 Ruth Hunegnaw
Ruth Hunegnaw Zuena Mushtaq
Zuena Mushtaq Gospel Enyindah-Asonye
Gospel Enyindah-Asonye Marjorie Robert-Guroff
Marjorie Robert-Guroff- Immune Biology of Retroviral Infection Section, Vaccine Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
HIV infected individuals have been shown to be pre-disposed to pulmonary infections even while receiving anti-retroviral therapy. Alveolar macrophages (AMs) play a critical role in lung innate immunity, but contradictory results have been reported regarding their functionality following HIV infection. Here, using the SIV rhesus macaque model, we document the effect of SIV infection on the phenotypic and functional properties of AMs. Following infection with SIVmac251, AMs in bronchoalveolar lavage (BAL) sampled over 2- to 20-weeks post-infection (wpi) were compared to those in BAL samples from naïve macaques. AM expression of proinflammatory cytokines TNF-α, IL-6, IL-1β, and chemokine RANTES drastically increased 2-wpi compared to AMs of naïve macaques (p < 0.0001 for all), but dropped significantly with progression to chronic infection. Phagocytic activity of AMs 2-and 4-wpi was elevated compared to AMs of naive animals (p = 0.0005, p = 0.0004, respectively) but significantly decreased by 12-wpi (p = 0.0022, p = 0.0019, respectively). By 20-wpi the ability of AMs from chronically infected animals to perform SIV-specific antibody-dependent phagocytosis (ADP) was also diminished (p = 0.028). Acute SIV infection was associated with increased FcγRIII expression which subsequently declined with disease progression. Frequency of FcγRIII+ AMs showed a strong trend toward correlation with SIV-specific ADP, and at 2-wpi FcγRIII expression negatively correlated with viral load (r = −0.6819; p = 0.0013), suggesting a contribution to viremia control. Importantly, PD-1 was found to be expressed on AMs and showed a strong trend toward correlation with plasma viral load (r = 0.8266; p = 0.058), indicating that similar to over-expression on T-cells, PD-1 expression on AMs may also be associated with disease progression. Further, AMs predominantly expressed PD-L2, which remained consistent over the course of infection. PD-1 blockade enhanced SIV-specific ADP by AMs from chronic infection indicating that the PD-1/PD-L2 pathway may modulate functional activity of AMs at that stage. These findings provide new insight into the dynamics of SIV infection leading to AM dysfunction and alteration of pulmonary innate immunity. Our results suggest new pathways to exploit in developing therapies targeting pulmonary disease susceptibility in HIV-infected individuals.
Introduction
Macrophages are found in tissues of infected individuals and play a continuous role in pathogenesis. Although earlier studies reported that macrophages were the major cell type initially infected by HIV (1), more recent studies have shown that CD4+ T cells are preferentially targeted by HIV/SIV at the site of transmission (2, 3). However, macrophages still serve as targets for the virus in vivo (4). They can sustain viral replication, disseminate virus, and serve as a viral reservoir post-infection (5–7). Cells of the macrophage/monocyte lineage vary greatly in phenotype, longevity, and in phagocytic, immunoregulatory, and secretory properties (8–11). Macrophages are categorized as classically (M1) or alternatively (M2) activated based on surface markers and functional role (12, 13). M1 macrophages mediate inflammatory responses against pathogens while M2 macrophages have anti-inflammatory properties, promoting tissue repair and remodeling (14). Alveolar macrophages (AM) in the lung uniquely express both M1 and M2 phenotypic markers, indicating ability to quickly respond to pathogens but also prevent immune activation in response to harmless antigens that enter the alveolar lumen (15).
AM express CD4 and chemokine receptors making them vulnerable to HIV infection (16). However, macrophages may be poorly susceptible to HIV induced cytopathic effects. Minimal consequences of HIV infection on the macrophage transcriptome were observed (17); in contrast expression of HIV Nef and gp120 envelope induced macrophage activation (18, 19). Indirect activation can also occur by exposure of uninfected macrophages to viral gene products or cytokines from other infected cells (20).
AM are important lung phagocytes (21), yet AM obtained from HIV-infected individuals have shown contradictory results regarding the impact of viral infection on phagocytosis. Some studies have shown impaired phagocytosis of opportunistic pathogens by AM (22–26); others have reported no change in AM phagocytic activity during infection (27, 28). Such variations in outcome may result from differences in length of infection or reliance on the use of monocyte derived macrophages (MDM) and infected cell lines which may not ideally represent clinical situations. Macrophages can utilize Fcγ receptors to internalize antibody-opsonized virions or infected cells, potentially leading to antibody-mediated clearance of infectious material (29). Antibody-dependent phagocytosis (ADP) by AM contributes to protection against viral infections such as influenza (30), West Nile (31), adenovirus (32), and SARS coronavirus (33). However, ADP-mediated protection by macrophages against HIV infection has not been observed.
Here we investigated the dynamics of SIV-related changes in AM activity and function by sampling bronchoalveolar lavage (BAL) from SIV infected rhesus macaques during acute and chronic infection. The AM response to SIV infection consisted of phenotypic changes and alterations in proinflammatory responses, ability to respond to gp120 antigen, and phagocytic activity. FcγRIIIb expression on AM was linked to SIV-specific ADP and viral control during acute infection. Novel results showed increased expression of Programmed Cell-Death-1 (PD-1) on AM from chronically infected macaques and positive correlations between PD-1 expressing AMs and SIV viremia. We believe this is the first report of PD-1 expression on AMs of SIV infected macaques. Our results suggest associations between PD-1 expression, macrophage dysfunction, and lack of viremia control.
Materials and Methods
Animals and Challenge
Forty-nine female Indian rhesus macaques used in this study were housed in the NCI Animal Facility, Bethesda, MD, under protocol VB-020. The NCI Facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and its Animal Care and Use Committee approved all animal experiments prior to study initiation. Standard practices followed recommendations made in the Guide for the Care and Use of Laboratory Animals of the United States, National Institutes of Health, and the Weatherall report. Thirty macaques were used as naïve controls and 19 macaques were challenged weekly intravaginally using a repeated low-dose of SIVmac251 (800 TCID50). Infection was confirmed for 14 of the macaques with plasma viral loads of ≥50 SIV RNA copies/ml as assessed by droplet digital PCR (Chung et al., manuscript in preparation). The remaining five macaques were infected intrarectally with a single high dose of SIVmac251 (4000 TCID50). Out of the 19 infected macaques, 9 were euthanized after 2 wpi and the remaining 10 infected macaques were monitored longitudinally for up to 20 wpi.
BAL Collection and Processing
BAL samples were obtained using standard techniques (34). Briefly, rhesus macaques were anesthetized, and an endotracheal tube was inserted through which sterile saline solution (10 ml/kg) was instilled. Suction was applied to recover the instilled fluid and the lung lavage was collected in sterile conical tubes. Cells were pelleted by centrifuging at 1600 RPM for 10 min at 4°C and washed in 25 mL cold PBS. Centrifugation was repeated, and cells were resuspended for counting. Cells showed ~90% viability as determined by trypan blue staining.
AM Enrichment and Stimulation
BAL cells were enriched for AMs by depleting EPCAM+, CD2+, and CD20+ cells. The following reagents were used: rabbit polyclonal anti-EPCAM (N3C3) (GeneTex, Irvine, CA); CD2 and CD20 MicroBeads for non-human primate (Miltenyi Biotec, Auburn, CA). Depletion was performed using MACS cell separation technology according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). Enriched AMs were stimulated for 6 h in 500 μl R-10 media (RPMI + 10% FBS + 5% Pen-Strep + 5% Anti-Anti) containing purified native HIV-1 envelope gp120 (Advanced Bioscience Laboratories, Inc., Rockville, MD) at a concentration of 200 nM or LPS (ThermoFisher Scientific, Waltham, MA) at 500 ng/ml concentration. For experiments assessing cytokine production by PD-1+ AMs, cells were stimulated with 10 μg/ml LPS in the presence of Golgistop (1 μl, BD Biosciences) for 18 h prior to intracellular staining.
AM Antibody Dependent Phagocytosis (ADP) Assay
ADP activity was measured as previously described (35). SIVmac251 gp120 was biotinylated with a Biotin-XX microscale protein labeling kit (Life Technologies, Grand Island, NY) and incubated with a 10-fold dilution of 1 μg Avidin coated Sky Blue fluorescent beads (0.8 μm diameter; Spherotech, Lake Forest, IL) overnight at 4°C. Enriched AMs were plated in a U-bottom 96 well plate at 40,000 cells/well and undiluted BAL-F was added. To determine phagocytic activity of AM independent of SIV specific antibody, BAL-F from naïve macaques was used. For SIV-specific ADP, autologous BAL-F from infected macaques was used. The bead-gp120 mixture was brought to a final 50-fold dilution in R-10 media and 50 μl was added to the cells and incubated for 3 h at 37°C. For PD-1 blocking experiments, BAL samples collected from macaques at 20 wpi were enriched for AMs as described above and incubated with either 10 μg/ml Ultraleaf purified anti-human PD-1 antibody (EH12.2H7, BioLegend) or 10 μg/ml mIgG1 isotype control (Sigma-Aldrich). Cells incubated with naïve BAL-F were used for normalization. In all cases, after 3 h of incubation, 70 μl of 2% paraformaldehyde was added for fixation. Fluorescent bead uptake was assessed using a BD Biosciences LSRII flow cytometer. Bead uptake specifically by AM was made possible by focusing on cells that were autofluorescent in the FITC channel. The phagocytic score of each sample was calculated by multiplying the frequency of bead-positive cells by the degree of phagocytosis measured as mean fluorescence intensity (MFI) and dividing by 104. Values were normalized to background values (cells and beads with either PBS or naïve BAL-F) by dividing the phagocytic score of the test sample by the phagocytic score of the background samples.
