 Teja Celhar1†
Teja Celhar1† Hao Kim Lu1†
Hao Kim Lu1† Lia Benso2Larissa Rakhilina2Hui Yin Lee1,3Shubhita Tripathi1,4Olga Zharkova1,5Wei Yee Ong3Hiroko Yasuga1,6Bijin Au3Damien Marlier7
Lia Benso2Larissa Rakhilina2Hui Yin Lee1,3Shubhita Tripathi1,4Olga Zharkova1,5Wei Yee Ong3Hiroko Yasuga1,6Bijin Au3Damien Marlier7 Lina Hsiu Kim Lim5
Lina Hsiu Kim Lim5 Thomas Paulraj Thamboo8John S. Mudgett2‡Matthew F. Mackey2‡Dennis M. Zaller2‡
Thomas Paulraj Thamboo8John S. Mudgett2‡Matthew F. Mackey2‡Dennis M. Zaller2‡ John E. Connolly3,9
John E. Connolly3,9 Anna-Marie Fairhurst1,3,4,6*
Anna-Marie Fairhurst1,3,4,6*- 1Singapore Immunology Network, A*STAR, Singapore, Singapore
- 2Merck & Co., Inc., Boston, MA, United States
- 3Institute of Molecular and Cell Biology, A*STAR, Singapore, Singapore
- 4Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 5Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 6School of Biological Sciences, Nanyang Technological University, Singapore, Singapore
- 7Tessa Therapeutics, Singapore, Singapore
- 8Department of Pathology, National University Hospital, Singapore, Singapore
- 9Institute of Biomedical Studies, Baylor University, Waco, TX, United States
The global increase in autoimmunity, together with the emerging autoimmune-related side effects of cancer immunotherapy, have furthered a need for understanding of immune tolerance and activation. Systemic lupus erythematosus (SLE) is the archetypical autoimmune disease, affecting multiple organs, and tissues. Studying SLE creates knowledge relevant not just for autoimmunity, but the immune system in general. Murine models and patient studies have provided increasing evidence for the innate immune toll like receptor-7 (TLR7) in disease initiation and progression. Here, we demonstrated that the kinase activity of the TLR7-downstream signaling molecule, interleukin-1 receptor associated kinase 4 (IRAK4), is essential for mild and severe autoimmune traits of the Sle1 and Sle1-TLR7 transgenic (Sle1Tg7) murine models, respectively. Elimination of IRAK4 signaling prevented all pathological traits associated with murine lupus, including splenomegaly with leukocyte expansion, detectable circulating antinuclear antibodies and glomerulonephritis, in both Sle1 and Sle1Tg7 mice. The expansion of germinal center B cells and increased effector memory T cell phenotypes that are typical of lupus-prone strains, were also prevented with IRAK4 kinase elimination. Analysis of renal leukocyte infiltrates confirmed our earlier findings of an expanded conventional dendritic cell (cDC) within the kidneys of nephritic mice, and this was prevented with IRAK4 kinase elimination. Analysis of TLR7 at the protein level revealed that the expression in immune cells is dependent on the TLR7-transgene itself and/or autoimmune disease factors in a cell-specific manner. Increased TLR7 protein expression in renal macrophages and cDCs correlated with disease parameters such as blood urea nitrogen (BUN) levels and the frequency of leukocytes infiltrating the kidney. These findings suggest that controlling the level of TLR7 or downstream signaling within myeloid populations may prevent chronic inflammation and severe nephritis.
Introduction
The immune system provides a crucial barrier against invasive pathogenic challenges. Defects can lead to opportunistic infections, cancer or the development of autoimmunity. Systemic lupus erythematosus (SLE) is the archetypical autoimmune disorder, whereby the immune system attacks itself in a systemic manner. It is complex and is driven by both genetic and environmental factors with no effective prevention or cure (1, 2).
SLE is characterized by the development of anti-nuclear antibodies (ANAs), particularly dsDNA- and RNA-reactive autoantibodies. These bind to self-antigens, resulting in the formation of immune complexes (ICs) which deposit in tissues, initiating, and promoting inflammation, tissue destruction, and often culminating in organ failure (3). The ICs in SLE patients contain U1 and Y1 RNA, with levels correlating with disease activity and the characteristic type-I interferon (IFN) signature (4). The primary candidate for RNA-associated IC activation is toll-like receptor (TLR)-7. TLR7 recognizes single-stranded RNA (ssRNA) and is central to host defense against invading viruses (5, 6). However, it also plays a fundamental role in the development and progression of autoimmunity. Immunological studies show that SLE sera can induce TLR7 expression in neutrophils and that these are primed for NETosis, which is also increased in SLE (7, 8). Furthermore, TLR7 mRNA expression is higher in PBMCs from SLE patients, with levels correlating with the expression of IFNα (9).
Genetic studies have demonstrated TLR7/TLR8 associations with SLE across multiple ethnicities (10, 11). The rs3853839 SNP in the 3′ untranslated region (3′ UTR) of TLR7 is associated with SLE in the Asian population (11–13). The risk allele of rs3853839 confers higher levels of the TLR7 transcript, most likely through an epigenetic microRNA mechanism (14). In a Mexican pediatric population, increased TLR7 gene copy number was associated with higher levels of TLR7 and IFNα mRNA (10). Since TLR7 is located on the X chromosome, the gene dosage can be affected by aneuploidies. Indeed, both males and females with increased numbers of the X chromosome have an increased incidence of SLE (15, 16). Additionally, it has been recently shown that TLR7 escapes from X chromosome inactivation, which results in a higher responsiveness to TLR7 ligands in females (17). This enhanced TLR7 expression likely contributes to the female bias in SLE and other autoimmune diseases.
Multiple murine models support a requirement for TLR7 in the development of ANAs and subsequent autoimmune pathology (18–20). A moderate increase of TLR7 through the BXSB-derived Y-autoimmune accelerator (Yaa) locus is essential for the rapid-onset of severe disease in Yaa-associated strains (21–24). Furthermore, we have shown that a ~2-fold upregulation of TLR7 in the mild NZM2410-derived Sle1 background (Sle1Tg7) is sufficient for the progression to severe disease (25). In other model systems, a higher copy of TLR7 permits the progression to severe autoimmunity in the absence of any additional susceptibility locus (26).
