrogen sulfide, p-cresol, phenolic and indolic derivates, which could influence the epithelial cells viability and proliferation, thus affecting gut barrier function and immune response. Some of these activities have been linked to the pathogenesis of IBD and other gastrointestinal diseases (22).
Several studies analyzed the effect of diet rich in protein (HPD) on gastrointestinal physiology and basic epithelial response. Rats consuming HPD had different microbiota composition and metabolic activity, altered colonocytes morphology and enzymatic pathways, more goblet cells and increased production of mucin when compared to rats consuming normoproteic diet (23–25). Induction of experimental inflammation in mice fed by HPD led to severe colitis with high mortality (26). Recently, study analyzing the macronutrient source and quantity found that high amount of dietary casein most significantly contributes to colitis sensitivity while psyllium fiber sensitivity decreases. In both these cases, microbiota composition, and density and gut barrier functions were the major mechanisms involved (27). Taken together these studies showed that various dietary proteins can change host's response to gut microbiota including fine tuning of mucosal immune system and eventually influence the susceptibility to experimental colitis.
Products of intestinal microbiota metabolism (such as short-chain fatty acids, SCFA) influence T cell maturation and activity and may regulate the host's immune system both directly and indirectly via other cells (28, 29). By attaching to the gut epithelium, microbes can regulate the balance in gut T cell response. Depending on the particular microbe, they may induce either regulatory T cells or pro-inflammatory Th17 cells (30, 31). In either case, mononuclear cells in the gut mucosa play crucial role in mucosal immune response and in sensitivity to intestinal inflammation. In fact, mononuclear cells such as macrophages residing in the gut mucosa contribute to the local tolerance and non-inflammatory environment, by producing IL-10 and PGE2 and not responding to bacterial lipopolysaccharide (32). Whereas, Ly6C+ blood monocytes are recruited to the mucosa in response to pro-inflammatory stimulation; outnumbering resident macrophages and producing large quantities of IL-1β and TNF-α (33, 34).
The aim of this study was to analyze how dietary protein can contribute to the IBD pathogenesis, with special focus on microbial and immunological mechanisms using two sets (animal and plant protein-based) of fully synthetic normo- and hyper-proteic diets. The gnotobiotic experiments were employed to decipher the importance of host-diet-microbiota interaction and its consequences for acute intestinal inflammation.
Materials and Methods
Animals
Immuno-competent BALB/c and immuno-deficient RAG2 knock-out mice on BALB/c background were obtained from breeding colony of the Institute of Physiology of the CAS and Institute of Microbiology of the CAS, respectively, and all experiments were performed under either conventional or germ-free conditions at the Institute of Microbiology of the CAS. Mice were fed with Maintenance diet for rats and mice No. 1324 (Altromin Spezialfutter, GmbH & Co. KG, Germany), unless stated otherwise. Different experimental groups were housed in separate cages. In addition to that, germ-free mice were housed in Trexler-type plastic isolators under sterile conditions, supplied with sterile water for several generations before the experiments started. This study was carried out in accordance with the recommendations of the ethics standards defined by the EU legislation on the use of experimental animals (2010/63/EU) and Czech animal welfare act. The protocol was approved by the Institute of Microbiology animal care and use committee (approval ID: 85/2015 and 108/2016).
Diets and Experimental Design
In most experiments, shortly after weaning, the mice were switched either to animal protein-based diet—control (aCD, 176 g/kg crude protein; Cat# C1000) and high-protein diet (aHPD, 514 g/kg crude protein; Cat# C1001) or plant protein-based diet—control (pCD, 173 g/kg crude protein; Cat# C 1000/110007) and high-protein diet (pHPD, 500 g/kg crude protein; Cat# C1001/1001127; all from Altromin Spezialfutter, GmbH & Co. KG). All these diets were prepared synthetically, containing either casein (animal) or wheat gluten (plant) as a protein source (Table 1). Mice were maintained on these diets for 3 weeks before the experiment started and then throughout the whole experiment. In some experiments, the long-time effect of the diet was analyzed by switching the diet already to the parental generation of the mice. We measured gut permeability for macromolecules in healthy mice fed with different diets. For this purpose, we treated mice orally with 440 mg/kg of body weight of FITC-labeled 3–5 kD molecule of dextran (Merck, Cat# FD4), and measured serum fluorescence 4 h later, as we published previously (35).
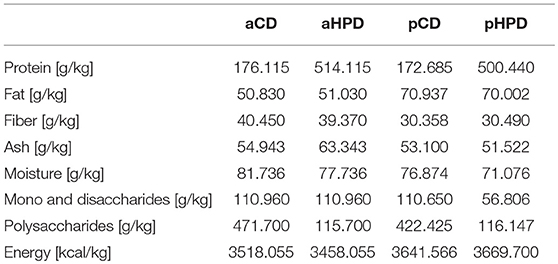
Table 1. Composition of the experimental diets.
Acute and Chronic Experimental Colitis
Acute and chronic experimental colitis was induced by 3% dextran sulfate sodium (DSS, 36–50 kDa; MP Biomedicals, CA, USA; Cat# 02160110) solution in sterile tap water ad libitum (36). Acute colitis was evaluated on the last day of the experiment (day 8) by using a disease activity index (DAI), colon length and histological scoring system, as described previously (37). DAI was determined as a mean of the following three parameters: weight loss (none 0, 5% 1 point, 5–10% 2 points, 10–15% 3 points, >15% 4 points) stool consistency (solid 0 points, loose stool that do not stick to the anus 2 points, and 4 points for liquid stools that stick to the anus), rectal bleeding (none 0, positive test for occult blood 2 points, and 4 points for gross bleeding). Occult blood in feces was evaluated with Fecal Occult Blood Test (Okult-viditest Rapid; Vidia, Vestec, Czech Republic), which is based on guaiacum reaction. Postmortem, the mesenteric lymph nodes, spleen, and colon were collected from each mouse for further analyses. Colon shortening is an indirect marker of colitis severity, so the entire colon was removed (from caecum to anus) and measured by placing it without tension on a ruler. Next, colon descendens was collected for histological analysis, as described previously. Briefly, tissues were fixed in 4% formalin, dehydrated in ethanol and embedded in paraffin. Four micrometer sections were rehydrated and stained by hematoxylin and eosin. Subsequent microscopic evaluation was made by experienced pathologist in blinded manner.
Chronic DSS colitis was induced by three cycles consisting of 5 days DSS and 9 days tap water. The analyses of colitis severity by DAI, colon shortening, and mucosal damage were performed as described above. The level of acute-phase protein haptoglobin was determined in mouse serum using the mouse haptoglobin ELISA Duoset (Bio-Techne, Minneapolis, MN, USA; Cat# DY4409).
Macrophages Depletion in vivo
Two days before DSS treatment, the mice were intraperitoneally injected with 200 μl of empty liposomes or liposomes loaded with clodronate (Liposoma BV, Amsterdam, The Netherlands; Cat# CP-010-010) to deplete blood monocytes and possibly some of tissue macrophages in spleen and colon (38). To maintain and promote the depletion through the whole DSS treatment period, we injected the mice every 3 days starting 2 days before the DSS introduction (day −2, day 1, and day 4).
Tissue Cell Cultures and Cytokines Measurement
Colon tissue was cultivated ex vivo as described earlier (37). Briefly, three millimeter punch biopsy from distal colons were collected, weighted and cultured in 500 μl of complete RPMI medium (Merck; Cat# R0883) containing 10% heat-inactivated fetal bovine serum (Biochrom GmbH, Germany; Cat# S 0115) and 1% Antibiotic-Antimycotic solution (Merck) in humidified incubator (37°C, 5% CO2) for 48 h. The supernatants were collected and stored at −20°C until analysis. Cytokines were measured in tissue culture supernatants using appropriate ELISA sets (Bio-Techne; Cat# DY410, DY406, DY401, and DY3626) according to the manufacturer's instructions.
