 Robert Pollmann1
Robert Pollmann1 Elias Walter2
Elias Walter2 Thomas Schmidt1Jens Waschke2
Thomas Schmidt1Jens Waschke2 Michael Hertl1
Michael Hertl1 Christian Möbs1
Christian Möbs1 Rüdiger Eming1*
Rüdiger Eming1*- 1Department of Dermatology and Allergology, Philipps-Universität Marburg, Marburg, Germany
- 2Institute of Anatomy and Cell Biology, Ludwig-Maximilians-Universität München, Munich, Germany
Pemphigus vulgaris (PV) is a rare blistering disease caused by IgG autoantibodies against the epidermal adhesion molecules desmoglein (Dsg)3 and Dsg1 providing a well-characterized paradigm of an antibody-mediated organ-specific autoimmune disease. In PV patients who have achieved clinical remission after B cell-depleting therapy, relapses often coincide with a reoccurrence of B cells and Dsg-specific autoantibodies. Here, we analyzed Dsg3-specific B cell subpopulations (i.e., total CD19+ B cells, CD19+CD27−B cells, CD19+CD27+ memory B cells, and CD19+CD27hiCD38hi plasmablasts) in peripheral blood of both PV patients (n = 14) at different stages of disease and healthy individuals (n = 14) by flow cytometry using fluorescently labeled recombinant human Dsg3 protein. Applying this approach, Dsg3-specific B cells could be detected at low frequencies (0.11–0.53% of CD19+ B cells) and numbers of Dsg3-specific memory B cells were significantly increased in PV patients in clinical remission receiving minimal immunosuppressive therapy. Finally, we confirmed in vitro that Dsg3-reactive memory B cells were able to produce anti-Dsg3 IgG autoantibodies upon ex vivo activation. Thus, monitoring of Dsg3-specific B cells in PV is of particular interest to further characterize the immunopathogenesis of PV.
Introduction
Pemphigus vulgaris (PV) is an autoimmune disease characterized by chronic and progressive erosive lesions of the mucosa, and blister formation at the suprabasilar layer of the skin caused by IgG autoantibodies (auto-ab) against adhesion molecules of the epidermis (1–3). The desmosomal cadherin desmoglein (Dsg)3 is the major autoantigen of PV with mucosal-dominant type while in patients with mucocutaneous PV, i.e., affecting mucosa and skin, auto-ab against Dsg1 are additionally found (4, 5). In the majority of PV patients, anti-Dsg IgG auto-ab correlate with disease activity (6, 7) and are therefore regularly used in routine diagnostics (8). The pathogenic relevance of auto-ab against Dsg3 has been shown in several in vitro and in vivo models by causing loss of keratinocyte cohesion (9–12), whereas a synergistic effect with other non-desmoglein autoantibodies is currently discussed (13, 14). Based on the well-described pathogenesis, the characterized autoantigens and the fact that Dsg-reactive IgG auto-ab are sufficient to cause blisters, PV is considered as a paradigm of an antibody-mediated organ-specific autoimmune disease. Moreover, PV serves as a model disease for the characterization of autoimmune mechanisms that finally lead to the generation of autoantigen-specific antibodies (15).
The B cell-depleting monoclonal anti-CD20 antibody rituximab leads to a marked decrease of Dsg3 auto-ab-titers paralleled by a fast clinical remission in the majority of PV patients (16–18), underlining the crucial role of continuous auto-ab production in PV by Dsg3-specific B cells, plasmablasts, and plasma cells.
Although the majority of patients achieve clinical remission after rituximab treatment, clinical relapses occur frequently in PV patients on long-term follow-up with reoccurrence of B cells and Dsg3 auto-ab in peripheral blood (19). This data suggests that Dsg3-specific B cells reappear at a certain time point during remission providing the base for a potential disease relapse. However, whether clinical relapses result from either Dsg3-specific B cells that have not been completely depleted by therapy or by de novo generated autoreactive B cells has not yet been fully elucidated. Genetic characterization of anti-Dsg3-IgG produced by B cells from PV patients indicates that patients with recurrent disease maintain a limited set of autoreactive Dsg3-specific B cell clones that persist over time (20). In contrast, using proteomic analysis of serum auto-ab, a recent study revealed a much more polyclonal and diverse pool of IgG auto-ab in PV (21).
