 Kentaro Izumi
Kentaro Izumi Katja Bieber
Katja Bieber Ralf J. Ludwig
Ralf J. Ludwig- 1Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
- 2Lübeck Institute of Experimental Dermatology, Center for Research on Inflammation of the Skin, University of Lübeck, Lübeck, Germany
Autoimmune bullous dermatoses (AIBDs) are a group of rare chronic inflammatory skin diseases, which clinically manifest as blisters and erosions of the skin and/or mucosa. Immunologically, AIBDs are characterized and caused by autoantibodies targeting adhesion molecules in the skin and mucosa. According to the histological location of the blistering, AIBDs are classified into the following two main subtypes: pemphigus (intraepidermal blistering) and pemphigoid (subepidermal blistering). Most AIBDs were potentially life-threatening diseases before the advent of immunosuppressive drugs, especially systemic steroid therapies, which suppress pathogenic immunological activity. Although there have been recent advancements in the understanding of the pathogenesis of AIBDs, glucocorticosteroids and/or adjuvant immunosuppressive drugs are still needed to control disease activity. However, the long-term use of systemic immunosuppression is associated with major adverse events, including death. Based on the growing understanding of AIBD pathogenesis, novel treatment targets have emerged, some of which are currently being evaluated in clinical trials. Within this article, we review the current clinical trials involving pemphigus and pemphigoid and discuss the rationale that lead to these trials. Overall, we aim to foster insights into translational research in AIBDs to improve patient care.
Introduction
Autoimmune bullous dermatoses (AIBDs) are a heterogeneous group of skin diseases that are characterized and caused by autoantibodies targeting adhesion molecules in the skin and/or mucous membranes. Depending on the targeted adhesion molecules and the location of the blistering, AIBDs are classified into the following two major types: pemphigus diseases with autoantibodies targeting desmosomal proteins (1) and pemphigoid diseases with autoantibodies targeting the structural proteins of dermal-epidermal junction (2). In pemphigus diseases, including pemphigus vulgaris (PV) and pemphigus foliaceus (PF), autoantibody binding leads to the disruption of epidermal adhesion, resulting in the clinical finding of flaccid blisters and the histological finding of intraepidermal blistering (1). In pemphigoid diseases, the linear deposition of autoantibodies along the dermal-epidermal junction causes subepidermal blistering, resulting in tense blisters. Based on the target molecules of the autoantibodies and the clinical manifestations, pemphigoid diseases are classified as bullous pemphigoid (BP), mucous membrane pemphigoid (MMP), epidermolysis bullosa acquisita (EBA), anti-laminin-γ1/p200 pemphigoid (p200), pemphigoid gestationis (PG), lichen planus pemphigoides (LPP), linear IgA bullous dermatosis (LAD), and dermatitis herpetiformis (DH), which is associated with gluten-sensitive enteropathy and characteristic granular IgA deposits in the upper dermis (3–5).
Although the advent of systemic steroid therapy significantly improved the prognosis of AIBDs, these groups of diseases are still potentially life-threatening, now mainly due to adverse events resulting from corticosteroid treatment (6). Due to the chronicity of AIBDs, the prolonged administration of systemic steroids is often needed to induce and maintain clinical remission, leading to various adverse effects such as cytopenia, diabetes mellitus, osteoporosis, hypertension, gastrointestinal ulcers, and infections due to immunosuppression (7). Furthermore, severe infection induced by an immunocompromised state is one of the most important causes of death during AIBD treatment (8, 9). Thus, the development of alternative treatment modalities that have fewer adverse events is urgently needed for the treatment of AIBD patients.
Based on the growing understanding of AIBD pathogenesis (1, 10, 11), novel therapeutic targets and/or treatment modalities have been identified (12–15). Some of those new treatments are currently being evaluated in clinical trials. To foster translational AIBD research, in this article, we summarize the current clinical trials involving pemphigus and pemphigoid diseases. For this purpose, we searched clinicaltrials.gov1 and the EU clinical trials register2 through December 2018 and selected clinical trials on pemphigus or pemphigoid disease with the status “recruiting (the study is currently recruiting participants),” “active, not recruiting (the study is ongoing, and participants are receiving an intervention or being examined, but not currently being recruited or enrolled),” and “completed (the study has ended normally, and participants are no longer being examined or treated),” and an initiation in 2013 or later (Tables 1, 2, Figure 1). To provide a more comprehensive overview, we list the trials before this time frame in Supplementary Tables 1, 2. In addition, we included one clinical trial that is registered with the Australia and New Zealand Clinical Trials Registry, which was recently presented at the 5th International Pemphigus and Pemphigoid Foundation Scientific Conference in Orlando (16).
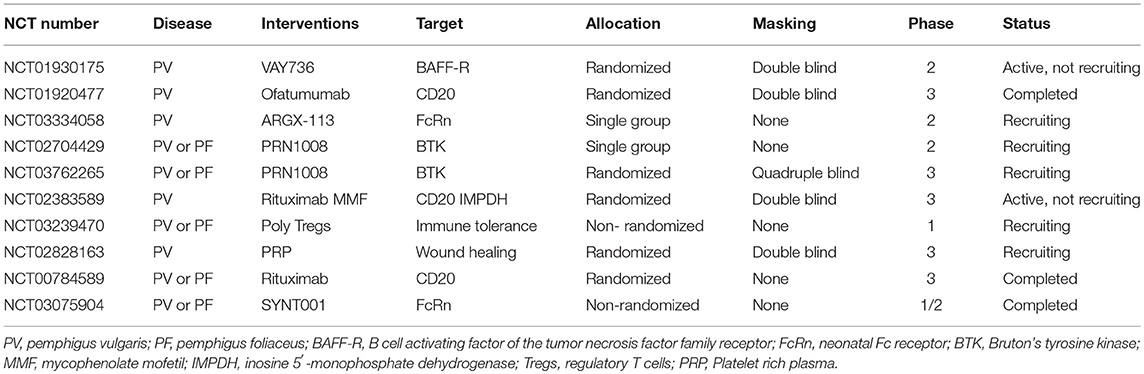
Table 1. Current clinical trials in pemphigus.
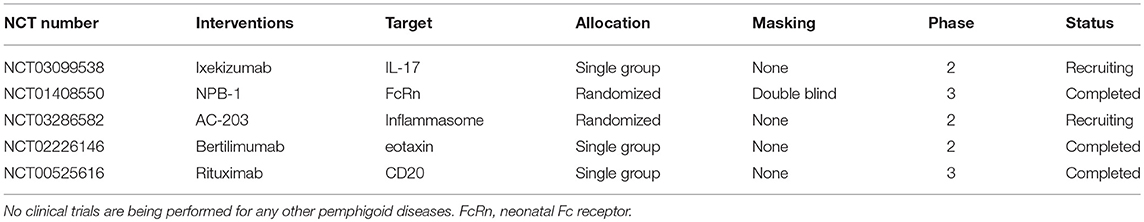
Table 2. Current clinical trials in bullous pemphigoid.
