 Stefan Audehm1
Stefan Audehm1 Manuel Glaser2†
Manuel Glaser2† Matteo Pecoraro3†
Matteo Pecoraro3† Eva Bräunlein1†Sabine Mall1
Eva Bräunlein1†Sabine Mall1 Richard Klar1Manuel Effenberger4Julian Albers1Henrique de Oliveira Bianchi1
Richard Klar1Manuel Effenberger4Julian Albers1Henrique de Oliveira Bianchi1 Janet Peper5
Janet Peper5 Nahid Yusufi6Dirk H. Busch4
Nahid Yusufi6Dirk H. Busch4 Stefan Stevanović5,7
Stefan Stevanović5,7 Matthias Mann3Iris Antes2
Matthias Mann3Iris Antes2 Angela M. Krackhardt1,8,9*
Angela M. Krackhardt1,8,9*- 1Klinik und Poliklinik für Innere Medizin III, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
- 2Center for Integrated Protein Science at the Department for Biosciences, Technische Universität München, Freising, Germany
- 3Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany
- 4Institut für Medizinische Mikrobiologie, Immunologie und Hygiene, Technische Universität München, Munich, Germany
- 5Eberhard Karls University Tübingen, Interfaculty Institute for Cell Biology, Tübingen, Germany
- 6Nuklearmedizin, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
- 7Partner Site Tübingen, German Cancer Consortium (DKTK), Tübingen, Germany
- 8Partner Site Munich, German Cancer Consortium (DKTK), Munich, Germany
- 9German Cancer Research Center (DKFZ), Heidelberg, Germany
Adoptive transfer of T cells transgenic for tumor-reactive T-cell receptors (TCR) is an attractive immunotherapeutic approach. However, clinical translation is so far limited due to challenges in the identification of suitable target antigens as well as TCRs that are concurrent safe and efficient. Definition of key characteristics relevant for effective and specific tumor rejection is essential to improve current TCR-based adoptive T-cell immunotherapies. We here characterized in-depth two TCRs derived from the human leukocyte antigen (HLA)-mismatched allogeneic repertoire targeting two different myeloperoxidase (MPO)-derived peptides presented by the same HLA-restriction element side by side comprising state of the art biochemical and cellular in vitro, in vivo, and in silico experiments. In vitro experiments reveal comparable functional avidities, off-rates, and cytotoxic activities for both TCRs. However, we observed differences especially with respect to cytokine secretion and cross-reactivity as well as in vivo activity. Biochemical and in silico analyses demonstrate different binding qualities of MPO-peptides to the HLA-complex determining TCR qualities. We conclude from our biochemical and in silico analyses of peptide-HLA-binding that rigid and high-affinity binding of peptides is one of the most important factors for isolation of TCRs with high specificity and tumor rejection capacity from the MHC-mismatched repertoire. Based on our results, we developed a workflow for selection of such TCRs with high potency and safety profile suitable for clinical translation.
Introduction
Adoptive T-cell transfer of immune receptor transgenic T cells has demonstrated high clinical potential especially for chimeric antigen receptors (CAR) (1). TCRs, however, are far behind CARs with respect to clinical translation. A number of candidate TCRs have been proposed (2–4), however, only a few clinical trials have been published (5, 6) likely explainable by the challenging selection process for suitable peptide antigens as well as equally potent and safe TCRs.
Diverse qualities of TCRs have been described to be essential and potentially predict anti-tumor reactivity in vivo as especially functional avidity and affinity of TCRs (7–11). In addition, slow dissociation half-life of TCRs from peptide-MHC complexes (p-MHC) has been reported to correspond with in vivo activity (12). There are less recommendations for high avidity TCRs deriving from the allogeneic or xenogeneic environment or selected by affinity maturation. This is especially important as these TCR may harbor an enhanced risk profile for crossreactivity and therefore toxicity (13, 14). Furthermore, the functionality and efficiency of transferred TCR cell-surface expression depend on the intrinsic quality of the TCR complex (15) and the T-cell specificity might be affected by the formation of mixed heterodimers composed of endogenous and transgenic TCR chains (16).
Suitable target epitope selection remains equally difficult despite the large number of possible human leukocyte antigen (HLA)-peptide ligands. Peptide-HLA binding affinity has been identified as particular important (17, 18). Candidate epitopes are often selected by prediction algorithms. Sequence- and stability-based p-MHC binding predictions are valuable tools (19, 20) to get approximated binding qualities (21) and are used for initial peptide-candidate screenings (22). In addition, combined approaches using sequence- and structure-based algorithms have been applied (23, 24). Peptide ligands to be used as target antigens in cancer can be also directly identified by immunopeptidomics (25, 26) to be potentially used in combination with in-silico p-MHC binding prediction (26, 27).
Defining priorities for the selection of epitope and TCR candidates, including the non-self-repertoire, would be an important step to foster clinical translation. We here present an in-depth characterization and comparison of two TCRs identified in a single HLA-mismatched allorestricted approach (sHLAm) recognizing two different peptides derived from myeloperoxidase (MPO) sharing the same restriction element HLA-B*07:02 (HLA-B7). One of the TCRs has been previously described as highly specific and tumor-reactive (27, 28). Identification and characterization of the second TCR are described in this manuscript. State of the art key experiments investigating functional qualities of TCR-transgenic TCR as well as in-depth target peptide characterization have been applied to address the question, which set of in vitro and in silico analyses may support straight forward selection of promising peptide and TCR candidates suitable for clinical translation.
Materials and Methods
Cell Lines and T Cells
The isolation of peripheral blood mononuclear cells (PBMC), the isolation of naïve CD8+ T cells and the culturing of target cell lines were realized as described previously (27, 28). For analyses of the MPO-specific TCRs the following cell lines were used: NB-4 (Cell Lines Service, CLS, Germany), SiG-M5 (DSMZ), K562 (ATCC CCL-243), KG1a (CLS), HL-60 (CLS), ML2 (The CABRI consortium), C1R (26), NSO-IL15 (kindly provided by S.R. Riddell), 293Vec-RD114 (BioVec Pharma), and variant types of lymphoblastoid cell lines (LCL) (kindly provided by Steve Marsh). Cell culturing was done as previously described (27). All cell lines were periodically tested for mycoplasma negative status by PCR and cell line authentication was done by flow cytometry-based analyses of cell surface markers and HLA-A*- and HLA-B* typing by next-generation sequencing (Center for Human Genetics and Laboratory Diagnostics, Munich, Germany).
Antibodies and HLA Multimers
Antibodies used for activation of T cells and flow cytometry: anti-hCD3 FITC [UCHT1; Becton Dickinson, Franklin Lakes, NJ, USA (BD)], anti-hCD4 APC/Pacific Blue (RPA-T4; BD), anti-hCD8 APC/V450 (RPA-T8; BD), anti-hCD62L PE, anti-hCD45RO PE (UCHL1; BD), anti-hCD45RA APC (HI100; BD), anti-mouse TCR-β chain (anti-TCRm) FITC/PE/APC (H57-597, BD), anti-hCD45 APC [J.33; Beckman Coulter (BC)], anti-hCD3 AF700 (UCHT1; BD), anti-hCD5 PECyTM5 (UCHT2; BD), anti-hCD4 V450 (RPA-T4; BD), anti-HLA-B7 PE (BB7.1; Merck). HLA multimers were synthesized as previously described (29).
Selection and Expansion of T Cells Specific for the MPO Ligands
Expansion of T cells specific for the HLA-B7 MPO2-ligand was performed as described previously (27). T cells were subsequently screened for functionality. Prior to the multimer sort, expanded T cells from the MPO2 single HLA-mismatch setting were co-incubated with lethally irradiated (30 Gy) C1R-B7 tumor cells to remove unspecific HLA-B7-alloreactive T cells (30). After 12 h of co-incubation, the T cells were stained with an anti-human CD137-APC antibody and depleted by MACS sorting (Miltenyi) according to the manufacturer instructions. Subsequently, peptide-specific T cells were sorted using HLA multimers on a flow cytometric cell sorter (MoFlo; Dako, Glostrup, Denmark). HLA-multimer positive T cells were cloned by limited dilution.
TCR Isolation and Retroviral TCR Transfer
The usage of TCR variable alpha and beta domains were determined by PCR followed by Sanger Sequencing as described previously (31). After in silico murinization and cysteine modification of the constant domains of the TCR and insertion of an additional cysteine bridge, the complete sequence was codon-optimized (Genscript, Piscataway Township, NJ, USA) and cloned by a bi-cistronic construct, consisting of both TCR chains into the pMP71 backbone. To produce retroviral supernatants, the embryonal kidney cell line 293Vec-RD114 (BioVec Pharma, Québec, Canada) was used and a retroviral transduction protocol for TCR- and HLA-allele transduction as described previously (32).
HLA-Vector Constructs Used for T-Cell Clone Isolation, Testing of Specificity, and HLA-Peptide Restriction
The different tumor cell lines without endogenous gene expression of MPO or the desired restriction element HLA-A1, HLA-A2, HLA-B7, HLA-B15, or HLA-B44, respectively, were transduced with the retroviral vector pMP71 containing the HLA of interest attached to green fluorescent protein (GFP) or MPO attached to Discosoma Red Fluorescent Protein (DsRed) Express II. The sequences were cloned into the vector as a bi-cistronic construct separated by the porcine teschovirus-1-derived peptide element P2A. For the generation of in vitro transcribed (IVT) mRNA the bi-cistronic constructs were cloned into the plasmid pcDNA3.1(–) (Thermo Fisher Scientific). HLA-allele retroviral transduced cell lines are indicated with the appendix e.g., “-B7”.
MPO, MPO2, and MPO5 Mini-Gene Design and Expression in Target Cell Lines
Gene fragments containing the MPO2 or MPO5 sequence in the center were amplified by designing primers resulting in oligonucleotides of an overall length of ~200 base pairs flanked by a start and a stop codon. The following primers were used for the cloning of MPO2- and MPO5-mini-gene: 5′-TACAGGCGGCCGCCACCATGACGGCGGTGAGGGC-CGC-3′, 5′-TAGTCGACGGGGCTGCGTCTGTTGTTGC-3′, and 5′TACAGGCGGCCGC-CACCATGCTGGCAGGGGACACCCG-3′, 5′-TAGTCGACGTACTTCCTCATGGCCGTTG3′ using the MPO gene derived from NB-4 cells as template. For the expression of mini-genes in HLA-B*07:02-positive target cells, amplified oligonucleotides were cloned into the retroviral vector MP71 containing also dsRed Express II as selection marker. Retroviral supernatant production and retroviral transduction was done as previously described (32).