To further examine the persistence of autoreactive peripheral blood B cells in pemphigus, we sought to characterize Dsg3-specific B cell subpopulations (i.e., mature naïve, memory, and plasmablasts) in PV patients at different stages of disease utilizing fluorescently labeled recombinant human Dsg3 (Dsg3-AF647) like it has been previously demonstrated for other antigens like tetanus toxin (22, 23). Our results show that (1) Dsg3-specific B cells can be detected at low frequencies in peripheral blood of pemphigus patients, (2) Dsg3-specific memory B cells were significantly increased especially in remitting patients receiving minimal therapy, and (3) isolated Dsg3-specific memory B cells from a PV patient secreted anti-Dsg3 IgG after in vitro stimulation. Thus, B cell monitoring with Dsg3-AF647 provides a novel and highly specific tool to investigate the persistence and distribution of autoreactive B cells in PV during the disease course.
Results
AF647-Labeled Dsg3 Detects Dsg3-Specific B Cell Clones
In this study we aimed at detecting Dsg3-specific B cells by flow cytometry using fluorescently labeled recombinant Dsg3-AF647 for staining of Dsg3-specific B cell receptors (BCR) as schematically shown in Figure 1A. The fluorescence labeling of recombinant Dsg3 did not functionally impair the interactions between Dsg3-AF647 and Dsg3 compared to homophilic binding of recombinant unlabeled human Dsg3 protein as determined by atomic force microscopy (AFM; Figure 1B). Furthermore, binding of Dsg3-AF647 to Dsg3 was reduced to the same extent compared to unlabeled Dsg3 after adding the monoclonal Dsg3-specific antibody AK23 (24) demonstrating the specificity of this interaction (Figure 1B). To test whether Dsg3-AF647 is capable of binding to Dsg3-specific B cells, the specificity of Dsg3-AF647 staining was evaluated by means of the monoclonal mouse B cell hybridoma (BCH) clone 2C10 producing anti-Dsg3 IgG together with the non-Dsg3-specific BCH clone 1F12 (Figure 1C). Production of anti-Dsg3 IgG by 2C10 was confirmed by ELISA with human Dsg3 protein (Figure 1D). Both BCH clones expressed surface IgG indicating the presence of a functional BCR (Figure 1C). However, only IgG+ cells from clone 2C10 showed a strong positive signal upon incubation with Dsg3-AF647 while no specific staining was observed with the control clone 1F12. Binding of Dsg3-AF647 to clone 2C10 was almost completely blocked by preincubation with unlabeled Dsg3 protein (Figure 1C). Of note, the mean fluorescence intensity of Dsg3-AF647 strongly correlated with surface IgG in clone 2C10 (Figure 1E) suggesting a BCR-dependent binding of Dsg3-AF647 to 2C10. In addition, fluorescently labeled human collagen VII (ColVII; noncollagenous domain 1, NC1 (1)) protein which is structurally not related to Dsg3 served as negative control and showed no specific staining in neither of the two BCH cell clones (Supplementary Figure 1B). Finally, we analyzed the sensitivity of Dsg3-AF647 staining by titrating the Dsg3-reactive clone 2C10 with bulk 1F12 cells followed by incubation with Dsg3-AF647 since in peripheral blood of PV patients Dsg3-specific B cells are expected at a low frequency. Our results revealed that Dsg3-specific B cells could be detected within a pool of non-Dsg3-reactive BCH at frequencies of even <1% (Figure 1F).