Cell Preparation and Flow Cytometry Analysis
Single cell suspensions from mesenteric lymph nodes (mLN) and spleens were prepared by mechanical disruption and passed through a 70 μm cell strainer (Becton Dickinson; Cat# 352350). After the washing (300 × g, 5 min, 4°C), red blood cells from spleens were lysed by 5 min incubation in RBC lysing buffer (1 mM EDTA, 150 mM NH4Cl, 10 mM KHCO3). The supernatant with lysed red blood cells was removed and cells were used for further analyses. Single cell suspensions from colons were prepared using the published protocol (39). Next, the cells were blocked by normal mouse serum and incubated with fluorochrome-conjugated antibodies recognizing extracellular epitopes (Supplementary Table 1). Then, the cells were treated with eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific; Cat# 00-5523-00) and stained for intracellular antigens (Supplementary Table 1). To distinguish viable and dead cells, Fixable Viability Dye—eFluor 780 (Thermo Fisher Scientific; Cat# 65-0865-18) was added to the staining mix before fixation. Data were obtained by measuring the samples on LSRII (BD Biosciences, CA) and the FlowJo software (Tree Star Inc., Ashland, OR; RRID: SCR_008520) was used for data analyses. Example of used gating strategy is shown on Supplementary Figure 7.
Real-Time PCR
Three millimeter punch biopsy from distal colons were collected, weighted, immediately immersed in RNAlater RNA Stabilization Reagent (Qiagen, Hilden, Germany; Cat# 76106) and stored at −20°C until RNA extraction. Next, the tissue was homogenized with ceramic beads (Lysing matrix D; MP Biomedicals, Santa Ana, CA, USA; Cat# 116913050) using FastPrep-24 (MP Biomedicals) and total RNA was isolated using TRI reagent (Zymo Reseaerch, Irvine, CA, USA; Cat# R2050). Next, the samples were treated with DNAse (TURBO DNA-free™ Kit; ThermoFisher Scientific; Cat# AM1907) according the manufacturer's instructions, and then reverse transcribed into cDNA with the SuperScript IV Reverse Transcriptase and the RNaseOUT™ Recombinant Ribonuclease Inhibitor (ThermoFisher Scientific; Cat# 10777019), in the presence of random primers (Generi Biotech s.r.o., Hradec Králové, Czech Republic). The resulting cDNA was used for quantitative PCR (CFX96 Touch™, Bio-Rad) with gb SG PCR Master Mix (Generi Biotech s.r.o.; Cat# 3005). Before use, each set of primers (Supplementary Table 2) and RT-PCR conditions were extensively optimized to ensure high efficiency of the reaction and to avoid primer dimers. The cycling parameters were as follows: 3 min at 95 °C, 40 cycles of 30 s at 94 °C, 40 s at 59 °C, and 50 s at 72 °C. Quantitative RT-PCR data were invariably normalized to the expression levels of the reference gene ribosomal protein S12 (Rps12) by means of the 2−ΔΔCt method.
Gut Microbiota Analysis
Stool samples were collected at three different time points: before switching to experimental diets (day −21), before disease induction (day 0) and at the termination of the experiment (day 8). Total DNA was extracted using MasterPure Complete DNA and RNA Purification Kit (Epicenter, Illumina Inc., Madison, WI, USA; Cat# MC85200) with repeated bead-beating in Lysing Matrix Y tubes using FastPrep homogenizer (both MP Biomedicals) and PCR inhibitors were removed using InhibitEx Tablets (Qiagen; Cat# 19590). DNA was then standardized using Qubit dsDNA High Sensitivity kit (Thermo Fisher Scientific). PCR targeting V3 and V4 regions of bacterial 16S was conducted using Kapa HiFi DNA polymerase (Kapa Biosystems, Wilmington, MA, USA), primers 341F (5′-CCTACGGGNGGCWGCAG- 3′) and 806R (5′-GGACTACHVGGGTWTCTAAT- 3′) (40) (Generi Biotech s.r.o.) and 10% BSA (Merck; Cat# 8894). Cycling conditions consisted of initial denaturation (94°C, 3 min) followed by 30 cycles of denaturation (94°C, 30 s), annealing (54.2°C, 45 s), extension (72°C, 75 s) and final extension (72°C, 10 min). For PCR targeting fungal ITS1 region, PPP Master Mix (Top-Bio, Vestec, Czech Republic; Cat# P126) with 18SF (5′-GTAAAAGTCGTAACAAGGTTTC-3′) and 5.8SR (5′-GTTCAAAGAYTCGATGATTCAC-3′) primers and 10% BSA was used (41). Cycling conditions were 95°C, 5 min; 35 cycles of 95°C, 30 s; 50°C, 30 s; 72°C, 60 s, and 72°C, 10 min. PCR triplicates were pooled and purified by SequalPrep Normalization Plate Kit (Thermo Fisher Scientific; Cat# A1051001). Samples within library were pooled and sequencing adaptors were ligated using TruSeq DNA PCR-free LT Sample Preparation Kit (Illumina, Madison, WI, USA; Cat# FC-121-3001). Ligated libraries were quantified with KAPA Library Quantification Kit (Kapa Biosystems, Wilmington, MA, USA; Cat# KK4824) and sequenced on MiSeq Illumina Platform using Miseq Reagent Kit v3 (Illumina; Cat# MS-102-3003) at The Genomics Core Facility, CEITEC (Brno, Czech Republic). Sequencing data were processed using QIIME version 1.9.1 (42). Quality filtering, chimera detection and read demultiplexing and read clustering were done as described previously (43). Fungal reads were in addition extracted for ITS1 region using ITSx package (44). Identification of representative sequences was done using RPD classifier (45) against bacterial GREENGENES database 13.8 (46) and fungal UNITE database 7.2 (UNITE Community (2017): UNITE QIIME release. Version 01.12.2017. UNITE Community. https://doi.org/10.15156/BIO/587481). Finally OTU table was produced. The data are available in the Sequence Read Archive (SRA) http://www.ncbi.nlm.nih.gov/sra under the submission number SUB4976849. Briefly, for microbiota analysis, Chao1 index was used to describe alpha diversity and Principle Coordinate Analysis (PCoA) based on Bray Curtis dissimilarity metrics was used to describe beta diversity. Next, Linear discriminant analysis effect size (LEfSe; RRID: SCR_014609) was used to determine the typical taxonomic profiles of communities in each group (42, 47). Functional composition of a bacterial metagenome was predicted by PICRUSt tool, using the 16S amplicon data (48).
Gut Microbiota Metabolomics
At day 0, one fecal pellet of ~25 mg from each mouse was collected and homogenized by vigorous vortexing in 500 μl of phosphate buffered saline (PBS; pH = 7.4). Next, the sample was centrifuged (13,000 × g for 10 min) to remove particulate matter and supernatant was transferred to a fresh microfuge tube and centrifuged again. The resulting supernatant was lyophilised at −58°C overnight, re-suspended in D2O (500 μl) containing 0.01% trimethylsilyl propionic acid as an internal standard and transferred to a 5-mm NMR tube. The NMR spectra were recorded on a 600 MHz Bruker Avance III spectrometer (Bruker BioSpin, Rheinstetten, Germany) equipped with a 5-mm TCI cryogenic probe head. More detailed description of experimental conditions of NMR analysis and corresponding processing steps is given in Supplementary information. Total protein content in fecal pellet filtrates was measured by the bicinchoninic acid (BCA) assay (Pierce™ BCA Protein Assay Kit; ThermoFisher Scientific; Cat# 23227) according to manufacturer's recommendations.
Statistical Analysis
One-way analysis of variance (ANOVA) with Tukey's multiple comparison test was used to compare multiple experimental groups. Two-way ANOVA with Bonferoni post-test was used in determination of significant weight and DAI changes. Differences between two groups were evaluated using an unpaired two-tailed Student's t-test. The data are presented as the mean ± standard deviation and differences were considered statistically significant at P ≤ 0.05. GraphPad Prism statistical software (version 5.0, GraphPad Software, RRID: SCR_002798) was used for analyses.
Results
Protein-Rich Diet of Animal Origin Worsen Acute Colitis While Protein-Rich Diet of Plant Origin Does Not
To analyze the effect of quantity and source of dietary protein on acute intestinal inflammation, we exposed mice fed with animal or plant protein-based diets to DSS. We found that the resulting colitis was significantly more severe in animals fed with diet rich in animal protein (aHPD) than in animals fed with diet containing normal amount of animal protein (aCD) (Figure 1A). On the other hand, mice fed with diet rich in plant protein (pHPD) were only marginally more susceptible to acute colitis than animals fed with diet containing normal amount of plant protein (pCD) (Figure 1B). Neither animal protein- (Figure 1C) nor plant protein-based diets (Figure 1D) influenced the proportions of regulatory T cells or Th17 cells in mesenteric lymph nodes. But unlike the increase in plant protein, increase in animal protein led to significant increase in colonic Ly-6Chi monocytes and their activation. The intestinal inflammation in aHPD mice was accompanied with increased colonic expression of anti-inflammatory TGF-β, pro-inflammatory cytokines (TNF-α and IL-1β) and inducible NO synthase. In the similar situation, pHPD generally decreased their expression. Neither aHPD nor pHPD influenced the colonic expression of IL-18 (Supplementary Figure 1). Furthermore, we found that neither aHPD nor pHPD significantly changed animal growth, gut permeability for macromolecules or gut cytokine production in healthy mice (just before the DSS exposure) (Supplementary Figure 2).