Functional Characterization of T-Cell Clones and TCR-Transgenic T Cells
The cytotoxic reactivity of T cells was tested by flow cytometry making use of changes in signal of the HLA-B7eGFP-transgenic target cells during co-cultivation for different time periods and effector to target ratios. For relative quantification of cytotoxicity, AccuCheck counting beads were used according to the manufacturer's protocol (Thermo Fisher Scientific) and the results were normalized to either non-TCR-transduced T cells or irrelevant T-cell clones. The supernatants of the co-cultivation were used to measure IFN-γ by ELISA (BD) or GM-CSF, IFN-γ, and lL-2 multiplexed by flow cytometry (Miltenyi Biotec) according to the manufacturer's instructions. The functional avidity of the different TCR was assessed as previously described (27). The 58 HLA-B7 restricted ligands used for the analysis of potential off-target reactivity of TCRF5.4 as tested in co-cultivation experiments, were synthesized (EPS221 synthesizer, Abimed, Langenfeld, Germany) following the 9-fluorenylmethyl-oxycarbonyl/tertbutyl (Fmoc/tBu) methodology. To define peptide residues essential for the recognition by TCRF5.4, amino acid substitution assays using alanine- and threonine variants of the MPO2 peptide (Genscript) were performed as described previously (32). Based on the acquired motif, the ScanProsite tool (SIB Swiss Institute of Bioinformatics) was used to identify proteins containing the same pattern. If not stated otherwise, the effector to target ratio was set to 1:1 for the co-incubation assays using 20,000 cells in each fraction.
UV-Mediated Peptide Exchange
Biotinylated HLA-B7 monomers loaded with the UV-cleavable epitope AARG(J)TLAM were created as previously described (33). Peptide-HLA stability was measured using the protocol on the basis of Rodenko et al. (34). In brief, HLA-B7 monomers loaded with the UV-sensitive peptide were exposed to UV light (366 nm) in the presence of graded amounts (0–200 μM) of MPO peptides. Afterward 1:10 dilutions of the UV-exchanged monomers were adhered via streptavidin to the bottom of a 96-well plate (F96 maxisorp nunc-immune plate, Thermo scientific) and a beta-2-microglobulin ELISA was performed. The OD measurements were performed on a Tecan Sunrise multi-well-reader using Magellan software (version 7.2).
Flow Cytometry-Based koff-Rate Determination of TCRF5.4 and TCR2.5D6
The koff-rate was analyzed as described previously (35). In brief, 5 ×106 TCR-transduced T cells were stained with the reversible TCR-specific multimer for 45 min and subsequently incubated with an anti-human CD8 eF450 antibody (Thermo Fisher Scientific) for 20 min. Washed T cells were then stained with 0.2 mg propidium iodide solution for 5 min. 100 μl of the stained TCR-transduced suspension (1–10 ×106 cell/ml) was added to 900 μl FACS buffer prepared in a koff-rate FACS tube under constant cool condition (qutools GmbH, Munich, Germany) for the whole measurement. After initiation of analysis in a CyAn ADP Lx 9 color flow cytometer (Beckman Coulter, Miami, US) for 30 s, 1 ml 2 mM D-biotin was injected into the koff-rate FACS tube. The measurement was stopped after recording events for a total of 15 min. Data were analyzed using FlowJo v9.5.2 software (FlowJo, LLC, Ashland) and a one-phase exponential decay curve fitting tool of GraphPad Prism (version 7.04, San Diego, USA).
Human AML Tumor Models and Adoptive T Cell Transfer
The immunocompromised NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mouse strain was used to establish an NB4 derived myeloid sarcoma model following the experimental procedure as described previously (28). In brief, 1 ×107 NB4-B7eGFP- or NB4-B15eGFP tumor cells were subcutaneously inoculated into the right or left flank, respectively. One day prior to intravenous injection of 2 ×107 TCR-transgenic CD8+ TCM (day 8), mice were irradiated with 1 Gy total body irradiation. Irradiated (80 Gy) human interleukin-15 producing murine NSO cells were injected intraperitoneally twice per week (27). The mice were maintained according to conventional institute guidelines and with the approval of local authorities.
Molecular Modeling
MPO5- and the respective variant-HLA-B7 complexes were simulated based on the crystal structure of the HLA-B7 complex bound to the peptide TPQDLNTML (Protein Data Bank (PDB) ID: 4U1H) (36) as template. The MPO2- and MPO2 variant-HLA-B7 complexes were modeled as follows: the MPO5-HLA-B7 protein conformations were directly modeled based on 4U1H; for the backbone conformation of the MPO2 peptide and its variants the crystal structure of the octamer HLA-B*08:01 complex bound to the peptide GGKKKYKL (PDB ID: 1AGD) (37) was taken as no experimental structure of an octamer-HLA-B7 complex was available. For this the HLA α1 and α2 subdomains of 4U1H and 1AGD were structurally aligned using PyMOL, version 1.5.0.4 (Schrödinger). Only the subdomains α1 and α2 of the HLA-B7 protein were considered for modeling. Mutations of the peptide sequences and conformational adaption of the highly flexible binding site residues E70 or R62 were conducted with IRECS (38, 39). Molecular dynamics (MD) simulations and system setup as well as MD analysis were performed with AmberTools16 and the Amber16 software package (40). The Amber ff14SB potential energy function and parameter set (41) were chosen to model the solute. The modeled complexes were simulated in a neutralized (Na+, Cl−), rectangular box of water molecules [TIP3P force field (42)] with a minimum solute distance of 14 Å to the box boundary. MD simulations were performed with the CUDA compatible GPU version of pmemd, applying periodic boundary conditions (43). Long-range electrostatic interactions were computed with the Particle Mesh Ewald (PME) method (43). A cutoff of 12 Å was used for the computation of non-bonded interactions. MD simulations were performed with a time step of 1 fs. The SHAKE algorithm (44) was applied to constrain bonds involving hydrogen atoms. Temperature and pressure were controlled applying the Berendsen thermostat and barostat (45), using coupling time constants of 0.5 ps (heat up) and 10.0 ps (simulations at 300 K), respectively, and a pressure relaxation time of 2.0 ps. Systems were heated up to room temperature in the NVT ensemble with a sequence of heat up MD simulations at stepwise increasing temperatures totaling up to 1.5 ns (0 K: 10 ps; 5 K, 10 K, 20 K, 50 K: 50 ps; 100 K, 200 K, 200 K: 100 ps; 200 K, 300 K: 200 ps; 300 K: 590 ps), using different restraint settings (0 K, 5 K, 10 K, 20 K: 2.39 kcal mol−1 Å−2 for all solute atoms; 50 K, 100 K, 200 K: 2.39 kcal mol−1 Å−2 for all backbone heavy atoms; 200 K: 0.24 kcal mol−1 Å−2 for all backbone heavy atoms; 200 K, 300 K: 0.24 kcal mol−1 Å−2 for all backbone heavy atoms of the HLA-B7 protein; 300 K: no restraints). The heated systems were equilibrated in the NPT ensemble for 1.5 ns at 300 K and 1 bar. All systems were simulated for 450 ns in total by performing three independent 150 ns MD simulations per system. Trajectory processing and analysis was performed with cpptraj (46). For the latter, MD frames were extracted from the three trajectories every 100 ps and clustered with respect to their conformation, using the average-linkage hierarchical agglomerative clustering approach (47), applying a minimum cluster distance of 0.75 Å and the best-fit coordinate RMSD of the peptide backbone heavy atoms as distance metric. Hydrogen bonds were calculated applying default settings of cpptraj. Peptide-HLA-B7 protein binding affinities were estimated applying the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) approach (48, 49) with the single trajectory protocol as implemented in MMPBSA.py (50) using default settings [GB model: OBC-II model (51), atomic radii: mbondi2 (51), no contributions from solvated ions, surface tension applied to the solvent-accessible surface area: 0.0072 kcal mol−1 Å−2 (52)]. Solute entropy contributions were neglected in the binding affinity estimates. For MM/GBSA free energy computations, snapshots from all three MD replica were combined, discarding the snapshots from the first 25 ns of each MD replicon. For the calculation of root mean square fluctuations (RMSF), each 150 ns-long MD replicon was split in six 25 ns bins and Cα-RMSFs were calculated for each bin separately with cpptraj. To yield the final RMSF values for each MD replicon, the single RMSF values of each bin were averaged. Electrostatic potentials were calculated using the PDB2PQR server (Version 2.0.0) and APBS (Version 1.3) (53, 54), keeping default settings. Figures of peptide-HLA-B7 complexes were generated with VMD (Version 1.9.2) (55).
Statistics
Statistical analyses of T-cell experiments were performed using GraphPad Prism software version 7.04. Results are presented as standard deviations (SD) of the mean. Samples were compared using the non-parametric Mann-Whitney's t-test and multiple t-test as indicated in the figure legends.
Results
Identification of a Novel HLA-B7 Allorestricted TCR Targeting a New MPO-Derived Epitope
We have previously identified a leukemia-associated HLA-B0702-restricted epitope derived from MPO (MPO466−474 named MPO5) by mass spectrometry (MS) as well as the TCR2.5D6 derived from the sHLAm T-cell repertoire with high peptide specificity and leukemic reactivity suitable for clinical application in the context of allogeneic stem cell transplantation (27). In our MS-based approach additional MPO epitopes presented on myeloid tumor cells have been identified (27) and selected for stimulation in sHLAm approaches to identify new MPO-specific TCRs. In fact, one novel TCR candidate (TCRF5.4) directed against the MPO145−152-peptide (MPO2, Table 1) with promising characteristics in early screening analyses was discovered. Shortly summarized, this TCR was identified after prior CD137 depletion of alloreactive T cells as previously described (30) followed by MPO2-multimer sorting and T-cell cloning (Supplementary Figures 1A,B). Specific T-cell clones were detected (Supplementary Figure 1C) expressing all the same TCR chains (Vα2 and Vβ1, TCRF5.4). CD8+ central memory T cells (TCM) transduced with an optimized and cysteine-modified TCRF5.4 construct (59, 60) displayed high transduction efficacy and TCR expression (Supplementary Figure 1D). TCRF5.4-transduced CD8+ TCM were highly specific for target cells either pulsed with or endogenously presenting the respective peptide after mini-gene transfer (Supplementary Figure 1E). Moreover, primary malignant cells derived from patients with myeloproliferative neoplasia (MPN) expressing MPO and HLA-B7 were well recognized (Supplementary Figure 1F). To exclude major cross-reactivity, we screened common HLA alleles expressed on different lymphoblastoid cell lines (LCL) pulsed with MPO2 and compared the reactivity to non-pulsed conditions (Supplementary Figure 1G; Supplementary Table 1). TCRF5.4-transgenic T cells showed alloreactivity against LCL8 carrying the HLA-alleles A*02:04/B*51:01. Co-incubation of TCRF5.4-transgenic T cells with C1R-A2 or Tapasin-1 deficient T2 (HLA-A2+/HLA-B51+) confirmed MPO2-independent HLA-B51 cross-reactivity (Supplementary Figure 1H; Supplementary Table 1). However, no recognition of TCRF5.4-transgenic T cells was seen against a set of 58 HLA-B7 restricted peptides (Supplementary Figure 1I). The TCRF5.4 peptide crossreactivity risk was tested on HLA-B7+ LCL1 cells by using alanine or threonine substitution variants (Ala/Thr-scan) of MPO2 (Supplementary Figures 1J,K). Interestingly, we observed different TCR-binding motifs for both amino acids substitution variants indicating a flexible binding pattern of the TCR toward the p-MHC. Applying the ScanProsite tool (61) for the combined Ala/Thr-scan motif (X-P-A-Q-X-X-V-X) revealed 297 hits outside the MPO source protein whereas only 1 hit was found if the motifs were analyzed separately.