The endosomal location of TLR7 is believed to be critical for the anti-viral response, in addition to preventing activation by self-RNA, which can lead to inflammation (27, 28). Stimulation with ssRNA activates a MyD88-dependent signaling cascade. In the early stages of activation, MyD88 recruits IL1R associated kinases (IRAK) 1, 2, and 4, forming the Myddosome (29). Phosphorylation of IRAK1 and IRAK2 leads to their dissociation from the complex and association and activation of TRAF6 and TAK1 (29–31). TAK1 activates MAPKs and subsequently triggers the activation of various transcription factors including interferon regulatory factors (IRF)3, IRF5, and IRF7, that have been previously described as risk factors for SLE (32–36), and NF-κB (29–31). This ultimately leads to the transcription and production of inflammatory cytokines and chemokines (37–39). The TAK1-dependent activation of NF-κB and IRF5 activation are mediated through the IRAK4 kinase domain (40). The IRAK4 kinase domain is necessary for the activation of IRF5 and for the TAK1-dependent activation of NF-κB. Additionally, IRAK4 also plays a structural, kinase-independent role, which can mediate TAK1-independent NF-κB activation (37, 41). The relative contribution of these pathways to the development of autoimmunity and severe inflammation is unclear.
A recent study investigated the role of IRAK4 in A20-binding inhibitor of NF-kB1 (ABIN1) mutant mice (42). ABIN1 is an ubiquitin sensing protein that interacts with death induced signaling complex (DISC) proteins to prevent FADD/caspase8 binding (43). Elimination of ABIN1 results in a TNF-dependent induction of programmed cell death, fetal liver apoptosis, anemia, hypoplasia, and early mortality (43). However, polyubiquitin-binding defective mutants of ABIN1 (ABIN1[D485N]) live longer, developing severe fibrosis in spleens, hearts, lungs, and intestine (44). These mice also develop murine lupus-like traits at a very young age (4 mos), which include splenomegaly with B cell and myeloid expansion, hypergammaglobulinemia with increases not only in IgG, but also in IgM, IgA and IgE, and ANAs (44). The ABIN1 mutant phenotypes are MyD88-dependent, possibly due to the loss of the humoral response and autoantibodies, as indicated in other models (19, 44). In addition, a mutation in IRAK1 or IRAK4 prevents the splenomegaly, ANA production, hypergammaglobulinemia and GN in ABIN1[D485N] mice (42). In a similar manner to the ABIN1 knockout, the phenotype is associated with TLR hyperactivation and increased TNF. However, it not known whether TNF reaches high systemic levels or if these lupus-traits are TNF dependent. Moreover, given the promiscuous nature of the activation of leukocytes, it is unclear as to the specific receptor dependency of disease and the role of IRAK4/1 on any given receptor. Furthermore, an assessment of leukocyte infiltration in the context of the diseased kidney would be helpful in understanding the regulatory roles of IRAK4 in this system.
Additionally, the role of TLR signaling and regulatory molecules was assessed in the BXSB lupus-prone model (45). The aggressive lupus-like phenotypes in the male are driven by a 2-fold increase in TLR7 expression due to the presence of the yaa locus (21, 23, 24). The females have normal TLR7 expression levels due to x-inactivation. Interestingly, Murphy et al. showed that female mice have increased expression of the TLR inhibitors, Tollip, and IRAK-M compared to their male yaa-expressing counterparts. In addition, the male mice had comparatively more phosphorylation following TLR4 or TLR7 ligation, despite having similar expression levels of IRAK1/4. Elimination of IRAK4 kinase activity in BXSB. IRAK4ki/ki F2 mice resulted in a reduction in splenomegaly and number of splenic F4/80+ macrophages, neutrophils, total B cells, plasmablasts, and total CD11c+ cells (45). Supporting these findings, in vivo administration of a selective inhibitor of IRAK4, BMS-986126, prevented the development of GN, plasma anti-dsDNA autoantibodies and reduced IFNa+pDCs in both NZB/W and MRLlpr mice, and splenic IL-6+CD11b+ myeloid cells in latter (46).
Taken together, these interesting investigations justify a more extensive analysis on leukocyte activation and expansion within the spleen itself, and in particular, the immunological events within the affected diseased kidney. Moreover, it is important to know if these suppressive effects of IRAK4 elimination on TLR7-dependent lupus-traits are due to the loss of susceptibility loci resulting from the introduction of B6-nonautoimmune background into the F2 progeny.
In these studies, we set out to characterize the role of IRAK4 kinase activity in the development of mild and severe TLR7-dependent systemic autoimmunity. In addition, we analyzed the renal leukocyte infiltrate and changes in protein levels of TLR7 in the context of pathology. We generated IRAK4 kinase dead (IRAK4KD/KD) mice and backcrossed these with Sle1 and Sle1Tg7 mice to generate Sle1IRAK4KD/KD and Sle1Tg7IRAK4KD/KD mice. The model in our studies uses a low copy TLR7-BAC transgenic system, therefore we were able to examine genomic vs. disease effects on TLR7 protein expression in individual leukocyte compartments. Our data shows that IRAK4 kinase activity is essential for the development of all known autoimmune traits in both models, including splenomegaly, leukocyte activation, antibody production, and renal leukocyte recruitment with accompanying kidney disease. TLR7 is known to play a critical role in the onset of lupus, yet studies reporting comparative expression at the protein level are limited (47, 48). We assessed TLR7 protein levels using high dimensional flow cytometry and determined that the expression was regulated by disease and / or the low copy TLR7-BAC transgene in a leukocyte and tissue specific manner. Importantly, blood urea nitrogen (BUN) levels and CD45+ leukocyte infiltration, which are indicators of kidney inflammation and damage, correlated with TLR7 protein expression in renal cDCs and macrophages. Taken together our data demonstrates a complex cross-regulation between TLR7 receptor expression across multiple leukocytes that needs further investigation. Moreover, this work shows that the TLR7 downstream signaling pathway may be a key target for therapeutic intervention in lupus nephritis.
Methods
Mice
Mice were bred in the Biological Resource Center, A*STAR in Singapore or at Taconic in New York, USA. B6.Sle1 (Sle1) mice bearing NZM2410-lupus susceptibility intervals and the generation of the conditional BAC-Tg7 for increased TLR7 expression have been previously described (25, 49). Breeding pairs for TLR7−/− mice were kindly provided by Prof. Shizuo Akira, Osaka University, Japan (5). The care and use of laboratory animals conformed to the NIH guidelines and all experimental procedures conformed to an IACUC approved animal protocol (#161176). Female mice were aged to 6–7 months to detect autoimmune traits.