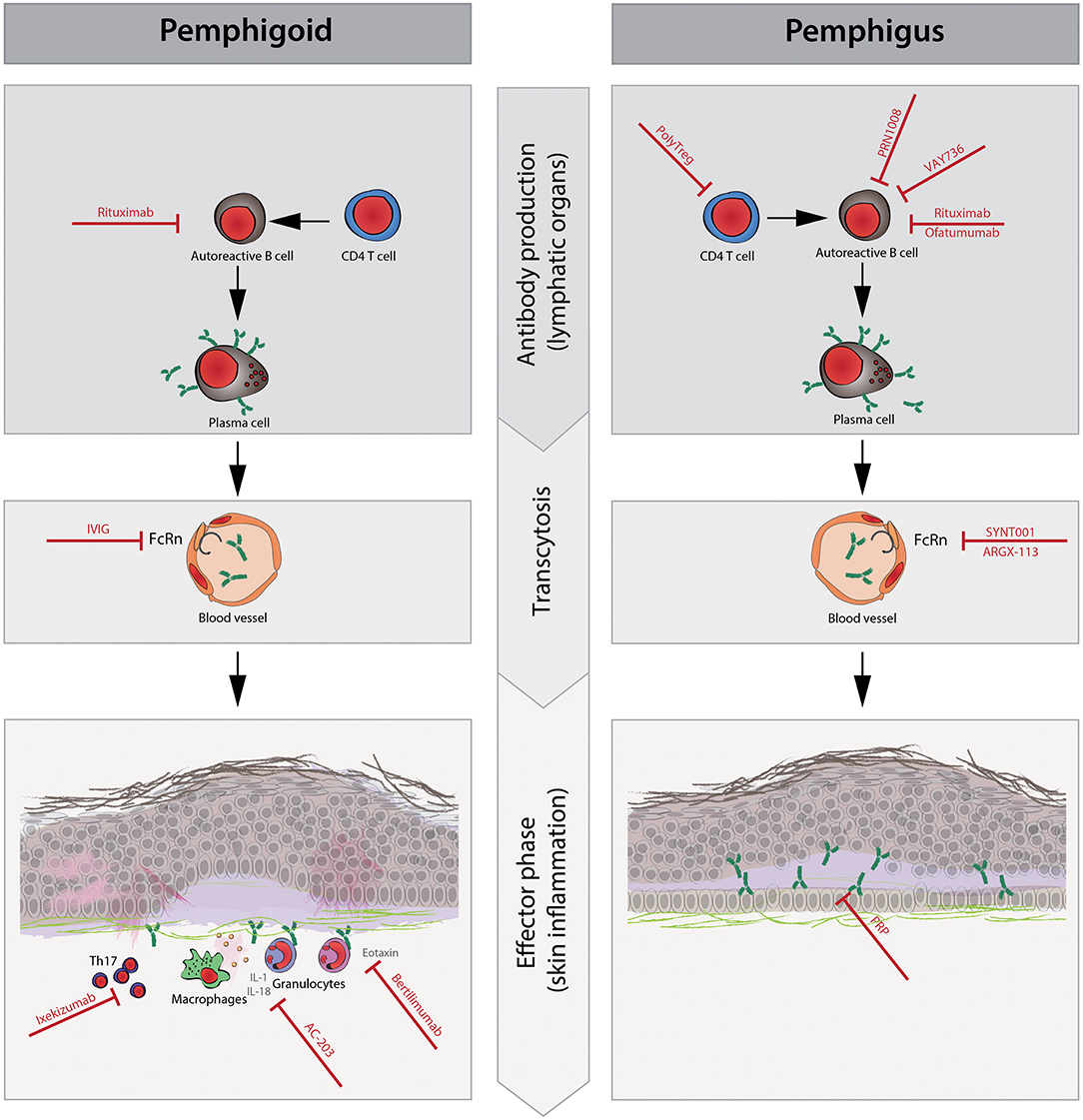
Figure 1. Schematic representation of the pathophysiology and new therapeutic targets of pemphigus and pemphigoid diseases. The pathophysiology of pemphigus and pemphigoid diseases consists of the following three phases: (1) CD4+T cells promote autoreactive B cell activation, proliferation, and differentiation to plasma cells that produce pathogenic autoantibodies. (2) Circulating pathogenic antibodies are transferred to the dermal epidermal junction or intracellular space of the epidermis. Neonatal Fc receptor (FcRn) plays a role in prolonging the half-life of IgG antibodies during this phase. (3) After the binding of pathogenic autoantibodies to target molecules, pro-inflammatory cells such as granulocytes and macrophages are recruited to the immune complex in lesional skin by chemokines (e.g., eotaxin). Then, granulocytes elicit reactive oxygen species (ROS), elastases, and proteases, resulting in tissue damage such as blisters and/or erythema, which are clinical symptoms in pemphigoid diseases but not in pemphigus diseases. Cytokines [e.g., interleukin (IL)-1beta and IL-18] and Th17 polarization are thought to enhance local inflammation. During the antibody production phase, rituximab and ofatumumab deplete autoreactive B cells to prevent their differentiation to plasma cells. PolyTregs act on CD4+ T cells, and VAY736 and PRN1008 act on autoreactive B cells, resulting in less activation of autoreactive B cells. In the transcytosis phase, intravenous immunoglobulin (IVIg), SYNT001, and ARGX-113 saturate FcRn, contributing to the shortened half-life of pathogenic autoantibodies. In the effector phase, ixekizumab restores Th17 polarization and suppresses inflammatory augmentation. AC-203 modulates cytokines such as IL-1beta and IL-18, contributing to decreased inflammation. The inhibition of eotaxin with bertilimumab ameliorates the recruitment of eosinophils to local inflammation sites in pemphigoid disease, especially bullous pemphigoid. Platelet-rich plasma (PRP) is thought to promote wound healing in erosions.
Pemphigus
Anti-CD20 in Pemphigus (Rituximab or Ofatumumab)
Rituximab is a human chimeric IgG1 monoclonal antibody targeting CD20, which is a cell surface marker expressed by B cells (17). Rituximab exerts its treatment effects via the depletion of B-cells following its binding to CD20. Regarding the treatment of pemphigus with rituximab, case reports have suggested the efficacy of rituximab as a second- or third-line therapy (18–20). There are two main rituximab regimens for pemphigus; one regimen, which is based on the lymphoma protocol, is composed of a total of 4 doses of 375 mg/m2 weekly infusions (21), while the other regimen, which is based on the rheumatoid arthritis protocol, consists of a total of two doses of 1,000 mg (or 500 mg) biweekly intravenous infusions (22). Prior to evaluating the efficacy of a single cycle regimen of rituximab in refractory pemphigus cases, a multicenter, single arm, phase 2/3 clinical trial was conducted in France (23). At 3 months after rituximab treatment, 18 (86%) of 21 cases achieved complete remission (CR). Furthermore, 18 (86%) of 21 cases maintained CR after a median follow-up of 34 months. In addition, 8 of these 18 cases did not receive systemic corticosteroid therapy. To evaluate the safety and efficacy of rituximab in a controlled clinical trial, a prospective, parallel-group, open-label, randomized, phase 3 clinical trial of rituximab as a first-line treatment for moderate to severe cases of pemphigus (NCT00784589) was initiated (Table 1). Patients with PV or PF were randomized to receive either oral prednisolone at 1.0 to 1.5 mg/kg/day that was tapered over 12–18 months or oral prednisolone at 0.5–1.0 mg/kg/day that was tapered over 3–6 months plus rituximab 1 g on days 0 and 14 and 0.5 g at months 12 and 18. The primary endpoint was the proportion of patients who achieved CR off-therapy at month 24. The results of this clinical trial were recently reported (24). Regarding the primary endpoint, 34% of patients in the prednisolone arm achieved CR at month 24. In the rituximab plus prednisolone arm, CR was reached by 89% of the patients; the difference was significant. Furthermore, fewer grade 3–4 adverse events were observed in the rituximab plus prednisone group than in the prednisone alone group (27 events in 16 out of 46 patients; mean 0.59 [SD 1.15] vs. 53 events in 29 out of 44 patients; mean 1.20 [1.25]). Overall, this trial demonstrated that the first-line treatment of pemphigus with rituximab and lower doses of prednisolone is more effective and safer compared to high-dose prednisolone treatment. Based on the results of this clinical trial, the Food and Drug Administration approved the expansion of health insurance coverage of rituximab for pemphigus vulgaris in the United States.
