 Emily Levy1,2
Emily Levy1,2 Robert Reger1
Robert Reger1 Filip Segerberg3†
Filip Segerberg3† Melanie Lambert3†
Melanie Lambert3† Caroline Leijonhufvud3†Yvonne Baumer1
Caroline Leijonhufvud3†Yvonne Baumer1 Mattias Carlsten1,3*‡
Mattias Carlsten1,3*‡ Richard Childs1‡
Richard Childs1‡- 1National Heart Lung and Blood Institute, National Institutes of Health, Bethesda, MD, United States
- 2The Department of Molecular Medicine, The George Washington University, Washington, DC, United States
- 3Department of Medicine, Huddinge, Center for Hematology and Regenerative Medicine, Karolinska Institutet, Stockholm, Sweden
Adoptive transfer of natural killer (NK) cells can induce remission in patients with relapsed/refractory leukemia and myeloma. However, to date, clinical efficacy of NK cell immunotherapy has been limited to a sub-fraction of patients. Here we show that steps incorporated in the ex vivo manipulation/production of NK cell products used for adoptive infusion, such as over-night IL-2 activation or cryopreservation followed by ex vivo expansion, drastically decreases NK cell surface expression of the bone marrow (BM) homing chemokine receptor CXCR4. Reduced CXCR4 expression was associated with dampened in vitro NK cell migration toward its cognate ligand stromal-derived factor-1α (SDF-1α). NK cells isolated from patients with WHIM syndrome carry gain-of-function (GOF) mutations in CXCR4 (CXCR4R334X). Compared to healthy donors, we observed that NK cells expanded from WHIM patients have similar surface levels of CXCR4 but have a much stronger propensity to home to BM compartments when adoptively infused into NOD-scid IL2Rgammanull (NSG) mice. Therefore, in order to augment the capacity of adoptively infused NK cells to home to the BM, we genetically engineered ex vivo expanded NK cells to express the naturally occurring GOF CXCR4R334X receptor variant. Transfection of CXCR4R334X-coding mRNA into ex vivo expanded NK cells using a clinically applicable method consistently led to an increase in cell surface CXCR4 without altering NK cell phenotype, cytotoxic function, or compromising NK cell viability. Compared to non-transfected and wild type CXCR4-coding mRNA transfected counterparts, CXCR4R334X-engineered NK cells had significantly greater chemotaxis toward SDF-1α in vitro. Importantly, expression of CXCR4R334X on expanded NK cells resulted in significantly greater BM homing following adoptive transfer into NSG mice compared to non-transfected NK cell controls. Collectively, these data suggest up-regulation of cell surface CXCR4R334X on ex vivo expanded NK cells via mRNA transfection represents a novel approach to improve homing and target NK cell-based immunotherapies to BM where hematological malignancies reside.
Introduction
Natural killer (NK) cells play an important role in the body's defense against cancer and have proven to be powerful effectors against hematological tumor targets in pre-clinical studies (1). During the last decade, several published studies have demonstrated that adoptive infusion of mature short-term cytokine activated allogeneic NK cells can induce remission in subgroups of patients with relapsed/refractory leukemia and myeloma (2, 3). Clinical trials are currently evaluating the safety and efficacy of the adoptive infusion of ex vivo expanded NK cells (NCT02074657; NCT02271711). Advantages of ex vivo expansion include the possibility of utilizing repeated infusions of large numbers of these highly cytotoxic effector cells. Despite decades of promising preclinical studies, at present, only a relatively small proportion of patients treated with adoptive NK cell infusions have shown a clearly beneficial response (4, 5). NK cell immunotherapy may in part be limited by host factors such as tumor burden and aggressiveness, immune-mediated rejection of the infused cells, and the inhibitory effects of suppressive cell populations such as regulatory T cells (5–7). Deeper biological insights into these factors as well as the development of novel strategies to overcome these limitations are needed before the full therapeutic potential of NK cell immunotherapy can be realized in cancer patients.
A still relatively unexplored aspect of cellular cancer immunotherapy is the potential of the infused cells to home to the cancer-affected organ in vivo (8). For instance, the potential of adoptively infused NK cells to home to bone marrow (BM) compartments where hematological malignancies reside is likely critical to obtain clinically meaningful antitumor effects in patients with leukemia or myeloma. Consistent with this theory, a recent analysis by Bjorklund et al. showed that high expression levels of the BM homing chemokine receptor CXCR4 on the infused NK cells was associated with increased probability of objective response in patients with relapsed/refractory acute myeloid leukemia (AML) or high-risk myelodysplastic syndrome (MDS) (5). These and previously published data showing that CXCR4 expression is reduced on the NK cell surface shortly after activation with IL-2 (9) have led our group to hypothesize that modification of NK cells to express higher levels of CXCR4, including CXCR4 receptor variants associated with a gain-of-function (GOF), may represent a method to improve the clinical efficacy of adoptive NK cell immunotherapy for hematological malignancies.
There has been recent increased interest in the use of a number of different strategies to genetically modify NK cells to augment their anti-tumor potential (10). We have previously shown that mRNA electroporation can be used as an approach to efficiently transfect ex vivo expanded NK cells using the FDA approved MaxCyte platform (11). Therefore, we explored whether mRNA electroporation of CXCR4, including the WHIM syndrome naturally occurring GOF mutated variant CXCR4R334X (12), could be utilized to improve NK cell in vitro migration toward its ligand stromal-derived factor-1α (SDF-1α) and in vivo homing to BM compartments in mice. We further sought to validate this strategy by testing the hypothesis that NK cells expanded ex vivo from WHIM patients with GOF CXCR4R334X would have superior homing to the bone marrow compared to NK cells expressing wild-type CXCR4.
Here we show that commonly utilized methods to ex vivo activate and/or expand NK cells for adoptive infusion, such as over-night IL-2 activation or cryopreservation followed by ex vivo expansion, drastically decrease NK cell surface expression of CXCR4 compared to non-manipulated NK cells from PBMCs. This attenuation in receptor expression intensity was associated with a marked reduction in their capacity to migrate toward SDF-1α in vitro. Further, we show that NK cells expanded from WHIM patients have similar surface levels of CXCR4 but a much stronger propensity to home to BM compartments when adoptively infused into NOD-scid IL2Rgammanull (NSG) mice. Finally, we show that ex vivo expanded human NK cells genetically engineered to express the GOF CXCR4R334X receptor variant have significantly enhanced in vitro migration capacity toward SDF-1α and superior BM homing following adoptive transfer into NSG mice compared to non-transfected NK cell controls. Collectively, these data suggest introduction of CXCR4R334X on the cell surface of ex vivo expanded NK cells via mRNA transfection represents a novel approach to improve their BM homing capacity and thereby augment NK cell-based immunotherapies against BM-residing malignancies such as leukemia and myeloma.
Materials and Methods
Cells Lines and Primary Cells
Peripheral blood mononuclear cells (PBMCs) from healthy donor blood were isolated using Lymphocyte Separation Medium (MP Biomedicals) (NIH protocol 99-H-0050 and 2006/229-31/3). NK cells were isolated by magnetic bead separation; either CD3 depleted and CD56 selected (Miltenyi) or by using an NK cell isolation kit (Miltenyi). NK cells were expanded for 14–18 days in G-Rex flasks (Wilson Wolf Manufacturing) with irradiated human EBV-LCL feeder cells at a ratio of one NK cell to 10 feeder cells (1:10 ratio). Cells were cultured in X-vivo 20 media (Lonza) containing 10% human AB serum (Sigma), 1% Glutamax (Gibco), and 500 U/ml IL-2. Fresh media was supplied to cells starting on day 5 of expansion, and then every 48 h until the cells were harvested for use in experiments. Cryopreserved WHIM PBMCs (NIH protocols NCT05-I-0213; NCT14-I-0185) were generously provided by Dr. David McDermott through an NIH Material Transfer Agreement in accordance with NIH human subject research policies. K562 and MOLM14 cell lines were obtained from ATCC. MM.1S was kindly provided by Dr. Irene Ghobrial at the Dana-Farber Cancer Institute. SMI-LCLs were established in the NHLBI/NIH by the Childs' lab. Cell lines were propagated in RPMI 1640 supplemented with 10% heat-inactivated FBS (Sigma-Aldrich).