AM Staining
Mouse anti-non-human primate or anti-human fluorochrome-conjugated monoclonal Abs (except where specified otherwise) known to cross-react with Rhesus macaque antigens were used in this study. The following antibodies were used for surface staining: BV711 anti-CD4 (L200), BV786 anti-CD45 (D058-1283), BUV395 anti-CD3 (SP34-2), BUV496 anti-CD16 (3G8), BV711 anti-CD32 (FLI8.26/8.26), BUV737 anti-CD64 (10.1), BUV805 anti-CD14 (M5E2), BV605 anti-CD80 (L307), PE anti-CD86 (B7.2) (from BD Biosciences, San Jose, CA); PE-Cy7 anti-CD206 (19.2), APC-Cy7 anti-CD11b (ICRF44), Blue Dead cell stain kit for cell viability (from ThermoFisher Scientific, Waltham, MA); AF700 anti-CD11b (ICRF44), BV650 anti-CD11c (3.9), APC-Fire anti-PD-1 (EH12.2H7), BV786 anti PD-L1 (29E.2A3), APC anti-PD-L2 (24F.10C12), PE-Cy5 anti-HLA-DR (L243), BV510 anti-CD163 (GH1/61) (from BioLegend, San Diego, CA). For intracellular staining, BB515 anti-RANTES (2D5), APC anti-MIP-1α (11A3), BV421 anti-MIP-1β (D21-1351) (BD Biosciences, San Jose, CA); AF700 anti-IL-6 (MQ2-13A5) (ThermoFisher Scientific, Waltham, MA); rat monoclonal Ax488 anti-IL-10 (JES3-97D), BV605 anti-TNF-α (Mab11) (BioLegend, San Diego, CA) were used. The Blue LIVE/DEAD™ viability dye (ThermoFisher Scientific, Waltham, MA) was used to exclude dead cells. For staining, surface antibodies were added to cells and incubated for 25 min at room temperature (RT). Cells were then washed with PBS prior to resuspending in BD Cytofix/Cytoperm™ and incubating for 15 min at RT. Cells were then washed with BD Permwash, and incubated with intracellular staining antibodies for 25 min at RT. Finally, cells were washed with PBS and staining data were acquired using the 5 laser BD FACSymphony (BD Biosciences, San Jose, CA). Fluorescence Minus One (FMO) and isotype controls were used to confirm the phenotype and cytokine expressing population of AMs (Figure S1). Data analyses were performed with FlowJo (version 10.5, TreeStar, Inc Ashland, OR) software.
Quantification of IgGs by ELISA
Antibodies in BAL-F were measured as follows: wells of Greiner high-binding area 96-well plates were coated overnight at 4°C with 100 ng/well of SIVmac251 gp120 (for determining gp120-specific IgG) or 50 ng/well of anti-rhesus IgG (Alpha Diagnostics) (for determining total IgG) in sodium bicarbonate buffer (pH 9.6) (Sigma-Aldrich, St. Louis, MO). Wells were blocked with 200 μl of 1% BSA diluent/blocking solution (KPL) in distilled water for 2 h at RT. BAL-F (50 μl) was added and incubated for 1 h at 37°C. Env-specific IgG derived from purified serum IgG obtained from SIVmac251-infected macaques and quantified as previously described (36) was used to generate a standard curve for Env-specific IgG. Purified rhesus IgG (Non-Human Primate Reagent Resource) was used to generate a standard curve for total IgG. Plates were washed 5 times with 1X wash solution (KPL). Horseradish peroxidase-labeled goat anti-monkey IgG (50 μl at a 1:10,000 dilution; Alpha Diagnostics) was added, and plates were incubated for 1 h at 37°C. After washing as described above, 50 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (KPL) was added for 10–20 min at RT. Color development was stopped with 50 μl 1 M Phosphoric acid (Sigma), and plates were read at 450 nm by using a Biotek plate reader. SIV gp120-specific antibody levels were expressed as ng gp120-specific IgG/μg total IgG.
Statistical Analysis
Statistical analysis was performed using one-way ANOVA or 2way ANOVA with the Tukey multiple comparisons test as indicated in the Figure legends. The Wilcoxon matched-pairs signed rank test was also used where indicated. Correlation analyses were performed by non-parametric Spearman correlations. Statistics were generated using GraphPad Prism.
Results
Identification and Phenotyping of AMs Over the Course of SIV Infection
Rhesus macaques were infected with SIVmac251 and BAL was collected at 2, 4, 8, 12, and 20-wpi. Nineteen macaques were sampled at 2-wpi and 10 for the subsequent time points. BAL samples were also obtained from 30 naïve macaques. AMs from BAL were characterized by flow cytometry as HLA-DRhi, CD11bint, and CD163+CD206+ (37) (Figure 1A). The mean frequency of AMs was >80% of leukocytes found in the lavage samples, consistent with other reports on BAL specimens from naive rhesus macaques (37). No significant change in the frequency of AMs was observed over the 20 weeks of infection (Figure 1B). Expression of CD11c and CD14 has been shown to identify AM phenotypes with changes in expression occurring during the fibrotic stage of lung disease (38) Here, no significant changes in AM expression of CD11c or CD14 over the time period studied were observed (Figures 1C,D). We also did not observe changes in the percentage of AMs expressing the costimulatory molecules, CD80 and CD86, indicating that any inflammatory responses recorded may not be associated with the expansion of these cell subsets (Figures 1E,F).
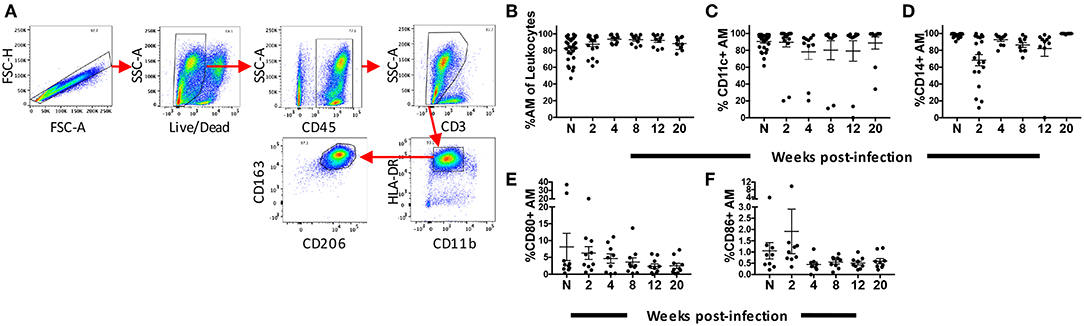
Figure 1. Identification and phenotyping of AMs over the course of SIV infection. BAL cells were collected from either naïve (n = 30) rhesus macaques or SIV infected macaques at weeks 2 (n = 19), 4 (n = 10), 8 (n = 10), 12 (n = 10), and 20 (n = 10). Recovered cells were stained and analyzed by flow cytometry. (A) Gating strategy used to identify AMs. Doublets and dead cells were excluded, and leukocytes were discriminated from epithelial cells by CD45 expression. Gates were extended to include highly granular and autofluorescent AMs. CD3 expression excluded lymphocytes. AMs were identified as FSChighSSChigh, CD11bintHLA-DRhighCD163+CD206+ (B) Frequency of CD163+CD206+ AMs in total BAL leukocytes. (C–F) Percentage of AMs expressing CD11c, CD14, CD80, and CD86, respectively. N = naïve.
