 Anthony Jaworowski
Anthony Jaworowski Anna C. Hearps
Anna C. Hearps Thomas A. Angelovich
Thomas A. Angelovich Jennifer F. Hoy
Jennifer F. Hoy- 1School of Health and Biomedical Sciences, RMIT University, Bundoora, VIC, Australia
- 2Department of Infectious Diseases, Alfred Hospital and Monash University, Melbourne, VIC, Australia
- 3Life Sciences Discipline, Burnet Institute, Melbourne, VIC, Australia
Combination antiretroviral therapy (ART) is effective at suppressing HIV viremia to achieve persistently undetectable levels in peripheral blood in the majority of individuals with access and ability to maintain adherence to treatment. However, evidence suggests that ART is less effective at eliminating HIV-associated inflammation and innate immune activation. To the extent that residual inflammation and immune activation persist, virologically suppressed people living with HIV (PLWH) may have increased risk of inflammatory co-morbidities, and adjunctive therapies may need to be considered to reduce HIV-related inflammation and fully restore the health of virologically suppressed HIV+ individuals. Cardiovascular disease (CVD) is the single leading cause of death in the developed world and is becoming more important in PLWH with access to ART. Arterial disease due to atherosclerosis, leading to acute myocardial infarction (AMI) and stroke, is a major component of CVD. Atherosclerosis is an inflammatory disease, and epidemiological comparisons of atherosclerosis and AMI show a higher prevalence and suggest a greater risk in PLWH compared to the general population. The reasons for greater prevalence of CVD in PLWH can be broadly grouped into four categories: (a) the higher prevalence of traditional risk factors e.g., smoking and hypertension (b) dyslipidemia (also a traditional risk factor) caused by off-target effects of ART drugs (c) HIV-related inflammation and immune activation and (d) other undefined HIV-related factors. Management strategies aimed at reducing the impact of traditional risk factors in PLWH are similar to those for the general population and their effectiveness is currently being evaluated. Together with improvements in ART regimens and guidelines for treatment, and a greater awareness of its impact on CVD, the HIV-related risk of AMI and stroke is decreasing but remains elevated compared to the general community. Monocytes are key effector cells which initiate the formation of atherosclerotic plaques by migrating into the intima of coronary arteries and accumulating as foam cells full of lipid droplets. This review considers the specific role of monocytes as effector cells in atherosclerosis which progresses to AMI and stroke, and explores mechanisms by which HIV may promote an atherogenic phenotype and function independent of traditional risk factors. Altered monocyte function may represent a distinct HIV-related factor which increases risk of CVD in PLWH.
Introduction
For PLWH at the present time, HIV infection is associated with different outcomes depending on access to care and treatment. In resource-constrained settings, those without access to antiretroviral therapy (ART), or with limited access to expensive, alternative ART that is required when first-line therapies fail, experience progressive disease with decreased CD4+ T cell counts ultimately leading to death from AIDS–related diseases. For PLWH who have access to effective antiretroviral regimens, current evidence suggests that life expectancy approaches that of HIV-negative individuals in the general population. Whilst the risk of AIDS and AIDS-related mortality may be low, long term health outcomes for these individuals depend on how soon after infection they are treated, with the best outcomes predicted for those who present early in infection with relatively preserved CD4 counts (1–5). Large cohort studies of persons from the United States, and from North America and Western Europe, have shown that the gap in life expectancy between PLWH and HIV-negative individuals has narrowed as a function of the year in which ART was initiated (2, 6). This is likely due to use of more potent and tolerable ART regimens and adoption of treatment guidelines recommending earlier commencement of ART at diagnosis of HIV.
Cardiovascular Disease in PLWH
Despite a reduction in AIDS-related mortality, HIV infection remains associated with an increased risk of age-related, inflammatory diseases which cause significant morbidity such as non-AIDS cancers, neurocognitive impairment and cardiovascular disease (CVD). Cardiovascular diseases, or diseases of the heart and vasculature are major causes of morbidity and mortality. They include ischaemic coronary artery disease (chronic angina and AMI), cerebrovascular disease (stroke and transient ischaemic attack), other heart diseases (including arrhythmias, inflammatory heart disease, valvular disease, rheumatic heart disease, and heart failure) and other cardiovascular diseases (peripheral vascular disease, aortic aneurysms, and hypertensive disease). The incidence of many of these conditions is increased in PLWH [for recent reviews see (7, 8)]. The incidence of CVD may be decreased following ART and the impact of ART will depend on the degree to which inflammation and immune activation are decreased following HIV virologic suppression. This review focuses on coronary artery disease (CAD) in well-resourced settings since it is the major cause of mortality in the general population and is an increasing cause of mortality in PLWH as the median age of this population is increasing. Specifically, we look at mechanisms that increase the risk of sub-clinical atherosclerosis which is elevated in these populations and leads to the major causes of death from CVD, i.e., AMI and ischaemic stroke, and focus on the role of activated monocytes as a likely effector contributing to these mechanisms.
Elevated Risk of CAD in virologically suppressed PLWH
In virologically suppressed individuals on stable and effective ART, CAD has become one of the leading causes of death (9). Early hospital record-based studies that compared the rate of AMI in large cohorts of HIV-positive and HIV-negative individuals presenting at the same US hospitals between 1996 and 2004, showed that the relative risk of AMI was 1.75-fold higher after adjusting for age, gender (sic), race, diabetes, hypertension, and dyslipidemia (10). Similarly, a study of US veterans enrolled since 2003 and followed-up until 2009, confirmed that the incidence of AMI was higher [HR = 1.48 after adjustment for Framingham risk score (FRS), co-morbidities and substance abuse] in HIV-positive veterans matched demographically to HIV-negative veterans (11). In both of these studies, participants were not restricted to those virologically suppressed by ART, and information on the proportion of successful virological suppression were not presented. Similar results were obtained in analyses of incidence of AMI in the French Hospital Database on HIV (FHDH-ANRS CO4) cohort (12) where standardized mortality ratios were 1.4 [95% confidence: 1.3–1.6] and 2.7 [1.8–3.9] compared to the general population for men and women, respectively. More recent studies have reported that the incidence is also higher when restricted to type 1 events, i.e., those associated with atherosclerotic plaque rupture or thrombosis, which are relevant as inflammatory co-morbidities in PLWH who are virologically suppressed on ART (13).
The higher rate of AMI reported in the above studies contrasts with a gradual decline of incidence in individuals enrolled via the Kaiser-Permanente health plan (California, USA) as a function of when HIV patients started ART. In those individuals who commenced ART between 2010 and 2011, the rate of AMI decreased to the point where the incidence was not statistically different from that in HIV-negative controls (14) and also no difference was observed in rates of AMI in PLWH who commenced ART at CD4 cell counts of ≥500/μL (15). Other studies report a reduction in AMI incidence over time as well; Masia et al reported a decrease in standardized incidence rates in PLWH relative to the general population in Spain when the intervals between 2006–2009 and 2010–2014 were compared (16). The observed reduction in AMI risk may be, in part, due to prescription of ART regimens with lower associations with CVD, adoption of guidelines for earlier initiation of ART, and increased awareness of traditional risk factors for CVD in PLWH. The more frequent interactions of PLWH within healthcare settings, and the consequent impact on CVD risk monitoring and treatment, is difficult to correct for in cohort analyses, and may be an important confounder in comparing CVD risk in HIV-positive and HIV-negative populations. Also, we have limited long term data on CVD risk in virologically-suppressed PLWH and the HIV-related CVD risk in virologically suppressed PLWH on ART for long periods of time remains to be established.
Why is CVD Risk Elevated in Virologically Suppressed HIV-Positive Individuals?
There are several potential and recognized factors that may elevate risk of CVD in PLWH.
Traditional Risk Factors
A higher prevalence of traditional risk factors for CVD such as smoking (17–19), diabetes (20, 21), dyslipidemia (including hypercholesterolemia, triglyceridemia, low HDL cholesterol and abnormal fat distribution) and hypertension (22, 23) is found in HIV-positive populations in well-resourced settings. As many of these factors are modifiable, reduction in CVD risk attributable to traditional risk factors is as effective in PLWH as strategies and treatments for the general population (24). Recently, Althoff and colleagues have used data from the North American AIDS Cohort Collaboration on Research and Design to estimate the contribution of population-attributable risk factors for AMI, in addition to other non-AIDS comorbidities, and concluded that considerable reduction in risk could be obtained by reducing total cholesterol levels and hypertension in PLWH (25).
Antiretroviral Drugs
The risk of AMI associated with ART use was robustly reported in the Data collection on adverse events of ARV Drugs (D.A.D) study (26) in which duration of ART was associated with an adjusted relative risk of AMI of 1.26 [1.12–1.41] per year. Specific drugs, or classes of drug, have also been associated with CV events; recent, but not cumulative use, of abacavir (27) and continued use of protease inhibitors (26, 28) including some recently-introduced combinations (29). The study by Ryom and colleagues using data from the D:A:D cohort, showed that cumulative use of the boosted protease inhibitor combination darunavir/ritonavir was associated with a 60% increase in baseline risk of CVD (using a composite endpoint that included AMI and stroke, and also sudden cardiac deaths and invasive cardiovascular procedures) over 5 years of use. In contrast, use of atazanavir/ritonavir was not associated with an increase in risk. These findings were not explained when analyses were controlled for effects of dyslipidemia or a protective effect of hyperbilirubinemia. Deleterious effects of antiretroviral drugs contribute to traditional CVD risk factors including increased circulating levels of cholesterol and triglycerides (26), visceral adiposity (30), reduced high-density lipoprotein-associated cholesterol (HDLc) levels (31), hypertension (32, 33) and metabolic syndrome (34–36) shown by studies linking the prevalence of these syndromes with years of ART (33). The hypothesized mechanism for the effect of current use of abacavir on AMI is via increased platelet reactivity, which reverses when abacavir is ceased (37, 38). Antiretroviral drugs probably mediate much of their effect on hypertension via their effects on other components of the metabolic syndrome such as diabetes, changes in HDL and low-density lipoprotein (LDL) levels and alterations in fat distribution which are themselves associated with hypertension; for a more complete discussion see (23, 33). Risks associated with ART can be ameliorated by avoidance of abacavir and specific protease inhibitors. Another factor is the effect that weight gain following ART initiation with various PI-based, integrase inhibitor-based and earlier non-thymidine analog-based regimens may have on CAD risk (39, 40). For further information, the reader is referred to a recent comprehensive review (41).