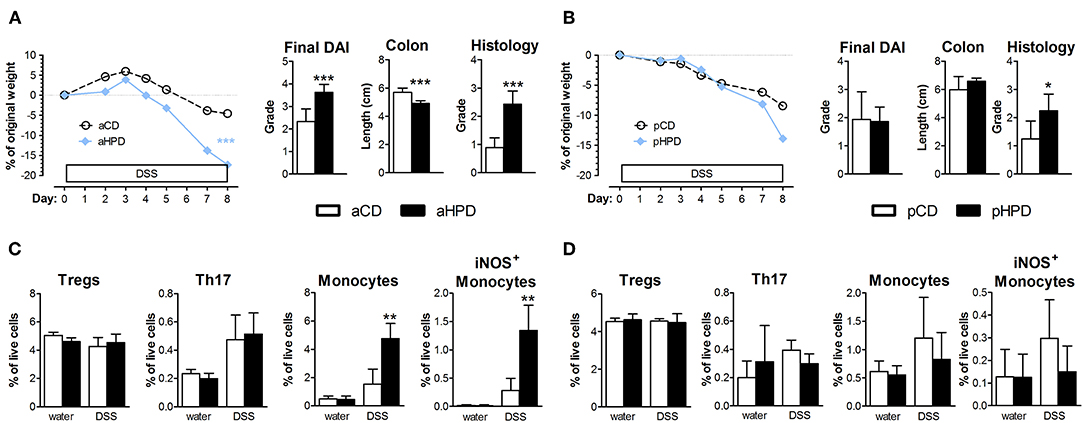
Figure 1. In immune-competent BALB/c mice, HPD of animal origin significantly worsens the severity of acute DSS-colitis (A) while HPD of plant origin does not (B). Colitis severity was measured by disease activity index (weight loss, stool consistency, and rectal bleeding), colon shortening and damage to colon mucosa. Neither aHPD (C) nor pHPD (D) changed the proportions of regulatory T cells (CD3+CD4+FoxP3+) or Th17 cells (CD3+CD4+RorγT+) in mesenteric lymph nodes, while monocytes (CD11clowLy-6Chi) were significantly increased and more activated (iNOS+) in colons of aHPD fed mice. The weight loss was analyzed by two-way ANOVA and other parameters by unpaired Student's t-test; *p < 0.05 **p < 0.01 ***p < 0.001. Data are from one representative experiment out of 6 (A,C) or 3 (B,D) independent experiments (n = 5–8).
Since short term dietary change may have different consequences than the whole life dietary change, we changed diet to mice before mating and induced acute DSS colitis in their offsprings. This way, we ensured that the mice were kept on the aCD or aHPD their entire life. Nevertheless, even in this setup, high amounts of animal protein significantly increased sensitivity to acute DSS colitis (Supplementary Figure 3).
The aHPD Worsens the Intestinal Inflammation in Chronic Colitis
Next, we induced chronic colitis in aCD and aHPD mice by repeated administration of DSS. We found that aHPD worsened the course of chronic DSS colitis. As shown on the weight curve, every cycle of DSS treatment led to a more pronounced inflammation in the aHPD-fed group (Figure 2).
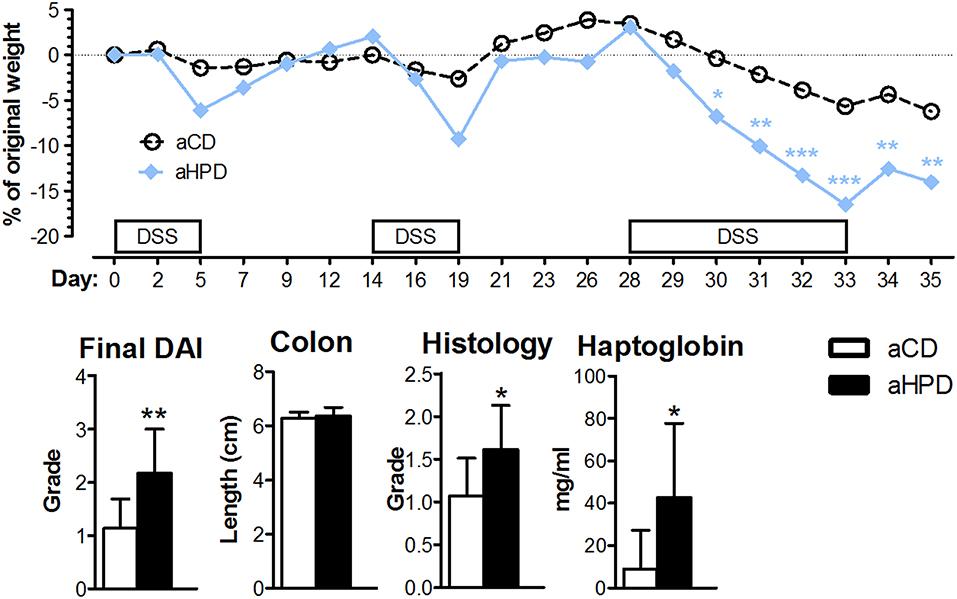
Figure 2. HPD of animal origin worsens the severity of chronic DSS-colitis. The weight loss was analyzed by two-way ANOVA and other parameters by unpaired Student's t-test; *p < 0.05 **p < 0.01 ***p < 0.001. Data are from one representative experiment out of 2 independent experiments (n = 5–8).
Gut Microbiota Is Significantly Altered in Both aHPD and pHPD-Fed Mice
Changing the diet from standard chow to synthetic diets, either plant or animal protein-based, did not significantly changed bacterial alpha diversity, but DSS-induced colitis led to significant decrease in bacterial alpha diversity regardless of the diet (Figure 3A). Interestingly, aHPD decreased bacterial diversity significantly more than aCD (p < 0.05), but there was no similar effect in plant-based diets. The PCoA plot showed clear clustering among different diets and treatments among bacteria (Figure 3B). First, there was a clear shift between day −21 and 0, induced by the diet change, which was more significant in both animal protein-based diets than in the plant protein-based ones. As expected, the protein source seemed to be more important for the bacterial community composition than the protein amount. This clustering became less clear at day 8, when DSS-induced colitis seemed to have the major impact on bacteria. There were particular bacterial taxa responsible for these shifts (Figure 3C). Animal protein-based diets led to increased relative abundance of genera Enterococcus, Streptococcus, Turicibater (only aCD) and Escherichia, and families Peptostreptococcaceae and Ruminococcaceaea. On the contrary, plant protein-based diets enriched OTUs belonging to families Bifidobacteriaceae and Desulfovibrionaceae in pCD and Coriobacteriaceae in pHPD and both showed increased abundance of lactobacilli and families Lachnospiraceae and Erysipelotrichaceae. DSS treatment resulted in significant shifts in genera Bacteroides and Parabacteroides no matter which protein source was used. In addition, we found significant increase of Escherichia coli also in all groups; most notably in both HPDs. It was enriched from 2.02, 1.01, 0.04, and 0.27 to 10.03, 38.03, 17.34, and 34.03% in aCD, aHPD, pCD, and pHPD, respectively. Compared to other groups, we found some specific changes caused by colitis, such as more OTUs assigned to genus Odoribacter and Akkermansia muciniphila in mice consuming aHPD, and genus Staphylococcus in both HPDs. In mice consuming plant protein-based diets, colitis increased the relative abundance of genus Enterococcus from 0.03 and 0.02% to 1.06 and 9.57% in pCD and pHPD, respectively.