Proinflammatory Cytokine and Chemokine Expression by AM Peaks at the Acute Phase of SIV Infection
Macrophages secrete proinflammatory cytokines and chemokines in response to HIV infection (39, 40). However, a clear dynamic of the cytokine and chemokine response by macrophages over the course of HIV/SIV infection has not been shown. Here we characterized cytokine levels in AM collected from SIV infected rhesus macaques between 2 and 20-wpi and compared them to levels in AMs from naïve macaques. A strong proinflammatory response was seen during the acute phase of infection. Expression of TNF-α, IL-6, and IL-1β in AMs was significantly increased by 2-wpi (p < 0.0001 for all; Figures 2A–C). By 4-wpi expression levels were significantly lower. Cytokine expression remained low until the 20-wpi time point except for IL-6 expression, which increased significantly by 12-wpi compared to 4-wpi (Figure 2B). The IL-6 expression level at 20-wpi was not significantly higher than levels seen in naïve populations. Similarly, a higher level of the chemokine RANTES was found in AMs from macaques at 2-wpi (Figure 2E). Expression decreased to levels comparable to naïve samples by 4-wpi and remained low until the 20-wpi time point (Figure 2E). MIP-1α expression did not change over the course of infection (Figure 2D). Overall, we observed a transient increase in the proinflammatory cytokine responses, which decreased to levels comparable to those of naïve macaques during the chronic phase of infection. To assess whether AMs were leaning toward an anti-inflammatory role during chronic infection, we evaluated the intracellular expression of IL-10 by AMs (Figure 2F). AMs play an important role in maintaining an anti-inflammatory environment in the lung (21, 41). Indeed, IL-10 expressing AMs were detected in naïve animals and the frequency of IL-10+ cells was significantly higher compared to that of acutely infected animals (Figure 2F). Interestingly, the percentage of IL-10 expressing AMs further decreased significantly as infection progressed into the chronic stage (Figure 2F). Thus, SIV infection was associated with a significant spike in the AM proinflammatory response by 2-wpi, which waned in chronic infection. In addition, the low proinflammatory cytokine response in chronic infection was not associated with an increase in IL-10-expressing AMs.
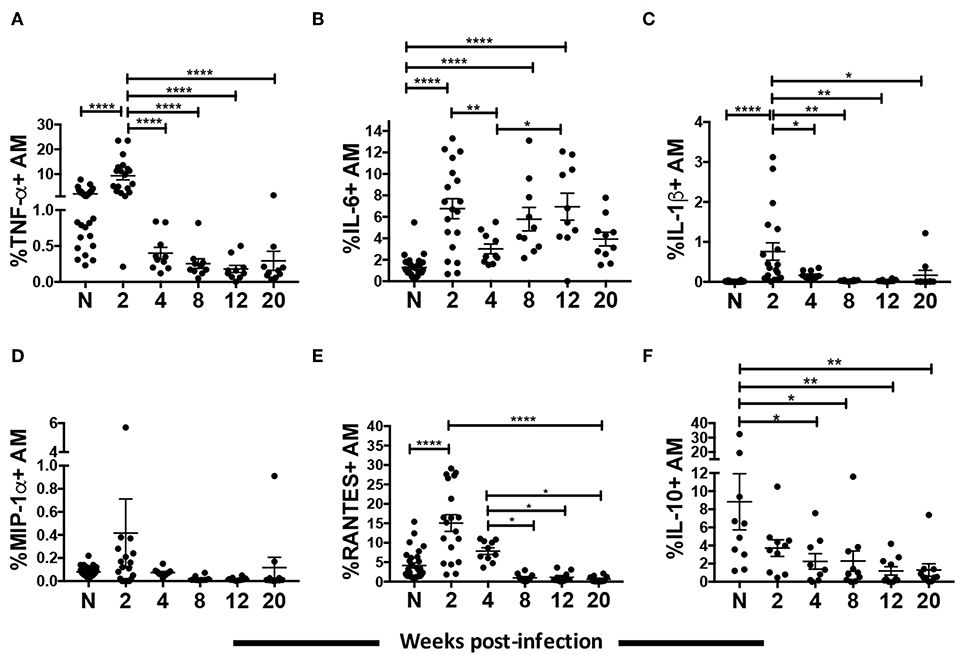
Figure 2. AM cytokine/chemokine expression over the course of SIV infection. BAL cells collected from naïve (A–E, n = 30; F, n = 10) or SIV infected rhesus macaques at 2 (A–E, n = 19; F, n = 10), 4, 8, 12, and 20 (n = 10) weeks post-infection were stained for intracellular expression of cytokines/chemokines. (A–F) Percentage of AMs expressing TNF-α, IL-6, IL-1β, MIP-1α, RANTES, and IL-10, respectively. Data are represented as mean ± SEM. Statistical differences were determined using one-way ANOVA and Tukey's multiple comparisons test. (*p < 0.05, **p < 0.01, ****p < 0.0001). N, naïve.
Chronic SIV Infection Is Associated With Diminished Response of AMs to gp120 and LPS Stimulation
To investigate AM activation, BAL cells obtained from naïve and acute and chronically infected macaques at weeks 2, 4, 8, 12, and 20 wpi, were incubated with native gp120 from R5 tropic SIV or LPS for 6 h, and intracellular expression of MIP-1β and IL-6 was assessed (Figures 3A,B). Both gp120 and LPS induced comparable levels of MIP-1β and IL-6 in AMs from naive and acutely-infected animals (Figures 3A,B). However, in chronically infected macaques gp120 did not induce intracellular IL-6 or MIP-1β expression in AMs (Figures 3A,B), and LPS did not stimulate expression of either factor to the level induced in naïve animals suggesting a dysfunctional state of the AMs (Figures 3C,D).
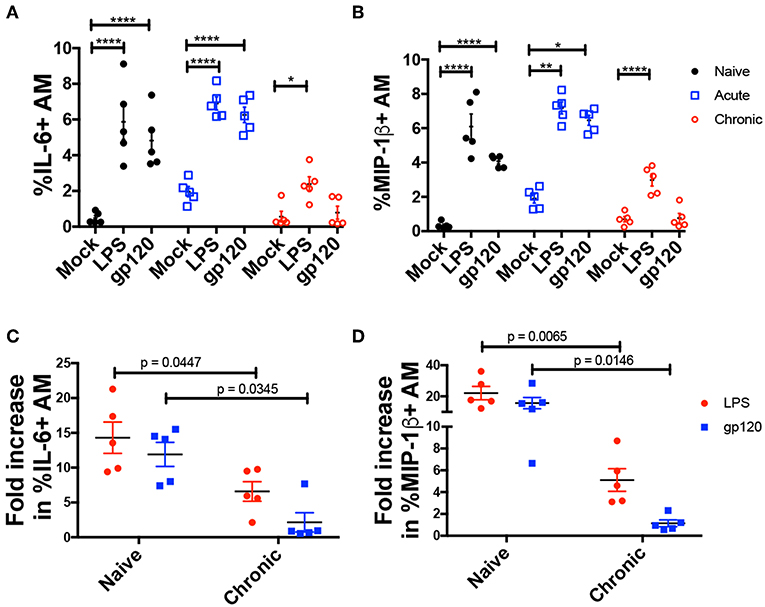
Figure 3. AMs from chronically infected macaques show diminished response to LPS and gp120 stimulation. BAL cells from 5 macaques at naïve or acute and chronic infection stages were cultured with LPS or native SIVmac251 gp120 protein for 6 h. Cells were then stained and analyzed by flow cytometry for cytokine/chemokine expression. (A,B) Percent of AMs expressing IL-6 and MIP-1β, respectively. (C,D) Fold increase in frequency of (C) IL-6+ and (D) MIP-1β+ AMs. Data are represented as mean ± SEM. Statistical differences were determined using 2way ANOVA and Tukey's multiple comparisons test. (*p < 0.05, **p < 0.01, ****p < 0.0001). N, naïve.
Changes in the Phagocytic Function of AM Over the Course of SIV Infection
To assess whether AMs contribute to clearance of SIV, we evaluated their phagocytic activity and ability to perform ADP over the course of SIV infection using BAL samples from 5 naïve animals and 5 SIV-infected macaques sampled between 2 and 20-wpi. While the BAL samples contained a high percentage of AMs (Figure 1B), the cells were further enriched for AMs using MACS by depleting CD2, CD20, and EPCAM positive cells (Figure 4A). To assess phagocytic capacity independent of SIV antibody, enriched cells were incubated with SIVmac251 gp120-coated fluorescent beads and BAL-F from naïve macaques prior to quantification of bead uptake. A significant increase in phagocytic activity was detected during the acute phase, 2 and 4-wpi (Figure 4B). At 8-wpi, phagocytosis decreased and was comparable to naïve cells. Activity further decreased significantly at the 12 and 20-wpi time points compared to the acute and naïve time points (Figure 4B).