HIV-Related Chronic Inflammation
Atherosclerosis is an inflammatory disease (42). A recent meta-analysis has confirmed an association of plasma markers of inflammation with CVD in PLWH (43) particularly interleukin 6 (IL-6), D-dimer and high-sensitivity C-reactive protein (hs-CRP), which are the most extensively studied biomarkers. However, in individual studies measuring various outcomes related to CVD, biomarkers of myeloid activation such as sCD14 and sCD163 have been more closely associated [(44–46) and discussed further below].
Monocytes/macrophages possess a full complement of pattern recognition receptors that promote inflammation by stimulating production of high levels of pro-inflammatory factors. Measuring their activation status, most conveniently via soluble plasma markers or cell surface markers on monocytes, is therefore a valuable approach to assessing inflammation and innate immune activation in individuals. Using cross sectional studies we reported that virologically suppressed PLWH have higher levels of circulating plasma biomarkers of inflammation and of myeloid activation compared to age-matched HIV-negative individuals, but similar to levels found in much older individuals (47, 48) which is consistent with findings from other laboratories (45, 49). We further estimated that the increase in immune activation was equivalent to an additional 2–4 years of aging in virologically suppressed individuals (50) and reasoned that this increased the risk of age-related inflammatory co-morbidities by a commensurate rate. We measured phenotypic markers on blood monocytes and reported their continued alteration in virologically suppressed individuals (47), however plasma biomarkers of monocyte/macrophage activation such as CXCL-10 correlate with monocyte subset and phenotypic alterations (51) and are a more convenient and robust measure in clinical studies.
The significance with respect to CAD of the extent to which immune activation and inflammation persist in PLWH on effective ART is evident from studies showing that measures of myeloid activation and inflammation are associated with non-calcified plaque (52, 53), coronary artery calcium (54, 55), carotid intima-media thickness (46) and predict cardiovascular events and death (56–59). Expression of tissue factor on monocytes, important for initiating platelet activation, is increased by TLR ligands such as LPS elevated in settings of HIV infection (60), although not at the concentrations found in the plasma of PLWH (61), and by thrombin (61). Schechter and colleagues further showed that tissue factor expression was restricted to CCR2+ classical monocytes (a discussion of human monocyte subsets is given below) although others have implicated CD16+ monocytes (62). While a discussion of late events in atherosclerosis and mechanisms of plaque rupture are outside the scope of this review, it is important to note that in a primate model of HIV, administration of an inhibitor of tissue factor activity on monocytes, Ixolaris, decreased immune activation (61). The association of monocyte activation markers and soluble myeloid activation biomarkers with incidence of CAD and with cardiovascular events is strong circumstantial evidence for a mechanistic role of activated monocytes in these processes. However, the mechanisms by which monocytes may contribute to CAD are best identified by functional comparisons of these cells present in PLWH and in well-matched HIV-negative individuals.
HIV-Specific Mechanisms
HIV produces pathogenic factors such as the Negative Regulatory Factor, Nef, that are potential mediators of HIV-related morbidities. HIV-infected foam cell macrophages have been detected in coronary arteries obtained post-mortem from HIV-positive individuals, and Nef has been shown to stimulate their formation by inhibiting reverse cholesterol efflux from macrophages (63), suggesting a direct mechanistic link. The significance of these observations is discussed in more detail below.
HIV-Associated Atherosclerosis
AMI follows thickening of the arterial wall by atherosclerotic plaque formation and eventual occlusion of coronary arteries. Ischaemic strokes result when unstable atherosclerotic plaques rupture and lodge in the brain, while peripheral artery disease shares similar initial events to CAD of plaque formation and narrowing of arteries at other sites. Both are significant co-morbidities elevated in PLWH. Atherosclerotic plaques identified in virologically suppressed PLWH are more likely to be non-calcified plaques considered to be unstable and high risk (64, 65).
As atherosclerosis underlies coronary artery diseases it is important to understand whether it is more prevalent in virologically suppressed HIV-positive individuals and how HIV impacts its development. Early studies reported increased pre-clinical atherosclerosis assessed by surrogate measures such as carotid artery intima media thickness (cIMT) in PLWH compared to HIV-negative controls (66, 67). More recently, Leon et al. reported that in a well-controlled virologically suppressed HIV-positive population with a low cardiovascular risk, as measured using the FRS, of <10%, 21% had sub-clinical atherosclerosis determined by carotid artery ultrasonography (68). In this group of patients, who had median FRS of 1% and all with an FRS <8%, traditional risk factors did not account for the high prevalence of sub-clinical atherosclerosis, but the authors found high IL-6 levels (>6.6 pg/mL) resulted in an odds ratio of 9 for sub-clinical atherosclerosis. These findings suggest an involvement of inflammation in heightened CVD risk in these subjects rather than the traditional risk factors accounted for by FRS (age, smoking, dyslipidemia, total and HDL cholesterol, systolic blood pressure).
An important factor underlying increased immune activation in PLWH is that most are cytomegalovirus (CMV) seropositive, and have an impaired ability to control CMV reactivation. CMV infection in PLWH has been associated with expansion of a unique population of adaptive natural killer cells which serve as a sensitive marker for the presence of CMV infection (69, 70). Using small cross-sectional studies of 93 PLWH and 37 healthy, HIV-negative controls, it has been shown that sub-clinical atherosclerosis (as measured by cIMT) is correlated with CD8 T cell responses to CMV antigen (pp65) (66). CMV infection in PLWH is also associated with expansion of CD8+ T cells expressing fractalkine receptor (CX3CR1; involved in endothelial homing and adhesion) and the PAR-1 receptor which can be activated by thrombin, although the impact of these cells on clinical atherosclerosis is unknown (71). Other studies have found correlations between CMV antibody (IgG) levels and carotid artery stiffness in HIV-positive women, although association of IgG with the prevalence of cardiovascular lesions was restricted to those who achieved virologic suppression (72). More recently, in a small cross-sectional study of 105 PLWH matched to 105 healthy, age- sex- and smoking-matched controls, a correlation was observed between CMV IgG levels and cIMT (73).
There are few well-controlled studies comparing the relative prevalence of atherosclerosis in well-managed virologically suppressed PLWH to appropriate HIV-negative controls. One such study found that the prevalence of coronary plaque as measured by Coronary Computed Tomography Angiography (CCTA) was higher in 78 HIV-positive men (of whom 95% were currently receiving cART and 81% were virologically suppressed) compared to 32 HIV-negative men recruited from the same clinics in Boston (59 vs. 34%, p = 0.02) (74). A similar larger study from the Multicentre AIDS Cohort Study confirmed these findings with a 1.25-fold higher prevalence of plaque after adjustment for cardiovascular risk factors (75). In contrast, studies from the Swiss HIV cohort have reported little or no increase in prevalence of high risk, non-calcified plaque and lower coronary atherosclerosis involvement than HIV-negative persons with a similar FRS (76). A smaller study demonstrated that HIV-positive women are more likely to have non-calcified plaque compared to HIV-negative women, after controlling for known cardiovascular risk factors, although prevalence of calcified plaque was not increased (52). The presence of non-calcified plaque is associated with higher levels of the macrophage activation marker sCD163 and with CVD in HIV-positive individuals (52, 53). A meta-analysis of 9 studies involving 1,229 HIV-positive individuals and 1,029 HIV-negative controls, concluded there was an increased prevalence of non-calcified plaque in PLWH as detected by CT (OR = 3.26 [1.30–8.18]) (77). Larger studies are required to measure the relative prevalence of non-calcified plaque in PLWH who commence ART with well-preserved CD4 counts.
Accumulation of Activated Macrophages in the Ascending Aortic Arch is Associated with CVD in HIV-Positive Individuals
[18F] FDG-PET is a technique which uses positron emission tomography to image uptake of [18F] 2-deoxyglucose into cells and tissues. There is evidence [for a summary, refer to papers listed in (44)], and it is assumed, that the main cell type that accumulates [18F] 2-deoxyglucose is the activated monocyte-derived macrophage, and that therefore the intensity of radioactivity detected by PET reflects accumulation and activation of monocytes to the activated endothelium. More specific macrophage tracers are being developed which leverage the fact that macrophages and dendritic cells express CD206, the mannose fucose-receptor: these include 99mTc-diethylenetriaminepentaacetic acid—mannosyldextran (Tilmanocept or LymphoSeek™) (78, 79) and radiolabeled anti-MFR monoclonal antibodies (80) but they have yet to be used extensively in studies of CVD risk in HIV.
In a proof-of-concept study, a greater uptake of [18F] FDG occurred in both the carotid arteries and the aorta of 9 PLWH compared to 5 HIV-negative participants (81). Similarly, in 27 virologically suppressed HIV-positive individuals without evidence of cardiovascular disease and age-, sex-, and CVD risk-matched to 27-HIV-negative individuals, the HIV-positive participants had greater accumulation of 18F radiolabel in the ascending aortic arch at levels comparable to those measured in 27 HIV-negative individuals with cardiovascular disease (44). These results provide evidence that HIV infection, even with successful virologic suppression, is associated with similar levels of arterial inflammation as found in individuals with pre-existing CVD. Follow-up studies from the same group showed that the arterial inflammation in a similar group of 41 HIV-positive patients was associated with higher numbers of high-risk coronary plaque (82).