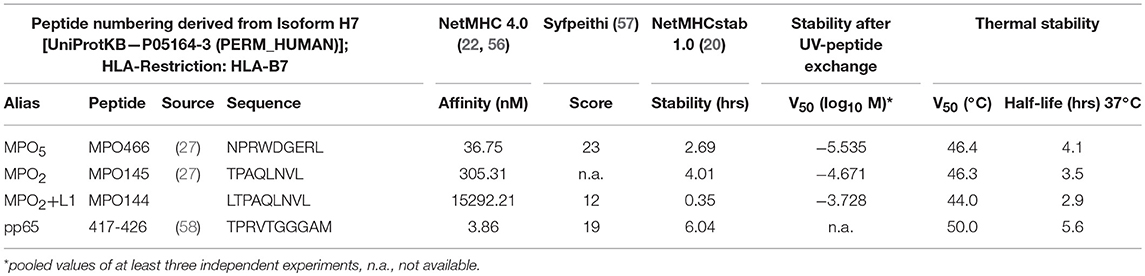
Table 1. Characteristics of analyzed p-MHC complexes.
Differences in Transgenic TCR Expression Between TCRF5.4 and TCR2.5D6 as Well as Quantitative and Qualitative Cytokine Secretion in vitro
Based on the qualities of TCRF5.4, the same source protein of recognized peptides and the same restriction element, we decided to compare this TCR to the recently characterized TCR2.5D6 to investigate which TCR may be superior and to define prospective criteria for rapid selection of most suitable allorestricted TCRs for adoptive transfer of TCR-transgenic T cells.
The TCR-transduction rates of recipient T cells differed in most but not all experiments approximately by 10% as the mean was 72.2% for TCRF5.4 and 82.3% for TCR2.5D6 (Figure 1A, left). The mean fluorescence intensity (MFI) of TCRF5.4 expression was also in many experiments inferior compared to TCR2.5D6 (Figure 1A, right). We observed that TCR2.5D6 is more efficiently expressed as nearly all positive TCR-signal is located on the surface of the T cells whereas TCRF5.4 is not fully expressed (Figure 1B).
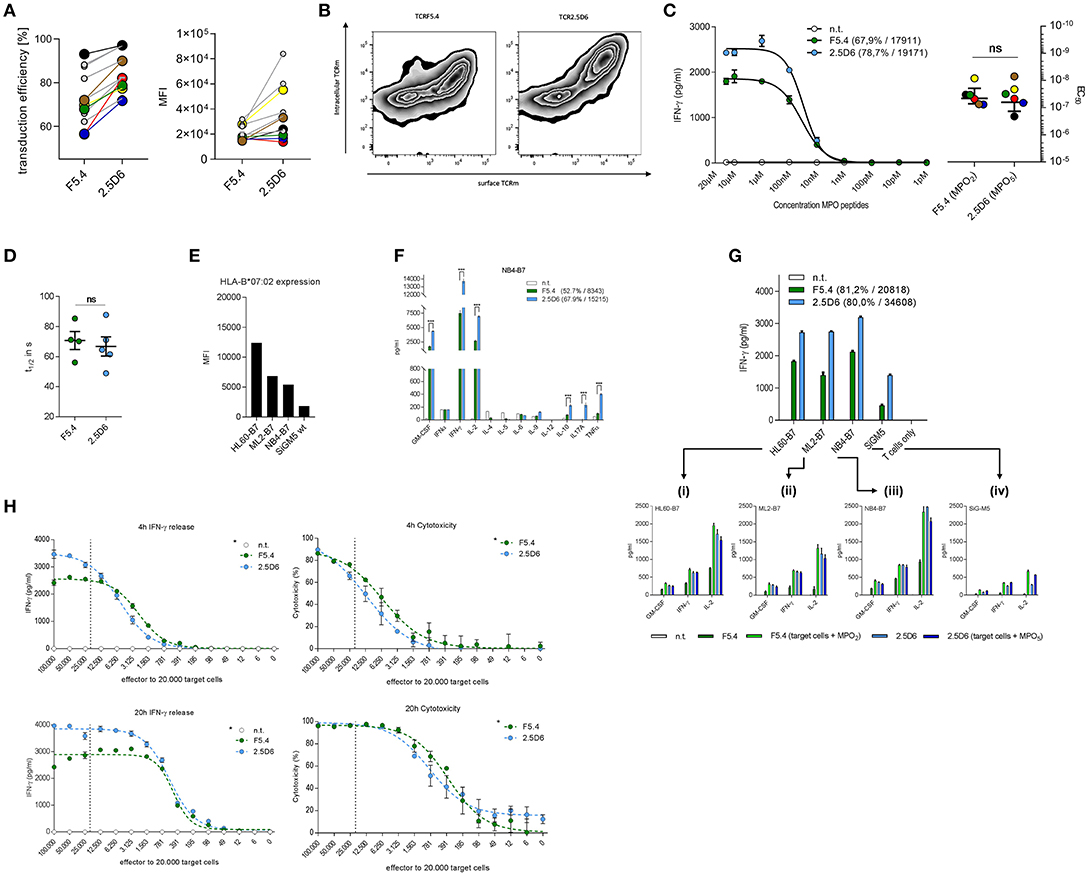
Figure 1. TCR surface expression and quality of cytokine release differs between TCRF5.4- and TCR2.5D6-transduced T cells. (A) Transduction efficiency measured by flow cytometry analysis of surface expression (left) and MFI (right) of TCRF5.4 (green) and TCR2.5D6 (blue). Connections between data points symbolize couples of the same recipient T cells used for TCR transduction (n = 11). Colored and bigger data points represent pairs of T cells that were also used for the functional avidity analyzes in (C). (B) Surface and intracellular anti-TCRm staining of both TCR (X-axis: surface TCRm signal, Y-axis: intracellular TCRm signal) measured by flow cytometry (n = 2). (C) Functional avidity of TCRF5.4- or TCR2.5D6-transduced PBMC analyzed in response to KG1a-B7 pulsed with graded amounts of MPO2 or MPO5 at an E/T ratio of 1:1 after 20 h of co-incubation. Half-maximal IFN-γ release was calculated using logarithmic dose-response fitting algorithm with variable slope (EC50) of GraphPad Prism. Mean ± SD of triplicates of one representative experiment are shown (n = 6). Colored data pairs represent T cell pairs also used in (A). (D) Flow cytometry based koff-rate measurements of TCRF5.4 (n = 4) and TCR2.5D6 (n = 5). The dissociation half-lives calculated by one-phase decay algorithm using GraphPad Prism are shown for both TCR. (E) MFI of HLA-B7 expression of selected AML cell lines measured by flow cytometry. (F) Multi-cytokine release of TCRF5.4 or TCR2.5D6 transduced PBMC in response to the HLA-B7-transgenic AML cell line NB4. NB4 is representatively shown for all 4 tested AML cell lines HL60, ML2, and SiG-M5. Standard deviations of the mean of triplicates are shown (n = 2 for each cell line) (G) IFN-γ secretion by TCRF5.4 or TCR2.5D6 transduced CD8+ TCM in response to AML-cell lines with endogenous MPO expression measured by ELISA. 10.000 effector cells were used in a ratio of 1:1. Standard deviations of the mean of triplicates are shown (n = 4). (Gi−iv) Cytokine secretion (GM-CSF, IFN-γ, and IL-2) measured by flow cytometry based multiplex analysis in response to selected AML cell lines is shown in triplicates either unpulsed or additionally pulsed with MPO2 or MPO5. (H) IFN-γ release (left panels) and cytotoxicity (right panels) of TCRF5.4- or TCR2.5D6-transduced CD8+ TCM against NB4-B7 cells analyzed for E/T titrations ranging from 5:1 to 0.0031:1 using a constant target cell amount of 20.000 for different periods of co-cultivation (4 and 20 h). The dashed line in each graph represents theoretically an E/T of 1:1. The percentage of killing was calculated using absolute counts of remaining NB4-B7 target cells normalized to non-transduced T cells by flow cytometry (n = 2, *81.2% transduction efficiency for TCRF5.4, 80.0% for TCR2.5D6, MFI TCRF5.4 = 20818, TCR2.5D6 = 34608). (C,G,H) Non-transduced TCM were used as negative controls and standard deviations of the mean of triplicates are shown if not otherwise stated. Transduction efficiency and MFI of both TCRs are either bracketed or referred by an asterisk (*) to the legend. IFN-γ ELISA was performed using supernatants of co-incubations (C,G,H) and multiplexed cytokine analysis by flow cytometry (F,Gi−iv). (D) Mann–Whitney test: ns: not significant (p > 0.05), (F) Multiple t-test with false discovery rate (FDR) of 1%, ***p < 0.001.
Subsequently we investigated the functional avidity of both TCRs toward p-MHC complexes on MPO-negative, HLA-B7-transgenic KG-1a cells. No significant difference in EC50 values was observed between both TCR-transduced T cells (Figure 1C, left). The colored data points in Figures 1A,C symbolize the same pairs of transgenic T cells of different TCR-transductions used for the measurements. Our data show that the EC50 values were comparable for both TCR independent of the differences in TCR expression. Similarly, koff-rate experiments analyzing the dissociation time of both TCRs from the p-MHC complex were also comparable for both TCR (Figure 1D). Thus, functional avidity and koff-rate analyses revealed similar results despite differences of TCR-expression and surface density levels.