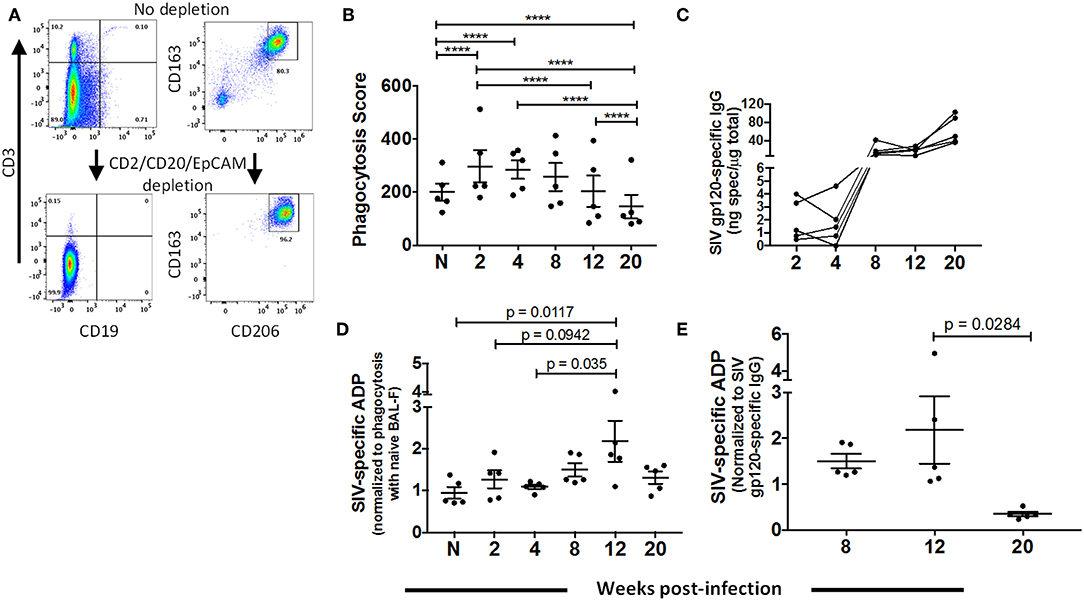
Figure 4. Dynamics of AM phagocytic activity over the course of SIV infection. BAL cells were collected from naïve (n = 5) or SIV-infected (n = 5) rhesus macaques at 2, 4, 8, 12, and 20 weeks PI for assay of phagocytic activity and ADP as described in Methods. (A) Representative gating showing depletion of lymphocytes (left panel) and percentage of AMs (right panel) from enriched (CD2+CD20+EPCAM+ depleted) and non-enriched live BAL cells. (B) Phagocytosis by AMs over the course of SIV infection. (C) SIVmac251 gp120 specific IgG in BAL-F over the course of infection for each sample. (D) AM SIV-specific ADP in the presence of autologous BAL-F normalized against the phagocytic activity in the presence of naïve BAL-F. (E) SIV-specific ADP score normalized against SIV gp120 specific IgG at the indicated time points. Data in (B), (D), and (E) are represented as mean ± SEM. Statistical differences were determined using 2 way ANOVA and Tukey's multiple comparisons test (****p < 0.0001).
Prior to evaluating ADP by AMs, we assessed SIV gp120-specific IgG in BAL-F (Figure 4C). Specific IgG was detected at 8-wpi with levels maintained or continuing to rise until 20-wpi. To determine SIV-specific ADP, AMs from naïve macaques or sampled over the course of SIV infection were incubated with antigen-coated beads along with autologous BAL-F. Phagocytosis scores were normalized against the score obtained using naïve BAL-F to highlight phagocytic activity associated with the presence of SIV-specific antibody (Figure 4D). Significantly higher phagocytic scores were detected at 12-wpi compared to those at naïve and 4-wpi time points, indicating that AMs could perform SIV-specific ADP (Figure 4D). To further assess ADP by AMs in chronic infection, phagocytic scores at chronic time points (Figure 4D) were normalized against SIV-specific antibody levels (Figure 4C). Results showed that in the presence of comparable antibody levels, AMs mediated ADP activity at 12-wpi. However, this activity was significantly compromised by 20 week PI (Figure 4E). Thus, AM are capable of gp120-specific ADP for a limited time period during chronic infection. This diminished functional capacity of AMs in the chronic stage further highlights the dysfunction observed upon LPS and gp120 stimulation (Figure 3).
Dynamics of FcγR Expression on AMs Over the Course of SIV Infection and Association With ADP
Three classes of FcγR are constitutively expressed on AMs with an active role in protecting against pathogens in the airway: high-affinity FcγRI (CD64) and low-affinity FcγRII (CD32) and FcγRIII (CD16) (42). FcγRIII occurs in two isoforms: FcγRIIIa (expressed mainly on NK cells and macrophages) and FcγRIIIb (expressed on neutrophils) (43–45). Infected MDMs have shown compromised expression of the γ signaling chain of FcγRIIIa, possibly leading to altered phagocytic activity (46). Monocytes from chronically infected individuals have also shown reduced expression of FcγRIIIa and diminished phagocytic activity (47). However, thus far no correlations have been found between ADP and HIV/SIV viral load, CD4 count or FcγR expression. Given the unique features of AMs compared to other tissue macrophages and monocytes, and the importance of phagocytic function in the lung, we investigated whether there was altered FcγR expression on AMs over the course of SIV infection and any association with ADP. BAL from naive macaques contained over 80% of FcγRI+ and FcγRII+ AMs (Figures 5B,C). Both the frequencies and MFI of FcγRII and FcγRI remained high and unchanged over the course of infection (Figures 5B,C,E,F). In contrast, the mean frequency of FcγRIII+ cells was 30% in naïve macaques. At 2-wpi there was a significant increase in FcγRIII+ AM frequency but levels significantly decreased by 8-wpi (Figure 5A). The low frequency of FcγRIII+ AMs was maintained until 20-wpi. A similar increase in AM FcγRIII MFI was not observed in the acute phase of infection compared to naïve animals (Figure 5D). However, expression levels were significantly reduced at the 8 and 20-wpi time points compared to levels at 2-wpi (Figure 5D) in concert with the decreased frequencies observed. The SIV-specific ADP observed at 12-wpi (Figure 4E) exhibited a strong trend toward positive correlation with the percentage of FcγRIII+ AMs at the same time point (Figure 5G). Further, the FcγRIII MFI at 2-wpi (Figure 5D) negatively correlated significantly with acute plasma viral load (Figure 5H). These data suggest that FcγRIII plays an important role in SIV-specific ADP and that its expression is associated with early viral load control.
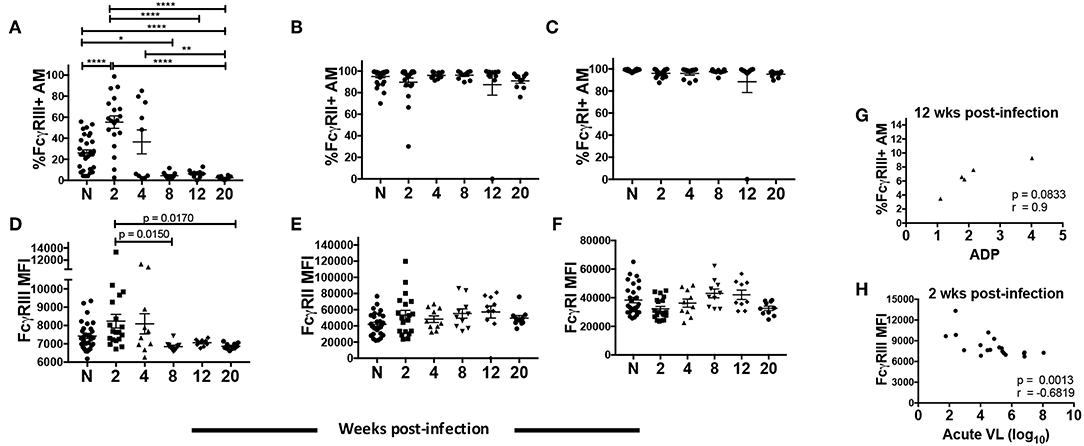
Figure 5. AM FcγR expression and role in SIV-specific ADP and viral load control. BAL cells collected from naïve (n = 30) or SIV infected rhesus macaques at 2 (n = 19), 4, 8, 12, and 20 (n = 10) weeks post-infection were stained. (A–C) Percentage of AMs expressing FcγRIII, FcγRII, and FcγRI, respectively. (D–F) MFI of FcγRIII, FcγRII, and FcγRI expression in AMs. (G) Phagocytic activity of AMs was measured in 5 infected macaques at 2, 4, 8, 12, and 20 wpi. Correlation of the frequency of FcγRIII+ AMs with SIV-specific ADP at 12-wpi (n = 5). (H) Surface expression of FcγRIII on AMs was assessed from a total of 19 acutely infected macaques. Correlation of AM FcγRIII expression at 2-wpi with acute viral load (n = 19). Data in (A–F) are represented as mean ± SEM. Statistical differences were determined using one-way ANOVA and Tukey's multiple comparisons test. (A–F) (*p < 0.05, **p < 0.01, ****p < 0.0001). Correlation statistics were generated using Spearman correlation. N, naïve.