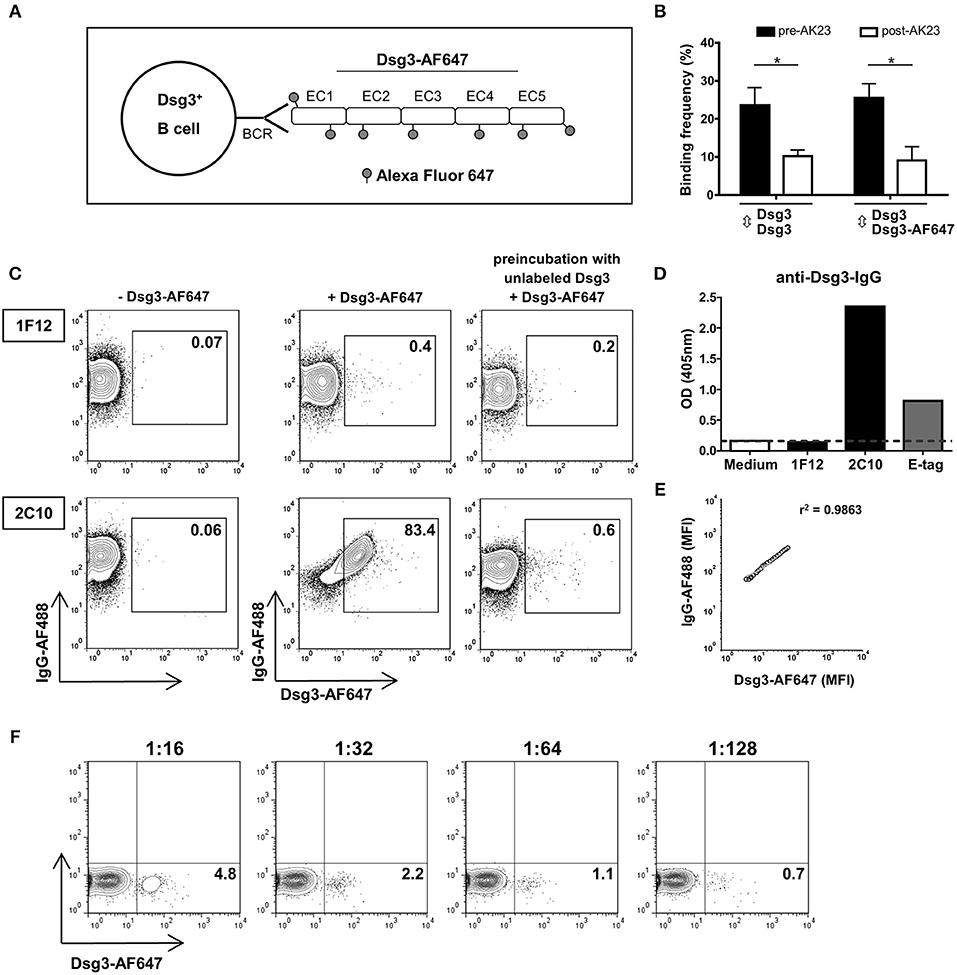
Figure 1. Detection of Dsg3-specific B cell hybridoma (BCH) using fluorescently labeled Dsg3. (A) Schematic drawing: the recombinant human extracellular domain (EC1-EC5) of Dsg3 was labeled with the fluorescent dye Alexa Fluor 647 (Dsg3-AF647) and was used for staining of Dsg3-specifc B cell receptors (BCR). (B) Binding of Dsg3-AF647 to Dsg3 ± addition of the monoclonal Dsg3-specific antibody AK23 was evaluated with atomic force microscopy. Cumulative data from 3 individual measurements with five replicates for each condition are presented as mean + SD. Statistical analysis was performed by multiple t-tests followed by Šidák correction. Differences between groups were considered statistically significant at p-values of <0.05 indicated as *. (C) Binding efficacy of Dsg3-AF647 to Dsg3-specific BCR was determined by staining of a Dsg3-specific BCH (2C10) and an unrelated BCH (1F12) together with anti-IgG antibody. Binding of Dsg3-AF647 was blocked by preincubation with unlabeled Dsg3. FACS plots shown are representative of three individual experiments. (D) Specificity of monoclonal BCH cells for Dsg3 was tested by ELISA. Anti-E-Tag served as positive and culture medium as negative control. (E) Correlation of mean fluorescence intensity (MFI) of Dsg3-AF647 with surface IgG. (F) Titration of Dsg3-specific BCH (2C10) cells in unrelated 1F12 cells in a calculated ratio of 1:16 (6.25%), 1:32 (3.13%), 1:64 (1.56%), and 1:128 (0.78%) representative of three individual experiments.
Dsg3-Specific B Cells Mainly Appear in the Memory B Cell Pool in PV Patients
Analysis of human Dsg3-specific B cell subpopulations (total CD19+ B cells, CD19+CD27− B cells, CD19+CD27+ memory B cells, and CD19+CD27hiCD38hi plasmablasts) was performed in peripheral blood of 14 clinically well-defined PV patients in either complete or partial clinical remission, or with relapsing disease (Supplementary Tables 1, 2). Individuals with no detectable anti-Dsg3 IgG auto-ab (Supplementary Figure 2) served as healthy control (HC) to determine any potential non-specific staining of Dsg3-AF647 in peripheral blood cells. Since a minor background staining with both Dsg3-AF647 and fluorescently labeled ColVII could also be observed in peripheral blood mononuclear cells of HC, only B cells with a high Dsg3-AF647 signal were considered to be Dsg3-specific (Supplementary Figure 3 for gating scheme). Specificity of Dsg3-AF647 staining in human peripheral blood was also indicated by a high Dsg3-AF647 signal that was only observed in CD19+ B cells but not in the CD19− population (Figure 2A).