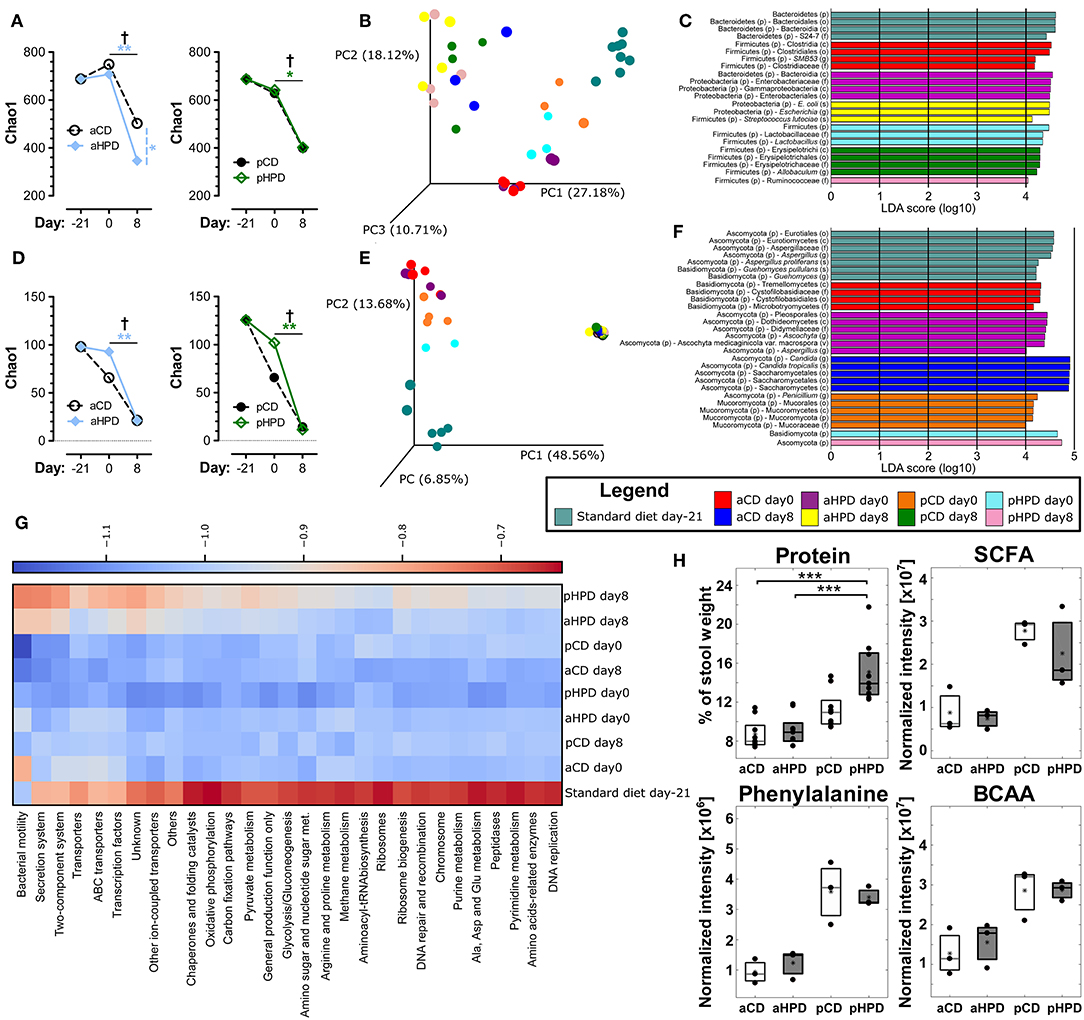
Figure 3. Gut microbiota, both bacterial and fungal, is profoundly changed by diet and intestinal inflammation. Comparison of impact of diet and gut inflammation on bacterial (A) and fungal (D) alpha diversity using Chao1 diversity index. Principal coordinates analysis (PCoA) plot using the Bray-Curtis distance metric shows the compositional differences induced by diet and inflammation at the diet switch (day −21) and before (day 0) and after the DSS treatment (day 8) for bacteria (B) and fungi (E). Each colored orb represents the microbiota composition in feces of one mouse. Each color represents one group of mice at the day −21, 0, or 8. Linear discriminant analysis effect size (LEfSe) for bacteria (C) and fungi (F) shows taxonomic units typical for each group. The first taxonomic unit represents phylum (p) and the second represents the lowest taxonomic rank revealed by the analysis, it being class (c)—order (o)—family (f)—genus (g)—species (s) or variety (v). Heatmap of bacterial gene functional predictions using the PICRUSt algorithm showing the changes in KEGG level-3 pathways (columns) and experimental groups (row) (G). Fecal protein content, as measured by BCA assay and amounts of SCFA, phenylalanine and BCAA, as measured by NMR-based metabolomics (H). The changes in alpha diversity were analyzed by ANOVA with Tukey's multiple comparison test; †p < 0.05 for changes in aCD or pCD and *p < 0.05 **p < 0.01 for the other comparisons. The metabolomic data were analyzed by Wilcoxon-Mann-Whitney test; *p < 0.05 **p < 0.01 ***p < 0.001 (n = 3). Boxplot legend: asterisk—mean value, black dots—metabolites' intensities in particular samples.
Similarly as in bacteria, there was some decreasing tendency in fungal alpha diversity in both CDs with the most striking decrease in fungal diversity during colitis induction (day 0 vs. 8) (Figure 3D). The beta-diversity shifts induced by diet and the effect of DSS-induced colitis were even more prominent for fungi (Figure 3E) than for bacteria. These changes were driven by marked shift in Ascomycota/Basidiomycota ratio before and after DSS treatment (from 62.49, 51.56, 55.28, and 327.03 to 3513.00, 5087.29, 3693.16, and 27738.64 for aCD, aHPD, pCD, and pHPD, respectively). Mice consuming animal protein-based diets showed increased relative abundance of families Cystofilobasidiaceae, Sporidiobolaceae, and Microbotryomycetes fam. incertae sedis. On the other hand, plant protein-based diets led to increased abundance of order Pleosporales and genera Piskurozyma, Cryptococcus, Ascochyta, and Aspergillus (Figure 3F). After DSS treatment, Candida tropicalis seemed to outgrow all other fungal species in all groups (from 0, 0, 0.3, and 1.6% to 94.9, 94.3, 93.7, and 88.5% in aCD, aHPD, pCD, and pHPD, respectively).
These taxonomic changes were reflected by the marked shifts in microbial function, as predicted by PICRUSt algorithm (Figure 3G). Almost all detected metabolic pathways or functional proteins seemed to be enriched in microbiota at day −21 when the mice were fed standard diet when compared to synthetic diets. The dietary change led to a tendency to reduce some pathways especially in plant protein-based diets at day 0 and partly also in aCD at day 8. Among proteins with the highest reduction belonged “bacterial motility proteins,” “secretion system” and “two-component system.” Interestingly, these pathways were slightly enriched after DSS treatment on day 8 in aHPD and pHPD. Moreover, overall pathways enrichment was visible at both HPDs on day 8 partly getting a pattern similar as at day −21.
Metabolomics Reveals Shifts Between Animal and Plant Protein Diets
We compared the fecal metabolite profile among healthy mice (day 0) on different diets to better understand the luminal environment that could affect the sensitivity to intestinal inflammation. We found that gluten was more resistant to digestion than casein, which led to an increase in stool protein content, especially in pHPD (Figure 3H). Next, we identified several distinct metabolites that may influence experimental colitis induction, using NMR-based approach. We found that while there were no major differences between matched normoproteic and hyperproteic diets, the animal and plant protein-based diets each led to production of different metabolites suggesting that the source, and not the amount, of protein was important. Total short-chain fatty acids (SCFA; acetate, propionate, butyrate) were increased in fecal content of mice fed with both gluten-based diets as compared to the casein-based ones. Nevertheless, there were no significant differences in SCFA related to protein amount. Similarly as SCFA, phenylalanine or total branched-chain amino acids (BCAA; valine, leucine, isoleucine) were also higher in plant protein-based diets and similar trends were also visible in glutamate, glutamine, citrate, threonine, and arabinose. Only cytidine showed any tendency for an increase in fecal content of animal protein fed mice (Supplementary Figure 4).
Both Microbiota and aHPD Are Needed for the Pro-inflammatory Effect of aHPD
First, we induced acute intestinal inflammation in germ-free (GF) mice on different diets, to find that both aCD and aHPD had similar severity of colitis (Figure 4A). Then we transferred gut microbiota from either healthy aCD-fed or healthy aHPD-fed mice to GF mice fed standard diet. Surprisingly, we found that microbiota transferred from aHPD-fed mice protected the mice from acute colitis (Figure 4B).