Antibodies and Flow Cytometry
Phenotype analysis were conducted antibodies targeting human CD56 (NCAM16.2), CD3 (UCHT1), CD57 (NK-1), CD2 (RPA2.10), DNAM-1 (DX11), NKp44 (p44-8), NKp30 (p30-15), NKp46 (9E2), TNF-α (Mab11), Granzyme B (Gb11), and IFN-γ (B27) from Becton Dickinson (BD) Biosciences (CA, USA). Antibodies targeting human CXCR4 (12G5), IgG2a isotype (MOPC173), KIR2DL/DS/2/3 (DX27), NKG2D (1D11), KIR3DL/DS1 (DX9), 2B4 (C1.7), and CD107a (H4A3) were purchased from Biolegend (CA, USA). Human LIR-1 (HP-F1) antibody was purchased from Lifespan Biosciences Inc (WA, USA). Live/Dead Aqua marker was purchased from Invitrogen (CA, USA). Flow cytometry was performed with a BD LSRII Fortessa cytometer and the data were analyzed with the FlowJo software (Treestar Inc.).
NK Cell Transfection
A MaxCyte GT instrument (MaxCyte) was used to introduce 4 μg mRNA/1 × 106 NK cells harvested 14–18 days after ex vivo expansion as previously reported (13). Custom-made mRNAs were obtained from TriLink Biotechnologies. Sequences that encode human CXCR4R334X and CXCR4WT were used (Supplementary Table 1).
Phenotyping
Ex vivo expanded NK cells from six healthy donors harvested on days 14–18 were transfected with CXCR4R334X- or CXCR4WT-coding mRNA, as described above. Non-transfected NK cells from each donor were used as controls. After culturing at 37°C, 6.5% CO2 for 24 h, NK cells were collected and stained for 30 min at 4°C with a viability marker and cell surface markers. Cells were washed twice and fixed in 1% paraformaldehyde (MP Biomedicals) in PBS prior to acquisition on a BD LSR II Fortessa (BD Biosciences).
Preparation of Cells for Scanning Electron Microscopy
Ten million expanded NK cells were electroporated (as described above) with either CXCR4R334X mRNA or mock-transfected with vehicle alone. Non-transfected NK cells and mock-transfected (vehicle only) from the same donor were used as controls. Eight hours following transfection, 1 × 106 NK cells were fixed in 4% glutaraldehyde (0.1M calcium chloride, 0.1M sodium cacodylate buffer, pH 7.2) over night at 4°C and prepared as described previously (14). Subsequently, cells were washed twice in 0.1M cacodylate and post-fixed using 1% OsO4 in 0.1M cacodylate buffer for 1 h. Cells were than dehydrated in an ethanol gradient series (30, 50, 70, 85, 95, and 100%). Afterwards samples were critically point dried with the Samdri-795 critical point dryer (Tousimis Research Corp, USA), mounted on aluminum stubs, and coated with 7.5 nm gold/palladium with an EMS 575-X sputter coater (Electron Microscopy Sciences, USA). Samples were imaged using the Hitachi S-3400N1 SEM at 7.5kV.
Degranulation and Cytokine Production Assay
Expanded NK cells transfected with mRNA, as described above, were co-cultured with K562, MOLM14, SMI-LCL or MM1.S tumor cells at a ratio of 1:1 in 37°C, 5% CO2. After 4 h, the cells were stained with the Live/Dead viability marker, CD56 antibody, and CD107a antibody. Cells were analyzed using an LSR Fortessa II flow cytometer (BD Biosciences). In the subsequent experiments, transfected NK cells were co-cultured with K562 only for 4 h at a ratio of 1:1 in 37°C, 5% CO2. Brefeldin A and Monensin (BD Biosciences) and CD107a antibody were added within the first hour of co-culture. Cells were first stained extracellularly and with the Live/Dead viability marker and CD56 antibody for 30 min at 4°C before fixation and permeabilization with Cytofix/Cytoperm solution (BD biosciences). Cells were then stained for intracellular expression of TNF-α and IFN-γ in Perm/Wash buffer (BD Biosciences) for 30 min at 4°C before they were washed and analyzed using an LSR II Fortessa flow cytometer (BD Biosciences).
In vitro Migration Assay
Migration assays were performed on cells 8 h after mRNA transfection using Corning Transwell® inserts. PBS containing 10% FBS and increasing concentrations of recombinant human SDF-1α (Biolegend) was added to the bottom chambers and 1 × 105 NK cells suspended in the same medium but without SDF-1α were added to the top chambers. The assay plate was placed in 37°C with 6.5% CO2 for 2 h. Cells were collected and quantified by the CyQuant kit and assay protocol (Invitrogen). Fluorescence intensity (excitation/emission: 480 nm/520 nm) was measured on a microplate spectrometer (Infinite® 200 PRO, Tecan). Cells plated straight to the bottom chamber were used as maximum control, and the proportion of migrated cells was calculated as a percent of total cells initially added to each well. CXCR4 blockade was achieved by pretreating NK cells for 30 min with 100 μM plerixafor (Mozobil® Sanofi Oncology) in 37°C prior to the migration assay.
Cellular Homing Assays in vivo
Eight to ten-week-old NOD-scid IL2Rgammanull (NSG) mice were purchased from Jackson Laboratories. NK cells from WHIM patients or healthy donors were expanded ex vivo for 19 days using irradiated EBV-LCL feeders as previously described and injected intravenously (i.v.) into NSG mice at a dose of 7 × 106 cells/animal. Animals received 100,000 IU of IL-2 intraperitoneally immediately following injection. Femur BM was harvested 24 h post-injection and human NK cells were identified based on CD56-positivety by flow cytometry. Ten million CXCR4R334X transfected or 10 × 106 non-transfected NK cells were injected i.v. into adult NSG mice. Cells were harvested from animals 24 h post-transfer from cardiac blood, liver, lungs, and the femur BM. Tissues were dissociated and analyzed by flow cytometry (Supplementary Figure 1). Human NK cells were identified based on positivity for MHC class I and human CD56.
In vivo Tracking of NK Cell Using Bioluminescence Imaging
Expanded NK cells were co-transfected with either 4 μg/1 × 106 cells of CXCR4R334X-coding mRNA and 4 μg/1 × 106 cells of luciferase-coding mRNA or 4 μg/1 × 106 cells of luciferase mRNA and equal volume of electroporation buffer. Eight million cells were injected i.v. into NSG mice. The biodistribution of the infused cells was tracked by bioluminescence imaging using the IVIS In Vivo Imaging System (PerkinElmer) 2 and 24 h post-cell infusion. Animals were injected intraperitoneally with 150 mg/kg D-luciferin Potassium salt (Gold Biotechnologies) suspended in PBS 10 min prior to imaging.
Statistical Analysis
Data were analyzed with Prism 7.0b software (GraphPad Software Inc.), using the two-tailed paired-T-test, the Welch's-T-test, the Wilcoxon sign-rank test, or the Mann-Whitney U-test to report significant differences between groups. The appropriate test was chosen based on data distribution, variances, and experimental set up. *P < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 were considered statistically significant, and ns, not significant.
Results
Impact of Cryopreservation, IL-2 Activation, and ex vivo Expansion on NK Cell Surface Expression of CXCR4 and Migration Toward the CXCR4 Ligand SDF-1α in vitro
CXCR4 was expressed on the surface of NK cells isolated from freshly collected blood samples from healthy donors, although expression levels were lower than T and B cells (Figure 1A). Given its relevance to our ongoing clinical trial evaluating the anti-tumor effects of adoptive transfer of ex vivo expanded NK cells, we next evaluated the impact of multiple different NK cell manipulations on CXCR4 expression including cryopreservation and thawing, IL-2 activation, and ex vivo expansion. Compared to NK cells within bulk PBMCs, isolated NK cells had a significant reduction in CXCR4 surface expression (p = 0.0002). There was an additional decrease in CXCR4 expression when isolated NK cells were rested overnight in IL-2-containing media compared to media without IL-2 (p = 0.003). This observation is consistent with prior reports of the effects of IL-2 on CXCR4 expression on NK, NK T, and T cells (9, 15). When freshly isolated NK cells were cryopreserved and then thawed, we observed a further substantial reduction in NK cell CXCR4 surface expression (p < 0.0001). Ex vivo expansion of the cryopreserved NK cells for 14 days using EBV-LCL feeder cells and IL-2 led to an increase of CXCR4 surface expression compared to their phenotype after thawing, however CXCR4 expression never fully recovered to the levels observed on the NK cells prior to isolation from the bulk PBMC population (Figure 1B).