Generation of the K213A Construct/Mouse
The IRAK4 K213A point mutant mice were generated by Taconic Biosciences Inc. on behalf of Merck & Co., Inc., Boston, MA, USA. In brief, a BAC clone containing murine C57BL/6J genomic sequences was used to generate the targeting vector, which contains the K213A point mutation in exon 5 and an FRT site flanked puromycin resistance gene cassette in intron 5 for positive selection. Correctly targeted ES cell clones were injected into blastocysts and the resulting chimeras crossed to FLP deleted mice to remove the puromycin resistance gene cassette. Heterozygous mice were further bred to generate homozygous and wild-type control mice in a 100% C57BL/6 NTac genetic background for experiments. The level of IRAK4KD/KD mRNA and protein expression in splenocytes of wild type, heterozygous, and homozygous mice was quantified using qPCR and Western Blotting, respectively. To confirm for the lack of IRAK4 kinase activity, GST-IRAK4KD/KD or GST/TEV-IRAK4WT/WT fusion proteins were generated and tested in vitro using an ULight kinase assay.
Assessment of Renal Disease
Albumin in urine was quantified by ELISA (Abcam, Cambridge, UK) and the concentrations were normalized to urine creatinine measured with a colorimetric assay as per manufacturer's instructions (Cayman Chemical, Ann Arbor, MI). Blood Urea Nitrogen (BUN) was assessed using the QuantiChrom Urea Assay Kit (BioAssay Systems). To detect GN, kidneys were fixed in formalin and embedded in paraffin. Three micrometer sections of formalin-fixed, paraffin-embedded kidney tissues were stained with hematoxylin and eosin and with periodic acid–Schiff (PAS). Microscopic morphologic analysis was performed by an independent pathologist according to the International Society of Nephrology / Renal Pathology Society (ISN/RPS) 2003 classification of lupus nephritis (50); Class I, normal by light microscopy; Class II, mesangial proliferation; Class III, focal endocapillary or extracapillary proliferation, and/or subendothelial deposits; Class IV, diffuse endocapillary or extracapillary proliferation and/or subendothelial deposits; Class V, membranous lupus nephritis; Class VI, advanced sclerosing lupus nephritis.
Serology
Sera were measured for circulating ANAs, dsDNA, anti-snRNP (1:200 dilution) and anti-RNA (1:100 dilution) using established ELISAs (25, 47, 51). HEp-2 staining was performed at a 1:200 dilution as per manufacturer's instructions (INOVA Diagnostics), with a goat-anti-mouse IgG-DyLight®488 secondary antibody (Abcam). HEp-2 were imaged at 100X using the Zeiss LSM 800 upright confocal microscope and processed with Zeiss Zen (Blue edition) software. HEp-2 staining patterns were evaluated by two independent investigators according to the International Consensus on Antinuclear Antibody (ANA) Patterns (ICAP). Luminex was used to quantify antibody subsets and subtypes including: IgA, IgM, IgG1, IgG2a/c, IgG2b, and IgG3 (Mouse Immunoglobulin Isotyping Magnetic Bead Panel, Merck Millipore). This panel is designed to detect IgG2a (from BALB/c mice), which cross-reacts with IgG2c from mice on the B6 background, which we have designated as IgG2a/c (52).
Fluorescent Microscopy
Frozen OCT embedded spleens were sectioned, fixed with 4% paraformaldehyde and stained with anti-CD4 (Ebioscience), anti-GL7 (BD Horizon) and anti-IgD (BD Pharmingen) antibodies. Confocal images of splenic germinal centers were collected as described above.
Cell Preparation and Flow Cytometry
Single cells suspension from both spleen and kidney were prepared as described previously (53). Splenocytes or kidney cells were resuspended in PBS with 1% (vol/vol) FBS (FACs wash), counted and labeled with the following antibody panels. Splenic B cells: CD45, CD19, B220, CD138, CD21, CD23, GL7, Fas, CD11b, and CD11c. In some cohorts a more comprehensive B cell panel was used, including CD93 and IgM to identify transitional T1 (CD93+sIgMhiCD23−), T2 (CD93+sIgMhiCD23+), and T3 (CD93+sIgMloCD23+) cells. Splenic T cells: CD45, CD3, CD4, CD8, CD44, CD62L, CD69, CXCR5, ICOS, and PD-1; splenic pDCs: CD11b, CD11c, mPDCA1, SiglecH, MHCII, CD86, CD9, CCR9, Gr-1, CD3, and B220. Splenic and kidney DCs & macrophages were stained with CD45, CD3, B220, CD8, CD11b, CD11c, Gr-1, F4/80, CD64, CD86, and MHCII. For the analysis of kidney B cells, T cells and pDCs and TLR7 expression in splenic and kidney subsets, the cells were stained with CD45, EpCAM, B220, CD44, CD62L, ICOS, CD138, CD4, CD8, NK1.1, Gr-1, mPDCA1, SiglecH, CD11b, CD11c, CD115, F4/80, MHC-II, and TLR7. All samples were stained with a live/dead fixable dye (Thermo Fisher Scientific, Waltham, MA). For intracellular TLR7 staining, the BD Cytofix/Cytoperm kit was used as per manufacturer instructions. Samples for the majority of panels were analyzed using a BD LSR Fortessa, a BD LSR II or BD Symphony with Flowjo 7.6. Antibodies were purchased from BD Biosciences, eBioscience, Biolegend, or Life Technologies.
Statistical Analyses
Statistical analysis was performed with Graph Pad Prism 7.0. Normal distribution of the data was assessed by the Kolmogorov-Smirnov test. Data comparing the 4 strains was analyzed using a 1-way ANOVA followed by the Bonferroni's multiple comparison tests for normally distributed data, while the Kruskal-Wallis test with post hoc Dunn's multiple comparisons test was used for non-parametric data. P values < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 for ANOVA. Additional 2 group comparisons were completed for single gene changes where indicated, using a two-tailed Student's t-test (normal distribution) or Mann-Whitney u-test (non-parametric); #P < 0.05, ##P < 0.01, ###P < 0.001 and ####P < 0.0001. Correlations were assessed by computing Pearson's coefficient r and a two-tailed t test was used to establish if the correlations were significant (P < 0.05). For the comparison of survival curves, the log-rank (Mantel-Cox) test was performed and differences were considered statistically significant at P < 0.05 (*).