In addition to rituximab, ofatumumab, a fully human anti-CD20 monoclonal antibody, has been demonstrated to be safe and effective for the treatment of autoimmune disorders other than pemphigus or pemphigoid (25). Preclinical studies suggested that ofatumumab shows a high affinity for CD20 and activates complement-dependent cytotoxicity (26). A double-blind, randomized, placebo-controlled, phase 3 clinical trial evaluating the efficacy of ofatumumab in pemphigus was completed in January 2018 (NCT01920477). This clinical trial enrolled moderate to severe PV patients with a history of at least failure of tapered steroid therapy. Participants received a stable dose of prednisone/prednisolone from a minimum of 20 mg/day up to a maximum of 120 mg/day or 1.5 mg/kg/day for 2 weeks prior to randomization. Thirty-five patients with PV were randomized to receive either ofatumumab or a placebo. Ofatumumab at 40 mg was subcutaneously injected at week 0 and week 4. From week 8, subjects were subcutaneously administered ofatumumab 20 mg every 4 weeks through week 56. The primary endpoints were the time to sustained CR on minimal steroid therapy (prednisone/prednisolone dose to <10 mg/day) and the duration of CR on minimal steroid therapy. The results of this study have thus far not been reported.
Similar to these two trials, another double-blind, randomized, phase 3 clinical trial of adjuvant rituximab vs. mycophenolate mofetil (MMF) as a therapy for pemphigus is ongoing (NCT02383589). In that study, 135 PV patients who are receiving standard systemic steroid treatment (oral prednisone 60–120 mg/day or equivalent) will randomly receive either (i) MMF and a rituximab-matched placebo or (ii) an MMF-matched placebo and rituximab. MMF is orally administered twice daily from day 1 to week 56. The initial dose of MMF is 500 mg, and the dose will be titrated to achieve a goal of 1,000 mg. Rituximab is administered at a dose of 1,000 mg intravenously on days 1, 15, 168, and 182. The primary endpoint is the proportion of patients who achieve sustained CR.
VAY736 in Pemphigus
B cell activating factor of the tumor necrosis factor family (BAFF) is a crucial cytokine for regulating B cell development in mice and humans (27, 28). BAFF functions by binding to the BAFF receptor (BAFF-R), B cell maturation antigen, and tumor necrosis factor receptor superfamily member (29). Although at physiological concentrations BAFF cannot rescue B cell apoptosis due to a strong B cell death signal, which is transduced via the B cell receptor (BCR) stimulated by autoantigens, at higher concentrations, BAFF causes the survival of autoreactive B cells, which contributes to the pathogenesis of autoimmune diseases (27, 30, 31). For example, elevated BAFF serum levels have been detected in various autoimmune diseases such as RA, systemic lupus erythematosus, Sjögren's syndrome, and systemic sclerosis (32–34). Taken together, BAFF is a likely therapeutic target for the treatment of autoimmune diseases. Regarding clinical translation, the anti-BAFF antibody belimumab was licensed for the treatment of systemic lupus erythematosus in 2011 (35). However, clinical trials of belimumab have not been conducted in pemphigus thus far. Alternatively, with regard to the inhibition of BAFF, its receptor can be blocked to achieve similar results. BAFF-R signaling drives B cell differentiation, proliferation and survival (36). VAY736 is a novel, defucosylated, human IgG1 monoclonal antibody targeting BAFF-R, providing both enhanced antibody-dependent cellular cytotoxicity-mediated depletion of B cells and the blockade of BAFF. To investigate the safety, tolerability and efficacy of VAY736 in PV, a randomized, placebo-controlled, double-blind, phase 2 clinical trial is currently ongoing (NCT01930175). In this trial, 16 mild-moderate PV patients will randomly receive either intravenous VAY736 or a placebo once. The primary endpoint is the efficacy of single cycle VAY736 administration in reducing Pemphigus Disease Area Index (PDAI) scores at week 12 compared to at the baseline. No results of this trial are currently available.
Anti-neonatal Fc Receptor (FcRn) in Pemphigus (SYNT001, ARGX-113)
FcRn plays an essential role in regulating host circulating IgG levels (37). FcRn protects IgG from intracellular digestion, leading to the prolongation of its half-life. This “IgG recycling system” is essential for host defense. However, it also maintains the concentration of circulating pathogenic IgG in various autoimmune diseases, including AIBD. Specifically, FcRn-deficient mice do not (or to a lesser extent) develop experimental AIBD after the injection of AIBD-inducing antibodies (10). Interestingly, saturation of the FcRn by the administration of high-dose human IgG (IVIg) reduces the pathogenic effects in antibody-transfer AIBD models. Thus, FcRn inhibition is one possible mode of action of IVIg therapy in AIBD (38, 39). Based on these observations, anti-FcRn targeting treatments have been developed. Currently, the following 2 clinical trials targeting FcRn are being conducted in pemphigus: SYNT001 (NCT03075904) and ARGX-113 (NCT03334058). SYNT001 is a humanized, deimmunized IgG4 monoclonal antibody that blocks the binding of FcRn to the Fc portion of IgG (40). ARGX-113 is a human IgG1-derived Fc-modified fragment with increased affinity for FcRn that reduces the circulating IgG concentration (41). In addition, both SYNT001 and ARGX-113 do not alter serum levels of albumin. As both trials are ongoing, no results have been published so far. Regarding NCT03075904, 16 PV or PF patients will be sequentially assigned to receive three different doses of intravenous SYNT001 weekly for either 5 or 14 weeks, and the primary endpoint is the count and percentage of adverse events (time frame; days 0–112 or days 0–175). At the 2018 pre-International Investigative Dermatology meeting in Orlando, the first results were reported for SYNT001. The infusion of SYNT001 in human subjects resulted in a rapid lowering of the circulating levels of IgG (mean total IgG reduction of 56% by day 30) with good safety and tolerability. Furthermore, 5 of the 7 subjects showed a reduction in disease activity by day 42 (42). Regarding NCT03334058, 12 newly diagnosed or relapsed PV patients will receive ARGX-113 intravenously. The primary endpoints are safety and tolerability up to 17 weeks.