The accumulation of [18F] FDG is evidence for activated macrophages accumulating in the arteries of the heart, and its association with plaque with low attenuation and positive remodeling emphasizes the clinical significance of this accumulation, but it does not provide a mechanism by which this occurs. In HIV-positive participants, higher accumulation of FDG was moderately associated with elevated plasma levels of sCD163, but not D-dimer or hs-CRP (44). This is consistent with a specific link with activated macrophages but not with generalized inflammation or thrombotic disease. Unfortunately, plasma markers of endothelial activation were not investigated. Taken together these studies suggest that atherosclerosis is elevated in PLWH receiving effective ART due to a greater accumulation of monocyte-derived macrophages in coronary arteries leading to formation of non-calcified, high risk plaques. Significantly, it has been reported that whereas treatment of PLWH with atorvastatin improved lipid profiles and reduced the prevalence of non-calcified plaque in these individuals after 12 months, there was no reduction in the accumulation of FDG measured by [18F] FDG-PET (83). This suggests that accumulation of activated monocyte-derived macrophages in the heart is caused by factors other than dyslipidemia and that it may represent an independent risk factor for CVD in PLWH. This may be specifically associated with myeloid cell activation. It is therefore important to understand how effective therapies used to reduce hypercholesterolemia are at reducing inflammation and monocyte activation that may contribute to CVD in PLWH. Initial reports from the SATURN-HIV trial showed that whereas treatment with the statin rosuvastatin was effective at reducing LDLc levels in HIV patients after 24 weeks, it was not effective in reducing inflammatory markers studied including well-established markers such as IL-6, hsCRP, and soluble TNF receptor isoforms (84). In contrast, lipoprotein-associated phospholipase A2 [an enzyme produced by myeloid cells and an independent predictor of CVD (85)] was significantly decreased in the statin arm compared to the placebo arm of the trial. Follow up studies of the same cohort indicated greater reduction in immune activation as assessed by various biomarkers: after 48 weeks, treatment with rosuvastatin reduced levels of the inflammatory marker IP-10 (interferon-γ-inducible protein-10 i.e., CXCL10) and the myeloid activation marker sCD14, as well as the proportion of non-classical monocytes expressing tissue factor (86). However, even after 48 weeks treatment with rosuvastatin, inflammatory markers IL-6, hsCRP, D-dimer and soluble TNF receptors were not reduced in this study (ibid). These data underscore the importance of monocyte activation in the mechanism leading to increased atherosclerosis and emphasizes the necessity of examining the functional properties of monocytes in these individuals to understand how their altered behavior might favor atherosclerotic plaque formation.
Initiation of Atherosclerotic Plaque Formation
Atherosclerotic plaques tend to form in arteries at sites of perturbed blood flow such as near bifurcations in arteries. The initiation of atherosclerotic plaque formation is accelerated by the presence of an activated endothelium at these sites. In individuals with traditional risk factors such as high levels of triglycerides, total cholesterol, or LDLc in plasma, lipid accumulates in the intimal layer beneath the endothelium where it is oxidized by reactive oxygen intermediates produced by activated endothelial cells and macrophages. The action of oxidative pathways derived from endothelial cells and macrophages produce different oxidized species in LDL particles (87, 88) but both lipid species have been found in atherosclerotic plaque-derived lipids, suggesting that both endothelial cells and macrophages participate in this pathway (89). Oxidized LDL (oxLDL) is a known risk factor for CVD which has been shown in one study of HIV-positive individuals (91% of whom received ART) (90) and in a second study (of patients with varying degrees of HIV viremia) (91) to be elevated in plasma, and is associated with subclinical atherosclerosis in this population (92, 93).
The Role of Monocyte Subsets in Atherosclerotic Plaque Formation
Monocytes initiate the process of atherosclerotic plaque formation following their transmigration from blood into the intima of arteries at sites of activated endothelium and cholesterol deposition (“fatty streaks”) (Figure 1). Here they mature into foam cells which are macrophages containing large numbers of lipid droplets (94). They also have important inflammatory roles in atherosclerotic plaque progression and rupture. The present review focuses on their role in atherosclerotic plaque initiation.
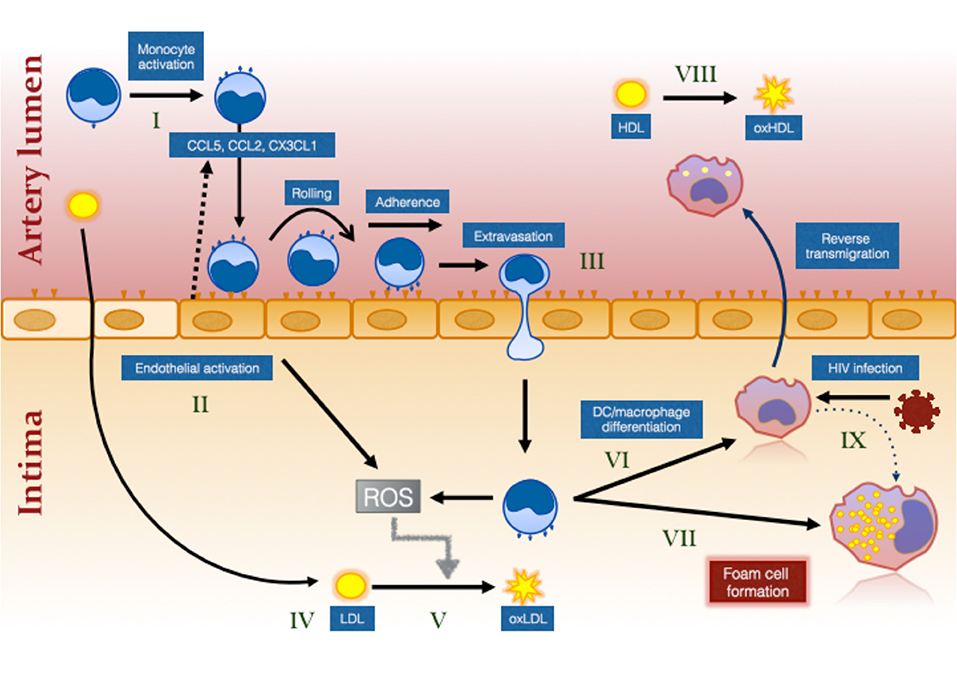
Figure 1. The effect of HIV infection on monocyte- and inflammation-mediated mechanisms of foam cell formation. HIV infection causes systemic monocyte activation (I) due to factors unresolved by ART including residual HIV viremia, CMV reactivation, elevated bacterial ligands and oxidative stress. Gut bacterial ligands (eg., lipopolysaccharide; LPS) activate classical and intermediate monocytes via CD14/TLR4 receptor while viral ligands activate non-classical monocytes via TLR7/8 (144). Unresolved inflammation also activates the endothelium (II) which secrete chemokines to attract different monocyte subsets (not illustrated) via specific chemokine receptors, CCR2, CCR5, and CX3CR1 (99). Intermediate monocytes in particular exhibit increased pro-atherogenic properties (127). Activation of the endothelium and of monocytes results in greater monocyte adherence, rolling, firm adhesion and extravasation, the last via either paracellular, or transcellular mechanisms (III) [reviewed in (145)]. Cholesterol accumulates in the intima due in part to ART-induced dyslipidemia and to increased traditional risk factors in PLWH (IV). This is oxidized by the activated myeloid and endothelial cells which produce reactive oxygen intermediates (ROS) (V). Monocytes that have migrated into the intima ingest LDL and oxLDL via LDL receptor and CD36/SR-A1/II (131), respectively and either mature into monocyte–derived dendritic cells (VI) and reverse migrate out of the intima or mature into immobile foam cells (VII). Changes in monocytes, including decreased ABCA1 expression (126), favors pathway (VII) over pathway (VI). Oxidation of HDL (VIII) that also occurs in virologically suppressed PLWH impairs the protective function of this lipoprotein and further increases the risk of atherosclerotic plaque formation. Direct infection of macrophages in the intima also induces an atherogenic phenotype promoting foam cell formation (IX).
Monocytes are currently classified into subsets based on the expression of CD14 which is the co-receptor for Toll-like receptor 4 (a pattern recognition receptor that recognizes lipopolysaccharide derived from gram-negative bacterial cell walls) and CD16 (the intermediate affinity IgG receptor, FcγRIII). The majority of monocytes in circulation are CD14hiCD16- classical monocytes which represent ~90% of the total in healthy individuals, while non-classical or patrolling monocytes (CD14dimCD16+) and intermediate monocytes (CD14hiCD16+) are minor subsets which each represent ~5% of circulating monocytes. It is likely that monocytes in circulation represent a continuum of classical monocytes newly emerging from the bone marrow and gradually maturing into intermediate then non-classical monocytes (95) or re-emerging from tissue sites such as the spleen (96, 97).
Monocyte subsets have different functional properties and must therefore be considered separately with respect to their roles in inflammation and atherosclerosis. Recruitment of monocytes to arteries at sites of inflammation is directed by chemokines released by activated endothelial cells, but the multiplicity of chemokines and their receptors makes it difficult to determine the role of individual chemokines. Mouse models have been particularly valuable in this context: using the ApoE knockout/western-type diet model of atherosclerosis, early experiments revealed the importance of the chemokine receptor CCR2 in atherogenesis (98) and subsequently that the murine equivalent of the classical monocyte subset (Ly6Chi CCR2+ monocytes) migrates in response to the chemokine CCL2 (99, 100). It was reported by Tacke et al that classical monocytes also migrate into atherosclerotic plaques in response to the fractalkine receptor, CX3CR1 which is unexpected since they do not express high levels of this receptor, and furthermore that non-classical monocytes (Ly6CloCX3CR1+) mainly migrate into atherosclerotic lesions in response to CCR5 but not CX3CR1 even though the latter is highly expressed in this subset (99). The recruited monocytes were shown in these studies to differentiate into lesional macrophages and therefore to potentially participate in disease progression. Neither study examined the migration of intermediate monocytes which are not a well-defined subset in mice. Support for the role of CCR5 in monocyte recruitment into atherosclerotic plaques has been obtained using the ApoE knockout mouse model of atherosclerosis, where it was shown that monocyte recruitment into plaques, and plaque progression, was reduced with treatment using the CCR5 antagonist Maraviroc, which is of relevance to PLWH who may be treated with ART regimens containing this drug (101). Following AMI, monocytes are also recruited into atherosclerotic plaques with distinct kinetics to co-ordinate left ventricular repair. In this setting, classical monocytes are recruited initially in response to CCR2 ligands, where they phagocytose necrotic debris and orchestrate pro-inflammatory responses (102). Non-classical monocytes are recruited later and participate in tissue repair, angiogenesis and extracellular matrix deposition, although in this study their migration was reported to be dependent on fractalkine receptor (ibid).