Results
Elimination of IRAK4 Kinase Activity Prevents Autoimmune Traits in Aged Sle1 and Sle1Tg7 Mice
We have previously shown that Sle1Tg7 mice develop severe autoimmune traits including splenomegaly and kidney disease at around 6 months of age (25, 51). In order to assess the role of IRAK4 kinase activity in the development of disease in this model, we generated IRAK4 kinase dead (IRAK4KD/KD) mice by engineering a mutation at position-213 from Lysine (K) to Alanine (A) using a plasmid with puromycin resistance as a selective marker (Figures S1A,B). This K213A IRAK4KD/KD mutant was independently confirmed to lack kinase activity in an in vitro enzymatic assay (Figure S1C). We also confirmed comparable expression of IRAK4 mRNA and protein in IRAK4KD/KD vs. wild-type splenocytes using qPCR and Western Blot, respectively (Figures S1D,E). We went on to breed these mice with Sle1Tg7 and aged 3 cohorts of female mice to ~6–7 months. Elimination of IRAK4 activity prevented both mild and severe splenomegaly exhibited by Sle1 and Sle1Tg7 mice, respectively (Figure 1A). Analyses of kidney function showed expected increases in BUN levels and urine albumin concentration in Sle1Tg7 mice compared to Sle1 controls, which were prevented in IRAK4 KD/KD mice (Figures 1B,C). Histopathology demonstrated that Sle1Tg7 kidneys had global endocapillary proliferation with cellular crescents in the glomeruli, while kidneys from Sle1, Sle1IRAK4KD/KD, and Sle1Tg7IRAK4KD/KD mice showed only mild segmental mesangial proliferation (Figure 1D). Accordingly, the development of class 3–4 GN was completely prevented in Sle1Tg7IRAK4KD/KD mice (Figure 1E). Consistent with these findings, the survival rate of Sle1Tg7 IRAK4KD/KD mice was 100% at 6–7 months whilst only 78.3% Sle1Tg7 mice survived (Figure 1F).
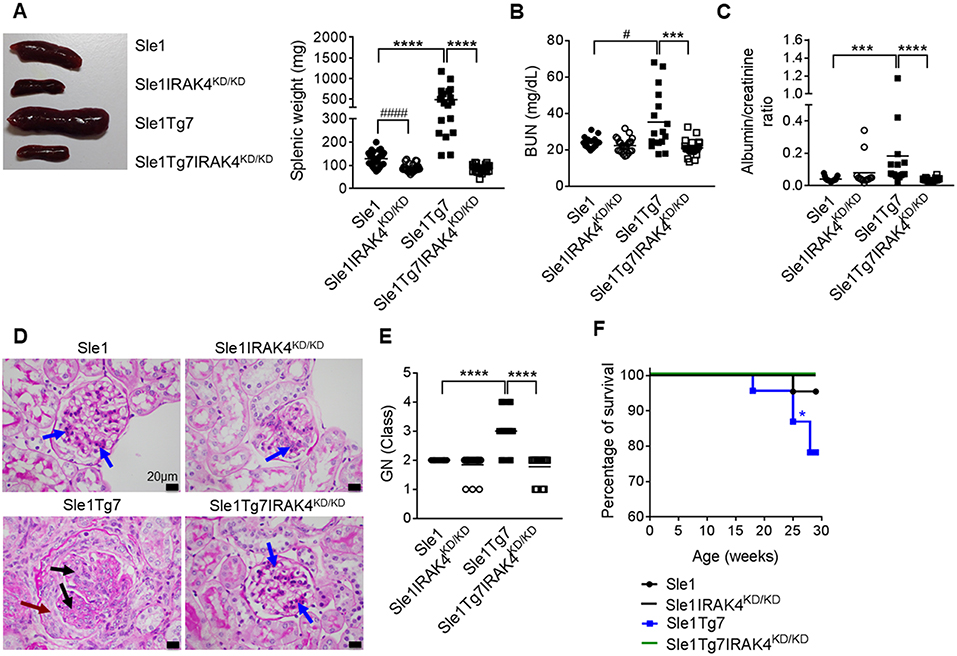
Figure 1. Prevention of spleen and kidney disease with the elimination of IRAK4 kinase activity. Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD female mice were aged to 6–7 months and analyzed for lupus disease parameters. (A) Photograph showing representative spleens from indicated strains and cumulative splenic weight (19–27 mice/group). Functional kidney assessment using (B) a blood urea nitrogen (BUN) assay in the same mice (17–23 mice/group) and (C) urine albumin levels normalized to creatinine (13–18 mice per group). Assessment of kidney disease class by PAS stain with (D) representative photomicrographs (original magnification x600, Black bars represent 20 μm), showing glomerular segments with mild mesangial proliferation (blue arrows), glomerular segments with endocapillary proliferation (black arrows) and cellular crescent (maroon arrow). (E) Cumulative data for kidney GN class for 17–19 mice/group (class I–IV represented as numbers 1–4). Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test) ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and non-parametric data, respectively, indicated by # (#P < 0.05, ####P < 0.0001). (F) Survival curves for each strain (22–23 mice per group); Sle1Tg7 mice survive significantly less than Sle1Tg7IRAK4KD/KD (*P < 0.05, Mantel-Cox test).
Circulating Autoantibodies and Immunoglobulin Titers Are Dependent on IRAK4 Kinase Activity
Characterization of autoantibodies with HEp-2 staining and fluorescent microscopy showed the presence of anti-nuclear autoantibodies (ANAs) in Sle1 mice, which were more pronounced in Sle1Tg7 mice, and prevented with IRAK4 kinase elimination (Figure 2A). Further analysis of HEp-2 staining patterns revealed an increase in the nucleolar and cytoplasmic reactive autoantibodies in Sle1Tg7 mice compared to controls (Figures 2B,D). The elimination of IRAK4 kinase activity resulted in Polar/Golgi-reactive antibodies in some Sle1IRAK4KD/KD and Sle1Tg7IRAK4KD/KD mice (Figure 2C), however their nature remains to be determined.