For the following characterization of both TCRs in co-incubation assays, we used MPO high expressing AML cell lines HL60, ML2, and NB4 (transgenic for HLA-B7) and SiG-M5 (endogenous HLA-B7 expression). HLA-B7 surface expression analysis revealed different levels of HLA-B7 within these cell lines (Figure 1E). First, we used the AML cell lines to investigate an extended cytokine profile of TCRF5.4- and TCR2.5D6-transduced PBMCs. Here we observed differences in the quantity but also quality of cytokine secretion between both TCRs (Figure 1F). The results, representatively shown for NB4, demonstrate increased amounts of IFN-γ, GM-CSF, and IL-2 for TCR2.5D6-transduced PBMCs as well as significantly enhanced secretion of IL-10, IL-17A, and TNFα compared to PBMCs transduced with TCRF5.4. Focusing on CD8+ T cells in subsequent assays, all four tumor cell lines with endogenous MPO expression were recognized consistently by TCRF5.4- and TCR2.5D6-transduced T cells, respectively (Figure 1G). Thereby, TCRF5.4-transduced CD8+ T cells released substantially lower amounts of IFN-γ, GM-CSF, and IL-2 compared to TCR2.5D6-transduced CD8+ T cells in experiments with equal TCR transduction rates but again reduced TCR expression levels. However, these differences could be compensated by additional MPO2 peptide pulsing (Figure 1Gi−iv). In contrast, pulsing of three out of four cell lines with peptide MPO5 did not have a major impact on cytokine secretion by TCR2.5D6-transgenic T cells. Our data indicate that HL60, ML2, and NB4 displayed saturating conditions of endogenously expressed MPO5 eliciting maximal reactivity of TCR2.5D6 while that was not the case for MPO2 and TCRF5.4. In case of SiG-M5 with lowest HLA-B7 expression, cytokine secretion was increased for both MPO-peptides after additional peptide-pulsing.
In subsequent analyses we investigated the impact of effector to target ratio titrations (E/T) on IFN-γ release and cytotoxic capacity of T cells transgenic for both TCR. Again, we observed differences at high E/T ratios with respect to quantity of IFN-γ release (Figure 1H, left panels). However, only minor differences were seen for cytotoxicity of both TCR-transduced T cells after 4 and 20 h of co-incubation (Figure 1H, right panels). At lower E/T ratios the IFN-γ secretion was comparable for both TCR and estimated half maximal E/T ratios for IFN-γ secretion as well as cytotoxicity were slightly favorable for TCRF5.4 at both time points.
TCR2.5D6 Shows Superior Tumor Control in vivo Compared to TCRF5.4
Subsequently, we tested both TCRs using the NB4-tumor model in immune deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (28) to assess whether TCRF5.4 could compete with TCR2.5D6 in vivo.
For initial in vivo experiments, we used TCR-transgenic CD8+ TCM and traced the growth of subcutaneous established tumors in mice that received either TCRF5.4-, TCR2.5D6- or not transduced T cells. The immunogenicity of the T-cell products was confirmed by functional in vitro tests prior to intravenous T-cell injection 8 days after tumor inoculation. The mice were sacrificed 7 days later for ex vivo analyses. Both TCR show highly comparable rejection capacity of NB4-B7+ tumors in comparison to control tumors (Supplementary Figure 2A) also reflected in significantly reduced tumor weights compared to the non-transduced T cell group (Supplementary Figure 2B). The percentage of CD3+ T cells found ex vivo within NB4-B7+ tumors was similar and significantly enhanced compared to the non-transduced T-cell group and NB4-B15+ control tumors (Supplementary Figure 2C). Investigation of T-cell distribution revealed slightly enhanced presence of TCRF5.4-T cells in blood and lung compared to TCR2.5D6-T cells (Supplementary Figure 2D).
To assess the long-term survival of mice, we performed transfer experiments with T-cell products that were comparable both in TCR-transduction rate and TCR surface density expression (Figure 2A). Analyzing T cells from this expanded transduction before injection confirmed results from previous experiments with respect to IFNγ secretion and cytotoxicity independent of the equal TCR transduction rate and expression level (Figure 2B). Using our in vivo mouse model, we observed a significant improved tumor control (Figure 2C) and median survival (Figure 2D) in TCR2.5D6-treated mice (46 days) compared to TCRF5.4 (28 days). Ex vivo analysis of tumors discovered a complete loss of HLA-B7eGFP expression of tumor cells within the TCR2.5D6-group, while the TCRF5.4-group retained the HLA-B7eGFP expression (Figure 2E). We analyzed again the T-cell distribution within each mouse (Figure 2F) and observed only few TCR-transgenic T cells in the BM, blood, lung, spleen or the tumor of the TCR2.5D6 group. In contrast, in the TCRF5.4 group, we detected TCR-transgenic T cells in the blood, lung and the spleen but not within the tumor. These results demonstrate superior tumor control of TCR2.5D6 T cells as well as different forms of tumor escape in both groups.
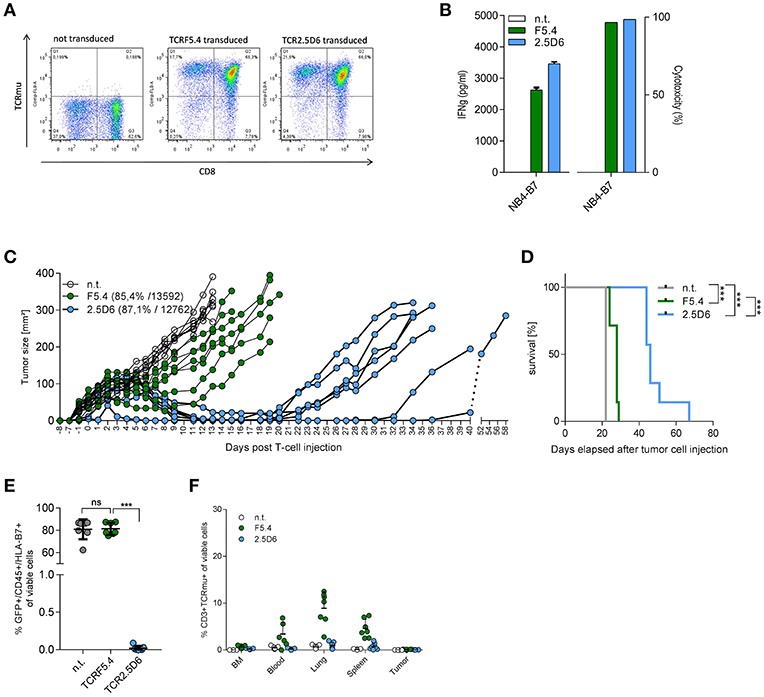
Figure 2. TCR2.5D6-transduced T cells show superior in vivo tumor killing efficacy. (A) FACS analysis of transduction efficacy for TCRF5.4- and TCR2.5D6-transduced T cells used for the long-term mouse experiment. The plots show pre-gated living CD3+ cells. X-axis represents the CD8 marker and Y-axis represents the TCRm+ T-cells. (B) Functionality of TCR-transduced T cells used for the long-term survival experiment of mice shown in (C) measured by IFN-γ release (right) and cytotoxicity (left) after co-cultivation for 24 h with NB4-B7 tumor cells in an E/T ratio of 1:1 in vitro. (C) Growth kinetic of NB4-B7eGFP tumors in NSG mice for a period of 58 days. Each line represents an individual tumor growth curve per mouse: n.t (n = 7), TCRF5.4 (n = 7), TCR2.5D6 (n = 7). (D) Kaplan Meier curve of mice shown in (C), ***p ≤ 0.0002. (E) Percentage of GFP+/HLA-B7+/NB4-B7eGFP tumors cell analyzed ex vivo by flow cytometry at individual time points after decease of mice shown in (C). Non-transduced (n = 7), TCRF5.4-transduced (n = 7) or TCR2.5D6-transduced T cells (n = 7), nsp ≥ 0.05, ***p = 0.0006. (F) Percentage of CD3+/TCRm+ living cells in BM, blood, lung, spleen, and tumor analyzed ex vivo by flow cytometry at individual time points after T-cell injection after decease of mice shown in (C). n.t (n = 7), TCRF5.4-transduced (n = 7) or TCR2.5D6-transduced T cells (n = 7). (C) Transduction efficiency and MFI of both TCRs are bracketed. (E) Significances are calculated by Mann-Whitney Test. (D) Survival statistics are calculated by Log-rank (Mantel-Cox) test.
Impact of p-MHC Interactions on T-Cell Recognition by Both TCRs
The discrepancy in tumor control observed for both TCR-transduced T-cell groups in vivo despite similar functional avidity and koff-rate was not simply explainable by differences in TCR expression as this was comparable within the long-term in vivo experiment. We therefore focused on peptide MHC (p-MHC) interactions potentially involved in differential function and investigated peptide affinity and HLA-stability of p-MHC binding for both MPO peptides in vitro as well as in silico. In case of the octamer MPO2 we performed additionally pulsing experiments with the N-terminal (LTPAQLNVL = MPO2+L1) and C-terminal (TPAQLNVLS = MPO2+S9) prolonged variant and detected IFN-γ secretion by TCRF5.4-transduced T cells in response to MPO2+L1 but not for MPO2+S9 (Figure 3A). Binding predictions of the MPO peptides were applied for either sequence-based (19, 22, 62) or stability-based predictions (20) (Table 1). In sequence-based binding affinity predictions, MPO2 was outperformed by MPO5. However, considering HLA-complex stability prediction the MPO2-MHC complex seemed to be more stable compared to the MPO5-MHC complex. Affinity and HLA-stability predictions for MPO2+L1 indicated weak performance in both cases.
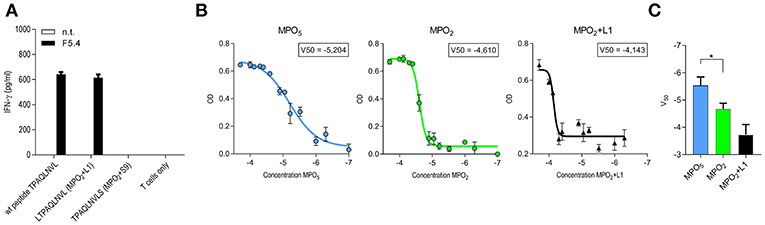
Figure 3. MPO5-HLA-B7 complex is superior in comparison to MPO2-HLA-B7 with respect to stability and binding quality. (A) IFN-γ release by TCRF5.4 together PBMC in response to HLA-B7+ LCL1 cell line pulsed with MPO2 or the non-americ counter partners LTPAQLNVL (MPO2+L1) or TPAQLNVLS (MPO2+S9), respectively. An E/T ratio of 1:1 was used. IFN-γ secretion of the supernatants after co-cultivation was measured by IFN-γ ELISA. Standard deviations of the mean of triplicates are shown (n = 2). (B) Beta-2-microglobulin (B2M)-ELISA of UV-mediated peptide exchanged HLA-B7 monomers. V50 values of a Boltzmann fitting for each MPO-peptide was determined by the optical density of B2M after degraded amounts of MPO-peptides were used for HLA-stabilization. Standard deviations of the mean of triplicates are shown (n = 3). (C) Pooled results of (B) for MPO5 (n = 4), MPO2 (n = 4), and MPO2+L1 (n = 3). Significance is calculated by Mann-Whitney Test: *p = 0.0286.