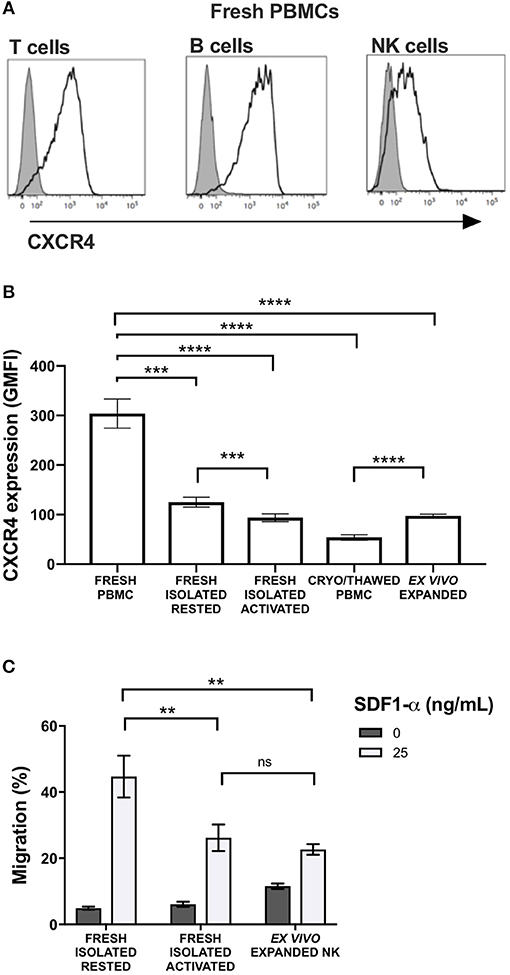
Figure 1. Commonly used methods to manipulate and prepare NK cells for adoptive infusion decrease surface expression of CXCR4 and reduce their ability to migrate toward SDF-1α in vitro. Surface expression of CXCR4 was evaluated by flow cytometry. (A) CXCR4 surface expression on fresh T cells, B cells and NK cells among PBMC. Representative histograms are shown (one representative donor). (B) CXCR4 intensity, as displayed as GMFI relative to FMO, on different NK cell preparations (n = 10 donors). (C) In vitro transwell migration of NK cells from populations in (B) toward SDF-1α (n = 10 donors). Statistical significance was evaluated with the paired t-test, two-tailed. Bar graphs present the mean and error bars report the SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Over-night IL-2 activated and ex vivo expanded NK cells, represents two prototypes of NK cell preparations currently being administered to humans in clinical trials (8). Compared to freshly isolated NK cells that had never been exposed to cytokines, over-night IL-2 activated and ex vivo expanded NK cells were less responsive to SDF-1α-induced in vitro migration (p = 0.0045 and 0.0095, respectively) (Figure 1C). As SDF-1α is known to be a critical ligand responsible for recruiting CXCR4-expressing cells into the BM (16–18), these data suggest that NK cell activation and/or ex vivo expansion of NK cells with IL-2 may impede the ability of these populations to home to BM compartments following adoptive transfer into patients. These data suggest reductions in NK cell CXCR4 expression that occur as a consequence of ex vivo manipulation approaches commonly used in most NK cell immunotherapy protocols, could have deleterious effects on this homing pathway. These observations are particularly relevant for patients who have BM-residing malignancies on clinical trials receiving adoptive NK cell infusions.
Ex vivo Expanded NK Cells From Patients With WHIM Syndrome Show Enhanced in vivo Bone Marrow Homing Compared to NK Cells Expanded From Healthy Donors
Individuals with WHIM syndrome harbor GOF mutations in the CXCR4 receptor (19), and despite having normal hematopoiesis, sequester leukocytes inside the BM niche which clinically manifests as severe leukopenia. Therefore, we hypothesized that ex vivo expanded NK cells expressing the GOF CXCR4 (CXCR4R334X) might have superior homing to the BM compared to ex vivo expanded NK cells expressing wild-type (WT) CXCR4. To test this hypothesis, we ex vivo expanded WT CXCR4-expressing NK cells from healthy donors and GOF CXCR4R334X-expressing NK cell from WHIM patients to evaluate the ability of the NK cells to migrate into BM compartments following adoptive transfer in immunodeficient mice. Although circulating NK cell numbers were low in the WHIM patients, we were able to successfully isolate and expanded their NK cells obtained from peripheral blood (Supplementary Table 2). The cell surface expression of CXCR4 on ex vivo expanded NK cells from 3 WHIM patients were higher (p = 0.04), although comparable with that of ex vivo expanded NK cells from a cohort of healthy donors (Figure 2A). Remarkably, when adoptively infused into NSG immunodeficient mice, NK cells expanded from WHIM patients had a much stronger propensity to home to BM compartments compared to NK cells expanded from healthy individuals (p < 0.0001) (Figures 2B–D). These data suggest strategies aimed at altering the function of the CXCR4 receptor on NK cells, rather than its overall surface density, may alone be sufficient to improve NK cell homing to the bone marrow.
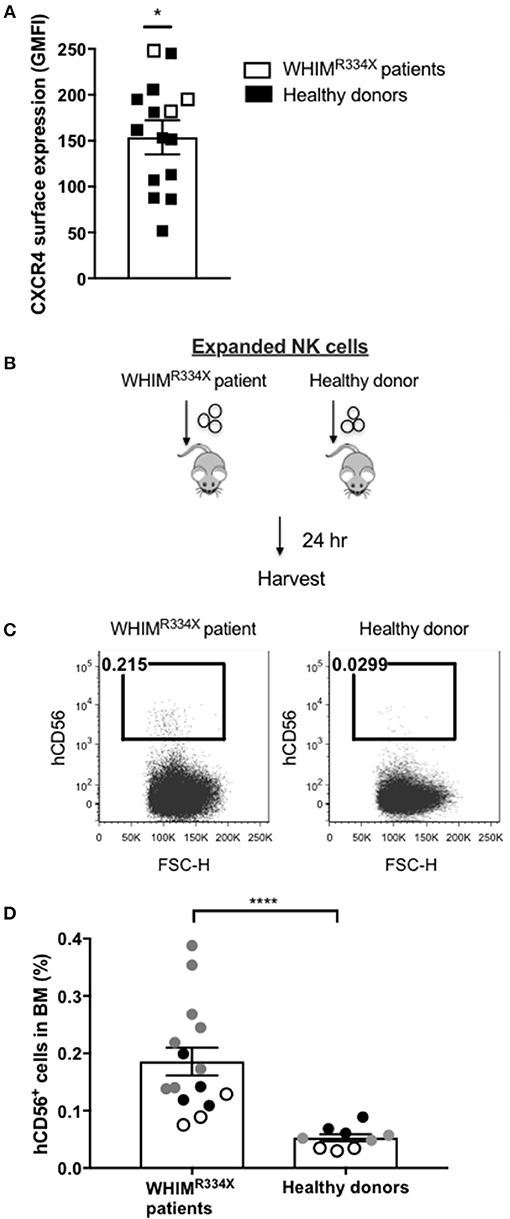
Figure 2. NK cells from patients with WHIM syndrome have similar intensity of CXCR4 cell surface expression but have significantly superior in vivo bone marrow homing compared to NK cells expanded from healthy donors. (A) Surface expression intensity of CXCR4 on expanded NK cells from patients with WHIM syndrome (white, n = 3 WHIM pts) vs. healthy donors (black, n = 9 donors). (B) Experiment schema illustrating injection of expanded human NK cells i.v. into NSG mice followed by a harvest 24 h later. (C) Proportion of CD56+ human NK cells in BM samples of mice injected with ex vivo expanded NK cells from healthy donors or from WHIM syndrome patients 24 h following infusion (one representative mouse from each group). (D) The fraction of human CD56+ cells recovered from the BM of mice injected with human NK cells (WHIM n = 15, healthy donors n = 9, data reported from 3 independent experiments; each experiment is represented by either white, gray or black circles). Statistical significance was evaluated by the Welch's T-test, two-tailed. Graphs present mean and error bars report SEM. *p < 0.05, ****p < 0.0001.