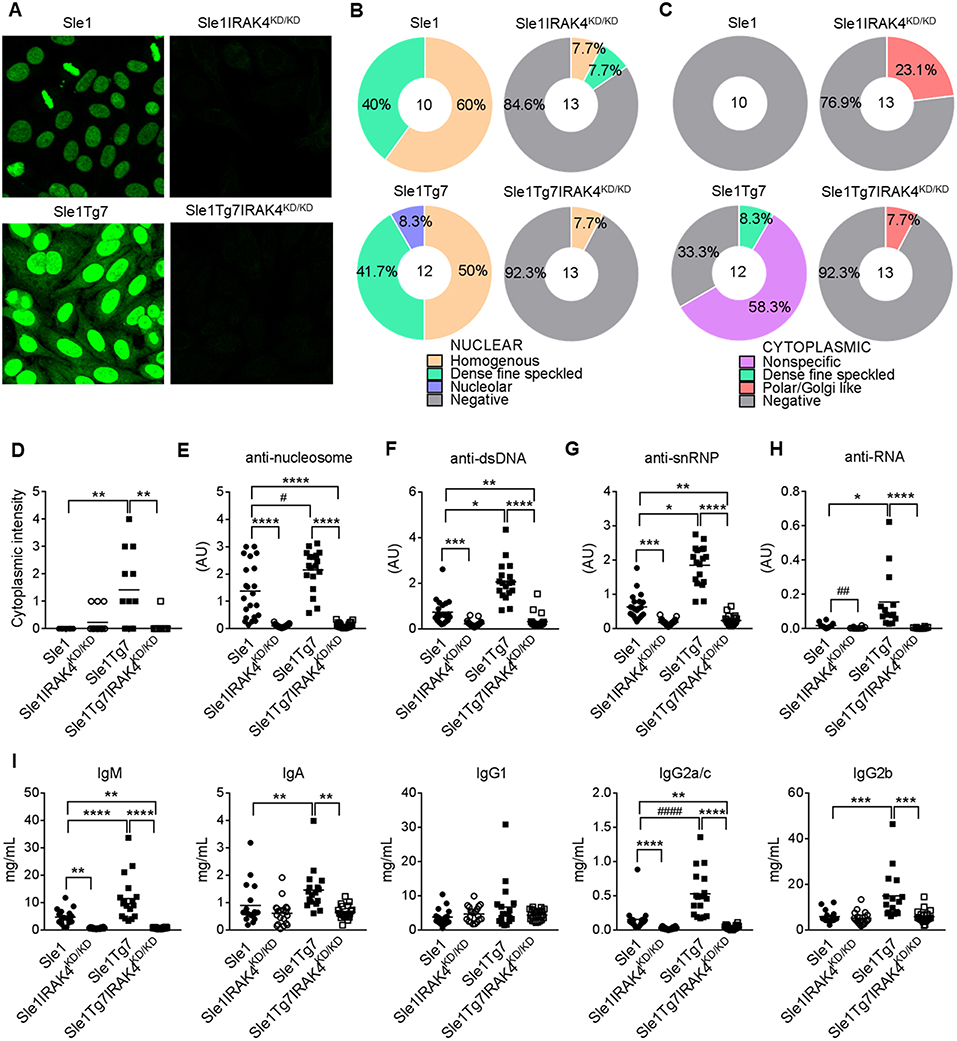
Figure 2. IRAK4 kinase activity is required for the production of autoantibodies and hypergammaglobulinemia observed in lupus-prone mice. (A) Representative microscopy images of HEp-2 staining patterns of serum autoantibodies from aged Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD mice. (B,C) Analysis of (B) nuclear and (C) cytoplasmic staining patterns determined by HEp-2 staining and microscopy. (D) Cytoplasmic intensity determined by HEp-2 microscopy. Serum for HEp-2 was from 3 independent cohorts with total 10–13 mice per group. (E–H) Analyses by ELISA of IgG autoantibodies with (E) nucleosome (dsDNA/histone), (F) dsDNA, or (G) snRNP or (H) RNA specificity in Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD mice (18–23 mice per group). (I) Luminex analysis of circulating antibody subclasses (IgM, IgA, IgG1, IgG2a/c, IgG2b, IgG3) across the 3 cohorts (data from 17–22 mice per group). Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and non-parametric data respectively, indicated by # (#P < 0.05, ##P < 0.01, ####P < 0.0001).
Our previously work has shown that Sle1Tg7 mice develop higher titers of anti-nucleosome, anti-dsDNA and anti-snRNP autoantibodies than their mild lupus-prone Sle1 counterparts (25) (Figures 2E–G). In these studies we also found that anti-RNA autoantibodies were increased in Sle1Tg7 mice (Figure 2H). Elimination of IRAK4 kinase activity prevented the development of all autoantibodies examined in both Sle1 and Sle1Tg7 mice (Figures 2E–H). Characterization of serum antibody isotypes showed increases in total IgM, IgA, IgG2a/c, and IgG2b in Sle1Tg7 mice compared to Sle1 controls and this was IRAK4 kinase dependent (Figure 2I). Interestingly IRAK4 was required for IgM and IgG2a/c levels in Sle1 mice, but not IgA or other IgG subtypes.
IRAK4 Kinase Activity Is Essential for the Development of Splenic Autoimmune Traits Observed in Sle1 and Sle1Tg7 Mice
Analyses of cell lineages using flow cytometry indicated that splenomegaly in Sle1Tg7 mice (Figure 1A) was characterized primarily by CD11b+ myeloid and CD4+ T cell expansion, as shown previously (Figure 3A and Table S1) (25, 51). This was prevented in IRAK4KD/KD mice. Consistent with our antibody data (Figure 2), plasma cells and germinal center (GC) B cells were increased in Sle1Tg7 mice compared to Sle1 controls and this was prevented with functional ablation of IRAK4 (Figures 3B,C). Moreover, the GC areas in Sle1IRAK4KD/KD and Sle1Tg7IRAK4KD/KD spleens were considerably smaller than in Sle1 and Sle1Tg7 counterparts (Figure 3D). Therefore, we compared the GCs to age-matched non-autoimmune prone C57BL/6J (B6) mice and found them to be equivalent (Figures S2A,B). The decreased frequency of marginal zone (MZ) CD21hiCD23− B cells identified previously in Sle1Tg7 and similar strains (25, 26, 51), was reduced but not completely restored with the elimination of IRAK4 kinase activity (Figure 3E). Additionally, we observed a higher percentage of age-related B cells (ABCs, B220+CD11c+CD11b+) in Sle1Tg7 mice compared to Sle1 controls which was prevented in IRAK4KD/KD mice (Figure 3F). Taken together, these finding suggest that the elimination of IRAK4 kinase activity prevents the progressive expansion, maturation and activation of B cells observed in the Sle1Tg7 strain.