To characterize the dysfunction observed in AMs from chronically infected macaques, we looked for parallels with T cell exhaustion. Since the initial description of T cell exhaustion in chronic viral infections (48, 49), common phenotypic and functional properties have been attributed to a dysfunctional state regardless of the type of pathogen. High and prolonged expression of inhibitory receptors are a key feature of T cell exhaustion in chronic viral infections (50). In particular, overexpression of the PD-1 receptor on CD8+ T-cells plays a major role in T cell dysfunction associated with HIV infection (51–53). Here, we identified PD-1+ AMs in rhesus macaque BAL and assessed changes in its expression on AMs from 6 macaques over the course of SIV infection (Figure 6A). When the frequency of PD-1+ AMs was assessed in naive macaques and compared to frequencies in SIV infected animals, five out of six SIV+ macaques showed an increase in frequency (Figure 6B). The PD-1+ AM frequency at 20-wpi was significantly different from both naïve and 2-wpi time points: p = 0.0419 and p = 0.0294, respectively. AMs at 20-wpi also displayed a significant increase in PD-1 MFI signifying increased expression of the receptor (Figure 6C). Furthermore, the percentage of PD-1+ AMs correlated positively with viral load at 20-wpi (Figure 6D). We also observed a strong trend toward correlation between PD-1 MFI and viral load at 20-wpi (Figure 6E), indicating an association between PD-1 expression in AMs and SIV disease progression.
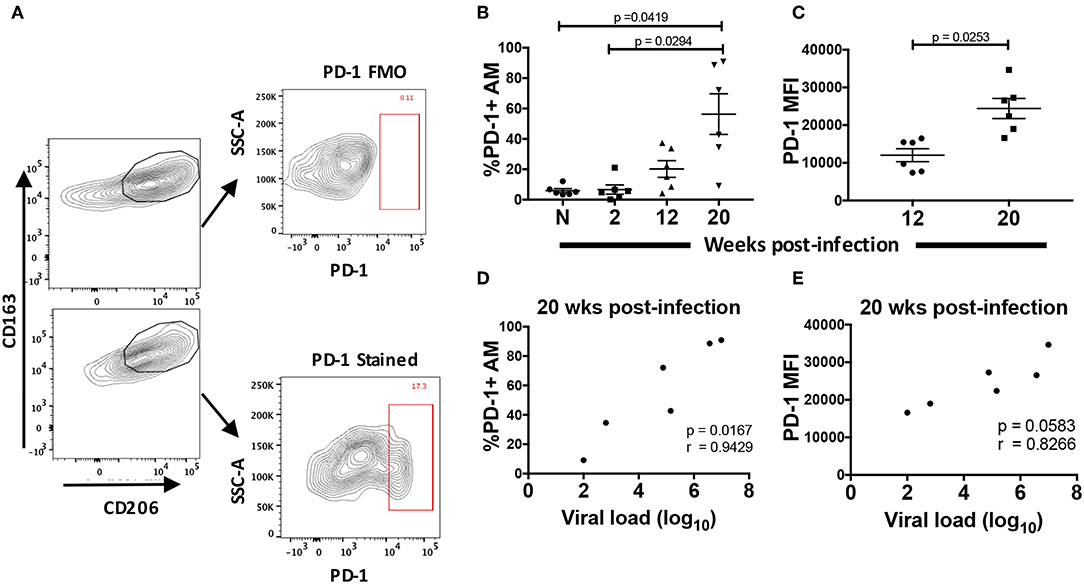
Figure 6. Surface expression of PD-1 is increased in chronically SIV-infected rhesus macaques. PD-1 expression was analyzed on AMs taken from 6 naïve macaques or at 2, 12, and 20 wpi. (A) Gating strategy showing a representative PD-1+ stained population. (B) Frequency of PD-1+ AMs over the course of SIV infection and (C) MFI of PD-1+ AMs at 12 and 20-wpi. (D) Correlation of percentage of PD-1+ AMs or (E) PD-1 MFI with viral load at 20-wpi. Data in (B) and (C) are represented as mean ± SEM. Statistical differences were determined using one-way ANOVA and Tukey's multiple comparisons test. Correlation statistics were generated using Spearman correlation. n = 6 macaques for all. (B–E) N, naïve.
Blockade of PD-1 Improved Phagocytic Activity of Macrophages
PD-1 has two well-known ligands via which inhibitory effects have been reported to occur: Programmed Death -Ligand 1 (PD-L1) and Programmed Death Ligand 2 (PD-L2). While PD-L1 has been shown to be expressed broadly in both hematopoeitic and non-hematopoeitic cells, a more restricted expression pattern has been documented for PD-L2, namely antigen presenting cells (APC) (54). In order to identify a potential role for the PD-1 pathway on AM function, we looked for expression of these ligands on AMs after SIV infection. AMs from naïve macaques predominantly expressed high levels of PD-L2 (Figure 7B). This expression decreased modestly, but not significantly, after SIV infection and remained consistent thereafter. On the other hand, we observed minimal expression (~10 fold lower than that of PD-L2) of PD-L1 on AMs from naïve macaques (Figure 7A). Further, PD-L1 expression significantly decreased over the course of SIV infection. These results indicate that AM dysfunction is more likely associated with increased PD-1 expression rather than changes in the expression of its ligands and may occur through the PD-1/PD-L2 pathway.
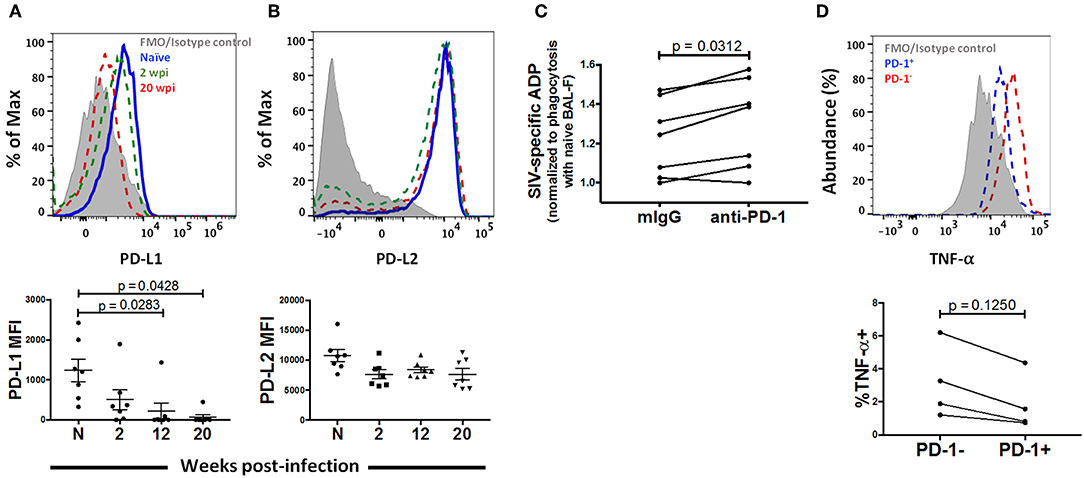
Figure 7. PD-1 blockade restores phagocytic activity in chronically infected macaques. Expression of (A) PD-L1 and (B) PD-L2 on AMs after 2, 12, and 20 wpi. n = 7. Paired one-way ANOVA with Tukey's multiple-comparisons correction. (C) Phagocytosis score of MACS sorted AMs treated with either mIgG or anti-PD-1 antibody. n = 7. Wilcoxon matched-pairs signed rank test. (D) AMs from macaques at 20 wpi were stimulated with LPS for 18 h and TNF-α expression was compared between the PD-1+ and PD-1− populations of AMs. n = 4. Wilcoxon matched-pairs signed rank test. N, naive.
To identify whether PD-1 blockade can rescue AM phagocytosis, we examined the phagocytic activity of AMs from 7 chronically infected macaques in the presence of PD-1 blocking antibody or mouse IgG as control. Interestingly, we found higher phagocytic scores in the PD-1 blockade group in 6 out of the 7 animals (p = 0.0312), indicating that PD-1 expression is likely mechanistically linked to impairment of phagocytic activity (Figure 7C). We further assessed cytokine expression of PD-1 expressing AMs. We stimulated AMs from chronically infected macaques with 10 μg/ml LPS and compared cytokine expression between PD-1+ and PD-1− populations. Although not significant, we found lower expression of TNF-α on PD-1 expressing cells compared to the PD-1− population (Figure 7D). Comparisons with additional cytokines and chemokines (IL-6, IL-1β and RANTES) are not reported here as expression of these cytokines could not be detected in the AMs from the infected macaques that were tested. Taken together, these data strongly suggest that PD-1 expression on AMs during SIV infection may be a marker of dysfunction.
Discussion
Our results show that in the SIV macaque model, the pro-inflammatory response of AMs to SIV infection peaks in the acute phase of infection and is diminished in the chronic phase as observed following stimulation with LPS or gp120. We report that AMs from SIV infected macaques can perform ADP early in infection, but this activity diminishes in the chronic phase. In novel findings, we report an elevated percentage of PD-1+ AMs in chronically infected macaques, which positively correlated with viral load. During the same period diminished inflammatory responses and ADP were observed.