To verify the stability results of all three MPO-peptides experimentally, we performed HLA-B7 UV-peptide exchange assays with titrated amounts of peptide added to a constant concentration of HLA-B7 monomers bound to a UV-sensitive peptide. Our data showed that MPO5 is superior to MPO2 with almost one log increased potential to rescue the HLA-B7 molecule (Figures 3B,C; Table 1) while MPO2+L1 had the lowest potential for HLA-stabilization. Thus, our results oppose the results of the NetMHC stability prediction. As a second experiment, we performed thermal shift assays for the MPO peptides to characterize their potential to prevent denaturation of the HLA-complex with ascending temperature (Supplementary Figure 3A; Table 1). Interestingly the MPO2- and MPO5-HLA complex heat-associated denaturation is very similar, as the half maximal melting temperature (V50) for both peptides, calculated by Boltzmann fitting, was almost identical (46°C). The MPO2+L1 complex is more sensitive to heating (44°C) while the pp65(417−426) peptide, taken as a control, with the highest predicted peptide affinity also has the highest temperature stability (50°C). However, the decay of the MPO HLA-complexes over time at constant 37°C revealed, similar to our UV-exchange and in silico sequence-based affinity prediction results, that MPO5 had a better stabilizing effect on the MHC complex (4.1 h) compared to MPO2 (3.5 h; Supplementary Figure 3B; Table 1). In contrast to stability predictions, we clearly see differences in peptide and HLA-complex dissociation at physiological temperature indicating that MPO5 has a longer koff-rate compared to MPO2.
To investigate whether the peptides are differentially presented on the surface of various target cell lines, we analyzed the surface presentation of both peptides directly on a panel of AML cell lines. We used a targeted MS approach using heavy labeled counterparts of the MPO-peptides as a reference within each sample. In fact, we observed large differences in the detection of both MPO peptides suggesting MPO2 to be markedly lower presented on the surface of all AML cells lines compared to MPO5 (Supplementary Figure 3C). In addition, there was a similar relative intensity of surface peptide presentation between the different cell lines with SiG-M5 showing the lowest peptide presentation corresponding to the lowest HLA-B7 density and recognition by both TCRs (Supplementary Figure 3C; Figure 1E). However, technical issues need to be considered as MPO2 may not be efficiently ionized based on its amino acid sequence. In contrast, the amino acid sequence of MPO5, especially the arginines at positions 3 and 8, favor the detection by MS. Thus, comparison of quantitative presentation of both peptides by MS may have limitations.
Modeling of MPO-Peptide HLA Ligands Confirms Stable HLA Peptide Binding and Reduced Fluctuation of MPO5 Within the Binding Cleft Correlating With Reduced Cross-Reactivity
For detailed analyses of MPO2/5-HLA-B7 binding properties, we generated structural models of the complexes and performed molecular dynamics (MD) simulations for a total time of 450 ns for each system. Figures 4A,B depict a representative MD conformation of MPO2/5-HLA-B7 complexes and Table 2 presents an alignment of both peptides according to structurally equivalent residue positions. Our results show that MPO2 features no structurally equivalent residue for W4 in MPO5 and that the TCR accessible residues are Q4, N6, V7 in MPO2, and W4, D5, E7, R8 in MPO5, respectively (Figures 4A,B; Table 2; violet residues). Hence, these residues are prone to affect TCR binding. Figures 4C,D highlight the steric and electrostatic properties of these solvent-exposed residues. All other MPO2/5 residues are oriented toward the HLA-B7 peptide binding cleft (Figures 4A,B; Table 2; orange residues). Of note, strong differences could be observed by analyzing the conformational stability of both peptides in the bound complex. MPO5 forms a stable complex structure throughout all MD simulations. MPO2, however, shows an overall stronger conformational flexibility, especially toward the N-terminus (Figures 4E,F), indicating a less stable binding to HLA-B7. This fits with our Molecular Mechanics/Generalized Born Surface Area (MM/GBSA)-based binding affinity estimations in which the affinity of MPO5 is stronger for HLA-B7 compared to MPO2 (Δ: −112.67 kcal mol−1 with σ = 20.55 and = 0.35, Δ: −88.02 kcal mol−1 with σ = 9.52 and = 0.16, σ: standard deviation, : standard error of the mean). The superior binding of MPO5 can be explained by stronger enthalpic stabilization of the complex as it forms on average 2.25 hydrogen bonds more than MPO2 (Table 3; upper region). MPO5 is especially stabilized via hydrogen bonds of N1, R3, W4, and R8 compared to equivalent residues of MPO2 (lower region of Table 3; Supplementary Table 2). Particularly R3 of MPO5 is anchored strongly at the bottom of a deep pocket of the HLA-B7 peptide binding cleft via hydrogen bonds of its guanidino group with HLA-B7 residues S97 and D114 (see Figure 4B; green inlay). MPO5 shows pronounced flexibility only at position G6 (Figure 4E), which is strongly reduced by Ala- or Thr-exchange at this position (G6A or G6T; Supplementary Figure 4B, yellow inlay). This leads to stronger p-MHC binding affinities (Δ: −112.94 kcal mol−1 with σ = 12.40 and = 0.20, Δ: −118.28 kcal mol−1 with σ = 11.51 and = 0.19) as well as to higher IFN-γ release of TCR2.5D6-transduced T cells (27). In contrast, MPO2 binding is characterized by high flexibility distributed over several residues (Figure 4E). This multi-flexibility of the peptide correlates to the reduced HLA-stability seen in our UV-exchange and thermal shift assays and the weaker p-MHC binding affinity and constitutes one conceivable explanation for the reduced surface presentation, compensable by external peptide pulsing (Figure 1G) as well as the inferiority of TCRF5.4 in our in vivo experiments. However, structural features that might explain why MPO2 still binds well enough, are (i) its residue L5 (Figure 4A, green inlay) as it sterically fits into the central space of the HLA-B7 peptide binding cleft, (ii) N6, which forms stable hydrogen bonds with E152 (Table 3; Supplementary Table 2), and (iii) the central residues Q4 and L5 are located deeper in the HLA-B7 peptide binding cleft, leading to a stronger stabilization of these residues via hydrogen bonds (Table 3; Supplementary Table 2). Further MD simulations of HLA-B7 complexes comprising Ala/Thr variants of MPO2/5 supporting the effects described here are presented in Supplementary Figures 4A,B indicating that also TCR cross-reactivity may be implicated by peptide binding. Summarized, our p-MHC modeling strongly promotes our experimental results and revealed reasons for the weaker MPO2-HLA-B7 binding quality and in turn why MPO5 is superior in this regard.
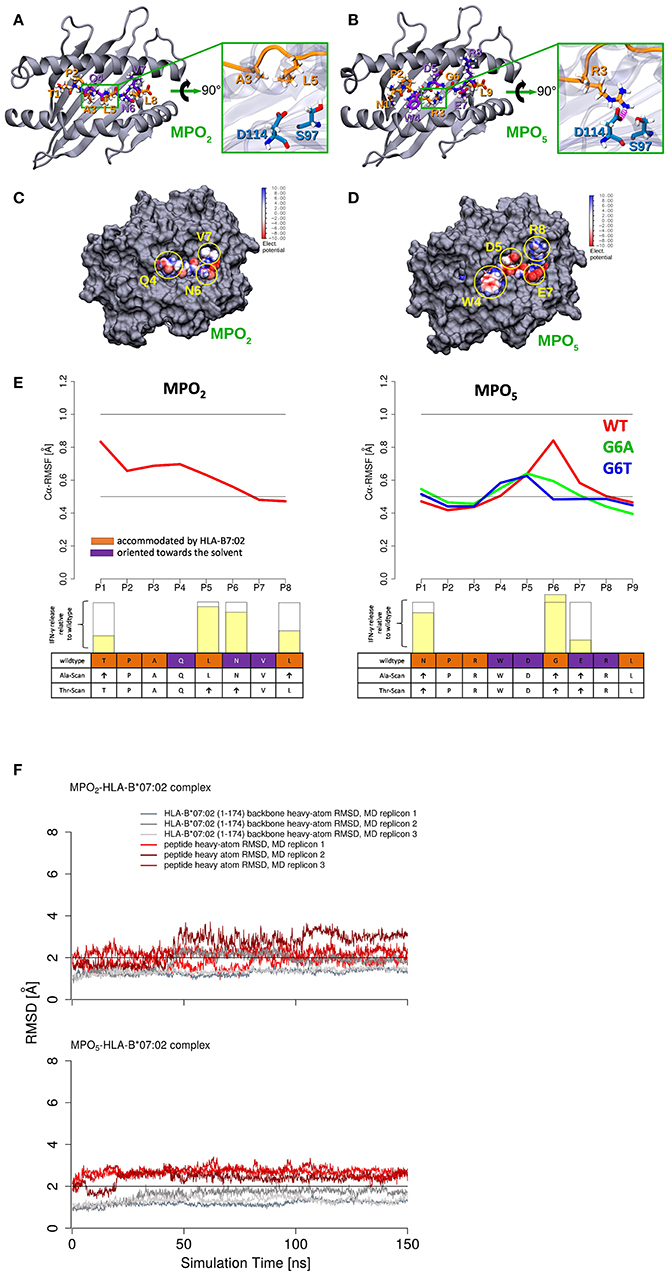
Figure 4. MPO2/5-HLA-B7 structure-based modeling revealed less fluctuation of MPO5 within the HLA-binding cleft compared to MPO2. (A,B) Representative conformation of MPO2/5 in the bound state. The HLA-B7 protein is shown as cartoon representation (gray), the bound peptides are depicted in stick representation and are highlighted: (hydrogen atoms: white, nitrogen atoms: blue, oxygen atoms: red, carbon atoms: orange or violet), solvent-exposed peptide residues (violet carbon atoms), peptide residues accommodated by the MHC peptide binding cleft (orange carbon atoms). In the green inlays, the HLA-B7 residues S97 and D114 (blue sticks) are shown for a direct comparison of major differences in the MPO2/5 HLA-B7 binding modes and anchoring to the HLA-B7 peptide binding cleft. Peptide backbones are shown in orange cartoon representation. (A) The green inlay depicts the MPO2 residues A3 and L5, accommodated by the MHC peptide binding cleft. (B) The green inlay depicts the MPO5 residue R3. Hydrogen bonds are highlighted in magenta. (C,D) Space filling models showing the TCR interface of the MPO2/5-HLA-B7 complexes. Protein is depicted in gray and the bound peptides are depicted as van der Waals (vdW) surface colored according to their electrostatic potential (white: neutral areas, red: negatively charged areas, blue: positively charged areas). Solvent-exposed and TCRF5.4- or TCR2.5D6-accessible residues of the MPO2/5-HLA-B7 complexes are highlighted with yellow circles, respectively. (E) Calculated root mean square fluctuations (RMSF) of Cα atoms of the HLA-B7-bound MPO2/5 peptides (red) and G6A (green)/G6T (blue) variants of MPO5 (average of three independent MD simulations). The individual TCR-recognition motif from the alanine- and threonine- (Ala/Thr)-scans is shown together with the IFN-γ release (transparent yellow bars) observed in the Ala- or Thr-scan (Supplementary Figures 1J,K) relative to individual wildtype IFN-γ secretion (empty bars) below the RMSF plots. The height of the empty bars reflects the maximal IFN-γ release elicited by the wildtype peptide, ↑ = interchangeable amino acid residues. The Ala/Thr-scan for MPO5 is described elsewhere (27). (F) Root mean square deviation (RMSD) of HLA-B7 protein backbone heavy atoms and MPO2/5 heavy atoms during MD simulations calculated with respect to the initial energy minimized peptide-HLA-B7 complex models.