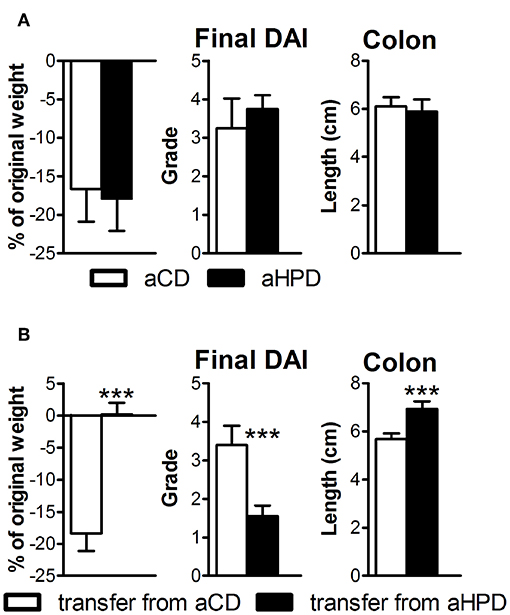
Figure 4. HPD of animal origin does not worsen the severity of acute DSS-colitis in germ-free animals (A), but microbiota transferred from aHPD fed mice mitigates colitis severity (B). The weight change shows percentage of weight gain/reduction between day 0 and 8 of DSS treatment. All parameters were analyzed by unpaired Student's t-test; ***p < 0.001 (n = 5–8).
The Pro-inflammatory Effect of aHPD Is Not Dependent on Adaptive Immunity
The lack of any differences between aCD or aHPD mice in regulatory T cells and Th17 cells (Figure 1C) suggested that adaptive immunity was not responsible for the pro-colitic effect of aHPD. To gauge the importance of adaptive immunity in the pro-inflammatory effect of aHPD, we repeated experiments with acute DSS colitis in mice lacking both T and B lymphocytes (RAG2−/−). Similarly as in immuno-competent BALB/c mice, aHPD-fed RAG2−/− mice were significantly more sensitive to the acute intestinal inflammation than aCD-fed mice (Figure 5A). Moreover, colons of aHPD mice produced more pro-inflammatory cytokines TNF-α, IL-6, and IL-33 as compared to aCD mice (Figure 5B). Although there was slightly higher weight loss in pHPD mice, they were not more sensitive to acute colitis than pCD mice (Supplementary Figure 5).
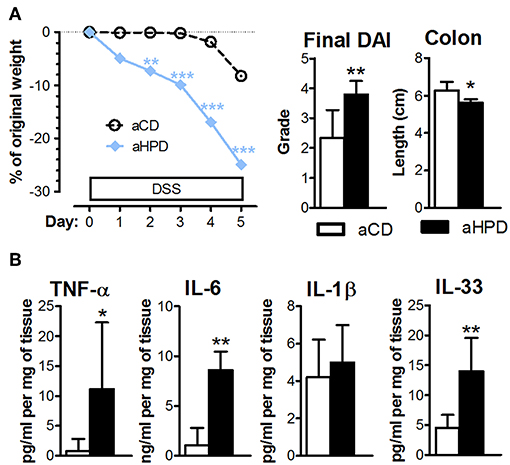
Figure 5. In immune-deficient RAG2−/− mice, aHPD worsens the severity of acute DSS-colitis (A), which is accompanied with the increase in TNF-α, IL-6, IL-1β, and IL-33 in the colon (B). The weight loss was analyzed by two-way ANOVA and other parameters by unpaired Student's t-test; *p < 0.05 **p < 0.01 ***p < 0.001. Data are from one representative experiment out of 3 independent experiments (n = 6–7).
Inflammation in aHPD Fed Mice Is Driven by Monocytes and Subsequent Pro-inflammatory Tuning of Innate Immunity
To analyze the importance of monocytes, we injected clodronate-containing liposomes (LIPO+) or empty liposomes (LIPO-) intraperitoneally every 3 days starting 2 days before the start of DSS. We found that this treatment abrogated the pro-colitic effects of aHPD in both BALB/c (Supplementary Figure 6) and RAG2−/− (Figure 6A) mice. In RAG2−/− mice, aHPD increased proportions of macrophages in spleen and neutrophils in colon (Figures 6B,C). LIPO+ treatment significantly reduced number of splenic Ly-6Clow macrophages and colonic Ly-6Chi monocytes and neutrophils in aHPD-fed mice (p < 0.001 in all cases). Interestingly, while clodronate liposomes were capable to markedly reduce the proportions of macrophages in the spleen, they did not have such effect on colonic macrophages (Figure 6C).
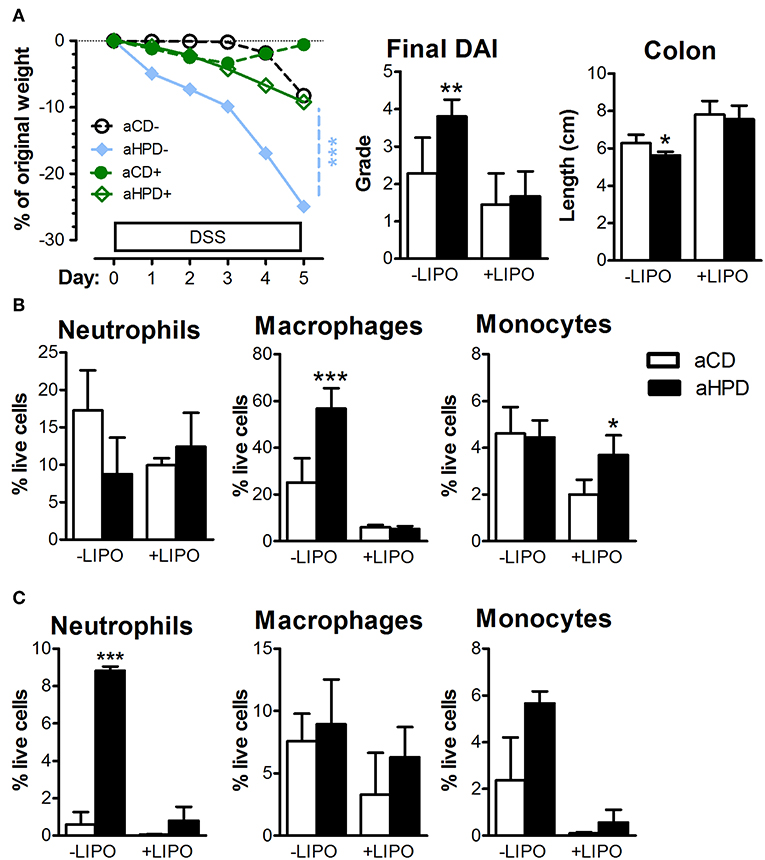
Figure 6. Intraperitoneal administration of clodronate liposomes is sufficient to mitigate the colitis in RAG2−/− (A) mice. Splenic (B) and colonic (C) populations of neutrophils, macrophages and monocytes as analyzed by flow cytometry in colitic RAG2−/− mice treated either with empty (–LIPO) or clodronate-loaded (+LIPO) liposomes. The weight loss was analyzed by two-way ANOVA and other parameters by unpaired Student's t-test; *p < 0.05 **p < 0.01 ***p < 0.001. Data are from one representative experiment out of 3 independent experiments (n = 6–7).
Discussion
Host's homeostasis is adjusted by continuous interactions between microbiota and immune system. Thus, change in microbiota caused by diet could lead to a shift in immune system reactivity and ultimately to changes in sensitivity to inflammatory diseases (49).
Here, we used animal model of ulcerative colitis to analyze how diet-microbiota-host interactions influence the sensitivity to intestinal inflammation. We found that HPD with protein of animal origin worsens acute colitis while HPD with protein of plant origin does not. Moreover, changing the diet 3 weeks before has similar outcome for the colitis as switching the diet already to the previous generation before conception. This suggests that increased sensitivity of mice fed aHPD is not a result of the slower adaptation of the organism to the fundamentally changed macronutrient ratio (13). While changes in dietary protein source in humans lead to profound changes in gut microbiota metabolism and host's gut mucosa transcription, casein-rich diet did not induce inflammation but influenced central homeostatic principles such as cell to cell signaling, cell cycle or death (50). This is in agreement with studies performed in IBD patients, where animal (especially milk) protein consumption correlated positively and consumption of plant protein negatively with incidence of Crohn's disease (20). In these cases, however, proteins in diets are often naturally accompanied with micronutrients with the ability to mitigate (51–53) or exacerbate (54–56) intestinal inflammation in animal models. To minimize the effect of these secondary bioactive molecules, we used well-matched and fully synthetic diets containing only one source of protein—casein or wheat gluten, respectively—in either normal (aCD/pCD) or high (aHPD/pHPD) amounts. The type and composition of other components was the same in all diets used. Diets of animal origin contain slightly more fiber (1% of total mass) and slightly less fat (2% of total mass). This, however, probably did not significantly influence the induction of colitis, because both fat and fiber content were similar between normo- and hyper-proteic diets of either type.