The percentage of AMs stayed consistent over the course of SIV infection (Figure 1B). Viral infections can lead to activation of macrophages and subsequent release of factors that recruit additional cells of the innate and adaptive immune system (55, 56). The lack of detection of macrophage recruitment to the lung may have resulted from a concurrent lymphocyte recruitment, making the relative number of macrophages appear consistent. Indeed, a higher percentage of lymphocytes in BAL of HIV-infected patients has been reported (57). Here, estimating actual AM numbers by multiplying cell count with percentage of AMs did not result in significant differences (Figure S2). Including absolute counting beads as part of the initial analysis might have provided more definitive answers. Variations in surface marker expression of AMs over 20 weeks of SIV infection were also not seen, except for changes in FcγRIII expression (Figures 1, 5). Phenotypic comparisons of AMs from the BAL of HIV-infected and uninfected individuals also have not shown differences (57).
Proinflammatory cytokines IL-6 and IL-1β, TNF-α, and chemokine RANTES was induced in AMs during acute infection (Figures 2A–E). The rapid secretion of cytokines and chemokines was expected as the innate immune system initiates lymphocyte recruitment to establish adaptive immunity prior to viral spread. Although the HIV/SIV infection rate of AMs may be low, macrophages respond to exposure to viral particles or virus-derived gene products such as Nef, Tat, and the gp120 envelope (18, 58). Following the acute phase, the elevated cytokine and chemokine responses by AMs were not sustained despite exposure to viral products and likely LPS from the gut (59). Further, stimulation with LPS or native gp120 ex vivo showed an impaired IL-6 and MIP-1β response of AMs from chronically infected macaques (Figure 3), indicating a potentially compromised inflammatory response against lung pathogens. This novel observation is consistent with data showing inhibition of TNF-α release by macrophages in response to Toll-like receptor (TLR)-4 stimulation during HIV infection, even though the expression level of TLR-4 remained unchanged (60). Desensitization of TLR ligands on AMs as a result of viral respiratory infections has also been reported (61). The β-Chemokines MIP-1α, MIP-1β, and RANTES can bind to the HIV-1 co-receptor CCR5 indicating a potential protective effect against entry of R5-tropic viruses (62–65). The pattern of chemokine expression here shows that any protective effect of β-chemokines expressed by AMs is likely limited to the acute phase of infection (Figures 2, 3).
The lowering of cytokine and chemokine responses after the acute phase of infection was not associated with increased IL-10 expression (Figure 2F). In fact, AMs of naïve macaques expressed the highest level of intracellular IL-10 (Figure 2F). Some studies have shown no induction of IL-10 secretion or mRNA in macrophage-tropic HIV-1 infected MDM compared to uninfected cells (66). Others have shown increased IL-10 secretion and mRNA levels for in vitro HIV-infected PBMC, monocytes and MDMs [reviewed in (67)]. Differences in virus tropism or target cells could account for the variations observed. Increased levels of IL-10 have been detected in the BAL-F of HIV infected patients (68), however the IL-10 in these BAL samples could have originated from other lymphocytes that upregulate IL-10 during HIV infection (69). In sum, our data indicate that SIV disease progression is not associated with increased AM IL-10 expression, suggesting that AMs are not switching to a more regulatory phenotype.
AMs are essential for lung microbial clearance. Despite reports that AM function is not affected by HIV-1 infection (27, 28), recent studies have shown that phagocytic activity can be impaired (26, 70). As viral load and activation status of macrophages can vary over the course of infection, here we tracked the phagocytic activity of AMs following SIV infection, providing new insights into the dynamics of AM function. AMs readily internalized antigen coated beads during acute infection, but this ability markedly decreased in chronically infected animals (Figure 4B). The initial phagocytic activity was non-specific, as no gp120-specific antibody was present, and was likely due to the rise in proinflammatory responses during acute infection (Figures 2A–E). ADP was observed in the chronic stage of infection when gp120-specific antibody levels increased in BAL-F (Figures 4C,D). However, a continued rise in gp120 antibody levels was not associated with increased ADP. In fact, decreased ADP activity was seen later in chronic infection. Therefore, even though the pattern of chemokine expressing AMs over the course of SIV infection (Figures 2A–E) suggested that frequencies returned to naive levels during the chronic phase of infection, the diminished functional activity observed suggested AM dysfunction.
FcγRIII expression was elevated during acute SIV infection but decreased as infection progressed (Figures 5A,D). These data are partly in contrast to data from HIV-infected MDMs that showed either elevated or no change in FcγR expression [reviewed in (6)]. The FcγRIII isoform expressed in macrophages (FcγRIIIa) consists of a ligand bindingα-chain associated with disulfide-linked γ-chains (71, 72). The γ signaling subunit of FcγRs has been shown to be downregulated as a consequence of HIV infection, resulting in aberrant downstream signaling required for phagocytosis (46, 73). The FcγRIII antibody used here recognizes the ligand binding FcγRIII α-chain (74); thus the loss of FcγRIII expression cannot be explained by downregulation of the γ-chain alone. Our data suggest that in spite of an initial increase in the frequency of FcγRIII+ AMs during acute infection (Figure 5A), prolonged HIV infection may lead to diminished expression of FcγRIII. This outcome may not have been observed previously as in vitro MDM infection experiments mainly mimic acute stages of infection. Nevertheless, although the frequency of FcγRIII+ AMs at 12-wpi had dwindled to <30% of the level observed in naïve animals, a correlative trend between the frequency of FcγRIII+ AMs and ADP was observed (Figure 5G) indicating the importance of this receptor in ADP activity mediated by AMs. Furthermore, FcγRIII MFI negatively correlated with acute viremia suggesting an important role for FcγRIII-mediated phagocytosis in initial viral load control (Figure 5H).
The PD-1/PD-L pathway plays a key role in negative regulation of adaptive immunity in HIV and other viral infections [reviewed in (75)], but few studies have explored the role of PD-1 in innate immunity, particularly by macrophages. PD-1 expression on T cells following immune activation and its role in T-cell exhaustion when highly expressed during HIV-1 and other chronic viral infections have been described (52, 76). Regarding macrophages, PD-1 expression has been linked to diminished ability to clear microbial invasion in septic mice (77), inhibition of phagocytic activity and tumor immunity (13), and inability to perform phagocytosis and intracellular killing in patients with tuberculosis (78). Unlike on T cells, PD-1 expression on macrophages in these studies described a single positive population. Here, we also identified a single PD-1+ population of AMs derived from chronically infected macaques (Figures 6C,D). The macaques were clear of lung infections at the time of BAL collection, suggesting PD-1 expression was only associated with SIV infection. Until now, no direct evidence has implicated PD-1 in AM dysfunction. Our data show a direct correlation between PD-1 and SIV viremia and suggest that in keeping with correlations described with T cells (53), disease progression can also be associated with PD-1 expression in AMs (Figure 6E).
While PD-1 ligands were found to be expressed on AMs from naïve macaques, PD-L1 expression levels were low and subsequently declined after SIV infection (Figure 7A). This result was unexpected and in contrast to a study on MDMs that showed up-regulation of PD-L1 and PD-L2 after exposure to inactivated HIV virions (79). However, the alveolar environment is indeed unique and it is unsurprising that AM responses to SIV infection differ from in vitro exposure of MDMs to virions. Rodriguez-Garcia et al. further indicated differential regulation of PD-L1 and PD-L2 by IL-10, whereby presence of IL-10 increased PD-L1 expression and its blockade increased PD-L2 expression (79). Analysis of IL-10 expression by AMs in our study showed a gradual decrease after SIV infection and may be one explanation as to why we observed decreased PD-L1 expression (Figure 2C). PD-L2 expression, however, remained high and did not increase with decreased IL-10 expression. Future analysis of the factors found in BAL-F could provide further insight into the dynamics of PD-L expression on AMs.
PD-1 blockade experiments have shown enhancement of SIV/HIV-specific responses, proliferative ability and cytokine production by exhausted PD-1 high T cells (80–82). In keeping with this, blockade of the PD-1/PD-L pathway has been reported to ameliorate phagocytic function in macrophages found in the tumor microenvironment and in active tuberculosis (78, 83). Here, we also found that blockade of PD-1 could significantly improve phagocytic activity further highlighting PD-1 as a factor playing a role in dysfunction of AMs (Figure 7C). In addition, although not significant potentially due to small sample size, we found lower abundance of TNF-α expressing PD-1+ AMs compared to the PD-1− population during chronic infection (Figure 7D). These data suggested that the presence of PD-1 on AMs is likely a factor in any inhibitory role exerted on AMs through the PD-1/PD-L pathway.
In summary, our longitudinal investigation has provided important new information about the consequences of SIV infection on AMs, and in novel findings, propose a role for PD-1, a well-recognized inhibitor of adaptive immune responses, on innate immunity against SIV infection.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
Forty-nine female Indian rhesus macaques used in this study were housed in the NCI Animal Facility, Bethesda, MD, under protocol VB-020. The NCI Facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and its Animal Care and Use Committee approved all animal experiments prior to study initiation. Standard practices followed recommendations made in the Guide for the Care and Use of Laboratory Animals of the United States, National Institutes of Health, and the Weatherall report.
Author Contributions
RH and MR-G: conceptualization. RH, GE-A, and MR-G: methodology. RH and ZM: investigation. RH, ZM, and TH: resources. RH: writing—original draft. RH, ZM, GE-A, TH, and MR-G: writing—review and editing. MR-G: supervision.