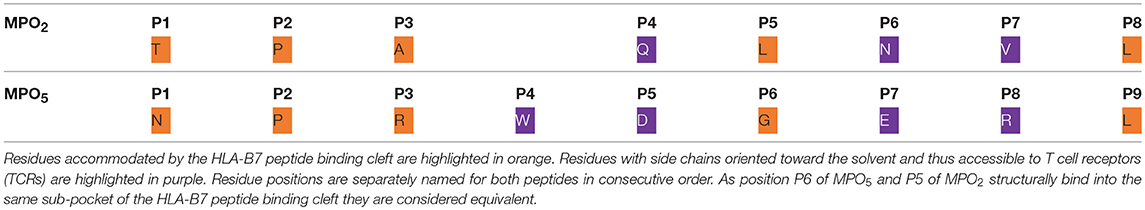
Table 2. Alignment of the MPO2 octamer and the MPO5 nonamer peptides according to their HLA-B7-bound structures.
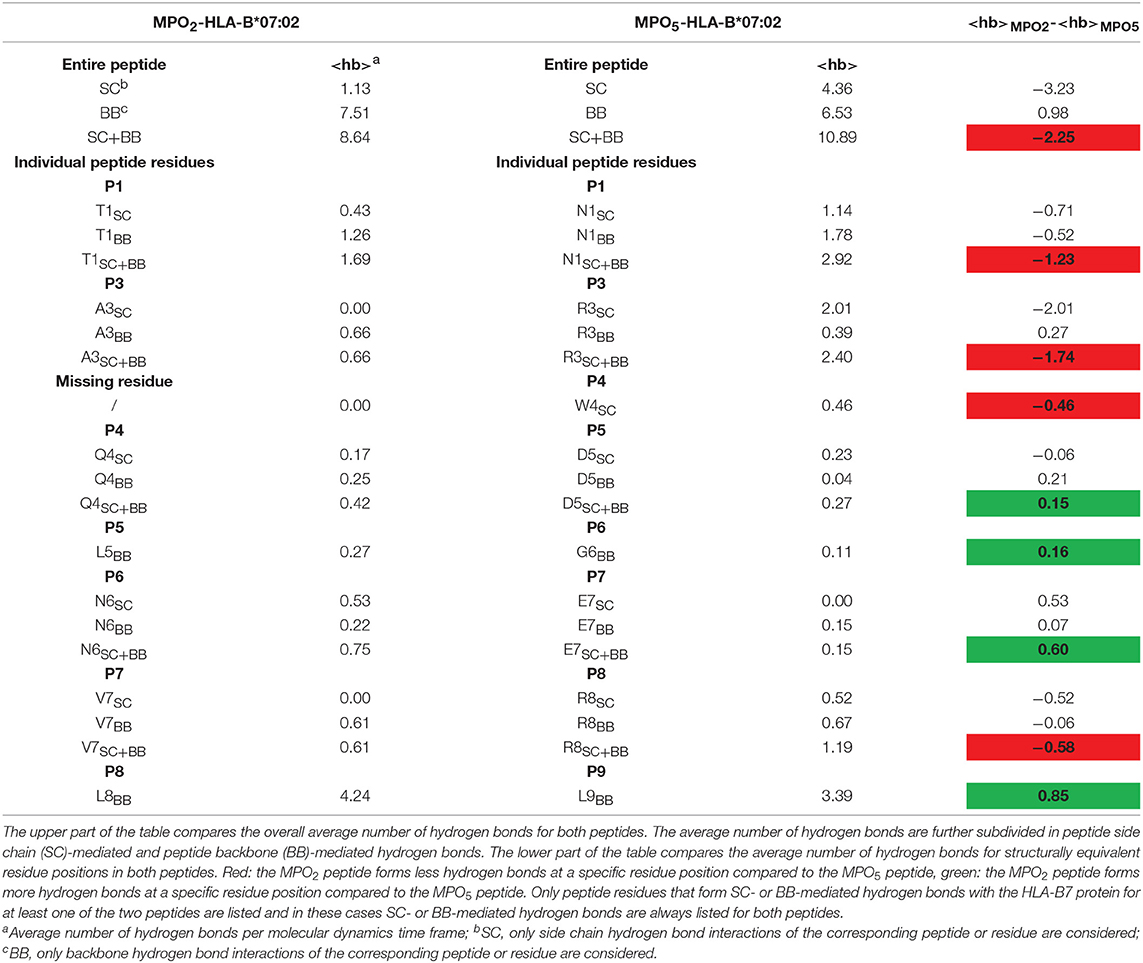
Table 3. Comparison of the average number of peptide-HLA-B7 binding cleft hydrogen bonds observed during the MD simulations for the bound MPO2/5 peptides.
Discussion
We here compared two MHC-mismatched allorestricted TCR with specificity for two different MPO-derived peptide ligands presented by HLA-B7 and identified by MS immunopeptidomic analyses of tumor samples derived from patients with MPN. Such TCR may be used in the context of allogeneic stem cell transplantation in a haplo- or sHLAm setting as well as alternatively within a conditioning regimen using TCR-transgenic T cells. A novel allorestricted TCR (TCRF5.4) demonstrating high specificity for a new HLA-B7-restricted MPO2-derived peptide was compared by in-depth functional characterization with the previously identified TCR2.5D6 (27) in order to define characteristics essential for effective tumor rejection. In fact, both TCRs differed strongly in their tumor rejection capacity in our long-term in vivo experiments using transduced T cells with comparable TCR expression demonstrating TCR2.5D6 to be clearly superior to TCRF5.4. The group receiving TCRF5.4-transgenic T cells was not able to completely reject the tumor and experienced an early relapse. In contrast, tumor eradication was more effective in the group of TCR2.5D6, although also these animals relapsed. However, tumor escape mechanisms were different in both groups. Whereas, tumors in mice treated with TCR2.5D6-transduced T cells lacked HLA-B7 and lost all TCR-transgenic T cells, TCRF5.4-transgenic T cells could be detected in all examined tissues except in tumors with preserved HLA-B7 expression. Loss of HLA-B7 indicates the immunogenic pressure induced by TCR2.5D6-transduced T cells. In contrast, although TCRF5.4-transgenic T cells have well engrafted, our experiments indicate that tumor escape relies more on peptide and/or TCR-intrinsic qualities resulting in T-cell exclusion from the tumor.
As in vivo experiments are highly laborious, it is desirable to define in vitro assays most effective in predicting in vivo activity. Numerous in vitro assays have been proposed to reflect strength of T-cell response correlating with tumor rejection capacity in vivo (8–10, 12, 17, 18, 63) although these analyses have been mainly tested in the autologous setting. In fact, many of these TCR-dependent functional assays showed comparable results for both TCR in our experiments and therefore failed to predict in vivo outcome in the allorestricted setting.
Focusing on differences between our allorestricted TCRs we observed in almost all TCR transductions reduced quantitative and qualitative surface expression for TCRF5.4. Although the quality of TCR expression is critical for T cell performances and endogenous TCR chains can influence the function and presentation of transgenic TCR (16), the TCRF5.4 represents a generally well-expressed TCR. Furthermore, for the detailed comparisons of both TCRs we used freshly isolated T cell subpopulations from different healthy donors for several reasons. (i) First, this minimize donor-dependent effects of TCR-transgenic T cells that would rule out general statements for the observed functional qualities. (ii) Second, the use of freshly isolated donor T cells ensures optimal T-cell fitness and reduces the impact of T-cell exhaustion because of freeze and thaw cycles, strenuous selection processes, and/or too long culture periods. (iii) Third, large numbers of TCR-transductions using different donor T cells give the opportunity to estimate possible donor-related heterogeneity in results or, as observed in our case, a reliable robustness and consistency of T cells transduced by TCRF5.4 and TCR2.5D6. Furthermore, TCR-transgenic T cells and HLA-transgenic tumor cell lines were used without additional adjustment of TCR- or antigen quality after transduction. Hence, our results include individual characteristics of both TCRs that would also be present in clinical settings in the T-cell products for adoptive transfers. However, for our significant long-term in vivo experiments we selected T-cell products for both TCR with comparable quantitative and qualitative TCR expression and still observed inferior tumor rejection capacity of TCRF5.4-transduced T cells. These results indicate that differences in TCR expression may not represent a decisive criterion for TCR selection in our case. Especially, we could not confirm any correlation of in vivo anti-tumor reactivity to dissociation half-lives of TCRs from their p-MHC complexes (12, 64) as well as functional avidity of TCRs toward the cognate MHC-peptide complexes (9). This could be explained by specific features of MHC-mismatched TCRs as they have alternative binding properties to the mismatched MHC (65) compensating for lower affinity of the peptide toward MHC as well as the TCR toward peptide.
Differences in quantitative and qualitative cytokine secretion, however, revealed to be the most significant indicator for reduced in vivo performance seen in almost all in vitro analyses with differing but also equal TCR expression potentially explaining inferiority in tumor rejection (66, 67). In contrast, TCRF5.4-transduced T cells were competitive to those transduced with TCR2.5D6 with respect to in vitro cytotoxicity being in line with previous observations that this functional quality may not represent the most sensitive test for T-cell responses even with differing TCR expression rates (68) indicating once more that TCR expression is not the most indicative factor for TCR functionality.