Transfection of Expanded NK Cells With CXCR4R334X-Coding mRNA Transiently Upregulates Cell Surface CXCR4 Leading to Improved Chemotaxis Toward SDF-1α in vitro Without Negatively Impacting NK Cell Viability, Phenotype, and Cytotoxic Function
We have previously shown that mRNA electroporation can be used as an approach to efficiently transfect ex vivo expanded NK cells using the FDA approved MaxCyte platform (11). Therefore, we explored whether mRNA electroporation of CXCR4, including the WHIM syndrome naturally occurring GOF mutated variant CXCR4R334X (12), could be utilized to improve NK cell in vitro migration toward its ligand SDF-1α and in vivo homing to BM compartments in mice. We further sought to validate this strategy by testing the hypothesis that NK cells expanded ex vivo from WHIM patients with GOF CXCR4R334X would have superior homing to the bone marrow compared to NK cells expressing wild-type CXCR4.
We recently reported that that mRNA electroporation using the MaxCyte GT platform can be used as an approach to efficiently transfect ex vivo expanded NK cells with transgenes that augment NK cell antibody-dependent cellular cytotoxicity (ADCC) and in vitro migration toward the CCR7 ligand CCL19 in vitro (11). Large scale electroporation with the MaxCyte GT instrument is clinically approved for investigational use and has been shown to be a safe and effective approach to genetically modify a variety of different cells (20–22). Therefore, we explored whether mRNA electroporation of CXCR4, including the WHIM syndrome naturally occurring GOF mutated variant CXCR4R334X, could be utilized to alter the expression of CXCR4 on the surface of expanded NK cells and improve their migration toward its ligand SDF-1α and in vivo homing to BM compartments in mice. CXCR4 mRNA electroporation efficiently augmented CXCR4 surface expression in an mRNA dose-dependent fashion (Figure 3A). Introduction of the GOF variant CXCR4R334X, or the wild-type protein CXCR4WT, did not reduce NK cell viability compared to unmodified or mock-modified cells (Figure 3B). Transfection of NK cells with either of the two mRNAs resulted in an increase of cell surface CXCR4 expression, which was transient and peaked at 4–8 h post-electroporation, then returned to baseline within 36–48 h (Figure 3C).
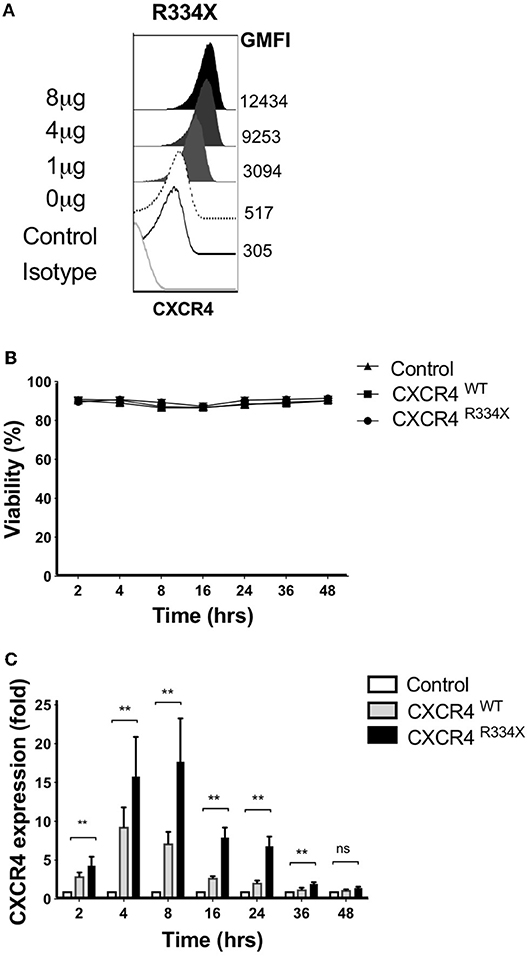
Figure 3. Ex vivo expanded NK cells transfected with CXCR4R334X mRNA have significantly upregulated surface expression of the CXCR4 receptor without impacting cellular viability. (A) Titration of increasing amounts of electroporated CXCR4R334X mRNA results in increased cell surface CXCR4 expression. (B) NK cell viability over time following mRNA transfection (n = 10 healthy donors). (C) CXCR4 surface expression kinetics over time reported as fold increase in CXCR4 on expanded NK cells transfected with either CXCR4R334X or CXCR4WT mRNA compared to their non-transfected control counterpart (n = 10 donors). Statistical significance was assessed by the Wilcoxon sign-rank test. Graphs present mean and error bars report SEM. **p < 0.01.
Expression of CXCR4R334X resulted in improved in vitro migration toward SDF-1α (Figure 4A). Compared to both control NK cells and NK cells electroporated with CXCR4WT-coding mRNA, NK cells engineered to express CXCR4R334X migrated more efficiently toward 25 and 50 ng/mL of SDF-1α (p = 0.0005 and 0.0039, respectively) (Figure 4A). A subsequent analysis revealed no differences in NK cell morphology between electroporated and non-electroporated NK cells (Supplementary Figure 2) and showed the migration potential for CXCR4R334X-modified NK cells, but not CXCR4WT-modified or unmodified cells, positively correlated with the expression intensity of cell surface CXCR4 (Supplementary Figure 3). This analysis further supports the advantage of utilizing GOF CXCR4-coding mRNA over wild-type CXCR4-coding mRNA, as the former not only results in higher cell surface expression but also appears to enhance the migration function of CXCR4 cell surface molecules to SDF-1 (Supplementary Figure 3). Importantly, we did not observe increased migration toward the non-CXCR4-binding chemokine CCL19 (Supplementary Figure 4), and pretreatment of NK cells with the specific CXCR4 agonist plerixafor abrogated the migration potential toward SDF-1α, regardless if the NK cells had undergone mRNA electroporation or not (Figure 4B). These findings confirm cellular chemotaxis assessed with this assay is dependent of the CXCR4/SDF-1α axis.
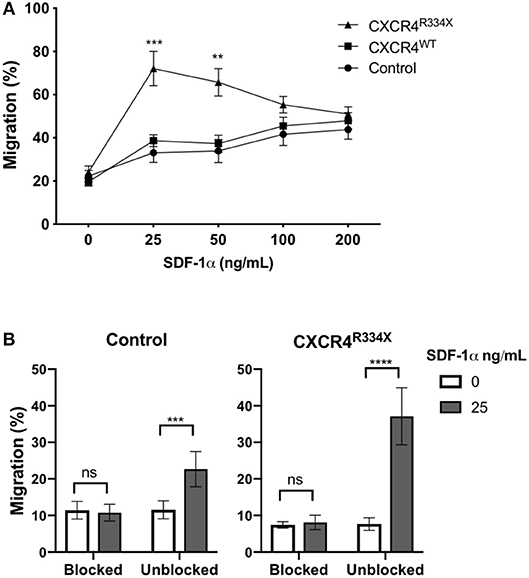
Figure 4. Transfection of ex vivo expanded NK cells with CXCR4R334X-coding mRNA but not with CXCR4WT-coding mRNA improves in vitro chemotaxis of cells toward SDF-1α. (A) In vitro migration of NK cells toward increasing concentration of SDF-1α (n = 10 donors, 4 independent experiments). (B) Migration toward SDF-1α when NK cells were pretreated with 100 μM of plerixafor to block the CXCR4 receptor (n = 9 donors, 1 experiment). Statistical significance was determined with the Student's paired t-test, two-tailed. Graphs present mean and error bars in panel A report SEM whereas error bars in (B) report SD. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Given the high transfection efficiency and increased in vitro migration of NK cells that had undergone CXCR4R334X mRNA electroporation, we next conducted studies to confirm that key features of these NK cell populations, including their phenotype and cytotoxic function, were preserved. As shown in Figure 5, there was no alterations in the activating or inhibitory receptor profiles of CXCR4 mRNA transfected NK cells compared to control non-transfected NK cells (Figure 5A). Importantly, expression of other surface markers that are critical for lymphocyte homing and transmigration through endothelium such as CD62L, integrin α4 (CD49d), and CD44 were unaltered by NK cell mRNA electroporation (Supplementary Figure 5). Additionally, NK cell cytokine production capacity (IFN-γ and TNF-α) and cytotoxic function against the gold standard NK cell tumor target K562 remained unaffected by mRNA transfection (Figures 5B–D, Supplementary Figures 6, 7) (23–25). Importantly, CXCR4 mRNA electroporated NK cells had preserved high cytotoxic function against leukemia, lymphoma, and multiple myeloma cell lines as assessed by CD107a degranulation (Figure 5E). Altogether, these data demonstrate that mRNA transfection of NK cells is an effective way to transiently introduce new CXCR4 molecules on the NK cell surface, and in NK cells transfected to express CXCR4R334X, improve NK cell chemotaxis toward SDF-1α without negatively impacting viability, phenotype or function.