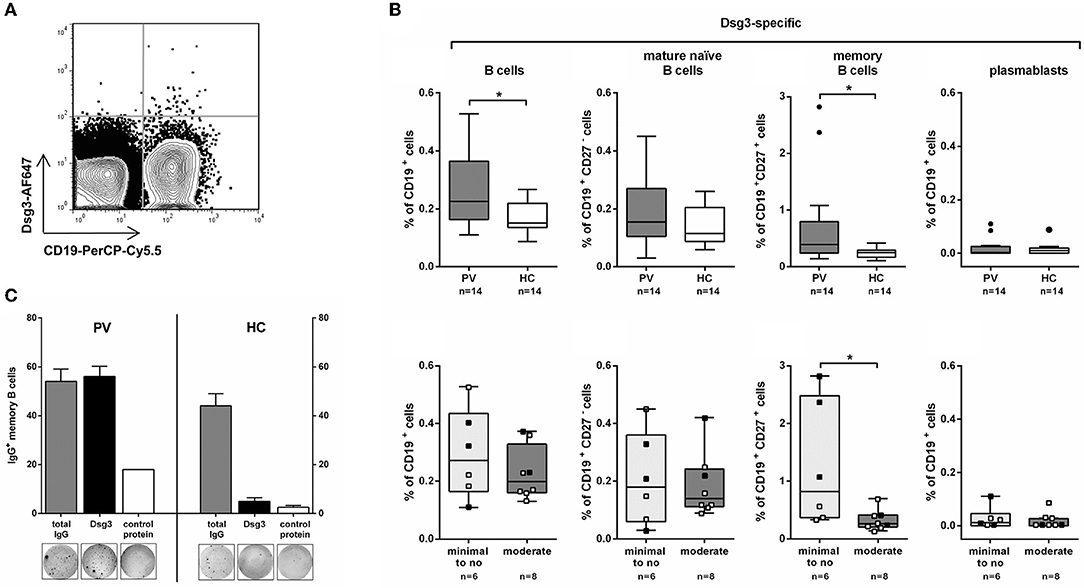
Figure 2. Dsg3-specific memory B cells are increased in remitting PV patients on minimal therapy. (A) Representative FACS plot showing staining for CD19+Dsg3-AF647+ B cells in one PV patient. (B) On the upper row: Dsg3-specific B cell populations (total CD19+, CD19+CD27− mature naïve, CD19+CD27+ memory, and CD19+CD27hiCD38hi plasmablasts) were analyzed in PV patients (n = 14) and healthy controls (HC; n = 14). On the lower row: PV patients were further subdivided based on their systemic treatment into minimal to no (n = 6) and moderate therapy (n = 8) showing highest numbers of Dsg3-specific CD19+CD27+ memory B cells on minimal therapy. ■: after Rituximab treatment; □: no Rituximab treatment. (C) CD19+CD27+Dsg3-AF647+ cells were isolated from peripheral blood of a PV patient and a healthy control by FACS sorting and stimulated with R848 and IL-2 to induce plasma cell differentiation. Dsg3-IgG-secreting and total IgG-secreting B cells were subsequently detected using ELISpot by seeding 250 cells per well on ELISpot plates coated either with recombinant human Dsg3 or human collagen VII as control protein. Statistical analysis was performed with two-tailed nonparametric Mann-Whitney-U-Test. Differences between groups were considered statistically significant at p-values of <0.05 indicated as *.
Dsg3-specific B cells were present in PV patients (0.11–0.53% of CD19+ B cells) at higher frequencies compared to HC (0.09–0.22% of CD19+ B cells; Figure 2B). The increase in Dsg3+ total CD19+ B cells was presumably due to the significantly higher number of Dsg3-specific CD19+CD27+ memory B cells in PV patients. On the other hand, the frequency of Dsg3-specific CD19+CD27− B cells (including mature naïve and CD27− memory B cells) did not differ between PV patients and HC (p = 0.3; see Supplementary Figure 3) for representative FACS plots). Dsg3-specific CD19+CD27hiCD38hi plasmablasts were only scarcely detected in peripheral blood of PV patients and HC, respectively. To determine whether Dsg3-specific memory B cells in patients with PV are capable of producing anti-Dsg3 IgG, we sorted CD19+CD27+Dsg3-AF647+ cells from a PV patient in clinical remission with persistent anti-Dsg3 IgG titers. We could demonstrate that in vitro stimulation of sorted CD19+CD27+Dsg3-AF647+ cells with toll-like receptor agonist R848 and interleukin-2 (IL-2) leads to plasma cell differentiation (according to Pinna et al. (25)) and that Dsg3-specific IgG-producing B cells were detected to a similar extent as IgG secreting cells in PV by ELISpot assay (Figure 2C). In contrast, although sorted Dsg3-AF647+ memory B cells from HC showed a marked IgG production, the majority of these cells did not possess the capacity to produce anti-Dsg3 IgG (Figure 2C).