PRN1008 in Pemphigus
B cell receptor signaling is a key player in B cell development and function. Bruton's tyrosine kinase (BTK) belongs to the Tec family of nonreceptor tyrosine kinases, and it is a vital component of B cell receptor signaling (43). BTK is predominantly expressed by B-lymphocytes from the pre-B cell stage to the mature B cell stage (44, 45). Based on the crucial role of BTK in B cell function, it has been identified as a potential target for the treatment of autoimmune disorders. Several studies have shown that ibrutinib, which is one of the BTK inhibitors under development, binds to BTK with high affinity, leading to the inhibition of B cell receptor signaling and resulting in the reduction of B cell activation involved in autoimmunity (46). In pemphigus treatment, a previous case report showed that ibrutinib improved clinical cutaneous lesions of paraneoplastic pemphigus complicated with chronic lymphocytic leukemia (47). However, there have been no case reports or series investigating the effects of this BTK inhibitor in PNP, PV and/or PF. PRN1008 is another BTK inhibitor that was evaluated in a phase 1 clinical trial that enrolled 80 healthy volunteers (Australian New Zealand Clinical Trials Registry No. ACTRN12614000359639). In that study, PRN1008 was considered safe. Regarding the pharmacokinetics and pharmacodynamics, BTK occupancy of more than 90% was observed within 4 h after dosing in both the single and multiple dose regimens and was closely correlated with the maximum plasma concentration (48). Based on the promising results from the phase 1 trial, an open-label, single-armed, phase 2 clinical trial of PRN1008 for PV treatment has been conducted (NCT02704429). In this trial, 27 pemphigus patients (including PV and PF) received PRN1008 orally for 12 weeks with a 12-week follow-up period. The primary efficacy endpoint was the initial control of disease activity during the first 4 weeks of therapy, during which new lesions cease to form and existing lesions begin to heal, without the need for prednisone-equivalent corticosteroid doses >0.5 mg/kg/day. More than 50% of patients have achieved control of disease activity within 4 weeks of starting PRN1008 thus far3. Furthermore, Principia has recently initiated a global, randomized, double-blind, placebo-controlled, pivotal phase 3 clinical trial (NCT03762265) in approximately 120 PV or PF patients to evaluate PRN1008 vs. a placebo. The primary endpoint is the proportion of participants who are in CR from week ≤ 29 to 37 with a prednisone dose of ≤ 5 mg/day.
Polyclonal Regulatory T Cells (PolyTregs) in Pemphigus
Among the T cell subtypes, regulatory T cells (Tregs) play an important role in regulating the immune system and preventing autoimmune disease development. Based on findings in animal models, including AIBD (49, 50), it is hypothesized that naturally occurring Tregs may be utilized for the treatment of autoimmune diseases and potentially replace the use of chronic immunosuppressive therapies that are associated with multiple adverse effects. A clinical trial of Treg adoptive therapy was started by treating graft vs. host diseases (GVHD) with expanded allogeneic Tregs (51). There has been a small study demonstrating the safe administration of autologous Tregs with decreased disease activity in patients with insulin-dependent diabetes (52). Subsequently, Brunstein et al. reported that HLA-matched umbilical cord blood-derived Tregs decreased the incidence of GVHD after double umbilical cord blood transplantation (53), indicating the potential efficacy and safety profile of the passive transfer of autologous Tregs in humans. The application of Tregs for lupus, cancer and organ transplantation has been addressed (54). The suppressive effects of Tregs have been reported by studies using an active PV mouse model (55). A recent study indicated that Treg induction via the anti-CD28 antibody reduces pathogenic IgG directing desmoglein (Dsg) 3 in the HLA-DRB1*04:02- transgenic PV mouse model (56). Although Tregs are expected to improve PV symptoms, there are no case reports or case series of autologous Treg injections in patients with PV to date. To evaluate the effects of Tregs on the manifestation of PV, a nonrandomized, open-label, phase 1 clinical trial is ongoing (NCT03239470). In this trial, 12 PV or PF patients will receive one infusion of autologous expanded Tregs (CD4+CD127lo/negCD25+) at one of the following doses: either 2.5 x 108 poly Tregs or 10 × 108 poly Tregs. The primary endpoint is the number of significant grade 3 or higher adverse events by week 52. The results from this study have thus far not been reported.
Comparison of Injections of Steroids to Autologous Platelet-Rich Plasma (PRP) in Oral Erosions in PV
Oral erosive lesions are a major hallmark of PV. These oral lesions cause severe pain, resulting in problems with eating and drinking (57). To improve oral lesions in PV, adjuvant topical or intralesional steroids are used, and based on evidence from case reports, the treatment is effective (58, 59). To date, however, no controlled clinical trial of the treatment of oral lesions in PV has been performed. PRP, which is concentrated plasma derived from autologous whole blood, is believed to promote wound healing (60). In a case report series, El-Komy et al. reported that six of seven PV patients showed improvement of their oral PDAI scores after PRP intralesional injection (61). To evaluate the effects of PRP in PV, an open-label, dose-escalation, multicenter phase 1 trial using autologous PRP has been conducted in adults with active PV (NCT02828163). This clinical trial was designed to compare the effects of PRP to those of intralesional steroid injection. Eleven PV patients received a 10 mg/mL triamcinolone injection on one side of the oral mucosa and a 1-mL PRP injection on the other side every 2 weeks for 3 months. The primary endpoint was the improvement of oral PV lesions in 3 months. Nine out of 11 participants completed the protocol, and 7 (78%) of those 9 patients showed improvement in oral PDAI and/or pain scores at the PRP injection sites. Although PRP resulted in clinical improvement, as in the previous study, there were no significant differences between PRP and intralesional steroid injections (62). Thus, autologous PRP might be used for the treatment of resistant oral erosions in pemphigus patients when intralesional steroid injection is contraindicated.
Future Potential Clinical Trials in Pemphigus
B cell depletion therapy using an anti-CD20 monoclonal antibody leads to short-term remission in the majority of pemphigus patients; however, we often observe relapsing disease after treatment. Although disease remission is related to the depletion of circulating Dsg3-specific B cells, the expansion of the same pathogenic B cell clone is observed during relapsing disease (63). Therefore, to maintain complete remission in PV, targeted removal of anti-Dsg3 memory B cells is essential. Recently, chimeric antigen receptor (CAR) technology was developed and resulted in novel treatments that led to the prolonged remission of refractory B cell leukemia and lymphoma. Adapted from that strategy, Dsg3 chimeric autoantibody receptor T cell (Dsg3-CAART) therapy has been reported to result in serological and histological improvements in experimental pemphigus mice without detectable off-target toxicity (15). A phase 1 clinical trial of Dsg3-CAART in PV patients is planned to investigate its safety and therapeutic potential.
Pemphigoid
Ixekizumab in BP
Th17 cells were first identified by their production of interleukin (IL)-17 (64, 65). A previous study indicated that the transfer of IL-17-producing Th17 cells into healthy SJL/J mice induced experimental autoimmune encephalomyelitis an animal model of MS (66). The prominent role of the Th17 polarization of T cells, as well as that of IL-17, in inflammation and autoimmunity was recognized after this observation. In addition to EAE, the Th17-IL-17 axis is also important in the pathogenesis of RA (67–69). Notably, neutralizing anti-IL-17 antibodies significantly decreased joint inflammation, cartilage destruction, and bone erosion in a collagen-immunized arthritis mouse model. Several recent studies suggest that Th17 cells and their cytokines also play roles in the pathogenesis of AIBD, including BP (70). More specifically, Le Jan et al. reported that IL-17A expression was significantly higher in both serum and lesional skin in BP patients than in normal healthy individuals (71). Furthermore, Chakievska et al. reported that IL-17A -/- mice were protected from the development of experimental BP induced by the transfer of anti-COL17 IgG (72). Taken together, these results indicate that IL-17A is a potential target for the treatment of BP. Ixekizumab is a recombinant fully humanized monoclonal antibody targeting IL-17 and is used for the treatment of psoriasis (73). However, there are neither case reports nor clinical studies reporting the effect of ixekizumab or other IL-17-targeting compounds on BP. To determine the efficacy of ixekizumab for the treatment of BP, an open-label, single-group phase 2 clinical trial is currently ongoing (NCT03099538). In this trial, twelve BP patients will receive subcutaneous injections of 160 mg ixekizumab on day 0, and then they will receive 80 mg of ixekizumab every 2 weeks until week 12. The primary endpoint is the median time from the start of treatment to the cessation of blister formation during the 12 weeks of therapy. There is no available report of the results of this trial to date.