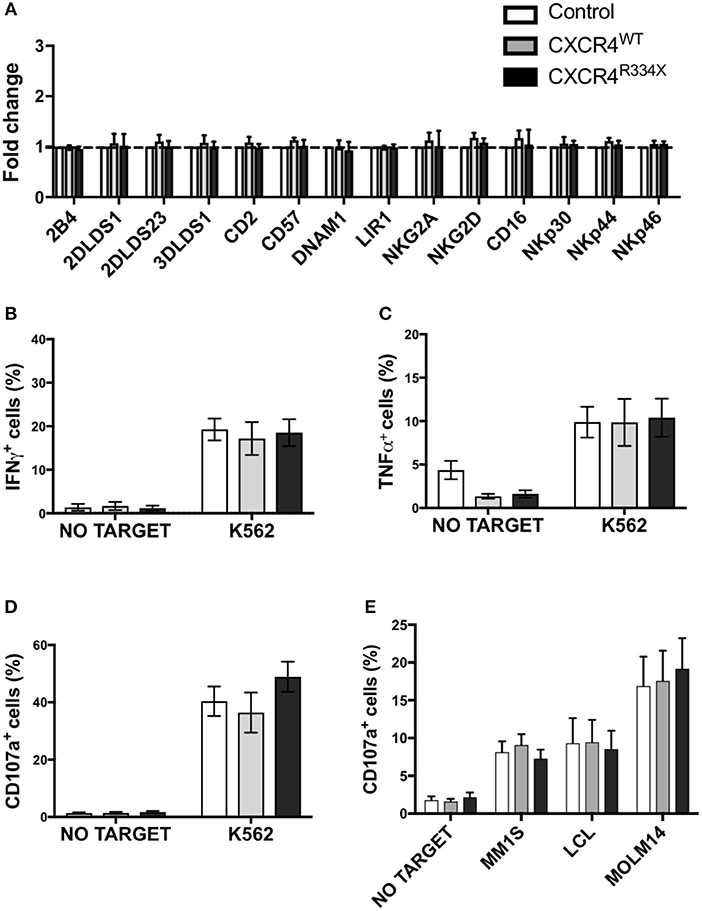
Figure 5. NK cells transfected with CXCR4 coding mRNAs have no alterations in NK cell inhibitory and activating receptors and have preserved anti-tumor cytotoxic function. (A) Inhibitory and activation NK cell receptor expression 24 h post-transfection with CXCR4-coding mRNAs compared to non-transfected control NK cells (n = 6 donors, 1 experiment). (B–D) NK cell intracellular TNF-α and IFN-γ, and surface CD107a expression following a 4-h co-culture with K562 cells (n = 6 donors, 1 experiment). (E) NK cell degranulation measured by NK cell surface upregulation of CD107a in response to co-culture with various tumor cell lines (n = 4 donors, 4 independent experiments). Statistical significance was determined with the Student's paired t-test, two-tailed. Graphs present mean and error bars report SEM.
Human NK Cells Transfected to Express CXCR4R334X Have Significantly Superior in vivo Homing to Bone Marrow Compartments Following Adoptive Transfer Into Immunodeficient Mice Compared to Control NK Cells
Because NK cells expanded from WHIM syndrome patients with CXCR4R334X had superior in vivo homing to the BM of immunodeficient mice, and because CXCR4R334X mRNA electroporated NK cells showed superior migration to SDF-1α in vitro, we next tested whether electroporation of CXCR4R334X in expanded NK cells from healthy human donors could be used as a strategy to improve their homing to the BM in NSG mice compared to non-transfected NK cell controls. As shown in Figure 6, significantly higher proportions of CXCR4R334X NK cells were recovered in the femur BM compared to controls (p = 0.0153). In contrast, there was a reduction in the proportion of CXCR4R334X NK cells recovered in the blood and lungs compared to controls (p = 0.0071) (Figures 6C,D). Similar frequencies of the two infused NK cell populations were recovered from liver (Figure 6E). Altogether, these differences in biodistribution patterns are consistent with transfection of the GOF CXCR4R334X receptor resulting in targeted NK cell homing to the BM. Finally, we used in vivo bioluminescent imaging to further confirm our findings: ex vivo expanded NK cells were transfected with either a combination of luciferase-coding and CXCR4R334X-coding mRNAs or luciferase-coding mRNA alone prior to injection into NSG mice. Bioluminescence imaging at 2 h post-infusion revealed that both cell fractions were initially trapped predominantly in the lungs, but at 24 h had assumed distinctly different anatomical localizations (Figure 7). Whereas, the mock-transfected NK cells (luciferase alone) were found in lungs, spleen, and to a low degree in the BM of the lower extremities, CXCR4R334X NK cells at 24 h were found mainly in BM niches including the skull, sternum, vertebrae, and bones of the upper and lower extremities. Taken altogether, these data establish that transfection of NK cells with CXCR4R334X-coding mRNA enhances their ability to enter BM compartments following adoptive transfer in vivo.
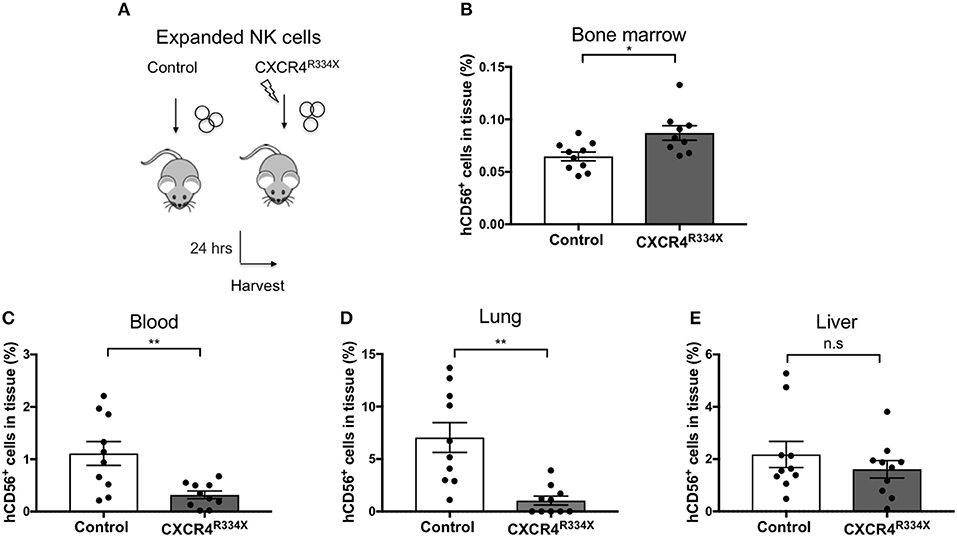
Figure 6. Ex vivo expanded NK cells transfected with CXCR4R334X-coding mRNA home significantly more efficiently to bone marrow compartments following adoptive infusion into NSG mice when compared to their non-transfected counterpart. (A) Experimental schematic illustrating the infusion of ex vivo expanded NK cells that have been transfected or not with CXCR4R334X-coding mRNA. (B–E) The frequency of recovered CD56+ human NK cells from the BM, blood, lung, and liver 24 h after infusion of the cells. Each dot represents one animal (n = 10 mice/group). Statistics were evaluated by the Welch's t-test, two-tailed. Graphs present mean and error bars report SEM. *p < 0.05, **p < 0.01.