There are no extensive data on how HIV infection, especially in settings of virologic suppression with ART, affect chemokine receptor expression on individual monocytes to modulate monocyte recruitment to atherosclerotic lesions. It is reported that HIV-infection alters proportions of CCR2- and CX3CR1-expressing monocytes in circulation, but this may be due to alterations in the distribution of monocyte subsets. In a cohort of virologically suppressed HIV-positive individuals with low cardiovascular risk, we have previously demonstrated an association between the proportion of CX3CR1+ CD16+ monocytes and cIMT, supporting a potential link between monocyte recruitment and atherosclerosis (103). We, and others, have reported that intermediate and non-classical CD16+ monocytes, are expanded in viremic PLWH (104–108) but no difference in subset proportions is seen in PLWH who are virologically suppressed (109). Recently, using a well-controlled longitudinal cohort of PLWH commencing ART, we showed that intermediate and non-classical monocyte proportions decreased rapidly on commencement of ART, reaching control levels within ~6 months of therapy (108). Taken together, these data suggest that when assessing the roles of individual subsets in CVD risk and progression, the impact of HIV infection and of ART on monocyte subsets must be considered.
Recruitment of monocytes to sites of atherosclerotic plaque formation is favored by increased expression of adhesion receptors such as ICAM-1 on activated endothelial cells (110). Systemic inflammation may also activate circulating monocytes to increase expression of adhesion receptors such as CD11b/CD18 and CD11d/CD18 (111) which recognize their cognate ligands such as ICAM-1 and VCAM-1 expressed on activated endothelium (112). Following attachment, monocytes transmigrate across the endothelium and enter the intima where they ingest deposited lipids via scavenger receptors including CD36 and scavenger receptor A1 and AII, the major receptors for oxLDL expressed on macrophages. Monocytes can reverse transmigrate and transfer lipid to acceptor molecules such as HDL, a process thought to maintain the health of the artery and regress atherosclerotic plaque development (113, 114) or they can differentiate into foam cells, which are large, immobile cells (115) characterized by lipid accumulation in lipid droplets and lysosomes, and are among the earliest pathogenic feature of atherosclerotic plaque formation. Lipids derived from oxLDL accumulate in lysosomes and in this intracellular compartment, unlike lipids found in lipid droplets, do not readily participate in cholesterol efflux pathways (116). Lysosomal foam cell lipids persist as a stable store, as data from the White Carneau Pigeon model of atherosclerosis indicates, after a change in diet (117). Thus, the differentiation/maturation pathway of the monocyte, which is dictated in part by the type of lipid ingested, may determine the initiation of atherosclerotic plaque development.
While models of atherosclerotic plaque development rightly emphasize the role of lipid accumulation in the artery wall as the initial stimulus activating the endothelium, attracting monocytes and acting as a substrate for foam cell formation, it is also clear that inflammatory mechanisms intersect this mechanism by independently activating monocytes and endothelial cells and oxidizing lipids by promoting oxidative stress pathways (118) (See Figure 1). However, as discussed above, PLWH may have elevated risk of atherosclerosis independent of traditional risk factors such as elevated total cholesterol and LDLc, and this increased risk may in part be accounted for by their increased levels of inflammation and monocyte activation. It is important to realize that foam cell formation by monocyte-derived macrophages may occur in response to toll-like receptor ligands including HIV ssDNA (119), rather than lipoprotein particles [reviewed in (118)] which is of relevance to HIV infection where there is evidence for elevated levels of circulating endotoxin as well as other TLR ligands derived from microbial translocation even in virologically suppressed individuals (47, 120, 121). Finally, as discussed further below, foam cells may be induced following alterations in macrophage cholesterol metabolism and efflux and metabolic changes in circulating monocytes may predispose them to differentiate into foam cells following transendothelial migration.
How Does HIV Infection Influence Monocytes to Promote CVD?
The association of plasma biomarkers of myeloid activation with CVD in HIV-positive individuals, especially where other markers of inflammation are not strongly associated (44, 53), suggest that activated monocytes or macrophages play a direct role in promoting atherosclerosis. However, while plasma levels of molecules released from activated myeloid cells such as sCD163 may be useful prognostic biomarkers, they do not provide information about the mechanistic link between CVD and particular myeloid cell types and their functions. For this, functional studies of cells obtained from PLWH are required.
Macrophages containing the major HIV antigen (p24 capsid protein) have been detected in coronary arteries present in autopsy material obtained from patients receiving ART (63). These cells were adjacent to the lipid core of the plaque and had morphological characteristics of foam cells. Using an in vitro infection model, Bukrinsky and colleagues showed that HIV infection of monocyte-derived macrophages impaired reverse cholesterol transport and down modulated the cholesterol transporter ATP Binding Cassette family member A type 1 (ABCA1) via a mechanism requiring the HIV accessory protein, Nef (122). This molecule is the major transporter through which cholesterol is effluxed from macrophages to acceptor molecules like apolipoprotein A1 in HDL particles. It was further shown that injecting Nef protein into mice to achieve circulating levels reported in viremic HIV+ individuals, exacerbated dyslipidemia and development of plaque in the high fat diet ApoE−/− mouse model of atherosclerosis. This work suggests a plausible mechanism for promoting atherosclerosis in PLWH, but its significance to their CVD risk depends on the prevalence of HIV-infected macrophages in coronary arteries and on the concentration of Nef circulating in virologically suppressed individuals, for which there is currently a paucity of information. It would be of interest to use modern techniques such as DNA- and RNAscope to evaluate HIV infection in resected coronary arteries and to accurately determine circulating levels of Nef protein in a current cohort of PLWH.
Given the persistence of monocyte activation in ART-treated PLWH and its association with CVD, we reasoned that monocytes present in PLWH may have pro-atherogenic functional properties induced via bystander mechanisms dependent on HIV infection, even if these cells are not infected with the virus. To address this, we used a static model of atherosclerotic plaque development in which a type 1 collagen matrix is overlaid with a monolayer of primary human endothelial cells (HUVEC) and monocytes freshly isolated from study subjects are added (123, 124). In this model, monocytes transmigrate across the HUVEC monolayer to enter the collagen matrix, and the fate of the monocytes is followed over the subsequent 48 h. This model has been used extensively to determine the reverse transmigration properties of monocytes and the endothelial cell-expressed molecules such as PECAM-1 governing extravasation (113, 125), but had not previously been used to compare monocyte behavior from clinical cohorts. Using this model, we have shown that monocytes isolated from virologically suppressed PLWH have a higher propensity to mature into foam cells than monocytes from age-matched HIV-negative individuals, and that plasma from these individuals contains factors that promote this functional phenotype (126). In this study, monocytes from PLWH also exhibited impaired reverse transmigration out of the collagen and across the HUVEC monolayer, which is possibly linked to their greater propensity to mature into immobile foam cells. These functional defects were accompanied with impaired cholesterol efflux and reduced ABCA1 expression at the mRNA level. We are currently examining monocytes isolated from a larger group of virologically suppressed PLWH with low/medium FRS, to determine whether we can detect the same monocyte atherogenic phenotype and, if so, whether interventions to reduce CV risk will improve their functional properties (Angelovich, Hearps, Trevillyan, Hoy, and Jaworowski unpublished).
Experiments we have conducted using this in vitro model have shown that the intermediate monocyte subset has the greatest propensity of the three monocyte subsets to differentiate into foam cells (127) and expresses the highest levels of intracellular TNF and IL-6, both in the steady state and in response to LPS (128). Thus, to the extent that the proportion of this monocyte subset is elevated in PLWH, this may exacerbate the pro-atherogenic properties of monocytes in these individuals.
It is interesting that monocytes isolated from virologically suppressed PLWH possess similar functional defects in cholesterol transport as the HIV-infected monocyte-derived macrophages described by Bukrinsky and colleagues, although there is no evidence to date that Nef is the circulating factor causing this functional change in the subjects we studied. Monocytes from relatively old but healthy HIV-negative individuals have similar properties (127) strengthening the view that there are similarities in monocyte inflammatory behavior induced by HIV and by healthy aging, and suggesting that mechanisms apart from Nef contribute to this phenotype.
The model we use to study monocyte atherogenicity in vitro involves activation of the endothelial monolayer with TNF, simulating an inflammatory milieu in vivo. TNF can decrease expression of ABCA1 (129) and via this mechanism promote foam cell formation, but in the above experiments it was removed from the culture medium before monocytes were added. Furthermore, ABCA1 expression was decreased in freshly isolated monocytes from HIV-positive subjects. The factors that reduce ABCA1 expression in monocytes from PLWH in vivo remain to be established. As a static model, this system does not provide reliable information about the adhesion and transmigration properties of monocytes ex vivo, only their behavior post migration. Measurements conducted under shear flow conditions will need to be made to address whether monocytes from virologically suppressed PLWH have abnormal endothelial adhesion and transmigration properties.
Abnormal Lipoproteins in PLWH may Influence Monocyte BEHAVIOR
It is important to consider that the pro-atherogenic phenotype of monocytes from virologically suppressed HIV-positive individuals revealed using the atherosclerotic plaque model is evident in the absence of added exogenous lipid or lipoprotein particles. However, we have used the same model to show that transmigrated monocytes can be induced to differentiate into foam cells by the addition of known atherogenic lipids such as oxLDL (124) and by LPS (127) into the culture medium. This may, in part, explain the pro-atherogenic properties of plasma from these individuals. LDL is oxidized (89) and taken up by macrophages via scavenger receptor A and CD36 (130–132) and since their expression is not regulated in a cholesterol-dependent manner, this promotes foam cell formation.