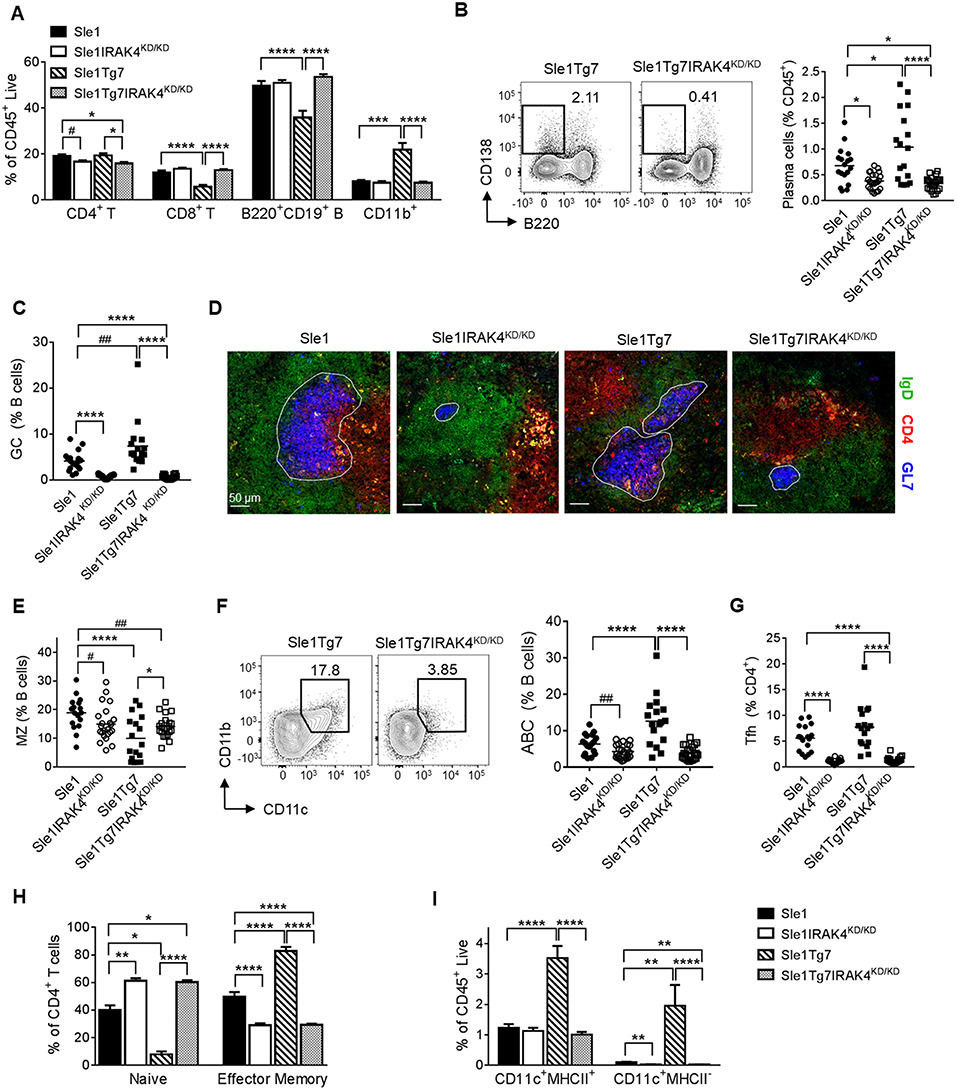
Figure 3. Functional elimination of IRAK4 prevents splenomegaly and associated leukocyte expansion observed in lupus-prone mice. Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD female mice were aged to 6–7 months and analyzed for characteristic autoimmune immunological traits. (A) Frequency of the indicated splenic leukocytes evaluated by flow cytometry. (B) Representative flow plots of plasma cells (CD138+B220−) from spleens of Sle1Tg7 and Sle1Tg7IRAK4KD/KD mice with cumulative data from all strains. (C) GC B cell frequency measured by flow cytometry as GL7+Fas+ cells, represented as % B cells (CD19+B220+). (D) Representative immunofluorescent images of splenic GC staining, 300 x scale bar represents 50 μm, with anti-GL7 (blue), anti-CD4 (red) and anti-IgD antibodies (green). (E) Cumulative frequency of marginal zone (MZ) B cells (CD21+CD23−) across all strains (represented as % of CD19+B220+ B cells). (F) Representative flow plots of age-related B cells (CD11b+CD11c+) with cumulative data (represented as % of CD19+B220+ B cells). (G) Cumulative data of CD4+ CXCR5+PD1+ T follicular helper (TFH) cells, and (H) the frequencies of naïve (CD44lowCD62Lhigh) and effector memory (CD44highCD62Llow) (represented as % of CD3+CD4+ T cells). (I) Frequencies of splenic CD11b+CD11c+MHC-II+ DCs and CD11b+CD11c+MHC-II− cells. Flow cytometry data is from 17 to 23 mice per group. Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and non-parametric data, respectively, indicated by # (#P < 0.05, ##P < 0.01).
Analyses of T cell subsets showed that CD4+ T follicular helper cells (TFH, CXCR5+ICOShi) were fewer in IRAK4 KD/KD mice compared to either Sle1 or Sle1Tg7 (Figure 3G). In a similar manner to the GCs, the TFH frequencies in Sle1IRAK4KD/KD spleens were comparable to non-autoimmune levels in B6 mice (Figure S2C). In addition, the increase in activation and expansion of effector memory CD4+ T cells characteristic of Sle1Tg7 was prevented in IRAK4KD/KD mice (Figure 3H and Table S1) (25, 51). Analyses of CD8+ T cells revealed similar reductions in effector memory cells, with concomitant increases in the naïve population, in IRAK4KD/KD mice (Table S1).
Analyses of myeloid subpopulations, cDCs, and CD8+ DCs were completed using the gating strategies in Figure S2D. The increase in the frequencies of eosinophils, PMNs, and both Gr1+ and Gr1− CD11b+ cells in Sle1Tg7 mice were completely prevented by IRAK4 KD/KD (Figure S2E and Table S1). The increase observed in Sle1Tg7 mice in cDCs, their possible precursors (Gr1−CD11c+MHCII−) and the associated changes in MHCII and CD86 expression, were also prevented by IRAK4KD/KD (Figure 3I, Figure S2F and Table S1. Analyses of CD8+ cDCs and plasmacytoid DCs (pDCs) frequencies showed a minor effect of disease or IRAK4KD/KD (Figures S2G,H, including pDC gating strategy).