Funding
This work was funded by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Josh Kramer and Matthew Breed, and William Magnanelli, Michelle Metrinko, and their staff in the NCI Animal Facility for expert care of the rhesus macaques and collection of all biopsy tissues; Katherine McKinnon and Sophia Brown (Vaccine Branch Flow Core, NCI) for their expert help with flow cytometry; Dr. Nancy Miller (DAIDS, NIAID) for providing the SIVmac251 challenge stock, originally developed by Dr. Ronald Desrosiers; and Dr. Hye Kyung Chung, Jigna Narola, and Harita Babbar (Advanced Bioscience Laboratories, Inc., Rockville, MD) for assessment of plasma SIV viral loads. Rhesus IgG was obtained from the NIH Non-Human Primate Reagent Resource.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01537/full#supplementary-material
References
1. Perno CF, Svicher V, Schols D, Pollicita M, Balzarini J, Aquaro S. Therapeutic strategies towards HIV-1 infection in macrophages. Antiviral Res. (2006) 71:293–300. doi: 10.1016/j.antiviral.2006.05.015
2. Haase AT. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu Rev Med. (2011) 62:127–39. doi: 10.1146/annurev-med-080709-124959
3. Stieh DJ, Matias E, Xu H, Fought AJ, Blanchard JL, Marx PA, et al. Th17 cells are preferentially infected very early after vaginal transmission of SIV in macaques. Cell Host Microbe. (2016) 19:529–40. doi: 10.1016/j.chom.2016.03.005
4. Honeycutt JB, Wahl A, Archin N, Choudhary S, Margolis D, Garcia JV. HIV-1 infection, response to treatment and establishment of viral latency in a novel humanized T cell-only mouse (TOM) model. Retrovirology. (2013) 10:121. doi: 10.1186/1742-4690-10-121
5. Balestra E, Perno CF, Aquaro S, Panti S, Bertoli A, Piacentini M, et al. Macrophages: a crucial reservoir for human immunodeficiency virus in the body. J Biol Regul Homeost Agents. (2001) 15:272–6.
6. Kedzierska K, Crowe SM. The role of monocytes and macrophages in the pathogenesis of HIV-1 infection. Curr Med Chem. (2002) 9:18931903. doi: 10.2174/0929867023368935
7. Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest. (2016) 126:1353–66. doi: 10.1172/JCI84456
8. Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. (1993) 7:19–24. doi: 10.1002/glia.440070106
9. Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol. (2012) 38:380–5. doi: 10.1165/rcmb.2007-0224RC
10. Parihar A, Eubank TD, Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun. (2010) 2:204–15. doi: 10.1159/000296507
11. Seljelid R, Eskeland T. The biology of macrophages: I. General principles and properties. Eur J Haematol. (1993) 51:267–75. doi: 10.1111/j.1600-0609.1993.tb01607.x
12. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
13. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
14. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
15. Mitsi E, Kamng'ona R, Rylance J, Solórzano C, Jesus Reiné J, Mwandumba HC, et al. Human alveolar macrophages predominately express combined classical M1 and M2 surface markers in steady state. Respir Res. (2018) 19:66. doi: 10.1186/s12931-018-0777-0
16. Lewin SR, Sonza S, Irving LB, McDonald CF, Mills J, Crowe SM. Surface CD4 is critical to in vitro HIV infection of human alveolar macrophages. AIDS Res Hum Retroviruses. (1996) 12:877–83. doi: 10.1089/aid.1996.12.877
17. Tsang J, Chain BM, Miller RF, Webb BL, Barclay W, Towers GJ, et al. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS. (2009) 23:2255–63. doi: 10.1097/QAD.0b013e328331a4ce
18. Porcheray F, Samah B, Léone C, Dereuddre-Bosquet N, Gras G. Macrophage activation and human immunodeficiency virus infection: HIV replication directs macrophages towards a pro-inflammatory phenotype while previous activation modulates macrophage susceptibility to infection and viral production. Virology. (2006) 349:112–20. doi: 10.1016/j.virol.2006.02.031
19. Carter CA, Ehrlich LS. Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol. (2008) 62:425–43. doi: 10.1146/annurev.micro.62.081307.162758
20. Twigg HL, Soliman DM, Day RB, Knox KS, Anderson RJ, Wilkes DS, et al. Lymphocytic alveolitis, bronchoalveolar lavage viral load, and outcome in human immunodeficiency virus infection. Am J Respir Crit Care Med. (1999) 159:1439–44. doi: 10.1164/ajrccm.159.5.9808031
21. Thepen T, Kraal G, Holt PG. The role of alveolar macrophages in regulation of lung inflammation. Ann N Y Acad Sci. (1994) 725:200–6. doi: 10.1111/j.1749-6632.1994.tb39802.x
22. Musher DM, Watson DA, Nickeson D, Gyorkey F, Lahart C, Rossen RD. The effect of HIV infection on phagocytosis and killing of Staphylococcus aureus by human pulmonary alveolar macrophages. Am J Med Sci. (1990) 299:158–63. doi: 10.1097/00000441-199003000-00003
23. Wehle K. Quantitative differences in phagocytosis and degradation of pneumocystis by alveolar macrophages in AIDS and Non-HIV patients in vivo. Cytopathology. (1993) 4:231–6. doi: 10.1111/j.1365-2303.1993.tb00093.x
24. Koziel H, Eichbaum Q, Kruskal BA, Pinkston P, Rogers RA, Armstrong MY, et al. Reduced binding and phagocytosis of Pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J Clin Invest. (1998) 102:1332–44. doi: 10.1172/JCI560
25. Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, et al. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol. (2014) 7:1116–26. doi: 10.1038/mi.2013.127
26. Collini PJ, Bewley MA, Mohasin M, Marriott HM, Miller RF, Geretti A-M, et al. HIV gp120 in the lungs of antiretroviral therapy-treated individuals impairs alveolar macrophage responses to Pneumococci. Am J Respir Crit Care Med. (2018) 197:1604–15. doi: 10.1164/rccm.201708-1755OC
27. Gordon SB, Molyneux ME, Boeree MJ, Kanyanda S, Chaponda M, Squire SB, et al. Opsonic phagocytosis of Streptococcus pneumoniae by alveolar macrophages is not impaired in human immunodeficiency virus–infected malawian adults. J Infect Dis. (2001) 184:1345–9. doi: 10.1086/324080
28. Elssner A, Carter JE, Yunger TM, Wewers MD. HIV-1 infection does not impair human alveolar macrophage phagocytic function unless combined with cigarette smoking. Chest. (2004) 125:1071–6. doi: 10.1378/chest.125.3.1071
29. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
30. He W, Chen C-J, Mullarkey CE, Hamilton JR, Wong CK, Leon PE, et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat Commun. (2017) 8:846. doi: 10.1038/s41467-017-00928-3
31. Chung KM, Thompson BS, Fremont DH, Diamond MS. Antibody recognition of cell surface-associated NS1 triggers Fc-γ receptor-mediated phagocytosis and clearance of west nile virus-infected cells. J Virol. (2007) 81:9551–5. doi: 10.1128/JVI.00879-07
32. Zsengellér Z, Otake K, Hossain S-A, Berclaz P-Y, Trapnell BC. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J Virol. (2000) 74:9655–67. doi: 10.1128/JVI.74.20.9655-9667.2000
33. Yasui F, Kohara M, Kitabatake M, Nishiwaki T, Fujii H, Tateno C, et al. Phagocytic cells contribute to the antibody-mediated elimination of pulmonary-infected SARS coronavirus. Virology. (2014) 454–5:157–68. doi: 10.1016/j.virol.2014.02.005
34. Kastello MD, Emmert AD, Denson RF, Kishimoto RA. Recovery of alveolar macrophages from rhesus and cynomolgus macaques by lung lavage. Am J Vet Res. (1979) 40:271–3.
35. Ackerman ME, Moldt B, Wyatt RT, Dugast A-S, McAndrew E, Tsoukas S, et al. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J Immunol Methods. (2011) 366:8–19. doi: 10.1016/j.jim.2010.12.016
36. Manrique M, Kozlowski PA, Wang S-W, Wilson RL, Micewicz E, Montefiori DC, et al. Nasal DNA-MVA SIV vaccination provides more significant protection from progression to AIDS than a similar intramuscular vaccination. Mucosal Immunol. (2009) 2:536–50. doi: 10.1038/mi.2009.103
37. Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda MJ. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol. (2014) 192:2821–9. doi: 10.4049/jimmunol.1302269
38. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
39. Fantuzzi L, Belardelli F, Gessani S. Monocyte/macrophage-derived CC chemokines and their modulation by HIV-1 and cytokines: a complex network of interactions influencing viral replication and AIDS pathogenesis. J Leukoc Biol. (2003) 74:719–25. doi: 10.1189/jlb.0403175
40. Alfano M, Graziano F, Genovese L, Poli G. Macrophage polarization at the crossroad between HIV-1 infection and cancer development. Arterioscler Thromb Vasc Biol. (2013) 33:1145–52. doi: 10.1161/ATVBAHA.112.300171
41. Bingisser R, Speich R, Zollinger A, Russi E, Frei K. Interleukin-10 secretion by alveolar macrophages and monocytes in sarcoidosis. Respiration. (2000) 67:280–6. doi: 10.1159/000029511
42. Jungi TW, Hafner S. Quantitative assessment of Fc receptor expression and function during in vitro differentiation of human monocytes to macrophages. Immunology. (1986) 58:131–7.