Our data support the recent study showing that high amounts of dietary protein accelerate carcinogenesis in AOM/DSS model of colon cancer by increase the colonic inflammation (57). Indeed, we confirmed that colons from colitic aHPD-fed mice produce more iNOS than colons of aCD-fed mice, but this difference seems to be more related specifically to casein than any protein. We found increased colonic expression of TGF-β, which forms one of the main regulatory feedback loop involving colonic macrophages and Tregs (33). Its increase during the more severe colitis in aHPD-fed mice may represent a local damage control and its suppressive effect on pro-inflammatory cells was probably counteracted by increased levels of pro-inflammatory cytokines, thus insufficient to regulate the inflammation in aHPD mice.
Main mechanism of acute DSS colitis is a damage to gut barrier function (58), but if it was compromised even before DSS exposure, it may accelerate colitis. We found that neither of the two high-protein diets significantly increased gut permeability for macromolecules in healthy mice, suggesting that the barrier was not compromised before the exposure to DSS. This is not in agreement with the study of Llewellyn et al. (27), who found that dietary casein increases gut permeability in healthy mice. We can speculate that this discrepancy was due to the better adaptation of our animals to the abrupt dietary change caused by the longer exposure (3 weeks instead of 1 week), different initial microbial colonization or differences in mouse strain (BALB/c instead of C57BL/6) used.
Diet is a major modulator of the gut intestinal microbiota thus can significantly influence the immune reactivity (14). There is a steep decrease in bacterial and fungal diversity as a result of DSS-induced colitis, which is typical feature shared with human UC (43). In the mice fed animal protein-based diets, we identified several bacterial genera, such as Enterococcus, Streptococcus and Peptostreptococcus, which have been formerly shown in association with IBD (59). Moreover, in colitic aHPD-fed mice, we found increased relative abundance of genera Odoribacter and Akkermansia which has been also linked to gastrointestinal diseases. Bacterial and fungal genera such as Streptococcus, Peptostreptococcus, Escherichia and Candida are known to express several virulence factors including proteolytic activity and biofilm formation that could improve their viability and adhesion in the gut and thus influence disease induction (60). While Candida mostly appeared after DSS course in all tested groups, streptococci, and peptostreptococci were present in the animal protein groups even before colitis induction and were massively outgrown with E. coli after DSS treatment. In our model, both Candida and E. coli were increased after feeding animal protein diet and thus they may facilitate detrimental effects of DSS treatment especially in aHPD group. These two microbes are often associated with diseased IBD patients (61, 62), and even their co-occurrence is necessary for severe forms of experimental colitis (63). Unfortunately, mechanisms of their cooperation are not yet known.
Prediction of proteins or metabolic pathways by PICRUSt analysis revealed some microbiota features that can be linked to increased susceptibility to gut inflammation. It has been shown that several strains of E. coli use flagella to improve their mobility in the gastrointestinal tract under acidic pH and in environment with higher levels of bile salts (64). Moreover, various transmembrane secretion systems are associated with pathobionts and pathogens in the gastrointestinal tract. Transport and secretion of metabolites including toxins can compromise the gut mucosa and even initiate inflammation (65). Two-component systems basically modulate microbial transcription in the way of quick adaptation to changing conditions thus having important role in bacterial survival. In our model, especially in HPDs, bacteria residing inflamed gut were equipped with these mechanisms improving their viability and promoting their possible pathogenicity.
By metabolomics analysis, we intended to define the conditions right before colitis induction and possibly connect them to the subsequent colitis course. Known for mostly beneficial effects, we determined the levels of SCFA. Though they are typically produced from saccharides, recent research has shown that also protein can serve as a substrate for their production (66). Nevertheless, when we compared animal and plant proteins intestinal digestion we found that animal protein derived the smallest levels of SCFA while plant protein showed quite high production. This could be associated with differences in endogenous digestibility of animal and plant proteins. In line with others (21, 22), our results are showing the later one is less digestible and thus available for further processing in the colon. Moreover, we found that plant protein diet led to milder colitis though some effect of DSS treatment was still visible. Since butyrate, as a main component of SCFA, has been shown to reduce experimental colitis in several studies we may speculate that its increased presence attenuated colitis also in our case (67). Potentially beneficial effects on colitis course could also have other metabolites that we found enriched after feeding plant protein diets. For instance, elevated phenylalanine could increase anti-inflammatory response via activation of epithelial calcium receptor (68), and similarly, increased glutamine could reduce NO and pro-inflammatory cytokines production (69). On the other hand, threonine supplementation has been shown to delay epithelial regeneration after colitis (70). Taken together, plant protein metabolites seem to have a potential to reduce colitis probably by a combined effect including strengthening of gut barrier and decreasing pro-inflammatory stimulation.
To further analyze the importance of gut microbiota in aHPD colitogenic effect, we used GF mice. Nevertheless, we did not find any significant differences in colitis severity between aCD- and aHPD-fed GF mice suggesting that the pro-inflammatory effect of aHPD is dependent on the presence of microbiota. While the absence of microbiota decreases the sensitivity of animals to acute DSS-induced colitis (71), it does not completely prevent it. This fact could even increase the sensitivity of our model to gauge the pro-colitic effect of aHPD. Next, we transferred gut microbiota from aCD or aHPD fed mice to GF mice that were not exposed to either diet and induced acute colitis in these mice. We found that mice transferred with microbiota from aHPD do not have significantly more severe colitis than these transferred with microbiota from aCD. This suggests that the both specific microbes and specific substrate (casein) needs to be present at the same time to increase sensitivity to intestinal inflammation. This is in agreement with the conclusions of the recent study of Llewellyn et al. (27). Interestingly, microbiota transferred from aHPD-fed mice made mice significantly less sensitive to colitis than microbiota from aCD. Unfortunately, we are not able to explain this counter-intuitive effect of aHPD-associated microbiota.
While they are not strictly needed for the acute DSS colitis to develop, (72), the absence or increased presence of T cells may influence its severity (71, 73). Therefore, we compared the proportion of regulatory T cells or Th17 cells in mesenteric lymph nodes of animals fed HPDs with their appropriate controls. Neither aHPD nor pHPD changed either of these T cell subsets, suggesting that T cells are not essential for the pro-inflammatory effect of aHPD. To test this hypothesis, we induced acute DSS colitis in immune-deficient RAG2−/− mice on different diets. Similarly as immune-competent BALB/c mice, aHPD-fed RAG2−/− mice are more sensitive to acute colitis. We found that severe colitis in aHPD-fed RAG2−/− mice is associated with high levels of homeostatic cytokine IL-33 and pro-inflammatory cytokines TNF-α and IL-6. While the former is produced by damaged epithelium signaling for repair (74), possibly secondary to a higher degree of epithelial damage in aHPD-fed mice, the latter are produced in high quantities by pro-inflammatory monocytes recruited from the blood (34).
We found that colonic monocytes were significantly increased in aHPD fed mice after colitis induction. To further study the effect of this mononuclear phagocytic system on aHPD-induced colitis sensitivity, we depleted macrophages in systemic compartment with clodronate (38). We found that this intervention abrogated the pro-colitic effect of aHPD and significantly decreased the numbers of colonic Ly-6Chi monocytes. Our results suggest that mainly monocytes recruited through blood stream are the drivers of aHPD-associated severe colitis whereas local macrophages have rather the anti-inflammatory regulating role. This is in line with findings that depletion of resident macrophages leads to more severe colitis (75). Interestingly, colonic neutrophils are decreased as well, and since they are not sensitive to a clodronate liposome-mediated depletion (38), this is probably caused by decrease in their recruitment due to milder colitis. Moreover, splenic monocytes in aCD fed mice were more effectively depleted by clodronate liposomes than these in aHPD fed mice probably because induction of phagocytosis in aCD fed mice, which makes them more susceptible to clodronate-induced cell depletion. In conclusion, we have shown that interactions between dietary protein of animal origin (casein) and gut microbiota increases sensitivity to intestinal inflammation. This pro-inflammatory effect is not dependent on adaptive immunity, but it is driven by activated Ly-6Chi monocytes and subsequent pro-inflammatory tuning of innate immunity. Our results suggest that IBD patients may benefit from changing the source of dietary protein and outline the possibility to use specific antibiotic or probiotic treatment aimed at gut microbiota.