Leukocyte Infiltration Associated With GN in Sle1Tg7 Mice Is Dependent on IRAK4 Kinase Activity
In a similar manner to our previous reports, we analyzed leukocyte infiltration into the kidney using flow cytometry (Figure S3A) (25, 51). We determined that the leukocyte expansion observed in Sle1Tg7 mice is dependent on IRAK4 kinase activity (Figures 4A,B). Moreover, CD4+ and CD8+ T cells from IRAK4KD/KD mice consisted of fewer effector memory and more naïve populations (Figure 4C and Figure S3B. Further characterization of the myeloid population revealed that the increases observed in PMNs and Gr1+ CD11b+ cells in Sle1Tg7 kidneys were IRAK4 kinase dependent (Figure 4D). CD11b+Gr1−CD11c+ cells were gated for F4/80 and MHCII to characterize renal macrophages (F4/80+CD11c+MHCII+), CD11b+ cDCs (F4/80−CD11c+MHCII+) and their putative precursor (CD11c+MHCII−) (Figure 4E) (51). Consistent with our earlier investigations, both the CD11b+ cDC and cDC-precursor populations were expanded with severe disease in Sle1Tg7 mice (51), and this was prevented with IRAK4 KD/KD (Figures 4E,F). Furthermore, IRAK4 kinase elimination increased the expression of MHCII on DCs and macrophages (Figure S3C).
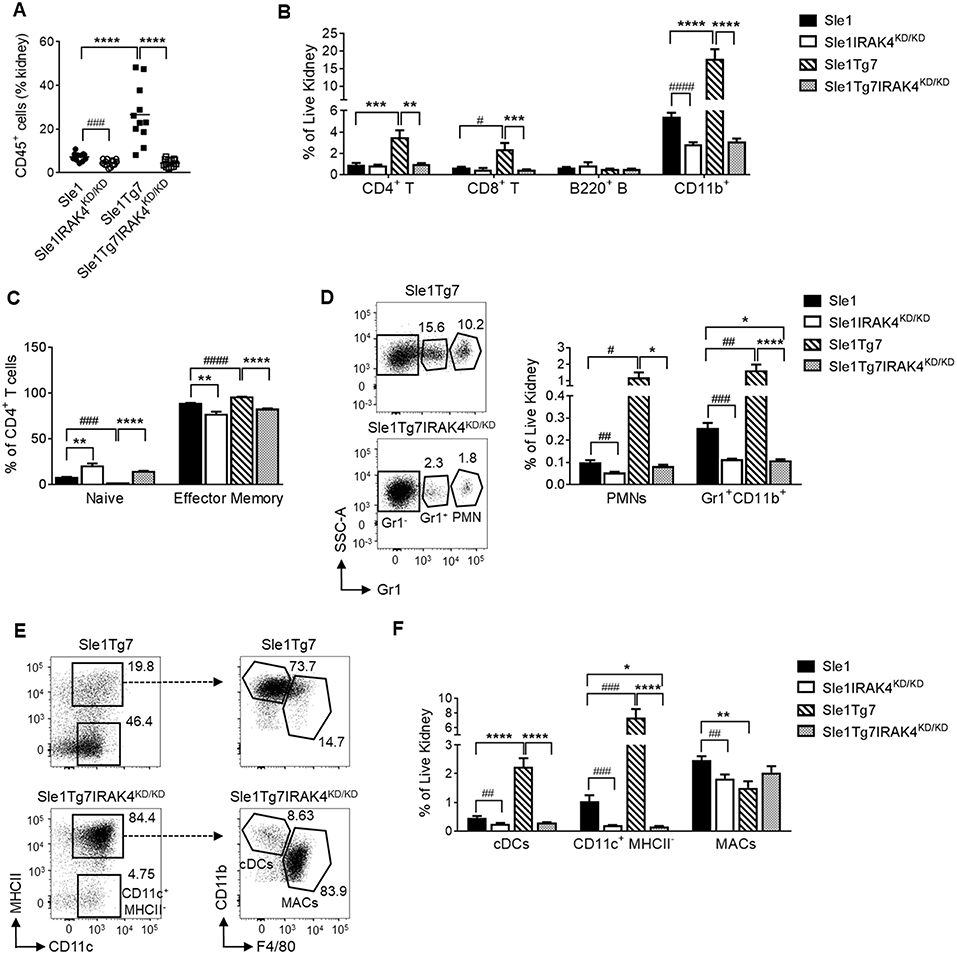
Figure 4. IRAK4 kinase activity is essential for renal leukocyte infiltration and activation of lupus prone mice. Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD female mice were aged to 6–7 months and analyzed for leukocyte infiltration and activation. Frequency of (A) total CD45+ leukocytes and (B) CD4+ T, CD8+ T, B220+ B and CD11b+ leukocyte subsets in the kidney by flow cytometry. (C) Cumulative data of the frequencies of naïve (CD44lowCD62Lhigh) and effector memory (CD44highCD62Llow) CD4+ T cells. Representative flow plots and cumulative data of the frequencies of (D) PMNs and Gr-1+ CD11b+ myeloid cells and (E) CD11b+CD11c+MHC-II+ cDCs, CD11c+ MHCII− cells and CD11b+CD11c+MHCII+F4/80+ macrophages (MACs) in the kidney. (F) Cumulative data of cells from (E) is shown. For (A,C) data from 11 to 14 mice/group is shown, while the rest is from 9 to 11 mice/group. Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and nonparametric data respectively, indicated by # (#P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001).
Additionally, we analyzed pDCs in the kidney using antibodies to B220, Siglec H and mPDCA1 (Figure S3A), which enabled a better identification than in our earlier study (51). We determined a significant increase of pDCs within the kidney, similar to previous observations (51) (Figure S3D).
Splenic TLR7 Expression Is Regulated by Both the TLR7-BAC and Disease State in a Leukocyte-Specific Manner
We have previously shown effective detection of intracellular TLR7 protein using flow cytometry with clone A94B10 (47). We used a 17-color panel to investigate all major leukocyte subtypes in spleens and kidneys (gating strategy for splenocytes in Figure S4A). TLR7 expression was undetectable to low in CD4+ T cells, CD8+ T cells, NK1.1+ cells, PMNs, and CD8+ DCs as shown previously (Figures S4B–D and data not shown) (47).
The mild autoimmunity of the Sle1 mice had no effect on TLR7 in B cell subsets, as indicated by a comparison with the B6 control (Figures 5A–C). Although the expression was low in both B cells and plasmablasts of Sle1 mice, levels were much higher in Sle1Tg7 mice (Figures 5A,B). This increase was largely prevented with the elimination of IRAK4 signaling. The fact that the elimination of IRAK4 kinase activity did not affect TLR7 levels in Sle1 mice reduces the likelihood of this being a factor when comparing the Sle1Tg7 and Sle1Tg7IRAKKD/ KD mice. A comparison of Sle1IRAKKD/KD and Sle1Tg7IRAKKD/KD mice, which have similar phenotypes and pathologies, shows that the TLR7-BAC transgene (Tg7) increases TLR7 expression in both B cells and plasmablasts. Since expression in Sle1Tg7 is higher than the respective Sle1Tg7IRAKKD/KD mice, it confirms that the inflammatory environment of severe autoimmunity and signaling through IRAK4 contributes to the increase in TLR7 expression (Figures 5A,B). Interestingly, TLR7 protein levels in plasma cells were only increased as a result of advanced autoimmunity and not the Tg7 itself (Figure 5C).