43. Selvaraj P, Carpen O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fc gamma receptor III (CD16) differ in membrane anchor and signal transduction. J Immunol. (1989) 143:3283–8.
44. Klaassen RJ, Ouwehand WH, Huizinga TW, Engelfriet CP, Borne von dem AE. The Fc-receptor III of cultured human monocytes. Structural similarity with FcRIII of natural killer cells and role in the extracellular lysis of sensitized erythrocytes. J Immunol. (1990) 144:599–606.
45. Perussia B, Ravetch JV. FcγRIII (CD16) on human macrophages is a functional product of the FcγRIII-2 gene. Eur J Immunol. (1991) 21:425–9. doi: 10.1002/eji.1830210226
46. Kedzierska K, Ellery P, Mak J, Lewin SR, Crowe SM, Jaworowski A. HIV-1 down-modulates γ signaling chain of FcγR in human macrophages: a possible mechanism for inhibition of phagocytosis. J Immunol. (2002) 168:2895–903. doi: 10.4049/jimmunol.168.6.2895
47. Dugast A-S, Tonelli A, Berger CT, Ackerman ME, Sciaranghella G, Liu Q, et al. Decreased Fc receptor expression on innate immune cells is associated with impaired antibody-mediated cellular phagocytic activity in chronically HIV-1 infected individuals. Virology. (2011) 415:160–7. doi: 10.1016/j.virol.2011.03.012
48. Gallimore A, Glithero A, Godkin A, Tissot AC, Plückthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. (1998) 187:1383–93. doi: 10.1084/jem.187.9.1383
49. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
50. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. (2009) 138:30–50. doi: 10.1016/j.cell.2009.06.036
51. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. (2006) 203:2281–92. doi: 10.1084/jem.20061496
52. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. (2006) 12:1198–202. doi: 10.1038/nm1482
53. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
54. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
55. Pribul PK, Harker J, Wang B, Wang H, Tregoning JS, Schwarze J, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. (2008) 82:4441–8. doi: 10.1128/JVI.02541-07
56. Huysentruyt LC, McGrath MS. The role of macrophages in the development and progression of AIDS-related non-Hodgkin lymphoma. J Leukoc Biol. (2010) 87:627–32. doi: 10.1189/jlb.0809564
57. Gordon SB, Jagoe RT, Jarman ER, North JC, Pridmore A, Musaya J, et al. The alveolar microenvironment of patients infected with human immunodeficiency virus does not modify alveolar macrophage interactions with Streptococcus pneumoniae. Clin Vaccine Immunol. (2013) 20:882–91. doi: 10.1128/CVI.00582-12
58. Choe W, Volsky DJ, Potash MJ. Induction of rapid and extensive beta-chemokine synthesis in macrophages by human immunodeficiency virus type 1 and gp120, independently of their coreceptor phenotype. J Virol. (2001) 75:10738–45. doi: 10.1128/JVI.75.22.10738-10745.2001
59. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
60. Tachado SD, Zhang J, Zhu J, Patel N, Koziel H. HIV Impairs TNF-α release in response to toll-like receptor 4 stimulation in human macrophages in vitro. Am J Respir Cell Mol Biol. (2005) 33:610–21. doi: 10.1165/rcmb.2004-0341OC
61. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolutionof respiratory influenza infection. J Exp Med. (2008) 205:323–9. doi: 10.1084/jem.20070891
62. Wu L, LaRosa G, Kassam N, Gordon CJ, Heath H, Ruffing N, et al. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J Exp Med. (1997) 186:1373–81. doi: 10.1084/jem.186.8.1373
63. Raport CJ, Gosling J, Schweickart VL, Gray PW, Charo IF. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1β, and MIP-1α. J Biol Chem. (1996) 271:17161–6. doi: 10.1074/jbc.271.29.17161
64. Nibbs RJB, Yang J, Landau NR, Mao J-H, Graham GJ. LD78β, a non-allelic variant of human MIP-1α (LD78α), has enhanced receptor interactions and potent HIV suppressive activity. J Biol Chem. (1999) 274:17478–83. doi: 10.1074/jbc.274.25.17478
65. Barmania F, Pepper MS. C-C chemokine receptor type five (CCR5): an emerging target for the control of HIV infection. Appl Transl Genomics. (2013) 2:3–16. doi: 10.1016/j.atg.2013.05.004
66. Dereuddre-Bosquet N, Clayette P, Martin M, Benveniste O, Fretier P, Jaccard P, et al. Lack of interleukin 10 expression in monocyte-derived macrophages in response to in vitro infection by HIV type 1 isolates. AIDS Res Hum Retroviruses. (1997) 13:961–6. doi: 10.1089/aid.1997.13.961
67. Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother. (2001) 12:133–50. doi: 10.1177/095632020101200301
68. Rizzardi GP, Marriott JB, Cookson S, Lazzarin A, Dalgleish AG, Barcellini W. Tumour necrosis factor (TNF) and TNF-related molecules in HIV-1+ individuals: relationship with in vitro Thl/Th2-type response. Clin Exp Immunol. (1998) 114:61–5. doi: 10.1046/j.1365-2249.1998.00672.x
69. Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood. (2009) 114:346–56. doi: 10.1182/blood-2008-12-191296
70. Staitieh BS, Ding L, Neveu WA, Spearman P, Guidot DM, Fan X. HIV-1 decreases Nrf2/ARE activity and phagocytic function in alveolar macrophages. J Leukoc Biol. (2017) 102:517–25. doi: 10.1189/jlb.4A0616-282RR
71. Ra C, Jouvin M-HE, Blank U, Kinet J-P. A macrophage Fcγ receptor and the mast cell receptor for IgE share an identical subunit. Nature. (1989) 341:752–4. doi: 10.1038/341752a0
72. Hibbs ML, Selvaraj P, Carpen O, Springer TA, Kuster H, Jouvin MH, et al. Mechanisms for regulating expression of membrane isoforms of Fc gamma RIII (CD16). Science. (1989) 246:1608–11. doi: 10.1126/science.2531918
73. Leeansyah E, Wines BD, Crowe SM, Jaworowski A. The mechanism underlying defective Fcγ receptor-mediated phagocytosis by HIV-1-infected human monocyte-derived macrophages. J Immunol. (2007) 178:1096–104. doi: 10.4049/jimmunol.178.2.1096
74. Tamm A, Schmidt RE. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for ligand binding. J Immunol. (1996) 157:1576–81.
76. Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. (2006) 80:11398–403. doi: 10.1128/JVI.01177-06
77. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U.S.A. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
78. Shen L, Gao Y, Liu Y, Zhang B, Liu Q, Wu J, et al. PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci Rep. (2016) 6:38362. doi: 10.1038/srep38362
79. Rodríguez-García M, Porichis F, de Jong OG, Levi K, Diefenbach TJ, Lifson JD, et al. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J Leukoc Biol. (2011) 89:507–15. doi: 10.1189/jlb.0610327
80. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. (2009) 458:206–10. doi: 10.1038/nature07662
81. Finnefrock AC, Tang A, Li F, Freed DC, Feng M, Cox KS, et al. PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination. J Immunol. (2009) 182:980–7. doi: 10.4049/jimmunol.182.2.980
82. Porichis F, Kwon DS, Zupkosky J, Tighe DP, McMullen A, Brockman MA, et al. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood. (2011) 118:965–74. doi: 10.1182/blood-2010-12-328070
Keywords: alveolar macrophage, SIV, rhesus macaque, antibody-dependent phagocytosis, PD-1, FcγRIII, bronchoalveolar lavage
Citation: Hunegnaw R, Mushtaq Z, Enyindah-Asonye G, Hoang T and Robert-Guroff M (2019) Alveolar Macrophage Dysfunction and Increased PD-1 Expression During Chronic SIV Infection of Rhesus Macaques. Front. Immunol. 10:1537. doi: 10.3389/fimmu.2019.01537
Received: 11 February 2019; Accepted: 19 June 2019;
Published: 03 July 2019.
Edited by:
Donald Sodora, Seattle Children's Research Institute, United StatesReviewed by:
Vijayakumar Velu, Emory University, United StatesIvona Pandrea, University of Pittsburgh, United States
Copyright © 2019 Hunegnaw, Mushtaq, Enyindah-Asonye, Hoang and Robert-Guroff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marjorie Robert-Guroff, Z3Vyb2ZmbUBtYWlsLm5paC5nb3Y=