NPB-01 in BP Unresponsive to Corticosteroids
With regard to pemphigus, the efficacy and safety of IVIg in refractory BP patients has been suggested by case reports and case report series (74–76). Based on these observations, several guidelines and consensus statements recommend the use of IVIg for refractory BP (77–79). Recently, Sasaoka et al. reported that IVIg reduced serum IL-6 concentrations, circulating pathogenic IgG levels, and skin inflammation disease activity in an active BP mouse model (80). Kamaguchi et al. reported that an anti-idiotypic antibody in IVIg significantly reduced the BP180 depletion of cultured keratinocytes stimulated with BP pathogenic IgG (81). To evaluate the effectiveness of IVIg in BP, a multicenter, randomized, double-blind, placebo-controlled, phase 3 clinical trial was conducted (NCT01408550). In this trial, 56 BP patients were parallelly assigned to receive either IVIg or a placebo. The IVIg group received an intravenous drip infusion of human IgG at 400 mg/kg/day for 5 consecutive days. The primary endpoint was the disease activity score (DAS) of skin lesions on day 15. The results of that study have recently been published (82). The DAS on day 15 was evaluated as the primary endpoint. Although the DAS on day 15 in the IVIg group (19.8 ± 22.2) was 12.5 points lower than that in the placebo group (32.3 ± 31.5), the difference between the groups was not significant (p = 0.089). However, a post hoc analysis of covariance using the DAS on day 1 as a covariate showed a significant difference between the IVIg and placebo groups (p = 0.041). Regarding the safety of IVIg in BP, the incidence of adverse drug reactions was 37.9% (n = 11/29) in the IVIg group vs. 18.5% (n = 5/27) in the placebo group. No significant difference in the incidence of adverse drug reactions was observed between the IVIg and placebo groups (p = 0.143). No patients in either group experienced any severe adverse drug reactions. Therefore, this report suggested that IVIg is a beneficial treatment modality for refractory BP cases treated with a moderate dose of systemic steroids.
Topical AC-203 in BP
The nucleotide-binding domain, leucine-rich repeat family, pyrin domain-containing 3 (NLRP3) inflammasome regulates the activation of caspase-1 and the release of pro-inflammatory cytokines such as IL-1β and IL-18 in macrophages (83–87). It is an essential innate immune sensor that is activated in response to various damage-associated molecular patterns (83, 84). Previous reports have suggested that dysregulation of the inflammasome attenuates several chronic diseases, including autoimmune diseases such as EAE (88) and systemic lupus erythematosus (89, 90). In addition, polymorphisms in both NLRP3 and caspase-1 recruitment domain-8 genes led to increased IL-1β production and are related to disease susceptibility and severity in RA (91). A recent study demonstrated that the expression levels of NLRP3 and inflammasome components are significantly higher in peripheral blood mononuclear cells from BP patients than in those from healthy donors (92). Furthermore, higher NLRP3 levels were positively correlated with BP disease activity. Although the precise mechanism by which NLRP3 contributes to pathogenesis in BP is still unclear, pharmacological modulation of the inflammasome pathway could be a novel therapeutic strategy in BP. However, in a mouse model of inflammatory EBA, caspase-1/11-deficient mice developed clinical disease manifestations identical to those of the wild-type controls after the injection of autoantibodies against type VII collagen (93).
To evaluate the therapeutic efficacy of caspase-1 inhibition in BP, AC-203, which is a topical ointment formulation of a modulator of the inflammasome and IL-1β pathways, was developed. To investigate the effects of AC-203 on BP, a randomized, open-label phase 2 clinical trial is currently conducted (NCT03286582). Forty BP patients received either topical AC-203 or clobetasol 0.05% topical ointment twice a day (time frame is 10 weeks). The primary endpoint is the incidence of adverse effects during the treatment period. No results of this trial are currently available.
Bertilimumab in BP
While the presence of eosinophils in the dermal infiltrate is one of the histological hallmarks of BP, their contribution to the pathogenesis of BP remains to be determined (94–97). Recent data, however, support the notion that eosinophils promote subepidermal blistering in BP (98). In line with the presence of eosinophils in the dermal infiltrate in BP, elevated expression levels of IL-5, eotaxin, and eosinophil colony-stimulating factor were detected in BP blister fluids (99). Furthermore, matrix metallopeptidase 9 is secreted by eosinophils in BP lesional skin (100), eosinophil cationic protein (ECP) is detected at the dermal-epidermal junction of lesional skin, and serum concentrations of ECP correlate with BP disease activity (101, 102). Based on these findings, eotaxin, which is released upon eosinophil activation (103), has emerged as a therapeutic target for the treatment of BP.
Bertilimumab is a fully human monoclonal antibody targeting eotaxin-1 (104). Therefore, the administration of bertilimumab is expected to impair eosinophil infiltration into the skin in BP. This hypothesis has been recently tested in an open-label, single arm, phase 2 clinical trial (NCT02226146). Eleven moderate to severe BP patients received 10 mg/kg of bertilimumab intravenously on days 0, 14, and 28 with approximately 13 weeks of follow-up. The primary endpoint was safety, including the incidence of adverse effects. The results of the study were presented at the 2018 American Academy of Dermatology annual meeting. Of the 11 subjects enrolled, 2 patients withdrew consent and 9 received bertilimumab. Bertilimumab was well tolerated in all 9 subjects, and no drug-associated serious adverse events were reported4 Preliminary analyses indicated that the subjects had an 81% decline in their Bullous Pemphigoid Disease Area Index (BPDAI) scores. Based on these results, bertilimumab has been granted fast track designation for the treatment of BP5.
Rituximab in BP
Rituximab has a well-documented efficacy in treating pemphigus (24). However, controlled clinical trials in pemphigoid diseases, including BP, are missing. Several case reports and case report series reported good outcomes of rituximab treatment for BP. Although the efficiency of rituximab for the treatment of BP varied among the studies, the overall CR rates were 60–70% and the relapse rates were approximately 20% in BP patients receiving rituximab therapy (105, 106). To evaluate the efficacy and safety of rituximab for the treatment of BP, an open-label, prospective, phase 3 clinical trial was conducted (NCT00525616). Eighteen BP patients received two intravenous injections of 1,000 mg of rituximab at 15-day intervals. The primary endpoint was clinical and biological disease control for up to 2 years. To date, no results of this trial are available.
Future Potential Clinical Trials in Pemphigoid
Spleen tyrosine kinase (SYK) is a nonreceptor cytoplasmic enzyme that is mainly expressed in hematopoietic cells and plays a pivotal role in regulating cellular responses to extracellular pathogens or antigen-immunoglobulin complexes (107). Because SYK acts downstream of activating Fc gamma receptor (FcγR), it has been considered a candidate drug target for antibody-induced diseases, such as pemphigoid diseases. In fact, a previous study has shown that SYK inhibition led to less activation of neutrophils stimulated with immune complexes and prevented the development of skin lesions in a preclinical EBA mouse model (108). In lesional skin in pemphigoid diseases, both eicosanoid leukotriene B4 (LTB4) and complement factor C5, the precursor of anaphylatoxin C5a, are found; however, their significance in the pathogenesis of pemphigoid diseases still needs to be elucidated. A previous preclinical mouse model of pemphigoid disease indicated a critical role of LTB4 in the recruitment of polymorph nuclear cells to the dermal epidermal junction (109). It is suggested that LTB4 may closely interact with C5a in the regulation of skin inflammation. Thus, inhibiting these two factors individually or in parallel might be effective for the treatment of pemphigoid diseases. These newly identified therapeutic targets should be addressed in future clinical trials.