HDL is normally a protective lipoprotein with respect to CVD since its major protein component, apolipoprotein A1, functions as a cholesterol acceptor from cells. However, HDL in plasma of PLWH exhibits an increased level of oxidation, and the levels of oxidized HDL (oxHDL) correlate with markers of systemic inflammation such as IL-6 and hsCRP (133). We have isolated HDL particles from plasma obtained from a limited number of virologically suppressed PLWH, incorporated the isolated particles into the atherosclerotic plaque model and showed that they promote foam cell formation by monocytes from healthy, HIV-negative individuals. This behavior was associated with defective redox properties of the HDL particles (134). Functional defects in HDL particles isolated from PLWH have been reported by others, specifically a decreased level of the enzymes Paraoxonase (PON) 1 and 3 and a decreased level of PON redox activity (135). PON enzymes have important anti-inflammatory properties: in the context of atherosclerosis they inhibit the oxidation of LDL particles (136–138) and reduce monocyte-attracting chemokine expression by the endothelium (139, 140). PON also inhibits cholesterol biosynthesis (141) and stimulates cholesterol efflux from macrophages (89), properties that reduce the propensity of macrophages to develop into foam cells. While human macrophages do not express PON1 or PON 3 these effects can be mediated by PON delivered via HDL particles (ibid).
The behavior of monocytes isolated from virologically suppressed PLWH and of monocytes from healthy subjects in response to HDL particles from PLWH are consistent with the observation of lower PON1 and 3 levels in these particles. As PON can be displaced from HDL particles by inflammatory proteins such as serum amyloid A in circumstances such as acute infection (142, 143) it will be critical to understand which factors promote loss of PON1 and 3 in chronic HIV infection.
Conclusions
While the relative risk of death from AMI has decreased significantly for PLWH and has approached that for comparable members of the general community, CVD will become an increasing cause of morbidity and mortality as the HIV-positive population ages. It is still unclear to what extent the risk of atherosclerosis is elevated in patients who commence ART at high CD4 counts, especially as individuals may be treated with ART for many decades, which is longer than follow up studies conducted to date. Monocytes are principal effectors that initiate atherosclerotic plaque formation at sites of endothelial activation and damage. The “decision” of monocytes to differentiate into foam cells rather than extravasate from the intima influences the chance that initiation and progression of atherosclerotic plaques occurs. Functional studies using freshly isolated monocytes from virologically suppressed HIV-positive individuals are consistent with a phenotype that promotes atherosclerosis and therefore suggest that therapies designed to target this phenotype will prove beneficial. Future studies should assess the impact of therapies that reduce inflammation including statins and agents that target pathogen-induced monocyte/macrophage activation to reduce the tendency of monocytes to migrate across activated endothelia and differentiate into foam cells. A better understanding of the transcriptional changes in monocytes in PLWH, especially those involved in cholesterol/lipid metabolism and accumulation, will inform pathways that need to be targeted to prevent this. Emerging technologies such as improvements to [18F] FDG-PET might be useful in assessing how successful interventions are at reducing the accumulation of activated macrophages in the heart. PLWH should be monitored for CV risk, encouraged to reduce modifiable risk factors and treated appropriately. The use of novel biomarkers such as oxLDL and oxHDL to improve risk prediction in the setting of HIV infection should also be considered.
Author Contributions
AJ conceived and wrote the manuscript. AH, JH, and TA contributed to the manuscript. All authors reviewed and approved the final manuscript.
Funding
This work was supported by NHMRC Project Grant 1108792 to AJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Grinsztejn B, Hosseinipour MC, Ribaudo HJ, Swindells S, Eron J, Chen YQ, et al. Effects of early versus delayed initiation of antiretroviral treatment on clinical outcomes of HIV-1 infection: results from the phase 3 HPTN 052 randomised controlled trial. Lancet Infect Dis. (2014) 14:281–90. doi: 10.1016/S1473-3099(13)70692-3
2. Marcus JL, Chao CR, Leyden WA, Xu L, Quesenberry CP Jr, Klein DB, et al. Narrowing the gap in life expectancy between HIV-infected and HIV-uninfected individuals with access to care. J Acquir Immune Defic Syndr. (2016) 73:39–46. doi: 10.1097/QAI.0000000000001014
3. Pinto AN, Grey P, Shaik A, Cooper DA, Kelleher AD, Petoumenos K. Early treatment of primary HIV infection is associated with decreased mortality. AIDS Res Hum Retroviruses. (2018) 34:936–941. doi: 10.1089/aid.2017.0284
4. Olson AD, Walker AS, Suthar AB, Sabin C, Bucher HC, Jarrin I, et al. Limiting cumulative HIV viremia copy-years by early treatment reduces risk of AIDS and death. J Acquir Immune Defic Syndr. (2016) 73:100–8. doi: 10.1097/QAI.0000000000001029
5. McManus H, O'Connor CC, Boyd M, Broom J, Russell D, Watson K, et al. Long-term survival in HIV positive patients with up to 15 Years of antiretroviral therapy. PLoS ONE. (2012) 7:e48839. doi: 10.1371/journal.pone.0048839
6. Antiretroviral Therapy Cohort, Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: a collaborative analysis of cohort studies. Lancet HIV. (2017) 4:e349–e356. doi: 10.1016/S2352-3018(17)30066-8
7. Barnes RP, Lacson JC, Bahrami H. HIV infection and risk of cardiovascular diseases beyond coronary artery disease. Curr Atheroscler Rep. (2017) 19:20. doi: 10.1007/s11883-017-0652-3
8. Pinto SM, da Silva M. Cardiovascular disease in the setting of human immunodeficiency virus infection. Curr Cardiol Rev. (2018) 14:25–41. doi: 10.2174/1573403X13666171129170046
9. Morlat P, Roussillon C, Henard S, Salmon D, Bonnet F, Cacoub P, et al. Causes of death among HIV-infected patients in France in 2010 (national survey): trends since 2000. AIDS. (2014) 28:1181–91. doi: 10.1097/QAD.0000000000000222
10. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. (2007) 92:2506–12. doi: 10.1210/jc.2006-2190
11. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. (2013) 173:614–22. doi: 10.1001/jamainternmed.2013.3728
12. Lang S, Mary-Krause M, Cotte L, Gilquin J, Partisani M, Simon A, et al. Increased risk of myocardial infarction in HIV-infected patients in France, relative to the general population. AIDS. (2010) 24:1228–30. doi: 10.1097/QAD.0b013e328339192f
13. Drozd DR, Kitahata MM, Althoff KN, Zhang J, Gange SJ, Napravnik S, et al. Increased risk of myocardial infarction in HIV-infected individuals in north america compared with the general population. J Acquir Immune Defic Syndr. (2017) 75:568–576. doi: 10.1097/QAI.0000000000001450
14. Klein DB, Leyden WA, Xu L, Chao CR, Horberg MA, Towner WJ, et al. Declining relative risk for myocardial infarction among HIV-positive compared with HIV-negative individuals with access to care. Clin Infect Dis. (2015) 60:1278–80. doi: 10.1093/cid/civ014
15. Silverberg MJ, Leyden WA, Xu L, Horberg MA, Chao CR, Towner WJ, et al. Immunodeficiency and risk of myocardial infarction among HIV-positive individuals with access to care. J Acquir Immune Defic Syndr. (2014) 65:160–6. doi: 10.1097/QAI.0000000000000009
16. Masia M, Padilla S, Garcia JA, Bernardino JI, Campins AA, Asensi V, et al. Decreasing rates of acute myocardial infarction in people living with HIV: a nationwide cohort study in Spain, 2004-2015. HIV Med 19:491–6. (2018). doi: 10.1111/hiv.12616
17. Mdodo R, Frazier EL, Dube SR, Mattson CL, Sutton MY, Brooks JT, et al. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: cross-sectional surveys. Ann Intern Med. (2015) 162:335–44. doi: 10.7326/M14-0954
18. Reynolds NR. Cigarette smoking and HIV: more evidence for action. AIDS Educ Prev. (2009) 21:106–21. doi: 10.1521/aeap.2009.21.3_supp.106
19. Rahmanian S, Wewers ME, Koletar S, Reynolds N, Ferketich A, Diaz P. Cigarette smoking in the HIV-infected population. Proc Am Thorac Soc. (2011) 8:313–9. doi: 10.1513/pats.201009-058WR
20. Brown TT, Li X, Cole SR, Kingsley LA, Palella FJ, Riddler SA, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS. (2005) 19:1375–83. doi: 10.1097/01.aids.0000181011.62385.91
21. Tien PC, Schneider MF, Cox C, Karim R, Cohen M, Sharma A, et al. Association of HIV infection with incident diabetes mellitus: impact of using hemoglobin A1C as a criterion for diabetes. J Acquir Immune Defic Syndr. (2012) 61:334–40. doi: 10.1097/QAI.0b013e31826bfc32
22. Schouten J, Wit FW, Stolte IG, Kootstra NA, van der Valk M, Geerlings SE, et al. Cross-sectional comparison of the prevalence of age-associated comorbidities and their risk factors between HIV-infected and uninfected individuals: the AGEhIV cohort study. Clin Infect Dis. (2014) 59:1787–97. doi: 10.1093/cid/ciu701
23. van Zoest RA, Wit FW, Kooij KW, van der Valk M, Schouten J, Kootstra NA, et al. Higher Prevalence of hypertension in HIV-1-infected patients on combination antiretroviral therapy is associated with changes in body composition and prior stavudine exposure. Clin Infect Dis. (2016) 63:205–13. doi: 10.1093/cid/ciw285
24. Smit MA, Michelow IC, Glavis-Bloom J, Wolfman V, Levine AC. Characteristics and outcomes of pediatric patients with ebola virus disease admitted to treatment units in liberia and sierra leone: a retrospective cohort study. Clin Infect Dis. (2017) 64:243–249. doi: 10.1093/cid/ciw725