Remitting PV Patients Show the Highest Number of Dsg3-Specific Memory B Cells
As some of the PV patients received systemic treatment (systemic corticosteroids, other adjuvant immunosuppressives, B cell depletion) they were subdivided into two groups with minimal to no therapy (including 6 remitting patients) or moderate therapy (including 8 patients with relapsing disease or partial remission; Supplementary Table 1). Interestingly, Dsg3-specific memory B cells were only increased in remitting PV patients receiving minimal therapy. On moderate treatment Dsg3-specific B cells were hardly detected similar to HC (Figure 2B). Here we could show that systemic immunosuppressive treatment seems to have an impact on the frequency of circulating autoreactive B cells in PV. Of note, in the group of remitting PV patients the highest numbers of Dsg3-specific memory B cells exceeding the levels observed in HC were particularly detected in patients who underwent previous B cell depletion (Patient 4, 8, 13 in Supplementary Table 1) suggesting a reconstitution of autoreactive memory B cells upon anti-CD20 antibody treatment (Supplementary Figure 4).
Discussion
In this study, we demonstrate by use of fluorescently labeled Dsg3 protein the detection of low-frequent Dsg3-specific autoreactive B cells in peripheral blood of PV patients. Flow cytometric analysis of B cell subpopulations with Dsg3-AF647 revealed significantly higher numbers of Dsg3-specific B cells in PV patients compared to HC, particularly within the memory B cell compartment.
Our present findings are in line with Nishifuji et al. (26) who detected circulating Dsg3-specific memory B cells in PV patients but not in HC using ELISpot analysis with a general low frequency (6.3–84.0 cells per 105 PBMC). This is similar to our study where we found low-frequent Dsg3-specific B cells in PV patients (0.11–0.53% of CD19+ B cells) even though the acquired percentages can only be compared to a limited extent due to the different assays used. In the work by Nishifuji et al., Dsg3-specific B cells spontaneously producing anti-Dsg3 IgG were exclusively observed in patients with the highest disease activity while Dsg3-specific memory B cells could only be detected after in vitro stimulation in a group of nine out of 14 patients (26). Of note, this group included patients with low or no disease activity pointing toward a persistence of Dsg3-specific memory B cells in patients in complete or partial remission which we could also observe in our study as Dsg3-specific CD19+CD27+ memory B cells were significantly increased in PV patients compared to HC. Since the generation of CD27+ memory B cells is thought to be induced in germinal centers and requires collaboration with T follicular helper (Tfh) cells (27), the presence of autoreactive memory B cells in PV patients points toward a defective Tfh cell tolerance checkpoint within the germinal center response in pemphigus as recently suggested by our group (28).
Most interestingly, the highest numbers of Dsg3-specific memory B cells were observed in 3 PV patients ~2 years after treatment with Rituximab (22–27 months; patient 4, 8, and 13; Supplementary Table 1), while increasing Dsg3-specific memory B cells could not be observed in a PV patient (patient 6) who experienced a relapse 35 months after rituximab (Supplementary Figure 4). Since disease relapses after rituximab treatment do often occur at a time point starting from 1 year after treatment (18, 19) this observation may point toward an ongoing reconstitution of autoreactive memory B cells. Those memory B cells would initially not produce autoantibodies however, upon immune activation with Dsg3 and other trigger factors, they possess the capability to rapidly differentiate into autoantibody-secreting plasma cells, hence providing the base for a potential disease relapse.
However, Dsg3-specific plasmablasts, representing antibody-secreting cells, were only scarcely detected in our study despite high anti-Dsg3 IgG titers in individual patients. This might be explained by their reduced expression of surface immunoglobulin which limits the capability for detecting these cells using fluorescently labeled Dsg3 (29). Furthermore, due to the restriction to peripheral blood for analysis of Dsg3-specific B cells we were not able to identify autoreactive plasma cells residing within the niches of lymphoid tissues or bone marrow that might account for the continuous secretion of autoreactive autoantibodies in PV. Nonetheless, clonal analysis of autoreactive B cells in PV showed that Dsg3-specific B cells in peripheral blood can persist in PV patients for many years during active disease, clinical remission or even after B cell-depleting therapy indicating the suitability of peripheral blood for monitoring autoreactive B cell responses (20). Whether these B cells are newly generated or persistent cells that were not completely removed by immunosuppressive therapy is still under investigation. Recent results showed that autoreactive B cells could persist in patients with PV but also in patients with lupus erythematosus (SLE) and to a much lower frequency in healthy individuals (30, 31).