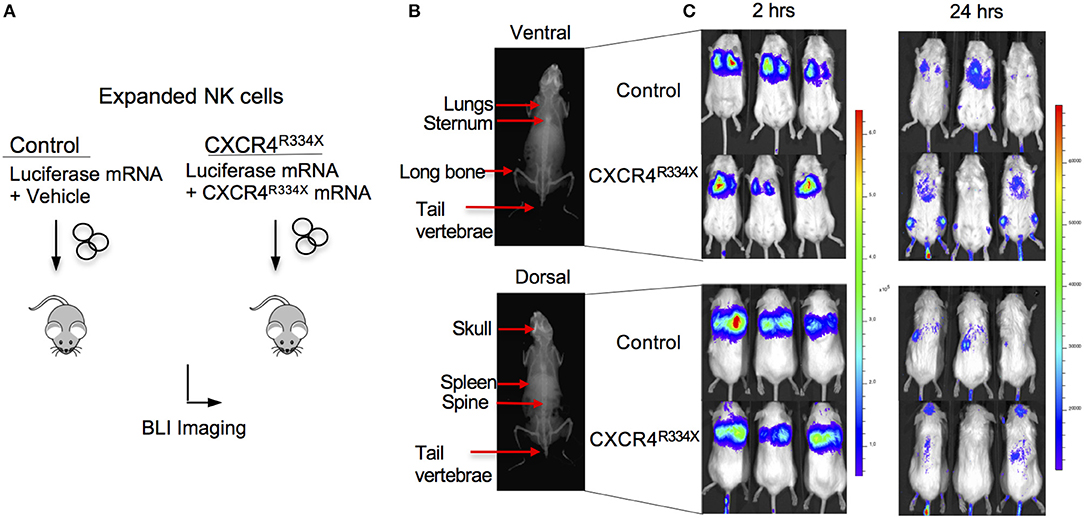
Figure 7. Ex vivo expanded NK cells transfected with CXCR4R334X-coding mRNA have superior trafficking to bone marrow compartments 24 h following adoptive transfer compared to their mock transfected counterpart. (A) Experimental schematic illustrating in vivo trafficking of infused NK cells transfected with mRNA coding for luciferase and CXCR4R334X or luciferase alone. (B) X-ray image of a mouse demonstrating ventral (top) and dorsal (bottom) anatomical views of NK cell distributions observed in (C). (C) Ventral (top) and dorsal (bottom) bioluminescence images of luciferase expressing NK cells 2 and 24 h after infusion into mice (n = 3 mice/group).
Discussion
The development of new methods to bolster the anti-tumor activity of adoptively infused immune cells is a pertinent topic of research within the scope of cancer immunotherapy. CAR-NK cells, checkpoint inhibition as well as bi- and tri-specific killer cell engagers are a few of the many exciting approaches that are currently being explored to improve NK cell-based immunotherapy for cancer (8). Although these and other approaches aimed at augmenting NK cell tumor killing capacity hold great promise, little research has focused on the importance, and potential for modifying NK cell homing to improve clinical outcomes. In this study, we explored mRNA transfection of NK cells to express CXCR4 (WT and CXCR4R334X variants) to enhance the trafficking of ex vivo expanded adoptively infused NK cells into BM niches. Our data show that highly efficient transfection with the naturally occurring GOF CXCR4 variant CXCR4R334X found in patients with WHIM syndrome can be achieved with electroporation using a clinically applicable platform without negatively impacting key NK cell phenotypic and functional characteristics. Establishing this method for modulating cellular homing lays the foundation for future studies exploring the therapeutic potential of adoptively infused CXCR4R334X-modified NK cells in patients who have BM residing malignancies.
With the specific aim of augmenting the efficacy of investigational NK cell-based cancer immunotherapies against leukemia and myeloma, we assessed the expression levels of the chemokine receptor CXCR4 on NK cells, a key receptor known to play a critical role for BM homing of multiple different cellular populations, perhaps best characterized in relation to hematopoietic stem cells (HSCs) (26–29). Further, we analyzed the impact of commonly utilized methods to isolate, process and activate NK cells on CXCR4 expression, including cryopreservation and thawing, culturing in IL-2, and ex vivo expansion. It is important to consider that cell handling methods and manipulation strategies used to prepare various cellular products for use in humans may have deleterious effects on the expression of cell surface chemokine receptors that play an important role in cellular homing. Nieto et al. reported ficoll treatment resulted in a decrease in expression of CCR2, CCR4, CCR5, and CXCR4 on monocytes and bulk lymphocytes (30). In this report, we show that isolation of NK cells from fresh PBMCs, as well as cryopreservation and thawing and ex vivo expansion leads to a significant reduction in surface expression of CXCR4. As reported previously for cytokine stimulated NK cells (9), we also observed that NK cells isolated from fresh PBMCs had a rapid reduction in cell surface CXCR4 expression following short-term IL-2 activation. As a consequence of these actions decreasing CXCR4 expression, migration capacity of NK cells toward the CXCR4 ligand SDF-1α decreased in vitro, which would be expected to reduce the potential of these cells to infiltrate BM compartments where malignancies such leukemia and myeloma reside. Such speculation is supported by an observation made in a recent phase I/II clinical trial of adoptive transfer of haploidentical NK cells to patients with relapsed/refractory leukemia or high-risk MDS (5). In this trial, patients treated with NK cell products that expressed high levels of CXCR4 were more likely to respond compared to those treated with NK cells expressing lower levels of this chemokine receptor. These data indirectly support the hypothesis that CXCR4 is a key receptor needed for adoptively infused NK cells to effectively target malignancies residing in the BM. Further, they establish CXCR4 as being a key candidate molecule to further study and manipulate in an effort to improve the ability of these populations to target leukemia and myeloma.
The CXCR4 receptor has long been known for its key role in chemotaxis of HSCs and other cells to the BM via signaling by its ligand SDF-1α, which is produced by BM stroma cells (31, 32). Targeted pharmacological inhibition of the CXCR4/SDF-1α interaction with plerixafor resulting in rapid mobilization of HSCs and other CXCR4-expressing cells from the BM into the circulation (33–37) and the observation that aberrant CXCR4 expression on tumor cells results in preferential BM metastasis (34, 38) highlight the singular importance that CXCR4 plays in cellular homing to the BM. The importance of this receptor in cellular trafficking to the BM was also highlighted in 2003 when GOF mutations in the CXCR4 gene were revealed as the mechanism for the sequestration of leukocytes in BM compartments of patients with WHIM syndrome (12). Since that discovery, several different families with distinct mutations in the CXCR4 gene have been described (19, 39). However, despite different mutations, WHIM syndrome patients have in common expression of CXCR4 receptors with a truncated C-terminus that impairs receptor internalization upon ligand binding and results in increased receptor signaling (39) compared to the wild type receptors (40). In this manuscript, we show for the first time that ex vivo expanded NK cells isolated from the circulation of patients with WHIM syndrome have stronger BM homing potential in vivo when infused into immunodeficient mice compared to NK cells expanded from healthy donors expressing WT CXCR4. Importantly, the observation that CXCR4 expression was not substantially more intense on NK cells expanded from WHIM patients compared to healthy controls suggest the improved BM homing capacity of these NK cells is related to augmented intracellular signaling previously shown to occur with the CXCR4R334X receptor (39). The non-canonical signaling of truncated GOF CXCR4 variants is well-characterized within the context of WHIM syndrome and Waldenström's macroglobulinemia, where the hyperfunctional phenotype of truncated CXCR4 results in sequestration of cells in BM niches (12, 33, 39, 41–44).