Conclusions
The clinical trials discussed here, which include several trials investigating novel therapeutic targets, demonstrate that translational research in pemphigus and pemphigoid is a fast-growing field. We thus expect that several novel treatments will be shortly available for the treatment of pemphigus and pemphigoid patients. Given the high, and thus far unmet, medical need in this field (110), this is highly encouraging and will hopefully improve the quality of life of the affected patients. In addition to the compounds and targets described here, several new targets have been recently identified in preclinical model systems, such as PDE4 or PI3Kδ inhibitors (111, 112). Hence, the preclinical pipeline is well developed and will contribute to the growing number of clinical trials in pemphigus and pemphigoid.
Author Contributions
KI and RL wrote the manuscript. KB prepared the figure. All authors read, commented on, and approved the final version of the manuscript.
Funding
This work was supported by research grants from the Deutsche Forschungsgemeinschaft: Clinical Research Unit Pemphigoid Diseases (KFO 303), the Excellence Cluster Precision Medicine in Chronic Inflammation (EXC 2167-390884018) and the Uehara Memorial Foundation.
Conflict of Interest Statement
During the last 3 years, RL has received research funding from Almirall, True North Therapeutics, UCB Pharma, ArgenX, TxCell, Topadur, Incyte and Admirx and fees for consulting or speaking from ArgenX, Immunogenetics, Novartis and Lilly.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00978/full#supplementary-material
Abbreviations
AIBDs, autoimmune bullous dermatoses; PV, pemphigus vulgaris; PF, pemphigus foliaceus; BP, bullous pemphigoid; EBA, epidermolysis acquisita; p200, anti-laminin-γ1/p200 pemphigoid; PG, pemphigoid gestationis; LPP, lichen planus pemphigoides; LAD, linear IgA bullous dermatosis; DH, dermatitis herpetiformis; RA, rheumatoid arthritis; MS, multiple sclerosis; CR, complete remission; MMF, mycophenolate mofetil; BAFF, B cell activating factor of the tumor necrosis factor family; BAFF-R, B cell activating factor of the tumor necrosis factor family receptor; BCR, B cell receptor; PDAI, Pemphigus Disease Area Index; FcRn, neonatal Fc receptor; IVIg, intravenous immunoglobulin; BTK, Bruton's tyrosine kinase; Tregs, regulatory T cells; GVHD, graft vs. host disease; Dsg, desmoglein; PRP, Platelet-rich plasma; CAR, chimeric antigen receptor; Dsg3-CAART, Dsg3 chimeric autoantibody receptor T cell; IL, interleukin; EAE, experimental autoimmune encephalomyelitis; DAS, disease activity score; NLRP3, nucleotide-binding domain, leucine-rich repeat family, pyrin domain-containing 3; BPDAI, bullous pemphigoid disease area index; ECP, eosinophil cationic protein; SYK, spleen tyrosine kinase; FcγR, Fc gamma receptor; LTB4, leukotriene B4.
Footnotes
1. ^https://clinicaltrials.gov/
2. ^2https://www.clinicaltrialsregister.eu/
3. ^3https://ir.principiabio.com/news-releases/news-release-details/principia-biopharma-reports-positive-prn1008-phase-2-top-line
4. ^4https://www.immunepharma.com/what-we-do/bertilimumab
5. ^5https://ir.immunepharma.com/press-releases/detail/168/bertilimumab-granted-fast-track-designation-for-the
References
1. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. (2017) 3:17026. doi: 10.1038/nrdp.2017.26
2. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
3. van Beek N, Zillikens D, Schmidt E. Diagnosis of autoimmune bullous diseases. J Dtsch Dermatol Ges. (2018) 16:1077–91. doi: 10.1111/ddg.13637
4. Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. (2012) 45:55–70. doi: 10.3109/08916934.2011.606447
5. Otten JV, Hashimoto T, Hertl M, Payne AS, Sitaru C. Molecular diagnosis in autoimmune skin blistering conditions. Curr Mol Med. (2014) 14:69–95. doi: 10.2174/15665240113136660079
6. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. (2002) 346:321–7. doi: 10.1056/NEJMoa011592
8. Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in patients with pemphigus. Acta Derm Venereol. (2017) 97:607–11. doi: 10.2340/00015555-2611
9. Cai SC, Allen JC, Lim YL, Chua SH, Tan SH, Tang MB. Mortality of bullous pemphigoid in Singapore: risk factors and causes of death in 359 patients seen at the National Skin Centre. Br J Dermatol. (2014) 170:1319–26. doi: 10.1111/bjd.12806
10. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol. (2017) 8:603. doi: 10.3389/fimmu.2017.00603
11. Liu Y, Li L, Xia Y. BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol. (2017) 8:1752. doi: 10.3389/fimmu.2017.01752
12. Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest. (2013) 123:800–11. doi: 10.1172/JCI60139
13. Dehner C, Rotzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am J Pathol. (2014) 184:2528–36. doi: 10.1016/j.ajpath.2014.05.016
14. Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight. (2017) 2:92253. doi: 10.1172/jci.insight.92253
15. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
16. Lee J, Werth VP, Hall RP 3rd, Eming R, Fairley JA, Fajgenbaum DC, et al. Perspective from the 5th international pemphigus and pemphigoid foundation scientific conference. Front Med. (2018) 5:306. doi: 10.3389/fmed.2018.00306
17. Coiffier B, Haioun C, Ketterer N, Engert A, Tilly H, Ma D, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood. (1998) 92:1927–32.
18. Herrmann G, Hunzelmann N, Engert A. Treatment of pemphigus vulgaris with anti-CD20 monoclonal antibody (rituximab). Br J Dermatol. (2003) 148:602–3. doi: 10.1046/j.1365-2133.2003.05209_10.x
19. Cooper HL, Healy E, Theaker JM, Friedmann PS. Treatment of resistant pemphigus vulgaris with an anti-CD20 monoclonal antibody (Rituximab). Clin Exp Dermatol. (2003) 28:366–8. doi: 10.1046/j.1365-2230.2003.01283.x
20. Goebeler M, Herzog S, Brocker EB, Zillikens D. Rapid response of treatment-resistant pemphigus foliaceus to the anti-CD20 antibody rituximab. Br J Dermatol. (2003) 149:899–901. doi: 10.1046/j.1365-2133.2003.05580.x
21. Salopek TG, Logsetty S, Tredget EE. Anti-CD20 chimeric monoclonal antibody (rituximab) for the treatment of recalcitrant, life-threatening pemphigus vulgaris with implications in the pathogenesis of the disorder. J Am Acad Dermatol. (2002) 47:785–8. doi: 10.1067/mjd.2002.126273
22. Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. (2013) 68:404–11. doi: 10.1016/j.jaad.2012.08.010
23. Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. (2007) 357:545–52. doi: 10.1056/NEJMoa067752
24. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
25. Castillo J, Milani C, Mendez-Allwood D. Ofatumumab, a second-generation anti-CD20 monoclonal antibody, for the treatment of lymphoproliferative and autoimmune disorders. Expert Opin Investig Drugs. (2009) 18:491–500. doi: 10.1517/13543780902832679
26. Lin TS. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers Med. (2010) 3:51–9.
27. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. (2002) 2:465–75. doi: 10.1038/nri844
28. Ng LG, Sutherland AP, Newton R, Qian F, Cachero TG, Scott ML, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol. (2004) 173:807–17. doi: 10.4049/jimmunol.173.2.807
29. Salzer U, Jennings S, Grimbacher B. To switch or not to switch–the opposing roles of TACI in terminal B cell differentiation. Eur J Immunol. (2007) 37:17–20. doi: 10.1002/eji.200636914
30. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. (2004) 20:441–453. doi: 10.1016/S1074-7613(04)00079-2
31. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. (1999) 190:1697–710.