In general, anti-Dsg3 IgG is found at a very low prevalence in healthy individuals (32), thus the low frequency of Dsg3-specific B cells in HC is suggestive of IgM+ B cells producing non-pathogenic, potentially cross-reactive natural IgM antibodies (33, 34). However, the observed signal for Dsg3-AF647 in HC may also be in part due to unspecific binding of the fluorescently labeled protein to B cells. Hence, inclusion of unspecific staining in samples from patients and controls was minimized to the best possible extent as only B cells with a very high Dsg3-AF647 signal were considered to be Dsg3-specific (Figure 2A).
To summarize, the present identification of peripheral blood Dsg3-reactive B cells in peripheral blood of PV patients provides further insights into the autoimmune B cell repertoire in PV. Monitoring of Dsg3-specific peripheral B cells with a special focus on Dsg3-specific CD19+CD27+ memory B cells might be a predictive tool to determine the effectiveness of therapeutic interventions in patients with PV. Particularly as recent studies suggest that clinical remission in PV upon therapy is associated with an increase of IL-10-secreting B cells downregulating B cell activation (18, 35), the extended flow cytometric analysis of Dsg3-specific IL-10-producing B cells in PV patients at different disease stages can provide further insights into the individual progression of disease.
Materials and Methods
Human Subjects
Peripheral blood from 14 PV patients (Supplementary Table 1) as well as 14 HC with no signs of autoimmune skin inflammation and no serum anti-Dsg3 IgG (Supplementary Figure 2) was drawn into citrate-phospate-dextrose-adenine (CPDA) anticoagulant. Each study participant gave written informed consent before inclusion in the study, which was approved by the Ethics Committee of the Medical Faculty of the Philipps-Universität, Marburg (Az. 20/14). The study was conducted in accordance with the Declaration of Helsinski.
Fluorescent Labeling of Recombinant Human Dsg3
Recombinant human Dsg3 (extracellular domain, aa 1-566), produced in the baculovirus expression system (36, 37), was fluorescently labeled using the AlexaFluor647 Labeling kit (Thermo Fisher Scientific, Schwerte, Germany) according to the manufacturers' protocol.
Detection of Dsg3-Specific B Cells
Peripheral blood mononuclear cells (PBMC) were isolated from peripheral blood using Lymphocyte Separation Medium (Capricorn, Ebsdorfergrund, Germany). Mouse BCH clones were cultured in RPMI-1640 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine (all Capricorn) and 10% FCS (Merck Millipore, Berlin, Germany). PBMC were washed twice with PBS + 1% FCS and 1 ×106 cells per sample were subsequently stained with Dsg3-AF647 together with mouse anti-human CD19-PerCP-Cy5.5 (HIB19), mouse anti-human CD27-PE (M-T271), mouse anti-human CD38-FITC (HIT2) and the respective isotype controls (all BD Biosciences, Heidelberg, Germany) or with goat anti-mouse IgG-AF488 (A-11029; Thermo Fisher Scientific, Waltham, MA, USA) for mouse BCH clones. After incubation for 20 min at 4°C cells were washed twice with PBS + 1% FCS and a minimum of 2.5 ×105 PBMC or 0.5 ×105 BCH per sample were acquired on a FACS Calibur (BD Biosciences). In a subset of experiments, sorting of cells was performed using FACS Aria III (BD Biosciences). Data analysis was performed using FlowJo 7.6 (TreeStar Inc., Ashland, USA).
ELISpot Assay
Dsg3-specific B cells (CD19+CD27+Dsg3-AF647+) were isolated from PBMC by FACS sorting. 2 ×103 cells were seeded in 96-well U-bottom microplates and cultured in RPMI-1640 +100 U/ml penicillin, +100 μg/ml streptomycin, +2 mM L-glutamine +10% FCS together with 2.5 μg/ml R848 (Mabtech AB, Nacka Strand, Sweden), and 1,000 U/ml human recombinant IL-2 (Roche, Mannheim, Germany) as previously described (25). After 5 days, cells were seeded at 250 cells/well in duplicates on an ELISpot plate coated with recombinant human Dsg3 or collagen VII and incubated for 20 h at 37°C in a humidified atmosphere containing 5% CO2. Detection of Dsg3-specific IgG+ spots was performed according to the manufacturer's protocol (Mabtech AB) and spots were counted automatically with an ELISpot plate reader (A.EL.VIS, Hanover, Germany).