25. Althoff KN, Gebo KA, Moore RD, Boyd CM, Justice AC, Wong C, et al. Contributions of traditional and HIV-related risk factors on non-AIDS-defining cancer, myocardial infarction, and end-stage liver and renal diseases in adults with HIV in the USA and Canada: a collaboration of cohort studies. Lancet HIV. (2019) 6:e93–e104. doi: 10.1016/S2352-3018(18)30295-9
26. Friis-Moller N, Sabin CA, Weber R, d'Arminio Monforte A, El-Sadr WM, Reiss P, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. (2003) 349:1993–2003. doi: 10.1056/NEJMoa030218
27. Dorjee K, Choden T, Baxi SM, Steinmaus C, Reingold AL. Risk of cardiovascular disease associated with exposure to abacavir among individuals with HIV: a systematic review and meta-analyses of results from 17 epidemiologic studies. Int J Antimicrob Agents. (2018) 52:541–53. doi: 10.1016/j.ijantimicag.2018.07.010
28. Mary-Krause M, Cotte L, Simon A, Partisani M, Costagliola D, Clinical Epidemiology Group from the French Hospital. Increased risk of myocardial infarction with duration of protease inhibitor therapy in HIV-infected men. AIDS. (2003) 17:2479–86. doi: 10.1097/00002030-200311210-00010
29. Ryom L, Lundgren JD, El-Sadr W, Reiss P, Kirk O, Law M, et al. Cardiovascular disease and use of contemporary protease inhibitors: the D:A:D international prospective multicohort study. Lancet HIV. (2018) 5:e291–e300. doi: 10.1016/S2352-3018(18)30043-2
30. Lake JE, Wohl D, Scherzer R, Grunfeld C, Tien PC, Sidney S, et al. Regional fat deposition and cardiovascular risk in HIV infection: the FRAM study. AIDS Care. (2011) 23:929–38. doi: 10.1080/09540121.2010.543885
31. Cotter AG, Satchell CS, O'Halloran JA, Feeney ER, Sabin CA, Mallon PW. High-density lipoprotein levels and 10-year cardiovascular risk in HIV-1-infected patients. AIDS. (2011) 25:867–9. doi: 10.1097/QAD.0b013e32834507f0
32. Fahme SA, Bloomfield GS, Peck R. Hypertension in HIV-infected adults: novel pathophysiologic mechanisms. Hypertension. (2018) 72:44–55. doi: 10.1161/HYPERTENSIONAHA.118.10893
33. Hatleberg CI, Ryom L, d'Arminio Monforte A, Fontas E, Reiss P, Kirk O, et al. Association between exposure to antiretroviral drugs and the incidence of hypertension in HIV-positive persons: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. HIV Med. (2018) 19:605–618. doi: 10.1111/hiv.12639
34. Wand H, Calmy A, Carey DL, Samaras K, Carr A, Law MG, et al. Metabolic syndrome, cardiovascular disease and type 2 diabetes mellitus after initiation of antiretroviral therapy in HIV infection. AIDS. (2007) 21:2445–53. doi: 10.1097/QAD.0b013e3282efad32
35. Calza L, Colangeli V, Magistrelli E, Rossi N, Rosselli Del Turco E, Bussini L, et al. Prevalence of metabolic syndrome in HIV-infected patients naive to antiretroviral therapy or receiving a first-line treatment. HIV Clin Trials. (2017) 18:110–117. doi: 10.1080/15284336.2017.1311502
36. Palacios R, Santos J, Gonzalez M, Ruiz J, Marquez M. Incidence and prevalence of the metabolic syndrome in a cohort of naive HIV-infected patients: prospective analysis at 48 weeks of highly active antiretroviral therapy. Int J STD AIDS. (2007) 18:184–7. doi: 10.1258/095646207780132415
37. Trevillyan JM, Arthur JF, Jing J, Andrews RK, Gardiner EE, Hoy JF. Effects of abacavir administration on structural and functional markers of platelet activation. AIDS. (2015) 29:2309–13. doi: 10.1097/QAD.0000000000000848
38. O'Halloran JA, Dunne E, Tinago W, Denieffe S, Kenny D, Mallon WG. Switching from abacavir to tenofovir disoproxil fumarate is associated with rises in soluble glycoprotein VI, suggesting changes in platelet-collagen interactions. AIDS. (2018) 32:861–866. doi: 10.1097/QAD.0000000000001783
39. McComsey GA, Kitch D, Sax PE, Tebas P, Tierney C, Jahed NC, et al. Peripheral and central fat changes in subjects randomized to abacavir-lamivudine or tenofovir-emtricitabine with atazanavir-ritonavir or efavirenz: ACTG study A5224s. Clin Infect Dis. (2011) 53:185–96. doi: 10.1093/cid/cir324
40. McComsey GA, Moser C, Currier J, Ribaudo HJ, Paczuski P, Dube MP, et al. Body composition changes after initiation of raltegravir or protease inhibitors: ACTG A5260s. Clin Infect Dis. (2016) 62:853–62. doi: 10.1093/cid/ciw017
41. Kumar S, Samaras K. The impact of weight gain during HIV treatment on risk of pre-diabetes, diabetes mellitus, cardiovascular disease, and mortality. Front Endocrinol. (2018) 9:705. doi: 10.3389/fendo.2018.00705
42. Ross R, Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
43. Vos AG, Idris NS, Barth RE, Klipstein-Grobusch K, Grobbee DE. Pro-inflammatory markers in relation to cardiovascular disease in HIV infection. a systematic review. PLoS ONE. (2016) 11:e0147484. doi: 10.1371/journal.pone.0147484
44. Subramanian S, Tawakol A, Burdo TH, Abbara S, Wei J, Vijayakumar J, et al. Arterial inflammation in patients with HIV. JAMA. (2012) 308:379–86. doi: 10.1001/jama.2012.6698
45. Burdo TH, Lentz MR, Autissier P, Krishnan A, Halpern E, Letendre S, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. (2011) 204:154–63. doi: 10.1093/infdis/jir214
46. Dirajlal-Fargo S, Sattar A, Kulkarni M, Funderburg N, McComsey GA. Soluble TWEAK may predict carotid atherosclerosis in treated HIV infection. HIV Clin Trials. (2017) 18:156–163. doi: 10.1080/15284336.2017.1366001
47. Hearps AC, Maisa A, Cheng WJ, Angelovich TA, Lichtfuss GF, Palmer CS, et al. HIV infection induces age-related changes to monocytes and innate immune activation in young men that persist despite combination antiretroviral therapy. AIDS. (2012) 26:843–53. doi: 10.1097/QAD.0b013e328351f756
48. Martin GE, Gouillou M, Hearps AC, Angelovich TA, Cheng AC, Lynch F, et al. Age-associated changes in monocyte and innate immune activation markers occur more rapidly in HIV infected women. PLoS ONE. (2013) 8:e55279. doi: 10.1371/journal.pone.0055279
49. Mendez-Lagares G, Romero-Sanchez MC, Ruiz-Mateos E, Genebat M, Ferrando-Martinez S, Munoz-Fernandez MA, et al. Long-term suppressive combined antiretroviral treatment does not normalize the serum level of soluble CD14. J Infect Dis. (2013) 207:1221–5. doi: 10.1093/infdis/jit025
50. Angelovich TA, Hearps AC, Maisa A, Martin GE, Lichtfuss GF, Cheng WJ, et al. Viremic and virologically suppressed HIV infection increases age-related changes to monocyte activation equivalent to 12 and 4 years of aging, respectively. J Acquir Immune Defic Syndr. (2015) 69:11–7. doi: 10.1097/QAI.0000000000000559
51. Wilson EM, Singh A, Hullsiek KH, Gibson D, Henry WK, Lichtenstein K, et al. Monocyte-activation phenotypes are associated with biomarkers of inflammation and coagulation in chronic HIV infection. J Infect Dis. (2014) 210:1396–406. doi: 10.1093/infdis/jiu275
52. Fitch KV, Srinivasa S, Abbara S, Burdo TH, Williams KC, Eneh P, et al. Noncalcified coronary atherosclerotic plaque and immune activation in HIV-infected women. J Infect Dis. (2013) 208:1737–46. doi: 10.1093/infdis/jit508
53. Burdo TH, Lo J, Abbara S, Wei J, DeLelys ME, Preffer F, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J Infect Dis. (2011) 204:1227–36. doi: 10.1093/infdis/jir520
54. Baker JV, Hullsiek KH, Singh A, Wilson E, Henry K, Lichtenstein K, et al. Immunologic predictors of coronary artery calcium progression in a contemporary HIV cohort. AIDS. (2014) 28:831–40. doi: 10.1097/QAD.0000000000000145
55. Longenecker CT, Jiang Y, Orringer CE, Gilkeson RC, Debanne S, Funderburg NT, et al. Soluble CD14 is independently associated with coronary calcification and extent of subclinical vascular disease in treated HIV infection. AIDS. (2014) 28:969–77. doi: 10.1097/QAD.0000000000000158
56. Grund B, Baker JV, Deeks SG, Wolfson J, Wentworth D, Cozzi-Lepri A, et al. Relevance of interleukin-6 and D-dimer for serious non-AIDS morbidity and death among HIV-positive adults on suppressive antiretroviral therapy. PLoS ONE. (2016) 11:e0155100. doi: 10.1371/journal.pone.0155100
57. Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS ONE. (2012) 7:e44454. doi: 10.1371/journal.pone.0044454
58. De Luca A, de Gaetano Donati K, Colafigli M, Cozzi-Lepri A, De Curtis A, Gori A, et al. The association of high-sensitivity c-reactive protein and other biomarkers with cardiovascular disease in patients treated for HIV: a nested case-control study. BMC Infect Dis. (2013) 13:414. doi: 10.1186/1471-2334-13-414
59. Nordell AD, McKenna M, Borges AH, Duprez D, Neuhaus J, Neaton JD, et al. Severity of cardiovascular disease outcomes among patients with HIV is related to markers of inflammation and coagulation. J Am Heart Assoc. (2014) 3:e000844. doi: 10.1161/JAHA.114.000844
60. Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. (2010) 115:161–7. doi: 10.1182/blood-2009-03-210179
61. Schechter ME, Andrade BB, He T, Richter GH, Tosh KW, Policicchio BB, et al. Inflammatory monocytes expressing tissue factor drive SIV and HIV coagulopathy. Sci Transl Med. (2017) 9:aam5441. doi: 10.1126/scitranslmed.aam5441
62. Funderburg NT, Zidar DA, Shive C, Lioi A, Mudd J, Musselwhite LW, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome. Blood. (2012) 120:4599–608. doi: 10.1182/blood-2012-05-433946
63. Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. (2006) 4:e365. doi: 10.1371/journal.pbio.0040365
64. Leber AW, von Ziegler F, Becker A, Becker CR, Reiser M, Steinbeck G, et al. Characteristics of coronary plaques before angiographic progression determined by Multi-Slice CT. Int J Cardiovasc Imaging. (2008) 24:423–8. doi: 10.1007/s10554-007-9278-9