Ethics Statement
This study was carried out in accordance with the recommendations of the ethics standards defined by the EU legislation on the use of experimental animals (2010/63/EU) and Czech animal welfare act. The protocol was approved by the Institute of Microbiology animal care and use committee (approval ID: 85/2015 and 108/2016).
Author Contributions
KK and MKv designed and carried out the experiments, analyzed the data and wrote the manuscript. SC, NG, AF, TH, PP, and ZJZ helped executing the experiments and analyzed the data. MC and MKu designed, performed and analyzed the metabolomics experiments. BS carried out the statistical analysis of metabolomic data. MKo performed and interpreted the bioinformatic analysis. HT-H helped in planning experiments and discussing important concepts. All authors critically revised the manuscript for important intellectual content and approved the final manuscript.
Funding
This study was supported by grants from Czech Science Foundation (16-06326S and 17-06632Y), grant from Ministry of Education, Youth and Sports of the Czech Republic (LO1509) and by Institutional Research Concept RVO: 61388971 (Institute of Microbiology of the CAS, v.v.i.).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Hana Cajthamlova, Zuzana Gajdarova, Pavel Rossmann, Linda Trejtnarova, and Barbora Drabonova for excellent technical assistance. We acknowledge the CF Genomics of CEITEC supported by the NCMG research infrastructure (LM2015091 funded by MEYS CR) for their support with obtaining scientific data presented in this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00919/full#supplementary-material
References
1. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. (2012) 336:1262–7. doi: 10.1126/science.1223813
2. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. (2011) 334:105–8. doi: 10.1126/science.1208344
3. Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. (2011) 332:970–4. doi: 10.1126/science.1198719
4. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. (2014) 505:559–63. doi: 10.1038/nature12820
5. Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell. (2014) 159:514–29. doi: 10.1016/j.cell.2014.09.048
6. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. (2014) 146:1489–99. doi: 10.1053/j.gastro.2014.02.009
7. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
8. Chassaing B, Aitken JD, Gewirtz AT, Vijay-Kumar M. Gut microbiota drives metabolic disease in immunologically altered mice. Adv Immunol. (2012) 116:93–112. doi: 10.1016/B978-0-12-394300-2.00003-X
9. Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. (2014) 124:4204–11. doi: 10.1172/JCI72331
10. Wang Y, Kasper LH. The role of microbiome in central nervous system disorders. Brain Behav Immun. (2014) 38:1–12. doi: 10.1016/j.bbi.2013.12.015
11. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. (2013) 13:800–12. doi: 10.1038/nrc3610
12. Blachier F, Beaumont M, Andriamihaja M, Davila AM, Lan A, Grauso M, et al. Changes in the luminal environment of the colonic epithelial cells and physiopathological consequences. Am J Pathol. (2017) 187:476–86. doi: 10.1016/j.ajpath.2016.11.015
13. Moschen AR, Wieser V, Tilg H. Dietary factors: major regulators of the gut's microbiota. Gut Liver. (2012) 6:411–6. doi: 10.5009/gnl.2012.6.4.411
14. El-Zaatari M, Kao JY. Role of dietary metabolites in regulating the host immune response in gastrointestinal disease. Front Immunol. (2017) 8:51. doi: 10.3389/fimmu.2017.00051
15. Persson PG, Ahlbom A, Hellers G. Diet and inflammatory bowel disease: a case-control study. Epidemiology. (1992) 3:47–52. doi: 10.1097/00001648-199201000-00009
16. Maconi G, Ardizzone S, Cucino C, Bezzio C, Russo AG, Bianchi Porro G. Pre-illness changes in dietary habits and diet as a risk factor for inflammatory bowel disease: a case-control study. World J Gastroenterol. (2010) 16:4297–304. doi: 10.3748/wjg.v16.i34.4297
17. Spooren CE, Pierik MJ, Zeegers MP, Feskens EJ, Masclee AA, Jonkers DM. Review article: the association of diet with onset and relapse in patients with inflammatory bowel disease. Aliment Pharmacol Ther. (2013) 38:1172–87. doi: 10.1111/apt.12501
18. Opstelten JL, de Vries JHM, Wools A, Siersema PD, Oldenburg B, Witteman BJM. Dietary intake of patients with inflammatory bowel disease: a comparison with individuals from a general population and associations with relapse. Clin Nutr. (2018). doi: 10.1016/j.clnu.2018.06.983. [Epub ahead of print]
19. Giovannucci E, Stampfer MJ, Colditz G, Rimm EB, Willett WC. Relationship of diet to risk of colorectal adenoma in men. J Natl Cancer Inst. (1992) 84:91–8. doi: 10.1093/jnci/84.2.91
20. Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am J Clin Nutr. (1996) 63:741–5. doi: 10.1093/ajcn/63.5.741
21. Gilbert JA, Bendsen NT, Tremblay A, Astrup A. Effect of proteins from different sources on body composition. Nutr Metab Cardiovasc Dis. (2011) 21 (Suppl 2):B16–31. doi: 10.1016/j.numecd.2010.12.008
22. Windey K, De Preter V, Verbeke K. Relevance of protein fermentation to gut health. Mol Nutr Food Res. (2012) 56:184–96. doi: 10.1002/mnfr.201100542
23. Andriamihaja M, Davila AM, Eklou-Lawson M, Petit N, Delpal S, Allek F, et al. Colon luminal content and epithelial cell morphology are markedly modified in rats fed with a high-protein diet. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G1030–1037. doi: 10.1152/ajpgi.00149.2010
24. Liu X, Blouin JM, Santacruz A, Lan A, Andriamihaja M, Wilkanowicz S, et al. High-protein diet modifies colonic microbiota and luminal environment but not colonocyte metabolism in the rat model: the increased luminal bulk connection. Am J Physiol Gastrointest Liver Physiol. (2014) 307:G459–470. doi: 10.1152/ajpgi.00400.2013
25. Lan A, Andriamihaja M, Blouin JM, Liu X, Descatoire V, Desclee de Maredsous C, et al. High-protein diet differently modifies intestinal goblet cell characteristics and mucosal cytokine expression in ileum and colon. J Nutr Biochem. (2015) 26:91–8. doi: 10.1016/j.jnutbio.2014.09.007
26. Lan A, Blais A, Coelho D, Capron J, Maarouf M, Benamouzig R, et al. Dual effects of a high-protein diet on DSS-treated mice during colitis resolution phase. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G624–G633. doi: 10.1152/ajpgi.00433.2015
27. Llewellyn SR, Britton GJ, Contijoch EJ, Vennaro OH, Mortha A, Colombel JF, et al. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology. (2018) 154:1037–46.e1032. doi: 10.1053/j.gastro.2017.11.030
28. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
29. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
30. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 Cell induction by adhesion of microbes to intestinal epithelial cells. Cell. (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
31. Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, et al. Critical role for the microbiota in CX3CR1(+) intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity. (2018) 49:151–163.e155. doi: 10.1016/j.immuni.2018.05.009
32. Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J Immunol. (2010) 184:6843–54. doi: 10.4049/jimmunol.0903987
33. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
34. Jones GR, Bain CC, Fenton TM, Kelly A, Brown SL, Ivens AC, et al. Dynamics of colon monocyte and macrophage activation during colitis. Front Immunol. (2018) 9:2764. doi: 10.3389/fimmu.2018.02764
35. Zakostelska Z, Kverka M, Klimesova K, Rossmann P, Mrazek J, Kopecny J, et al. Lysate of probiotic Lactobacillus casei DN-114 001 ameliorates colitis by strengthening the gut barrier function and changing the gut microenvironment. PLoS ONE. (2011) 6:e27961. doi: 10.1371/journal.pone.0027961
36. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. (1990) 98:694–702. doi: 10.1016/0016-5085(90)90290-H