32. Matsushita T, Hasegawa M, Matsushita Y, Echigo T, Wayaku T, Horikawa M, et al. Elevated serum BAFF levels in patients with localized scleroderma in contrast to other organ-specific autoimmune diseases. Exp Dermatol. (2007) 16:87–93. doi: 10.1111/j.1600-0625.2006.00485.x
33. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J Clin Invest. (2002) 109:59–68. doi: 10.1172/JCI14121
34. Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemicimmune-based rheumatic diseases. Arthritis Rheum. (2001) 44:1313–9. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S
35. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. (2011) 377:721–31. doi: 10.1016/S0140-6736(10)61354-2
36. Dörner T, Posch M, Wagner F, Hüser A, Fischer T, Mooney L, et al. THU0313 Double-blind, randomized study of VAY736 single dose treatment in patients with primary sjögren's syndrome (PSS). Ann Rheumatic Dis. (2016) 75(Suppl. 2):300–1. doi: 10.1136/annrheumdis-2016-eular.5840
37. Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. (2006) 116:2142–2151. doi: 10.1172/JCI27821
38. Iwata H, Vorobyev A, Koga H, Recke A, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. (2018) 13:153. doi: 10.1186/s13023-018-0896-1
39. Ishii N, Hashimoto T, Zillikens D, Ludwig RJ. High-dose intravenous immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Clin Rev Allergy Immunol. (2010) 38:186–195. doi: 10.1007/s12016-009-8153-y
40. Blumberg L, Jumphries JE, Lasseter KC, Blumberg RS. SYNT001: a humanized IgG4 monoclonal antibody that disrupts the interaction of FcRn and IgG for the treatment of IgG-mediated autoimmune diseases. Blood. (2017) 130(Suppl. 1):3483.
41. Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. (2018) 128:4372–86. doi: 10.1172/JCI97911
42. Werth VP CD, Blumberg L, Humphries J, Blumberg R, Hall R. FcRn blockade with SYNT001 for the treatment of pemphigus. J Invest Dermatol. (2018) 138:S92. doi: 10.1016/j.jid.2018.03.546
43. Satterthwaite AB, Li Z, Witte ON. Btk function in B cell development and response. Semin Immunol. (1998) 10:309–16. doi: 10.1006/smim.1998.0123
44. de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, et al. The Bruton's tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. (1993) 23:3109–14. doi: 10.1002/eji.1830231210
45. Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. (2001) 23:436–46. doi: 10.1002/bies.1062
46. Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. (2012) 14:R243. doi: 10.1186/ar4086
47. Lee A, Sandhu S, Imlay-Gillespie L, Mulligan S, Shumack S. Successful use of Bruton's kinase inhibitor, ibrutinib, to control paraneoplastic pemphigus in a patient with paraneoplastic autoimmune multiorgan syndrome and chronic lymphocytic leukaemia. Australas J Dermatol. (2017) 58:e240–2. doi: 10.1111/ajd.12615
48. Smith PF, Krishnarajah J, Nunn PA, Hill RJ, Karr D, Tam D, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton's tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. (2017) 83:2367–76. doi: 10.1111/bcp.13351
49. Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. (2018) 142:1831–42.e1837. doi: 10.1016/j.jaci.2018.04.006
50. Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M, et al. Regulatory T cells suppress inflammation and blistering in pemphigoid diseases. Front Immunol. (2017) 8:1628. doi: 10.3389/fimmu.2017.01628
51. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
52. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
53. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
54. Zwang NA, Leventhal JR. Cell therapy in kidney transplantation: focus on regulatory T cells. J Am Soc Nephrol. (2017) 28:1960–72. doi: 10.1681/ASN.2016111206
55. Yokoyama T, Matsuda S, Takae Y, Wada N, Nishikawa T, Amagai M, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. (2011) 23:365–73. doi: 10.1093/intimm/dxr020
56. Schmidt T, Willenborg S, Hunig T, Deeg CA, Sonderstrup G, Hertl M, et al. Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp Dermatol. (2016) 25:293–8. doi: 10.1111/exd.12919
57. Zand N, Mansouri P, Fateh M, Ataie-Fashtami L, Rezaee Khiabanloo S, Safar F, et al. Relieving pain in oral lesions of pemphigus vulgaris using the non-ablative, non-thermal, CO2 laser therapy (NTCLT): preliminary results of a novel approach. J Lasers Med Sci. (2017) 8:7–12. doi: 10.15171/jlms.2017.02
58. Mignogna MD, Fortuna G, Leuci S, Adamo D, Dell'Aversana Orabona G, Ruoppo E. Adjuvant triamcinolone acetonide injections in oro-pharyngeal pemphigus vulgaris. J Eur Acad Dermatol Venereol. (2010) 24:1157–65. doi: 10.1111/j.1468-3083.2010.03610.x
60. Chicharro-Alcantara D, Rubio-Zaragoza M, Damia-Gimenez E, Carrillo-Poveda JM, Cuervo-Serrato B, Pelaez-Gorrea P, et al. Platelet rich plasma: new insights for cutaneous wound healing management. J Funct Biomater. (2018) 9:10. doi: 10.3390/jfb9010010
61. EL-Komy MH, Hassan AS, Abdel Raheem HM, Doss SS, EL-Kaliouby M, Saleh NA, et al. Platelet-rich plasma for resistant oral erosions of pemphigus vulgaris: a pilot study. Wound Repair Regen. (2015) 23:953–5. doi: 10.1111/wrr.12363
62. El-Komy MHM, Saleh NA, Saleh MA. Autologous platelet-rich plasma and triamcinolone acetonide intralesional injection in the treatment of oral erosions of pemphigus vulgaris: a pilot study. Arch Dermatol Res. (2018) 310:375–81. doi: 10.1007/s00403-018-1824-x
63. Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, et al. Persistence of anti-desmoglein 3 IgG(+) B-cell clones in pemphigus patients over years. J Invest Dermatol. (2015) 135:742–9. doi: 10.1038/jid.2014.291
64. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. (2009) 361:888–98. doi: 10.1056/NEJMra0707449
65. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. (2008) 9:650–7. doi: 10.1038/ni.1613
66. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
67. Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. (2014) 35:493–500. doi: 10.1016/j.tips.2014.07.006
68. Takahashi H, Tsuji H, Hashimoto Y, Ishida-Yamamoto A, Iizuka H. Serum cytokines and growth factor levels in Japanese patients with psoriasis. Clin Exp Dermatol. (2010) 35:645–9. doi: 10.1111/j.1365-2230.2009.03704.x
69. Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. (2002) 51:102–4. doi: 10.1007/BF02684010
70. Toosi S, Bystryn JC. Potential role of interleukin-17 in the pathogenesis of bullous pemphigoid. Med Hypotheses. (2010) 74:727–8. doi: 10.1016/j.mehy.2009.10.038
71. Le Jan S, Plee J, Vallerand D, Dupont A, Delanez E, Durlach A, et al. Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol. (2014) 134:2908–17. doi: 10.1038/jid.2014.263
72. Chakievska L, Holtsche MM, Kunstner A, Goletz S, Petersen BS, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun. (2018) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
73. Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. (2016) 375:345–56. doi: 10.1056/NEJMoa1512711
74. Engineer L, Ahmed AR. Role of intravenous immunoglobulin in the treatment of bullous pemphigoid: analysis of current data. J Am Acad Dermatol. (2001) 44:83–8. doi: 10.1067/mjd.2000.112288
75. Jolles S. A review of high-dose intravenous immunoglobulin (hdIVIg) in the treatment of the autoimmune blistering disorders. Clin Exp Dermatol. (2001) 26:127–31. doi: 10.1046/j.1365-2230.2001.00779.x
76. Ahmed AR. Intravenous immunoglobulin therapy for patients with bullous pemphigoid unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. (2001) 45:825–35. doi: 10.1067/mjd.2001.116337
77. Venning VA, Taghipour K, Mohd Mustapa MF, Highet AS, Kirtschig G. British association of dermatologists' guidelines for the management of bullous pemphigoid 2012. Br J Dermatol. (2012) 167:1200–14. doi: 10.1111/bjd.12072
78. Mydlarski PR, Ho V, Shear NH. Canadian consensus statement on the use of intravenous immunoglobulin therapy in dermatology. J Cutan Med Surg. (2006) 10:205–21. doi: 10.2310/7750.2006.00048
79. Ahmed AR, Dahl MV. Consensus statement on the use of intravenous immunoglobulin therapy in the treatment of autoimmune mucocutaneous blistering diseases. Arch Dermatol. (2003) 139:1051–9. doi: 10.1001/archderm.139.8.1051
80. Sasaoka T, Ujiie H, Nishie W, Iwata H, Ishikawa M, Higashino H, et al. Intravenous IgG reduces pathogenic autoantibodies, serum IL-6 levels, and disease severity in experimental bullous pemphigoid models. J Invest Dermatol. (2018) 138:1260–7. doi: 10.1016/j.jid.2018.01.005
81. Kamaguchi M, Iwata H, Mori Y, Toyonaga E, Ujiie H, Kitagawa Y, et al. Anti-idiotypic antibodies against BP-IgG prevent type XVII collagen depletion. Front Immunol. (2017) 8:1669. doi: 10.3389/fimmu.2017.01669
82. Amagai M, Ikeda S, Hashimoto T, Mizuashi M, Fujisawa A, Ihn H, et al. A randomized double-blind trial of intravenous immunoglobulin for bullous pemphigoid. J Dermatol Sci. (2017) 85:77–84. doi: 10.1016/j.jdermsci.2016.11.003
83. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
84. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
85. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. (2013) 39:432–41. doi: 10.1016/j.immuni.2013.08.037
86. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
87. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. (2009) 10:241–7. doi: 10.1038/ni.1703
88. Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. (2010) 184:4610–4. doi: 10.4049/jimmunol.1000217
89. Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. (2014) 26:475–81. doi: 10.1097/BOR.0000000000000088
90. Duan Z, Zhang Y, Zeng Z, Pan F. Comment on “Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus”. J Immunol. (2012) 189:499; author reply 499–500. doi: 10.4049/jimmunol.1290031
91. Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Soderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology. (2008) 47:415–7. doi: 10.1093/rheumatology/kem372
92. Fang H, Shao S, Cao T, Lei J, Dang E, Zhang J, et al. Increased expression of NLRP3 inflammasome components and interleukin-18 in patients with bullous pemphigoid. J Dermatol Sci. (2016) 83:116–23. doi: 10.1016/j.jdermsci.2016.04.009
93. Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol. (2015) 194:3656–63. doi: 10.4049/jimmunol.1402688
94. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2018) 54:26–51. doi: 10.1007/s12016-017-8633-4
95. Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol. (2017) 177:141–51. doi: 10.1111/bjd.15114
96. van Beek N, Luttmann N, Huebner F, Recke A, Karl I, Schulze FS, et al. Correlation of serum levels of IgE autoantibodies against BP180 with bullous pemphigoid disease activity. JAMA Dermatol. (2017) 153:30–8. doi: 10.1001/jamadermatol.2016.3357
97. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med. (2018) 5:201. doi: 10.3389/fmed.2018.00201
98. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol. (2018) 138:1032–43. doi: 10.1016/j.jid.2017.11.031
99. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. (2000) 143:112–6. doi: 10.1046/j.1365-2133.2000.03599.x
100. Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. (1994) 93:2022–30. doi: 10.1172/JCI117196
101. Borrego L, Maynard B, Peterson EA, George T, Iglesias L, Peters MS, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. (1996) 148:897–909.
102. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol. (2017) 26:1248–52. doi: 10.1111/exd.13382
103. Honda K, Chihara J. Eosinophil activation by eotaxin–eotaxin primes the production of reactive oxygen species from eosinophils. Allergy. (1999) 54:1262–9.
104. Ding C, Li J, Zhang X. Bertilimumab Cambridge antibody technology group. Curr Opin Investig Drugs. (2004) 5:1213–8.
105. Amber KT, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
106. Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horvath B. Effectiveness and safety of rituximab in recalcitrant pemphigoid diseases. Front Immunol. (2018) 9:248. doi: 10.3389/fimmu.2018.00248
107. Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci. (2014) 35:414–22. doi: 10.1016/j.tips.2014.05.007
108. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-genome expression profiling in skin reveals SYK as a key regulator of inflammation in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:249. doi: 10.3389/fimmu.2018.00249
109. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:1104–13. doi: 10.1016/j.jid.2016.12.021
110. Lamberts A, Yale M, Grando SA, Horvath B, Zillikens D, Jonkman MF. Unmet needs in pemphigoid diseases: an international survey amongst patients, clinicians and researchers. Acta Derm Venereol. (2018) 99:224–5. doi: 10.2340/00015555-3052
111. Koga H, Kasprick A, Lopez R, Auli M, Pont M, Godessart N, et al. Therapeutic effect of a novel phosphatidylinositol-3-kinase delta inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
Keywords: autoimmune blistering skin disease, pemphigus, pemphigoid, clinical trial, treatment
Citation: Izumi K, Bieber K and Ludwig RJ (2019) Current Clinical Trials in Pemphigus and Pemphigoid. Front. Immunol. 10:978. doi: 10.3389/fimmu.2019.00978
Received: 29 January 2019; Accepted: 16 April 2019;
Published: 03 May 2019.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Raghavendra Rao, Manipal Academy of Higher Education, IndiaPilar Iranzo, Hospital Clínic de Barcelona, Spain
Cassian Sitaru, Freiburg University Medical Center, Germany
Jun Yamagami, Keio University Hospital, Japan
Branka Marinovic, University Hospital Centre Zagreb, Croatia
Copyright © 2019 Izumi, Bieber and Ludwig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, cmFsZi5sdWR3aWdAdWtzaC5kZQ==