65. Thomsen C, Abdulla J. Characteristics of high-risk coronary plaques identified by computed tomographic angiography and associated prognosis: a systematic review and meta-analysis. Eur Heart J Cardiovasc Imaging. (2016) 17:120–9. doi: 10.1093/ehjci/jev325
66. Hsue PY, Hunt PW, Sinclair E, Bredt B, Franklin A, Killian M, et al. Increased carotid intima-media thickness in HIV patients is associated with increased cytomegalovirus-specific T-cell responses. AIDS. (2006) 20:2275–83. doi: 10.1097/QAD.0b013e3280108704
67. Grunfeld C, Delaney JA, Wanke C, Currier JS, Scherzer R, Biggs ML, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS. (2009) 23:1841–9. doi: 10.1097/QAD.0b013e32832d3b85
68. Leon R, Reus S, Lopez N, Portilla I, Sanchez-Paya J, Giner L, et al. Subclinical atherosclerosis in low Framingham risk HIV patients. Eur J Clin Invest. (2017) 47:591–599. doi: 10.1111/eci.12780
69. Guma M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis. (2006) 194:38–41. doi: 10.1086/504719
70. Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, et al. Chronic HIV-1 viremia reverses NKG2A/NKG2C ratio on natural killer cells in patients with human cytomegalovirus co-infection. AIDS. (2010) 24:27–34. doi: 10.1097/QAD.0b013e3283328d1f
71. Mudd JC, Panigrahi S, Kyi B, Moon SH, Manion MM, Younes SA, et al. Inflammatory Function of CX3CR1+ CD8+ T cells in treated HIV infection is modulated by platelet interactions. J Infect Dis. (2016) 214:1808–16. doi: 10.1093/infdis/jiw463
72. Parrinello CM, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, et al. Cytomegalovirus immunoglobulin G antibody is associated with subclinical carotid artery disease among HIV-infected women. J Infect Dis. (2012) 205:1788–96. doi: 10.1093/infdis/jis276
73. Knudsen A, Kristoffersen US, Panum I, Hansen YB, Skottrup PD, Hasbak P, et al. Coronary artery calcium and intima-media thickness are associated with level of cytomegalovirus immunoglobulin G in HIV-infected patients. HIV Med. (2019) 20:60–62. doi: 10.1111/hiv.12672
74. Lo J, Abbara S, Shturman L, Soni A, Wei J, Rocha-Filho JA, et al. Increased prevalence of subclinical coronary atherosclerosis detected by coronary computed tomography angiography in HIV-infected men. AIDS. (2010) 24:243–53. doi: 10.1097/QAD.0b013e328333ea9e
75. Post WS, Budoff M, Kingsley L, Palella FJ Jr, Witt MD, Li X, et al. Associations between HIV infection and subclinical coronary atherosclerosis. Ann Intern Med. (2014) 160:458–67. doi: 10.7326/M13-1754
76. Tarr PE, Ledergerber B, Calmy A, Doco-Lecompte T, Marzel A, Weber R, et al. Subclinical coronary artery disease in Swiss HIV-positive and HIV-negative persons. Eur Heart J. (2018) 39:2147–2154. doi: 10.1093/eurheartj/ehy163
77. D'Ascenzo F, Cerrato E, Calcagno A, Grossomarra W, Ballocca F, Omede P, et al. High prevalence at computed coronary tomography of non-calcified plaques in asymptomatic HIV patients treated with HAART: a meta-analysis. Atherosclerosis. (2015) 240:197–204. doi: 10.1016/j.atherosclerosis.2015.03.019
78. Surasi DS, O'Malley J, Bhambhvani P. 99mTc-Tilmanocept: a novel molecular agent for lymphatic mapping and sentinel lymph node localization. J Nucl Med Technol. (2015) 43:87–91. doi: 10.2967/jnmt.115.155960
79. Zanni MV, Toribio M, Wilks MQ, Lu MT, Burdo TH, Walker J, et al. Application of a Novel CD206+ macrophage-specific arterial imaging strategy in HIV-infected individuals. J Infect Dis. (2017) 215:1264–1269. doi: 10.1093/infdis/jix095
80. Varasteh Z, Mohanta S, Li Y, Lopez Armbruster N, Braeuer M, Nekolla SG, et al. Targeting mannose receptor expression on macrophages in atherosclerotic plaques of apolipoprotein E-knockout mice using (68)Ga-NOTA-anti-MMR nanobody: non-invasive imaging of atherosclerotic plaques. EJNMMI Res. (2019) 9:5. doi: 10.1186/s13550-019-0474-0
81. Yarasheski KE, Laciny E, Overton ET, Reeds DN, Harrod M, Baldwin S, et al. 18FDG PET-CT imaging detects arterial inflammation and early atherosclerosis in HIV-infected adults with cardiovascular disease risk factors. J Inflamm. (2012) 9:26. doi: 10.1186/1476-9255-9-26
82. Tawakol A, Lo J, Zanni MV, Marmarelis E, Ihenachor EJ, MacNabb M, et al. Increased arterial inflammation relates to high-risk coronary plaque morphology in HIV-infected patients. J Acquir Immune Defic Syndr. (2014) 66:164–71. doi: 10.1097/QAI.0000000000000138
83. Lo J, Lu MT, Ihenachor EJ, Wei J, Looby SE, Fitch KV, et al. Effects of statin therapy on coronary artery plaque volume and high-risk plaque morphology in HIV-infected patients with subclinical atherosclerosis: a randomised, double-blind, placebo-controlled trial. Lancet HIV. (2015) 2:e52–63. doi: 10.1016/S2352-3018(14)00032-0
84. Eckard AR, Jiang Y, Debanne SM, Funderburg NT, McComsey GA. Effect of 24 weeks of statin therapy on systemic and vascular inflammation in HIV-infected subjects receiving antiretroviral therapy. J Infect Dis. (2014) 209:1156–64. doi: 10.1093/infdis/jiu012
85. Packard CJ, O'Reilly DS, Caslake MJ, McMahon AD, Ford I, Cooney J, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med. (2000) 343:1148–55. doi: 10.1056/NEJM200010193431603
86. Funderburg NT, Jiang Y, Debanne SM, Labbato D, Juchnowski S, Ferrari B, et al. Rosuvastatin reduces vascular inflammation and T-cell and monocyte activation in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. (2015) 68:396–404. doi: 10.1097/QAI.0000000000000478
87. Bhadra S, Arshad MA, Rymaszewski Z, Norman E, Wherley R, Subbiah MT. Oxidation of cholesterol moiety of low density lipoprotein in the presence of human endothelial cells or Cu+2 ions: identification of major products and their effects. Biochem Biophys Res Commun. (1991) 176:431–40. doi: 10.1016/0006-291X(91)90942-Z
88. Jialal I, Freeman DA, Grundy SM. Varying susceptibility of different low density lipoproteins to oxidative modification. Arterioscler Thromb. (1991) 11:482–8. doi: 10.1161/01.ATV.11.3.482
89. Aviram M, Rosenblat M. Macrophage-mediated oxidation of extracellular low density lipoprotein requires an initial binding of the lipoprotein to its receptor. J Lipid Res. (1994) 35:385–98.
90. Zidar DA, Juchnowski S, Ferrari B, Clagett B, Pilch-Cooper HA, Rose S, et al. Oxidized LDL levels are increased in HIV infection and may drive monocyte activation. J Acquir Immune Defic Syndr. (2015) 69:154–60. doi: 10.1097/QAI.0000000000000566
91. Duong M, Petit JM, Martha B, Galland F, Piroth L, Walldner A, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials. (2006) 7:41–7. doi: 10.1310/7381-M1YD-RTV5-4RYT
92. Parra S, Coll B, Aragones G, Marsillach J, Beltran R, Rull A, et al. Nonconcordance between subclinical atherosclerosis and the calculated Framingham risk score in HIV-infected patients: relationships with serum markers of oxidation and inflammation. HIV Med. (2010) 11:225–31. doi: 10.1111/j.1468-1293.2009.00766.x
93. Nou E, Lu MT, Looby SE, Fitch KV, Kim EA, Lee H, et al. Serum oxidized low-density lipoprotein decreases in response to statin therapy and relates independently to reductions in coronary plaque in patients with HIV. AIDS. (2016) 30:583–90. doi: 10.1097/QAD.0000000000000946
94. Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. (2010) 7:77–86. doi: 10.1038/nrcardio.2009.228
95. Cappellari R, D'Anna M, Bonora BM, Rigato M, Cignarella A, Avogaro A, et al. Shift of monocyte subsets along their continuum predicts cardiovascular outcomes. Atherosclerosis. (2017) 266:95–102. doi: 10.1016/j.atherosclerosis.2017.09.032
96. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
97. Tak T, Drylewicz J, Conemans L, de Boer RJ, Koenderman LJ, Borghans AM, et al. Circulatory and maturation kinetics of human monocyte subsets in vivo. Blood. (2017) 130:1474–7. doi: 10.1182/blood-2017-03-771261
98. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. (1998) 394:894–7. doi: 10.1038/29788
99. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/JCI28549
100. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/JCI29950
101. Cipriani S, Francisci D, Mencarelli A, Renga B, Schiaroli E, D'Amore C, et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation. (2013) 127:2114–24. doi: 10.1161/CIRCULATIONAHA.113.001278
102. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
103. Westhorpe CL, Maisa A, Spelman T, Hoy JF, Dewar EM, Karapanagiotidis S, et al. Associations between surface markers on blood monocytes and carotid atherosclerosis in HIV-positive individuals. Immunol Cell Biol. (2014) 92:133–8. doi: 10.1038/icb.2013.84
104. Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet. (1997) 349:692–5. doi: 10.1016/S0140-6736(96)10178-1
105. Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. (1995) 25:3418–24. doi: 10.1002/eji.1830251232
106. Jaworowski A, Kamwendo DD, Ellery P, Sonza S, Mwapasa V, Tadesse E, et al. CD16+ monocyte subset preferentially harbors HIV-1 and is expanded in pregnant Malawian women with Plasmodium falciparum malaria and HIV-1 infection. J Infect Dis. (2007) 196:38–42. doi: 10.1086/518443
107. Amirayan-Chevillard N, Tissot-Dupont H, Capo C, Brunet C, Dignat-George F, Obadia Y, et al. Impact of highly active anti-retroviral therapy (HAART) on cytokine production and monocyte subsets in HIV-infected patients. Clin Exp Immunol. (2000) 120:107–12. doi: 10.1046/j.1365-2249.2000.01201.x
108. Hearps AC, Agius PA, Zhou J, Brunt S, Chachage M, Angelovich TA, et al. Persistence of activated and adaptive-like NK Cells in HIV(+) individuals despite 2 years of suppressive combination antiretroviral therapy. Front Immunol. (2017) 8:731. doi: 10.3389/fimmu.2017.00731
109. Jaworowski A, Ellery P, Maslin CL, Naim E, Heinlein AC, Ryan CE, et al. Normal CD16 expression and phagocytosis of Mycobacterium avium complex by monocytes from a current cohort of HIV-1-infected patients. J Infect Dis. (2006) 193:693–7. doi: 10.1086/500367