In contrast, our data support that peptide presentation on the surface of tumor cells may represent a key factor for effective tumor rejection. In fact, although the MPO antigen expression in this work was every time identically for both TCR conditions as the targeted tumor cell lines were the same, the MPO-derived target peptide presentation for MPO2 and MPO5 was obviously different. Our results indicate a reduced peptide binding of MPO2 to HLA-B7 when comparing both peptides by either UV exchange assays, thermal shift assays or predicted HLA-affinity suggesting that reduced peptide binding toward HLA-B7 plays a major role in inferior tumor rejection. Although the absolute quantification of peptides on the surface of target cells by MS harbors major limitations, our MS data may support an inferior MHC presentation of MPO2 compared to MPO5 due to its highly reduced detection. Our data thereby confirm data from Engels et al. highlighting the role of peptide binding to tumor rejection (18), here in an MHC-mismatched setting.
To further deepen our understanding of p-MHC binding, we applied structure-based modeling and atomistic MD simulations and realized the potential of molecular modeling and simulation to rationalize the results of our peptide stability and amino acid substitution assays on a molecular level. We thereby not only confirmed UV exchange assays as well as sequence-based affinity prediction but also discovered MPO5 to be more rigid within the peptide binding cleft due to better stabilization of amino acid residues by hydrogen bonds at individual positions in comparison to MPO2. This finding, in fact, may be decisive for a high specificity of a selected TCR as more flexible amino acid residue positions of the peptide may impede stable binding of the TCR to peptide MHC. Indeed, we can explain by Cα atoms fluctuations especially for the MPO5 peptide that the binding quality of respective amino acid residues in their HLA-binding pocket favors a more stable peptide binding. The high flexibility in position 6 is markedly reduced after exchange by alanine or threonine indicating stabilization of the peptide toward HLA-B7. Importantly, reactivity of the TCR does not depend on the glycine within the wildtype peptide and is even enhanced after stabilization of the peptide by alanine or threonine. In contrast, the enhanced fluctuation observed for several amino acid residues of the MPO2 peptide correlates with overall less formed hydrogen bonds between the p-MHC complex compared to MPO5 and more flexibility of p-MHC recognition within the alanine and threonine scan. Taken together, our data extend previous publications (69) and indicate, that for cross-reactivity testing at least two different amino acids for amino acid substitution assays are necessary to assess possible TCR-binding motifs.
Moreover, there seems to be a correlation between the described flexibility of p-MHC binding as analyzed by structure-based modeling and the cross-reactivity of TCRF5.4 as identified by its observed flexibility to amino acids exchanges of the peptide, the high number of peptides predicted to be recognized by TCRF5.4 by ScanProsite as well as its MHC-cross-reactivity toward HLA-B51 which is in contrast to the observations made for TCR2.5D6. In this regard the rigidness evaluation of peptide-HLA binding might have the potential to serve as an appropriate tool to predict cross-reactivity. Although structure-based modeling of p-MHC complexes can provide valuable insights into the molecular basis of TCR-p-MHC recognition, this strongly depends on the availability of experimental structural data of p-MHC either of the HLA to be investigated or of close homologs. As the latter is often the case for many MHC proteins, homology modeling approaches can nowadays be applied to construct MHC protein models for most HLA-alleles. Bound peptides can be modeled via side chain placement/mutation algorithms using MHC-bound peptide backbone coordinates from X-ray structures as template. As peptide binding is determined by the structural and chemical characteristics of the binding cleft of the specific MHC allele, binding features have to be analyzed on an allele-specific basis. For this, standard analysis approaches, which are readily available for most MD-simulation packages, can be applied, such as hydrogen bond analysis (for, e.g., hydrophilic binding pockets) or residue contact maps (for, e.g., hydrophobic binding pockets) or combinations thereof.
Although the number of TCRs studied here is limited and more TCR candidates need to be tested to further prove our conclusions, our integrated view in TCR functionality and p-MHC binding suggests the quality and quantity of cytokine release and the characteristics of p-MHC binding as most relevant factors to be analyzed for a fast and efficient TCR identification procedure in the allogeneic MHC-mismatched repertoire. We conclude that an interdisciplinary approach between in silico-, T-cell-, and structural analyses is most promising to address the challenges of identification of TCR with high efficacy and safety profiles. We propose a curtailed procedure to identify allorestricted TCRs presumably also applicable to be used for other settings as for autologous TCR specific for other tumor-associated antigens and neoantigens (Figure 5). The final aim of this workflow is to identify efficiently TCR candidates for clinical translation to enable personalized immunotherapies with high tumor reactivity and a favorable safety profile.
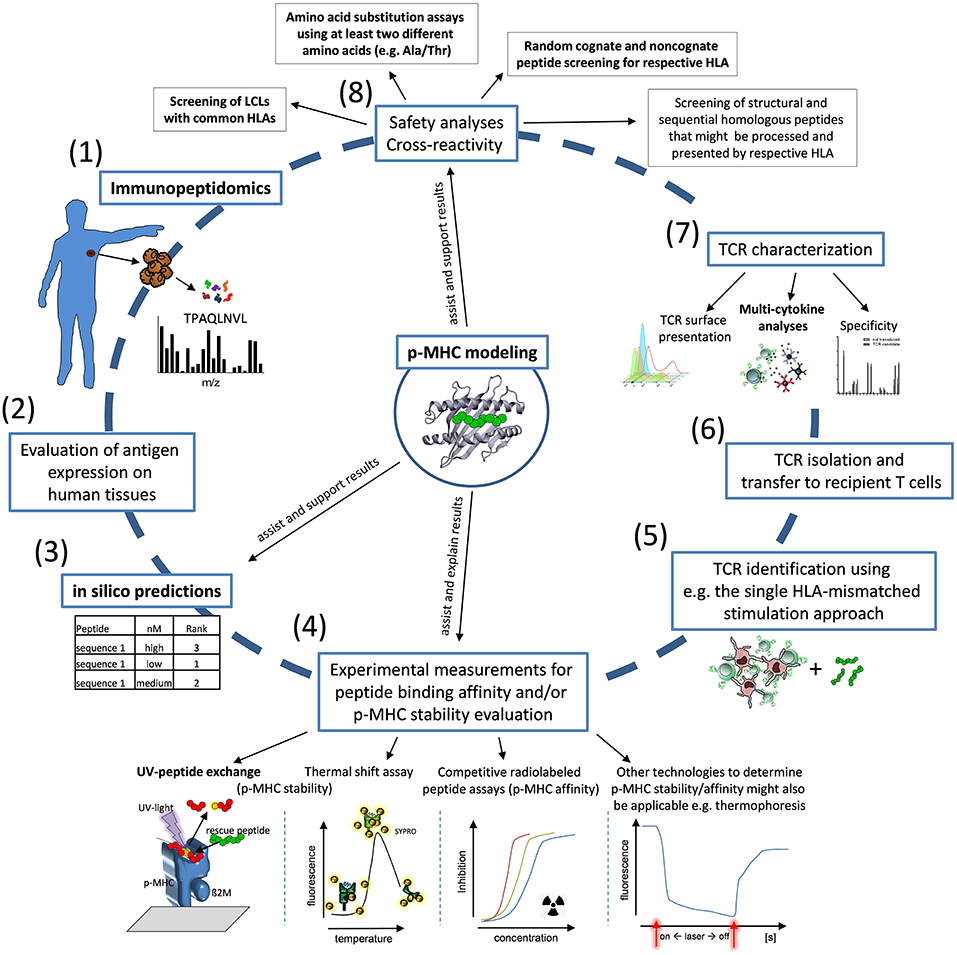
Figure 5. Workflow for identification of allo-MHC-restricted tumor-reactive TCRs. Proposed workflow for TCR identification: (1) Immunopeptidomics of patient samples and tumor material; (2) Evaluation of antigen expression on human tissues; (3,4) in silico prediction of candidate peptides for first ranking and experimental validation by HLA-affinity and/or HLA-stability assays in combination with p-MHC modeling; (5,6) TCR identification and TCR isolation; (7) TCR characterization with special interest to TCR surface presentation, cytokine secretion and specificity; (8) Characterization of the safety profile of TCR-transgenic T cells using LCL cell lines, amino acid substitution experiments and screening of homologous peptides in combination with structure-based modeling approaches. Critical assays for the identification of suitable TCR and peptides used in our analyses are highlighted in bold characters.
Ethics Statement
This study was carried out in accordance with the recommendations of conventional institute guidelines. The protocol was approved by the local authorities.
Author Contributions
SA and AK: study design and conception together with RK, EB, and SM. SM and SA: in vivo characterization studies and animal experiments. RK, EB, and SA: in vitro characterization studies. MP and MM: mass spectrometry studies. MG and IA: protein modeling and in silico MPO-peptide characterization. ME and DB: TCR koff-rate measurements, provision of HLA-multimers. SM, SA, HB, NY, and EB: experimental support in subcutaneous and i.v. injection of mice, maintenance of mice. JP and SS: experimental support UV-exchange assay, provision of UV-sensitive HLA-monomers. SA, MP, MG, IA, and AK: manuscript writing. SA, EB, RK, SM, JA, ME, MP, JP, MG, HB, NY, SS, DB, MM, IA, and AK: manuscript preparation and review process. AK: study supervision.
Funding
This work was supported by grants to AK from Deutsche Krebshilfe (110281) and Wilhelm Sander-Stiftung (2015.030.1), as well as to AK and IA from Deutsche Forschungsgemeinschaft (DFG; SFB824/C10, SFB1035/A10, and CIPSM) and the Technical University of Munich and International Graduate School of Science and Engineering (IGSSE).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Stephanie Rämisch and Ciara Utsch for excellent technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01485/full#supplementary-material
Abbreviations
B2M, beta-2-microglobulin; EC50, half maximal effective concentration; IVT, in vitro transcribed; koff-rate, dissociation half-live of TCR from p-MHC complex; MD simulation, molecular dynamics simulation; MFI, mean fluorescence intensity; RMSD, root mean square deviation; RMSF, root mean square fluctuations; sHLAm, single HLA-mismatched approach; TCM, T cells enriched for the central memory phenotype; TCRm, mouse TCR-β chain.
References
1. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
2. Xue SA, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A, et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood. (2005) 106:3062–7. doi: 10.1182/blood-2005-01-0146
3. Jahn L, Hombrink P, Hagedoorn RS, Kester MG, van der Steen DM, Rodriguez T, et al. TCR-based therapy for multiple myeloma and other B-cell malignancies targeting intracellular transcription factor BOB1. Blood. (2017) 129:1284–95. doi: 10.1182/blood-2016-09-737536
4. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. (2017) 130:606–18. doi: 10.1182/blood-2016-08-732636
5. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
6. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914–21. doi: 10.1038/nm.3910
7. Speiser DE, Kyburz D, Stubi U, Hengartner H, Zinkernagel RM. Discrepancy between in vitro measurable and in vivo virus neutralizing cytotoxic T cell reactivities. Low T cell receptor specificity and avidity sufficient for in vitro proliferation or cytotoxicity to peptide-coated target cells but not for in vivo protection. J Immunol. (1992) 149:972–80.
8. Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci USA. (1996) 93:4102–7. doi: 10.1073/pnas.93.9.4102
9. Zeh HJ III, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. (1999) 162:989–94.
10. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA. (2013) 110:6973–8. doi: 10.1073/pnas.1221609110
11. Presotto D, Erdes E, Duong MN, Allard M, Regamey PO, Quadroni M, et al. Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front Immunol. (2017) 8:1564. doi: 10.3389/fimmu.2017.01564
12. Nauerth M, Weissbrich B, Knall R, Franz T, Dossinger G, Bet J, et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci Transl Med. (2013) 5:192ra187. doi: 10.1126/scitranslmed.3005958
13. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. (2013) 5:197ra103. doi: 10.1126/scitranslmed.3006034
14. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
15. Heemskerk MH, Hagedoorn RS, van der Hoorn MA, van der Veken LT, Hoogeboom M, Kester MG, et al. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. (2007) 109:235–43. doi: 10.1182/blood-2006-03-013318
16. Sommermeyer D, Neudorfer J, Weinhold M, Leisegang M, Engels B, Noessner E, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. (2006) 36:3052–9. doi: 10.1002/eji.200636539
17. Chen W, Khilko S, Fecondo J, Margulies DH, McCluskey J. Determinant selection of major histocompatibility complex class I-restricted antigenic peptides is explained by class I-peptide affinity and is strongly influenced by nondominant anchor residues. J Exp Med. (1994) 180:1471–83. doi: 10.1084/jem.180.4.1471
18. Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell. (2013) 23:516–26. doi: 10.1016/j.ccr.2013.03.018
19. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. (1999) 50:213–9. doi: 10.1007/s002510050595
20. Jorgensen KW, Rasmussen M, Buus S, Nielsen M. NetMHCstab - predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology. (2014) 141:18–26. doi: 10.1111/imm.12160
21. Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. (1994) 152:163–75.
22. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. (2016) 32:511–7. doi: 10.1093/bioinformatics/btv639
23. Antes I, Siu SW, Lengauer T. DynaPred: a structure and sequence based method for the prediction of MHC class I binding peptide sequences and conformations. Bioinformatics. (2006) 22:e16–24. doi: 10.1093/bioinformatics/btl216
24. Ranganathan S, Tong JC. A practical guide to structure-based prediction of MHC-binding peptides. In: Flower DR, editor. Immunoinformatics: Predicting Immunogenicity In Silico. Totowa, NJ: Humana Press (2007). p. 301–8. doi: 10.1007/978-1-60327-118-9_22
25. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. (2012) 18:1254–61. doi: 10.1038/nm.2883
26. Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. (2016) 7:13404. doi: 10.1038/ncomms13404
27. Klar R, Schober S, Rami M, Mall S, Merl J, Hauck SM, et al. Therapeutic targeting of naturally presented myeloperoxidase-derived HLA peptide ligands on myeloid leukemia cells by TCR-transgenic T cells. Leukemia. (2014) 28:2355–66. doi: 10.1038/leu.2014.131
28. Mall S, Yusufi N, Wagner R, Klar R, Bianchi H, Steiger K, et al. Immuno-PET imaging of engineered human T cells in tumors. Cancer Res. (2016) 76:4113–23. doi: 10.1158/0008-5472.CAN-15-2784
29. Knabel M, Franz TJ, Schiemann M, Wulf A, Villmow B, Schmidt B, et al. Reversible MHC multimer staining for functional isolation of T-cell populations and effective adoptive transfer. Nat Med. (2002) 8:631–7. doi: 10.1038/nm0602-631
30. Wehler TC, Nonn M, Brandt B, Britten CM, Grone M, Todorova M, et al. Targeting the activation-induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T-cell lines. Blood. (2007) 109:365–73. doi: 10.1182/blood-2006-04-014100
31. Schuster IG, Busch DH, Eppinger E, Kremmer E, Milosevic S, Hennard C, et al. Allorestricted T cells with specificity for the FMNL1-derived peptide PP2 have potent antitumor activity against hematologic and other malignancies. Blood. (2007) 110:2931–9. doi: 10.1182/blood-2006-11-058750
32. Liang X, Weigand LU, Schuster IG, Eppinger E, van der Griendt JC, Schub A, et al. A single TCR alpha-chain with dominant peptide recognition in the allorestricted HER2/neu-specific T cell repertoire. J Immunol. (2010) 184:1617–29. doi: 10.4049/jimmunol.0902155
33. Bakker AH, Hoppes R, Linnemann C, Toebes M, Rodenko B, Berkers CR, et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc Natl Acad Sci USA. (2008) 105:3825–30. doi: 10.1073/pnas.0709717105
34. Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. (2006) 1:1120–32. doi: 10.1038/nprot.2006.121
35. Nauerth M, Stemberger C, Mohr F, Weissbrich B, Schiemann M, Germeroth L, et al. Flow cytometry-based TCR-ligand Koff -rate assay for fast avidity screening of even very small antigen-specific T cell populations ex vivo. Cytometry A. (2016) 89:816–25. doi: 10.1002/cyto.a.22933
36. Kloverpris HN, Cole DK, Fuller A, Carlson J, Beck K, Schauenburg AJ, et al. A molecular switch in immunodominant HIV-1-specific CD8 T-cell epitopes shapes differential HLA-restricted escape. Retrovirology. (2015) 12:20. doi: 10.1186/s12977-015-0149-5
37. Reid SW, McAdam S, Smith KJ, Klenerman P, O'Callaghan CA, Harlos K, et al. Antagonist HIV-1 Gag peptides induce structural changes in HLA B8. J Exp Med. (1996) 184:2279–86. doi: 10.1084/jem.184.6.2279
38. Hartmann C, Antes I, Lengauer T. IRECS: a new algorithm for the selection of most probable ensembles of side-chain conformations in protein models. Protein Sci. (2007) 16:1294–307. doi: 10.1110/ps.062658307
39. Hartmann C, Antes I, Lengauer T. Docking and scoring with alternative side-chain conformations. Proteins. (2009) 74:712–26. doi: 10.1002/prot.22189
40. Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, et al. AMBER 16. San Francisco, CA: University of California (2016).
41. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. (2015) 11:3696–713. doi: 10.1021/acs.jctc.5b00255
42. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. (1983) 79:926. doi: 10.1063/1.445869
43. Salomon-Ferrer R, Götz AW, Poole D, Grand SL, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. (2013) 9:3878–88. doi: 10.1021/ct400314y
44. Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. (1977) 23:327–41. doi: 10.1016/0021-9991(77)90098-5
45. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. (1984) 81:3684–90. doi: 10.1063/1.448118
46. Roe DR, Cheatham TEI. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. (2013) 9:3084–95. doi: 10.1021/ct400341p
47. Kaufman L, Rousseeuw PJ. Finging Groups in Data: An Introduction to Cluster Analysis New York, NY: Wiley (1990).
48. Srinivasan J, Cheatham TE III, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate–DNA helices. J Am Chem Soc. (1998) 120:9401–9. doi: 10.1021/ja981844+
49. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. (2000) 33:889–97. doi: 10.1021/ar000033j
50. Miller BR III, McGee TD Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. (2012) 8:3314–21. doi: 10.1021/ct300418h
51. Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. (2004) 55:383–94. doi: 10.1002/prot.20033
52. Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. (1990) 112:6127–9. doi: 10.1021/ja00172a038
53. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. (2001) 98:10037–41. doi: 10.1073/pnas.181342398
54. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. (2004) 32:W665–667. doi: 10.1093/nar/gkh381
55. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. (1996) 14:27–38. doi: 10.1016/0263-7855(96)00018-5
56. Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. (2008) 24:1397–8. doi: 10.1093/bioinformatics/btn128
57. Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. (1995) 41:178–228. doi: 10.1007/BF00172063
58. Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, et al. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. (1996) 70:7569–79.
59. Scholten KB, Kramer D, Kueter EW, Graf M, Schoedl T, Meijer CJ, et al. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol. (2006) 119:135–45. doi: 10.1016/j.clim.2005.12.009
60. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. (2007) 109:2331–8. doi: 10.1182/blood-2006-05-023069
61. de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. (2006) 34:W362–5. doi: 10.1093/nar/gkl124
62. Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. (2003) 12:1007–17. doi: 10.1110/ps.0239403
63. Gottschalk RA, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan-Bonnet G, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proc Natl Acad Sci USA. (2012) 109:881–6. doi: 10.1073/pnas.1119763109
64. Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol. (2001) 2:229–34. doi: 10.1038/85286
65. Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, et al. How a single T cell receptor recognizes both self and foreign MHC. Cell. (2007) 129:135–46. doi: 10.1016/j.cell.2007.01.048
66. Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. (2013) 494:361–5. doi: 10.1038/nature11824
67. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature. (2017) 545:98–102. doi: 10.1038/nature22311
68. Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. (1996) 183:1917–21. doi: 10.1084/jem.183.4.1917
Keywords: T-cell receptor (TCR), peptide-MHC modeling (p-MHC modeling), adoptive T-cell transfer therapy, TCR cell therapy, TCR identification, TCR characterization, trimolecular complex (TCR-p-MHC), target antigen characterization
Citation: Audehm S, Glaser M, Pecoraro M, Bräunlein E, Mall S, Klar R, Effenberger M, Albers J, Bianchi HO, Peper J, Yusufi N, Busch DH, Stevanović S, Mann M, Antes I and Krackhardt AM (2019) Key Features Relevant to Select Antigens and TCR From the MHC-Mismatched Repertoire to Treat Cancer. Front. Immunol. 10:1485. doi: 10.3389/fimmu.2019.01485
Received: 27 November 2018; Accepted: 13 June 2019;
Published: 28 June 2019.
Edited by:
Loretta Tuosto, Sapienza University of Rome, ItalyReviewed by:
Reno Debets, Erasmus Medical Center, Erasmus University Rotterdam, NetherlandsAmir Sharabi, Beth Israel Deaconess Medical Center, Harvard Medical School, United States
Copyright © 2019 Audehm, Glaser, Pecoraro, Bräunlein, Mall, Klar, Effenberger, Albers, Bianchi, Peper, Yusufi, Busch, Stevanović, Mann, Antes and Krackhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela M. Krackhardt, YW5nZWxhLmtyYWNraGFyZHRAdHVtLmRl; orcid.org/0000-0002-4752-0543
†These authors have contributed equally to this work