Our study is the first to harness the potential of GOF CXCR4R334X to improve the capacity of NK cells to home to BM compartments following adoptive transfer. As shown in our experiments, only the introduction of CXCR4R334X mRNA lead to an improvement in vitro in migration to SDF-1α. Somewhat unexpectedly, we observed that electroporation of NK cells resulting in a substantial increase in surface WT CXCR4 did not augment their migration capacity to SDF-1α. These data suggest acquisition of the mutated GOF CXCR4, rather than just the level of cell surface expression is a stronger determinant for augmenting the migration capacity of CXCR4 to SDF-1α. Nevertheless, we did observe a significant correlation between CXCR4 expression intensity on CXCR4R334X-engineered NK cells and migration capacity, highlighting the importance of achieving nominal receptor density of this mutated receptor on genetically manipulated NK cells. Importantly, we confirmed that NK cell in vitro migration to SDF-1α, including the augmented migration observed to occur following introduction of CXCR4R334X, could be completely abrogated by blocking with CXCR4 receptor antagonist plerixafor, establishing the stand alone importance of the CXCR4/SDF-1α axis in NK cell homing and migration. Taken together, our findings suggest that the GOF properties associated with the acquisition of surface expressed CXCR4R334X receptor play a key role in improving the capacity of NK cell migration toward SDF-1α. Importantly, this improvement in chemotaxis critical to BM homing following electroporation with CXCR4R334X mRNA did not occur at the expense of any reduction in NK cell viability or alteration in NK cell phenotype and function.
Whereas, genetic manipulation of primary T cells has been successful for several decades, the efficacy of gene engineering NK cells has until recently been limited (10). New technologies and the optimization of well-established approaches have now allowed for the broader exploration of genetic engineering of NK cells (10). In this regard, developing approaches to direct and target NK cells to tumors and tumor-bearing tissues would be expected to complement and potentiate other strategies focused on augmenting tumor recognition and cytotoxic function.
The striking observation that expression of the CXCR4R334X receptor on expanded NK cells leads to a distinct propensity toward homing to BM compartments in vivo following adoptive transfer into mice establishes a platform to test whether genetic manipulation to express CXCR4R334X can be used as a strategy to improve NK cell killing of BM residing cancers. However, questions remain regarding optimizing the dynamics of NK cell trafficking in relation to the time dependent kinetics of surface expression of CXCR4R334X receptor observed using this mRNA-based approach. Further, it is unknown whether the transient expression of CXCR4R334X, which lasts no longer than 48 h following mRNA electroporation, would be sufficient to mediate efficient clearance of BM-residing tumors. Nevertheless, as NK cells are naturally programmed to rapidly kill target cells without prior sensitization, the mRNA approach that we employed may provide a sufficient improvement in homing to facilitate the clearance of BM-residing tumor cells while simultaneously recruiting other critical immune cells to the tumor site (45). Depletion of receptors on the NK cell surface that compete for homing outside the BM, introduction of adhesion molecules or the use of drugs or radiation prior to cell infusion to prime BM compartments may be additional avenues explore to augment NK cell homing to BM compartments. Our data was generated using a xeno murine model to evaluate cellular homing of human cells. It is important to consider the cross reactivity of chemokine receptors with their cognate ligands may differ in humans compared to other species. Therefore, future clinical studies will need to be performed to validate our findings in humans. Finally, studies in tumor bearing animal models exploring the potential of adoptively infused NK cells genetically modified to express CXCR4R334X should be conducted as they could provide insights allowing for the optimal utilization of this strategy in humans with cancer. Within this context, it will be important to exclude the possibility that the function of these genetically modified NK cells could be suppressed by mesynchymal stromal cell populations expressing high levels of SDF-1α within the BM tumor niche. Additionally, as highlighted by studies of lymphocyte trafficking into tumor infiltrated BM compartments, a potential problem with tumor-bearing animal models is the fact that immense tumor growth in the BM compartment can destroy stromal cells and suppress their ability to produce SDF-1α, dampening the potential for CXCR4-mediated migration and infiltration into the BM (46, 47). The development of models that identify the proper combination and timing of cytoreductive therapy, including chemotherapy, irradiation, and antibody therapy prior to NK cell infusion to re-establish the SDF-1α gradient could resolve or at least reduce this issue.
Collectively, data from our study suggest up-regulation of cell surface CXCR4R334X on ex vivo expanded NK cells represents a novel approach to improve homing and target NK cell-based immunotherapies to BM where hematological malignancies reside. Importantly, our results provide a foundation for future studies aimed at bolstering the homing of NK cells to BM compartments for improved tumor eradication following adoptive NK cell infusions in patients with any BM-residing malignancies, including patients with solid tumors that reside or are metastatic to the BM. Although editing their migration potential as a stand-alone approach may improve their efficacy, combining methods that improve BM homing with strategies that augment the anti-tumor function of NK cells, may have the best chance to improve the efficacy of future adoptive NK cell immunotherapy trials in humans with cancer.
Contribution to the Field
The processing of NK cell grafts for cancer immunotherapy often triggers reduced expression of the bone marrow homing chemokine receptor CXCR4, leading to decreased in vitro migration toward the chemokine SDF-1α. This perturbation may dampen the potential of adoptive NK cell therapy against bone marrow-residing malignancies such as acute myeloid leukemia and myeloma. Using an animal model, we here show that ex vivo expanded NK cells from WHIM patients who somatically carry the natural occurring gain-of-function variant of CXCR4, CXCR4R334X, have a significantly increased propensity to home into the bone marrow compared to expanded NK cells from healthy donors carrying wildtype CXCR4. Furthermore, we show that healthy donor NK cells genetically engineered using an FDA-approved transfection instrument to express CXCR4R334X have significantly improved bone marrow homing potential compared to their unmodified counterparts. Our manuscript is the first proof-of-concept study to utilize gain-of-function CXCR4 to enhance bone marrow homing of primary human NK cells and will set the stage for a new line of research focusing on directing adoptively infused immune cells to the cancer. This approach is complementary to the waste majority of ongoing research focusing on improving tumor recognition and cytotoxicity of adoptively infused immune cells.
Data Availability
This manuscript contains previously unpublished data. The name of the repository and accession number are not available.
Ethics Statement
This study was carried out in accordance with the recommendations of the Animal Care User Committee at the NIH under protocol H-0111R1. This study was also carried out in accordance with the recommendations of Internal Review Board (IRB) at the NIH; PBMC from WHIM patient was collected at the NIH under NIH protocols NCT05-I-115 0213; NCT14-I-0185; from healthy donors under NIH protocol 99-H-0050 and under Etikprövningsnämnden (EPN) 2006/229-31/3. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocols were approved by the IRB and EPN, respectively.
Author Contributions
EL and MC wrote the manuscript. EL, RR, RC, and MC have designed experiments and analyzed the data. EL, FS, RR, ML, CL, and YB have performed experiments. All authors have critically reviewed the paper.