Atomic Force Microscopy
Cell-free AFM measurements were performed on a NanoWizard 3 AFM (JPK-Instruments, Berlin, Germany) as previously described (38) (see Supplementary Material for detailed description).
Detection of Anti-Dsg IgG
The presence of IgG auto-ab against Dsg1 or Dsg3 in blood of PV patients and HC was evaluated by anti-Dsg1- and anti-Dsg3-ELISA (Euroimmun, Lübeck, Germany) according to the manufacturer's protocol.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 6.02 (GraphPad Software Inc., La Jolla, USA). Cumulative data are displayed as box plots with median. For group comparisons two-tailed nonparametric Mann-Whitney-U-Test was applied. Data from AFM experiments were statistically evaluated by multiple t-tests followed by Šidák correction. Differences between the groups were considered as statistically significant at p values <0.05.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
Each study participant gave written informed consent before inclusion in the study, which was approved by the Ethics Committee of the Medical Faculty of the Philipps-Universität, Marburg (Az. 20/14). The study was conducted in accordance with the Declaration of Helsinski.
Author Contributions
RP performed the experiments, analyzed the data, and wrote the manuscript. EW performed and analyzed the AFM experiments. TS, JW, MH, and CM participated to the study design, contributed to the manuscript, and supervised the study. RE conceived the study, recruited pemphigus patients, and revised the manuscript.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) FOR 2497 to TS, JW, MH, CM, and RE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01375/full#supplementary-material
Abbreviations
AF647, Alexa Fluor 647; AFM, atomic force microscopy; auto-ab, autoantibodies; BCH, B cell hybridoma; BCR, B cell receptor; COLVII, type VII collagen; Dsg, desmoglein; HC, healthy controls; PV, pemphigus vulgaris.
References
1. Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a comprehensive review on pathogenesis, clinical presentation and novel therapeutic approaches. Clin Rev Allergy Immunol. (2018) 54:1–25. doi: 10.1007/s12016-017-8662-z
2. Spindler V, Waschke J. Pemphigus-A disease of desmosome dysfunction caused by multiple mechanisms. Front Immunol. (2018) 9:136. doi: 10.3389/fimmu.2018.00136
4. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. (1991) 67:869–77. doi: 10.1016/0092-8674(91)90360-B
5. Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. (1999) 40:167–70. doi: 10.1016/S0190-9622(99)70183-0
6. Harman KE, Seed PT, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM. The severity of cutaneous and oral pemphigus is related to desmoglein 1 and 3 antibody levels. Br J Dermatol. (2001) 144:775–80. doi: 10.1046/j.1365-2133.2001.04132.x
7. Schmidt E, Dähnrich C, Rosemann A, Probst C, Komorowski L, Saschenbrecker S, et al. Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol. (2010) 19:458–63. doi: 10.1111/j.1600-0625.2010.01069.x
8. Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. (2018) 5:296. doi: 10.3389/fmed.2018.00296
9. Hofrichter M, Dworschak J, Emtenani S, Langenhan J, Weiß F, Komorowski L, et al. Immunoadsorption of desmoglein-3-specific IgG abolishes the blister-inducing capacity of pemphigus vulgaris IgG in neonatal mice. Front Immunol. (2018) 9:1935. doi: 10.3389/fimmu.2018.01935
10. Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen. (Dsg3) produced by baculovirus. J Clin Invest. (1994) 94:59–67. doi: 10.1172/JCI117349
11. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol. (2017) 8:603. doi: 10.3389/fimmu.2017.00603
12. Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen. (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. (1997) 137:1091–102. doi: 10.1083/jcb.137.5.1091
13. Amber KT, Valdebran M, Grando SA. Non-desmoglein antibodies in patients with pemphigus vulgaris. Front Immunol. (2018) 9:1190. doi: 10.3389/fimmu.2018.01190
14. Sinha AA, Sajda T. The evolving story of autoantibodies in pemphigus vulgaris: development of the “super compensation hypothesis”. Front Med. (2018) 5:218. doi: 10.3389/fmed.2018.00218
15. Hertl M, Veldman C. Pemphigus—paradigm of autoantibody-mediated autoimmunity. Skin Pharmacol Appl Skin Physiol. (2001) 14:408–18. doi: 10.1159/000056375
16. Joly P, Mouquet H, Roujeau J-C, D'Incan M, Gilbert D, Jacquot S, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. (2007) 357:545–52. doi: 10.1056/NEJMoa067752
17. Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. (2008) 128:2850–8. doi: 10.1038/jid.2008.172
18. Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. (2013) 5:175ra30. doi: 10.1126/scitranslmed.3005166
19. Ahmed AR, Shetty S. A comprehensive analysis of treatment outcomes in patients with pemphigus vulgaris treated with rituximab. Autoimmun Rev. (2015) 14:323–31. doi: 10.1016/j.autrev.2014.12.002
20. Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, et al. Persistence of anti-desmoglein 3 IgG(+) B-cell clones in pemphigus patients over years. J Invest Dermatol. (2015) 135:742–9. doi: 10.1038/jid.2014.291
21. Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang H-Y, et al. Proteomic analysis of pemphigus autoantibodies indicates a larger, more diverse, and more dynamic repertoire than determined by B cell genetics. Cell Rep. (2017) 18:237–47. doi: 10.1016/j.celrep.2016.12.013
22. Frölich D, Giesecke C, Mei HE, Reiter K, Daridon C, Lipsky PE, et al. Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J Immunol. (2010) 185:3103–10. doi: 10.4049/jimmunol.1000911
23. Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. (2005) 105:1614–21. doi: 10.1182/blood-2004-07-2507
24. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. (2003) 170:2170–8. doi: 10.4049/jimmunol.170.4.2170
25. Pinna D, Corti D, Jarrossay D, Sallusto F, Lanzavecchia A. Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur J Immunol. (2009) 39:1260–70. doi: 10.1002/eji.200839129
26. Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. (2000) 114:88–94. doi: 10.1046/j.1523-1747.2000.00840.x
27. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
28. Hennerici T, Pollmann R, Schmidt T, Seipelt M, Tackenberg B, Möbs C, et al. Increased frequency of T follicular helper cells and elevated interleukin-27 plasma levels in patients with pemphigus. PLoS ONE. (2016) 11:e0148919. doi: 10.1371/journal.pone.0148919
29. Perez-Andres M, Paiva B, Nieto WG, Caraux A, Schmitz A, Almeida J, et al. Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry B Clin Cytom. (2010) 78(suppl. 1):S47–60. doi: 10.1002/cyto.b.20547
30. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci USA. (2008) 105:9727–32. doi: 10.1073/pnas.0803644105
31. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. (2007) 26:205–13. doi: 10.1016/j.immuni.2007.01.009
32. Prüßmann W, Prüßmann J, Koga H, Recke A, Iwata H, Juhl D, et al. Prevalence of pemphigus and pemphigoid autoantibodies in the general population. Orphanet J Rare Dis. (2015) 10:63. doi: 10.1186/s13023-015-0278-x
33. Lobo PI. Role of natural autoantibodies and natural IgM anti-leucocyte autoantibodies in health and disease. Front Immunol. (2016) 7:198. doi: 10.3389/fimmu.2016.00198
34. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. (2003) 301:1374–7. doi: 10.1126/science.1086907
35. Kabuto M, Fujimoto N, Tanaka T. Increase of interleukin-10-producing B cells associated with long-term remission after i.v. immunoglobulin treatment for pemphigus. J Dermatol. (2016) 43:815–8. doi: 10.1111/1346-8138.13295
36. Müller R, Svoboda V, Wenzel E, Gebert S, Hunzelmann N, Müller H-H, et al. IgG reactivity against non-conformational NH-terminal epitopes of the desmoglein 3 ectodomain relates to clinical activity and phenotype of pemphigus vulgaris. Exp Dermatol. (2006) 15:606–14. doi: 10.1111/j.1600-0625.2006.00451.x
37. Müller R, Dahler C, Möbs C, Wenzel E, Eming R, Messer G, et al. T and B cells target identical regions of the non-collagenous domain 1 of type VII collagen in epidermolysis bullosa acquisita. Clin Immunol. (2010) 135:99–107. doi: 10.1016/j.clim.2009.12.010
Keywords: autoimmunity, pemphigus vulgaris, desmoglein 3, B cells, flow cytometry
Citation: Pollmann R, Walter E, Schmidt T, Waschke J, Hertl M, Möbs C and Eming R (2019) Identification of Autoreactive B Cell Subpopulations in Peripheral Blood of Autoimmune Patients With Pemphigus Vulgaris. Front. Immunol. 10:1375. doi: 10.3389/fimmu.2019.01375
Received: 10 March 2019; Accepted: 30 May 2019;
Published: 14 June 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Maryam Daneshpazhooh, Tehran University of Medical Sciences, IranRudolf Armin Manz, Universität zu Lübeck, Germany
Donna Culton, University of North Carolina at Chapel Hill, United States
Copyright © 2019 Pollmann, Walter, Schmidt, Waschke, Hertl, Möbs and Eming. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rüdiger Eming, ZW1pbmdAbWVkLnVuaS1tYXJidXJnLmRl