110. Meerschaert J, Furie MB. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J Immunol. (1995) 154:4099–112.
111. Aziz MH, Cui K, Das M, Brown KE, Ardell CL, Febbraio M, et al. The upregulation of integrin alphaDbeta2 (CD11d/CD18) on inflammatory macrophages promotes macrophage retention in vascular lesions and development of atherosclerosis. J Immunol. (2017) 198:4855–67. doi: 10.4049/jimmunol.1602175
112. Meisel SR, Shapiro H, Radnay J, Neuman Y, Khaskia AR, Gruener N, et al. Increased expression of neutrophil and monocyte adhesion molecules LFA-1 and Mac-1 and their ligand ICAM-1 and VLA-4 throughout the acute phase of myocardial infarction: possible implications for leukocyte aggregation and microvascular plugging. J Am Coll Cardiol. (1998) 31:120–5. doi: 10.1016/S0735-1097(97)00424-5
113. Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. (2004) 101:11779–84. doi: 10.1073/pnas.0403259101
114. Randolph GJ. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. (2008) 19:462–8. doi: 10.1097/MOL.0b013e32830d5f09
115. Zerbinatti CV, Gore RW. Uptake of modified low-density lipoproteins alters actin distribution and locomotor forces in macrophages. Am J Physiol Cell Physiol. (2003) 284:C555–61. doi: 10.1152/ajpcell.00177.2002
116. Dhaliwal BS, Steinbrecher UP. Cholesterol delivered to macrophages by oxidized low density lipoprotein is sequestered in lysosomes and fails to efflux normally. J Lipid Res. (2000) 41:1658–65.
117. Jerome WG, Lewis JC. Early atherogenesis in White Carneau pigeons: effect of a short-term regression diet. Exp Mol Pathol. (1990) 53:223–38. doi: 10.1016/0014-4800(90)90046-G
118. Angelovich TA, Hearps AC, Jaworowski A. Inflammation-induced foam cell formation in chronic inflammatory disease. Immunol Cell Biol. (2015) 93:683–93. doi: 10.1038/icb.2015.26
119. Bernard MA, Han X, Inderbitzin S, Agbim I, Zhao H, Koziel H, et al. HIV-derived ssRNA binds to TLR8 to induce inflammation-driven macrophage foam cell formation. PLoS ONE. (2014) 9:e104039. doi: 10.1371/journal.pone.0104039
120. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
121. Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. (2009) 199:1177–85. doi: 10.1086/597476
122. Jennelle L, Hunegnaw R, Dubrovsky L, Pushkarsky T, Fitzgerald ML, Sviridov D, et al. HIV-1 protein Nef inhibits activity of ATP-binding cassette transporter A1 by targeting endoplasmic reticulum chaperone calnexin. J Biol Chem. (2014) 289:28870–84. doi: 10.1074/jbc.M114.583591
123. Muller WA, Weigl SA. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. (1992) 176:819–28. doi: 10.1084/jem.176.3.819
124. Angelovich TA, Hearps AC, Maisa A, Kelesidis T, Jaworowski A. Quantification of monocyte transmigration and foam cell formation from individuals with chronic inflammatory conditions. J Vis Exp 128:e56293. (2017). doi: 10.3791/56293
125. Muller WA, Randolph GJ. Migration of leukocytes across endothelium and beyond: molecules involved in the transmigration and fate of monocytes. J Leukoc Biol. (1999) 66:698–704. doi: 10.1002/jlb.66.5.698
126. Maisa A, Hearps AC, Angelovich TA, Pereira CF, Zhou J, Shi MD, et al. Monocytes from HIV-infected individuals show impaired cholesterol efflux and increased foam cell formation after transendothelial migration. AIDS. (2015) 29:1445–57. doi: 10.1097/QAD.0000000000000739
127. Angelovich TA, Shi MD, Zhou J, Maisa A, Hearps AC, Jaworowski A. Ex vivo foam cell formation is enhanced in monocytes from older individuals by both extrinsic and intrinsic mechanisms. Exp Gerontol. (2016) 80:17–26. doi: 10.1016/j.exger.2016.04.006
128. Hearps AC, Martin GE, Angelovich TA, Cheng WJ, Maisa A, Landay AL, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell. (2012) 11:867–75. doi: 10.1111/j.1474-9726.2012.00851.x
129. Field FJ, Watt K, Mathur SN. TNF-alpha decreases ABCA1 expression and attenuates HDL cholesterol efflux in the human intestinal cell line Caco-2. J Lipid Res. (2010) 51:1407–15. doi: 10.1194/jlr.M002410
130. Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. (1993) 268:11811–6.
131. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
132. Nicholson AC, Expression of CD36 in macrophages and atherosclerosis: the role of lipid regulation of PPARgamma signaling. Trends Cardiovasc Med. (2004) 14:8–12. doi: 10.1016/j.tcm.2003.09.004
133. Kelesidis T, Jackson N, McComsey GA, Wang X, Elashoff D, Dube MP, et al. Oxidized lipoproteins are associated with markers of inflammation and immune activation in HIV-1 infection. AIDS. (2016) 30:2625–33. doi: 10.1097/QAD.0000000000001238
134. Angelovich TA, Hearps AC, Oda MN, Borja MS, Huynh D, Homann S, et al. Dysfunctional high-density lipoprotein from HIV+ individuals promotes monocyte-derived foam cell formation in vitro. AIDS. (2017) 31:2331–6. doi: 10.1097/QAD.0000000000001642
135. Siegel MO, Borkowska AG, Dubrovsky L, Roth M, Welti R, Roberts AD, et al. HIV infection induces structural and functional changes in high density lipoproteins. Atherosclerosis. (2015) 243:19–29. doi: 10.1016/j.atherosclerosis.2015.08.036
136. Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. (2004) 95:764–72. doi: 10.1161/01.RES.0000146094.59640.13
137. Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, et al. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. (2004) 45:993–1007. doi: 10.1194/jlr.R400001-JLR200
138. Aviram M, Billecke S, Sorenson R, Bisgaier C, Newton R, Rosenblat M, et al. Paraoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities: selective action of human paraoxonase allozymes Q and R. Arterioscler Thromb Vasc Biol. (1998) 18:1617–24. doi: 10.1161/01.ATV.18.10.1617
139. Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, et al. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. (1995) 96:2882–91. doi: 10.1172/JCI118359
140. Mackness B, Mackness M. Anti-Inflammatory properties of paraoxonase-1 in atherosclerosis. In: Reddy ST, editor. Paraoxonases in Inflammation, Infection and Toxicology, New York, NY: Humana Press (2009). p. 143–51. doi: 10.1007/978-1-60761-350-3_13
141. Rozenberg O, Shih DM, Aviram M. Human serum paraoxonase 1 decreases macrophage cholesterol biosynthesis: possible role for its phospholipase-A2-like activity and lysophosphatidylcholine formation. Arterioscler Thromb Vasc Biol. (2003) 23:461–7. doi: 10.1161/01.ATV.0000060462.35946.B3
142. Rohrer L, Hersberger M, von Eckardstein A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr Opin Lipidol. (2004) 15:269–78. doi: 10.1097/00041433-200406000-00006
143. Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr OpinLipidol. (2007) 18:427–34. doi: 10.1097/MOL.0b013e3282364a17
144. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. (2010) 33:375–86. doi: 10.1016/j.immuni.2010.08.012
Keywords: HIV, monocytes/macrophages, atherosclerosis, foam cells/macrophages, inflammation
Citation: Jaworowski A, Hearps AC, Angelovich TA and Hoy JF (2019) How Monocytes Contribute to Increased Risk of Atherosclerosis in Virologically-Suppressed HIV-Positive Individuals Receiving Combination Antiretroviral Therapy. Front. Immunol. 10:1378. doi: 10.3389/fimmu.2019.01378
Received: 27 February 2019; Accepted: 31 May 2019;
Published: 19 June 2019.
Edited by:
Shannon Marie Murray, Nova Southeastern University, United StatesReviewed by:
Nicholas Funderburg, The Ohio State University, United StatesSuresh Pallikkuth, University of Miami, United States
Netanya Sandler Utay, The University of Texas Medical Branch at Galveston, United States
Copyright © 2019 Jaworowski, Hearps, Angelovich and Hoy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Jaworowski, QW50aG9ueS5qYXdvcm93c2tpQHJtaXQuZWR1LmF1