Funding
Funding for these studies was provided by the DIR NHLBI, the extremely generous contributions from the Dean R. O'Neal and Edward Rancic Fellowship, supporting researchers in the Childs' laboratory, and United States Public Health Service Commissioned Corps for key support for RADM Richard Childs as well as the Swedish Society for Medical Research (SSMF), the Wallenberg Clinical Fellow Award, Swedish Research Council, Swedish Pediatric Cancer Foundation, Radiumhemmets Research Foundation, the Clas Groschinsky Foundation and the Clinical Scientist Training Programme at Karolinska Institutet for the Carlsten lab.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Keyvan Keyvanfar for running the flow facility at the Hematology Branch, NHLBI, NIH, Xin Tian for statistical support from the OBR/NIH, Dr. David McDermott and Dr. Philip Murphy of NIAID/DIR for generously providing WHIM patient cells samples, and Iyadh Douagi for running the flow facility at HERM, Department of Medicine, Huddinge, KI. We would like to acknowledge the NHLBI Electron Microscopy core, as well as the animal care and user committee and the animal facility staff at the NIH for making these experiments possible. Additionally, we acknowledge the Institute of Biomedical Sciences at George Washington University for supporting EL's standing as a doctoral candidate.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01262/full#supplementary-material
References
1. Carlsten M, Malmberg K-J, Ljunggren H-G. Natural killer cell-mediated lysis of freshly isolated human tumor cells. Int J Cancer. (2009) 124:757–62. doi: 10.1002/ijc.24082
2. Alici E, Sutlu T, Björkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. (2008) 111:3155–62. doi: 10.1182/blood-2007-09-110312
3. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
4. Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. (2018) 67:483–94. doi: 10.1007/s00262-017-2100-1
5. Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin Cancer Res. (2018) 24:1834–44. doi: 10.1158/1078-0432.CCR-17-3196
6. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
7. Williams R, Cooley S, Bachanova V, Waldmann T, Blazar B, Weisdorf D, et al. Role of recipient CD8+ T cell exhaustion in the rejection of adoptively transferred haploidentical NK cells. Blood. (2016) 128:502. Available online at: http://www.bloodjournal.org/content/128/22/503
8. Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov. (2015) 14:487–98. doi: 10.1038/nrd4506
9. Beider K, Nagler A, Wald O, Franitza S, Dagan-Berger M, Wald H, et al. Involvement of CXCR4 and IL-2 in the homing and retention of human NK and NK T cells to the bone marrow and spleen of NOD/SCID mice. Blood. (2003) 102:1951–8. doi: 10.1182/blood-2002-10-3293
10. Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. (2015) 6:266. doi: 10.3389/fimmu.2015.00266
11. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. (2016) 7:105. doi: 10.3389/fimmu.2016.00105
13. Levy ER, Carlsten M, Childs RW. mRNA transfection to improve NK cell homing to tumors. Methods Mol Biol. (2016) 1441:231–40. doi: 10.1007/978-1-4939-3684-7_19
14. Baumer Y, McCurdy S, Weatherby TM, Mehta NN, Halbherr S, Halbherr P, et al. Hyperlipidemia-induced cholesterol crystal production by endothelial cells promotes atherogenesis. Nat Commun. (2017) 8:1129. doi: 10.1038/s41467-017-01186-z
15. Campbell GR, Loret EP, Spector SA. HIV-1 clade B tat, but not clade C tat, increases X4 HIV-1 entry into resting but not activated CD4+T cells. J Biol Chem. (2010) 285:1681–91. doi: 10.1074/jbc.M109.049957
16. Bernardini G, Sciumè G, Bosisio D, Morrone S, Sozzani S, Santoni A. CCL3 and CXCL12 regulate trafficking of mouse bone marrow NK cell subsets. Blood. (2008) 111:3626–34. doi: 10.1182/blood-2007-08-106203
17. McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol. (1999) 213:442–56. doi: 10.1006/dbio.1999.9405
18. Möhle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. (1998) doi: 10.1073/pnas.91.6.2305
19. McDermott DH, Gao JL, Liu Q, Siwicki M, Martens C, Jacobs P, et al. Chromothriptic cure of WHIM syndrome. Cell. (2015) 160:686–99. doi: 10.1016/j.cell.2015.01.014
20. Li L, Allen C, Shivakumar R, Peshwa MV. Large volume flow electroporation of mRNA: clinical scale process. Methods Mol Biol. (2013) 969:127–38. doi: 10.1007/978-1-62703-260-5_9
21. Koh S, Shimasaki N, Bertoletti A. Redirecting T cell specificity using T cell receptor messenger RNA electroporation. Methods Mol Biol. (2016) 1428:285–96. doi: 10.1007/978-1-4939-3625-0_19
22. Selmeczi D, Hansen TS, Met Ö, Svane IM, Larsen NB. Efficient large volume electroporation of dendritic cells through micrometer scale manipulation of flow in a disposable polymer chip. Biomed Microdevices. (2011) 13:383–92. doi: 10.1007/s10544-010-9507-1
23. Nagel JE, Collins GD, Adler WH. Spontaneous or natural killer cytotoxicity of k562 erythroleukemic cells in normal patients. Cancer Res. (1981) 41:2284–8.
24. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural Killer (NK) cell–mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. (1998) 188:2375–80. doi: 10.1084/jem.188.12.2375
25. Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J Immunol. (1996) 156:907–15.
26. Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. (1999) 283:845–8. doi: 10.1126/science.283.5403.845
27. Kahn J, Byk T, Jansson-Sjostrand L, Petit I, Shivtiel S, Nagler A, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. (2004) 103:2942–9. doi: 10.1182/blood-2003-07-2607
28. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. (2005) 201:1307–18. doi: 10.1084/jem.20041385
29. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
30. Nieto JC, Cantó E, Zamora C, Ortiz MA, Juárez C, Vidal S. Selective loss of chemokine receptor expression on leukocytes after cell isolation. PLoS ONE. (2012) 7:e31297. doi: 10.1371/journal.pone.0031297
31. Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2mnullmice. Leukemia. (2002) 16:1992–2003. doi: 10.1038/sj.leu.2402684
32. Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. (2002) 195:1145–54. doi: 10.1084/jem.20011284
33. McDermott DH, Lopez J, Deng F, Liu Q, Ojode T, Chen H, et al. AMD3100 is a potent antagonist at CXCR4(R334X) , a hyperfunctional mutant chemokine receptor and cause of WHIM syndrome. J Cell Mol Med. (2011) 15:2071–81. doi: 10.1111/j.1582-4934.2010.01210.x
34. Freed J, Shaffer CV, Moore CC. A novel CXCR4 pathway is required for migration of metastatic breast cancer cells. Cancer Res. (2015) 75:4043. doi: 10.1158/1538-7445.AM2015-4043
35. Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol. (2015) 6:429. doi: 10.3389/fimmu.2015.00429
36. Pantin J, Purev E, Tian X, Cook L, Donohue-Jerussi T, Cho E, et al. Effect of high-dose plerixafor on CD34+ cell mobilization in healthy stem cell donors: results of a randomized crossover trial. Haematologica. (2017) 102:600–9. doi: 10.3324/haematol.2016.147132
37. Girbl T, Lunzer V, Greil R, Namberger K, Hartmann TN. The CXCR4 and adhesion molecule expression of CD34+ hematopoietic cells mobilized by “on-demand” addition of plerixafor to granulocyte-colony-stimulating factor. Transfusion. (2014) 54:2325–35. doi: 10.1111/trf.12632
38. Yang P, Liang S-X, Huang W-H, Zhang H-W, Li X-L, Xie L-H, et al. Aberrant expression of CXCR4 significantly contributes to metastasis and predicts poor clinical outcome in breast cancer. Curr Mol Med. (2014) 14:174–84. doi: 10.2174/1566524013666131121115656
39. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. (2003) 34:70–4. doi: 10.1038/ng1149
40. Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. (2005) 105:2449–57. doi: 10.1182/blood-2004-06-2289
41. Lagane B, Chow KYC, Balabanian K, Levoye A, Harriague J, Planchenault T, et al. CXCR4 dimerization and2-arrestin mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. (2008) 112:34–44. doi: 10.1182/blood-2007-07-102103
42. Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, et al. Regulation of human chemokine receptors CXCR4: role of phosphorylation in desensitization and internalization. J Biol Chem. (1997) 272:28726–31. doi: 10.1074/jbc.272.45.28726
43. Gómez-Moutón C, Fischer T, Peregil RM, Jiménez-Baranda S, Stossel TP, Nakamura F, et al. Filamin A interaction with the CXCR4 third intracellular loop regulates endocytosis and signaling of WT and WHIM-like receptors. Blood. (2015) 125:1116–25. doi: 10.1182/blood-2014-09-601807
44. Bhandari D, Robia SL, Marchese A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol Biol Cell. (2009) 20:1324–39. doi: 10.1091/mbc.E08-03-0308
45. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 1022–37.e14. doi: 10.1016/j.cell.2018.01.004
46. Ponzetta A, Benigni G, Antonangeli F, Sciumè G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. (2015) 75:4766–77. doi: 10.1158/0008-5472.CAN-15-1320
Keywords: NK cells, homing, mRNA transfection, CXCR4, immunotherapy, WHIM
Citation: Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, Carlsten M and Childs R (2019) Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4R334X. Front. Immunol. 10:1262. doi: 10.3389/fimmu.2019.01262
Received: 14 February 2019; Accepted: 17 May 2019;
Published: 05 June 2019.
Edited by:
Ignacio Melero, University of Navarra, SpainReviewed by:
Daniel Olive, Aix Marseille Université, FranceAlessandro Poggi, Ospedale San Martino (IRCCS), Italy
Julian Pardo, Fundacion Agencia Aragonesa para la Investigacion y el Desarrollo, Spain
Copyright © 2019 Levy, Reger, Segerberg, Lambert, Leijonhufvud, Baumer, Carlsten and Childs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattias Carlsten, bWF0dGlhcy5jYXJsc3RlbkBraS5zZQ==
†These authors have contributed equally to this work
‡These authors share last authorship