37. Kverka M, Zakostelska Z, Klimesova K, Sokol D, Hudcovic T, Hrncir T, et al. Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol. (2011) 163:250–9. doi: 10.1111/j.1365-2249.2010.04286.x
38. Van Rooijen N. The liposome-mediated macrophage 'suicide' technique. J Immunol Methods. (1989) 124:1–6. doi: 10.1016/0022-1759(89)90178-6
39. Couter CJ, Surana NK. Isolation and flow cytometric characterization of murine small intestinal lymphocytes. J Vis Exp. (2016) 111:e54114. doi: 10.3791/54114
40. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. (2013) 41:e1. doi: 10.1093/nar/gks808
41. Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. (2013) 498:367–70. doi: 10.1038/nature12171
42. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. (2010) 7:335–6. doi: 10.1038/nmeth.f.303
43. Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, et al. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol. (2017) 23:4548–58. doi: 10.3748/wjg.v23.i25.4548
44. Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. (2013) 4:914–9. doi: 10.1111/2041-210X.12073
45. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
46. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. (2006) 72:5069–72. doi: 10.1128/AEM.03006-05
47. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
48. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. (2013) 31:814–21. doi: 10.1038/nbt.2676
49. Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. (2011) 8:110–20. doi: 10.1038/cmi.2010.67
50. Beaumont M, Portune KJ, Steuer N, Lan A, Cerrudo V, Audebert M, et al. Quantity and source of dietary protein influence metabolite production by gut microbiota and rectal mucosa gene expression: a randomized, parallel, double-blind trial in overweight humans. Am J Clin Nutr. (2017) 106:1005–19. doi: 10.3945/ajcn.117.158816
51. Luceri C, Guglielmi F, Lodovici M, Giannini L, Messerini L, Dolara P. Plant phenolic 4-coumaric acid protects against intestinal inflammation in rats. Scand J Gastroenterol. (2004) 39:1128–33. doi: 10.1080/00365520410007908
52. Sanchez-Fidalgo S, Cardeno A, Villegas I, Talero E, de la Lastra CA. Dietary supplementation of resveratrol attenuates chronic colonic inflammation in mice. Eur J Pharmacol. (2010) 633:78–84. doi: 10.1016/j.ejphar.2010.01.025
53. Liu B, Lin Q, Yang T, Zeng L, Shi L, Chen Y, et al. Oat beta-glucan ameliorates dextran sulfate sodium (DSS)-induced ulcerative colitis in mice. Food Funct. (2015) 6:3454–63. doi: 10.1039/C5FO00563A
54. Seril DN, Liao J, Ho KL, Warsi A, Yang CS, Yang GY. Dietary iron supplementation enhances DSS-induced colitis and associated colorectal carcinoma development in mice. Dig Dis Sci. (2002) 47:1266–78. doi: 10.1023/A:1015362228659
55. Constante M, Fragoso G, Calve A, Samba-Mondonga M, Santos MM. Dietary heme induces gut dysbiosis, aggravates colitis, and potentiates the development of adenomas in mice. Front Microbiol. (2017) 8:1809. doi: 10.3389/fmicb.2017.01809
56. Miles JP, Zou J, Kumar MV, Pellizzon M, Ulman E, Ricci M, et al. Supplementation of low- and high-fat diets with fermentable fiber exacerbates severity of DSS-induced acute colitis. Inflamm Bowel Dis. (2017) 23:1133–43. doi: 10.1097/MIB.0000000000001155
57. Tak KH, Ahn E, Kim E. Increase in dietary protein content exacerbates colonic inflammation and tumorigenesis in azoxymethane-induced mouse colon carcinogenesis. Nutr Res Pract. (2017) 11:281–9. doi: 10.4162/nrp.2017.11.4.281
58. Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. (1999) 48:137–43. doi: 10.1538/expanim.48.137
59. Roche-Lima A, Carrasquillo-Carrion K, Gomez-Moreno R, Cruz JM, Velazquez-Morales DM, Rogozin IB, et al. The presence of genotoxic and/or pro-inflammatory bacterial genes in gut metagenomic databases and their possible link with inflammatory bowel diseases. Front Genet. (2018) 9:116. doi: 10.3389/fgene.2018.00116
60. Klimesova K, Jiraskova Zakostelska Z, Tlaskalova-Hogenova H. Oral bacterial and fungal microbiome impacts colorectal carcinogenesis. Front Microbiol. (2018) 9:774. doi: 10.3389/fmicb.2018.00774
61. Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD. Gut. (2017) 66:1039–48. doi: 10.1136/gutjnl-2015-310746
62. Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. (2018) 67:574–87. doi: 10.1136/gutjnl-2017-314903
63. Sovran B, Planchais J, Jegou S, Straube M, Lamas B, Natividad JM, et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome. (2018) 6:152. doi: 10.1186/s40168-018-0538-9
64. Jubelin G, Desvaux M, Schuller S, Etienne-Mesmin L, Muniesa M, Blanquet-Diot S. Modulation of enterohaemorrhagic Escherichia coli survival and virulence in the human gastrointestinal tract. Microorganisms. (2018) 6:E115. doi: 10.3390/microorganisms6040115
65. Green E, Mecsas J. Bacterial secretion systems: an overview. In: Kudva I, Cornick N, Plummer P, Zhang Q, Nicholson T, Bannantine J, Bellaire B, editors. Virulence Mechanisms of Bacterial Pathogens. Washington, DC: ASM Press (2016). p. 215–39. doi: 10.1128/microbiolspec.VMBF-0012-2015
66. Macfarlane GT, Gibson GR, Beatty E, Cummings JH. Estimation of short-chain fatty acid production from protein by human intestinal bacteria based on branched-chain fatty acid measurements. FEMS Microbiol. Lett. (1992) 101:81–8. doi: 10.1111/j.1574-6968.1992.tb05764.x
67. Vieira EL, Leonel AJ, Sad AP, Beltrao NR, Costa TF, Ferreira TM, et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J Nutr Biochem. (2012) 23:430–6. doi: 10.1016/j.jnutbio.2011.01.007
68. Owen JL, Cheng SX, Ge Y, Sahay B, Mohamadzadeh M. The role of the calcium-sensing receptor in gastrointestinal inflammation. Semin Cell Dev Biol. (2016) 49:44–51. doi: 10.1016/j.semcdb.2015.10.040
69. Gong ZY, Yuan ZQ, Dong ZW, Peng YZ. Glutamine with probiotics attenuates intestinal inflammation and oxidative stress in a rat burn injury model through altered iNOS gene aberrant methylation. Am J Transl Res. (2017) 9:2535–47.
70. Gaifem J, Goncalves LG, Dinis-Oliveira RJ, Cunha C, Carvalho A, Torrado E, et al. L-threonine supplementation during colitis onset delays disease recovery. Front Physiol. (2018) 9:1247. doi: 10.3389/fphys.2018.01247
71. Hudcovic T, Stepankova R, Cebra J, Tlaskalova-Hogenova H. The role of microflora in the development of intestinal inflammation: acute and chronic colitis induced by dextran sulfate in germ-free and conventionally reared immunocompetent and immunodeficient mice. Folia Microbiol. (2001) 46:565–72. doi: 10.1007/BF02818004
72. Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. (1994) 107:1643–52. doi: 10.1016/0016-5085(94)90803-6
73. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity. (2015) 42:815–25. doi: 10.1016/j.immuni.2015.04.015
74. Seo DH, Che X, Kwak MS, Kim S, Kim JH, Ma HW, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci Rep. (2017) 7:851. doi: 10.1038/s41598-017-00840-2
Keywords: dietary protein, microbiota, colitis, germ-free, macrophage
Citation: Kostovcikova K, Coufal S, Galanova N, Fajstova A, Hudcovic T, Kostovcik M, Prochazkova P, Jiraskova Zakostelska Z, Cermakova M, Sediva B, Kuzma M, Tlaskalova-Hogenova H and Kverka M (2019) Diet Rich in Animal Protein Promotes Pro-inflammatory Macrophage Response and Exacerbates Colitis in Mice. Front. Immunol. 10:919. doi: 10.3389/fimmu.2019.00919
Received: 31 December 2018; Accepted: 09 April 2019;
Published: 26 April 2019.
Edited by:
Cecil Czerkinsky, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Silvia Melgar, University College Cork, IrelandStefania De Santis, Istituto Nazionale Biostrutture e Biosistemi, Italy
Copyright © 2019 Kostovcikova, Coufal, Galanova, Fajstova, Hudcovic, Kostovcik, Prochazkova, Jiraskova Zakostelska, Cermakova, Sediva, Kuzma, Tlaskalova-Hogenova and Kverka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miloslav Kverka, a3ZlcmthQGJpb21lZC5jYXMuY3o=