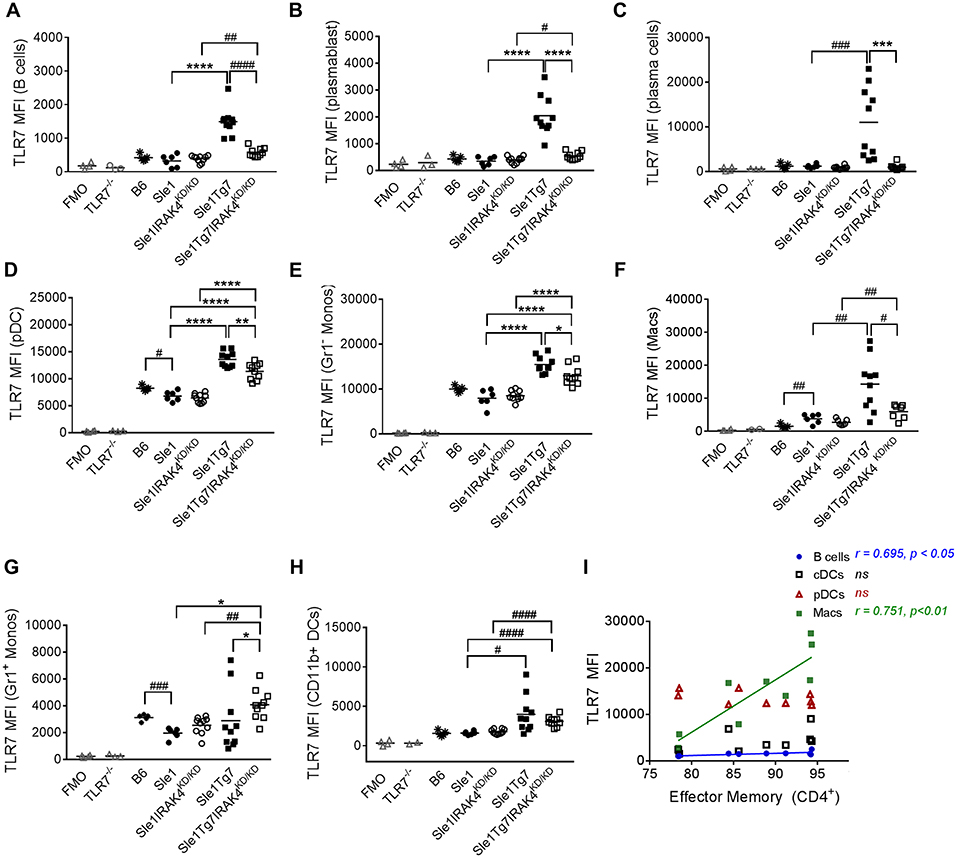
Figure 5. Expression of TLR7 in splenocytes is modulated by disease and Tg7 in a leukocyte specific manner. Splenocytes from 6 to 7months old B6, Sle1, Sle1Tg7, Sle1IRAK4KD/KD, and Sle1Tg7IRAK4KD/KD female mice were analyzed for intracellular TLR7 expression using flow cytometry. TLR7−/− mice and fluorescent minus one (FMO) controls were used to determine specificity. (A) Cumulative data of TLR7 expression in B cells, (B) plasmablasts, (C) plasma cells, and (D) CD115+Gr1− monocytes (Gr1− Monos), (E) F4/80+CD11blo macrophages (Macs), (F) B220+SiglecH+mPDCA1+ pDCs, (G) CD115+Gr1+ monocytes (Gr1+ Monos), (H) CD11b+CD11c+MHCII+ DCs (CD11b+DCs), across all strains. (I) Correlation of TLR7 expression in B cells, CD11b+ cDCs, pDCs and macrophages from Sle1Tg7 mice with effector memory CD4+ T frequencies in the spleen. Data from 6 to 10 mice per group is shown. Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and non-parametric data, respectively, indicated by # (#P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001). Correlations were performed by calculating Pearson's correlation coefficients (r) and their significance assessed by a two-tailed t-test.
pDCs have been implicated in the development of SLE. Surprisingly, TLR7 levels were lower in pDCs from Sle1 mice compared to B6 controls (Figure 5D). Furthermore, disruption of IRAK4 signaling did not affect TLR7 expression in these mild autoimmune mice. The increased expression in both Sle1Tg7 and Sle1Tg7IRAK4KD/KD mice also demonstrates regulation of expression by both the BAC-Tg7 and the inflammatory milieu of severe pathology, congruent to B cell findings.
TLR7 levels in Gr− (CD115+) monocytes and macrophages were augmented by the Tg7 and severe disease (Figures 5E,F). However, Gr1+ “inflammatory” monocyte TLR7 expression was only increased with the Tg7 (Figure 5G), independently of disease/signaling through IRAK4, with similar findings in CD11b+cDCs (Figure 5H). Interestingly, only macrophage TLR7 expression clearly correlated with CD4+ T cell effector memory frequency (Figure 5I). A positive association was also detected in B cells, however the biological relevance remains unclear, given the comparatively low TLR7 expression in this population.
Kidney Leukocyte TLR7 Expression Is Regulated by the BAC, IRAK4, and Disease
Our earlier studies have shown a critical role for renal CD11b+ cDCs in driving CD4+ T cell activation, inflammatory cytokine release and subsequent inflammation in Sle1Tg7 kidneys (51). In contrast to our results in the spleen, TLR7 levels in renal CD11b+ cDCs were observably higher in Sle1 mice compared to B6 counterparts (p = 0.05; Figure 6A). In addition, the expression was further augmented with the increased disease severity of Sle1Tg7 mice (Figure 6A). Our data also showed that these disease associated increases in TLR7 were dependent on IRAK4 kinase activity. Given that our earlier findings showed that renal and splenic cDCs were of similar ontogeny (51), this suggests that the inflammatory environments play an enormous impact in TLR7 expression and there are significant differences between the largely immunological environment of the spleen and the mixed tissue and immune environment of the kidney.
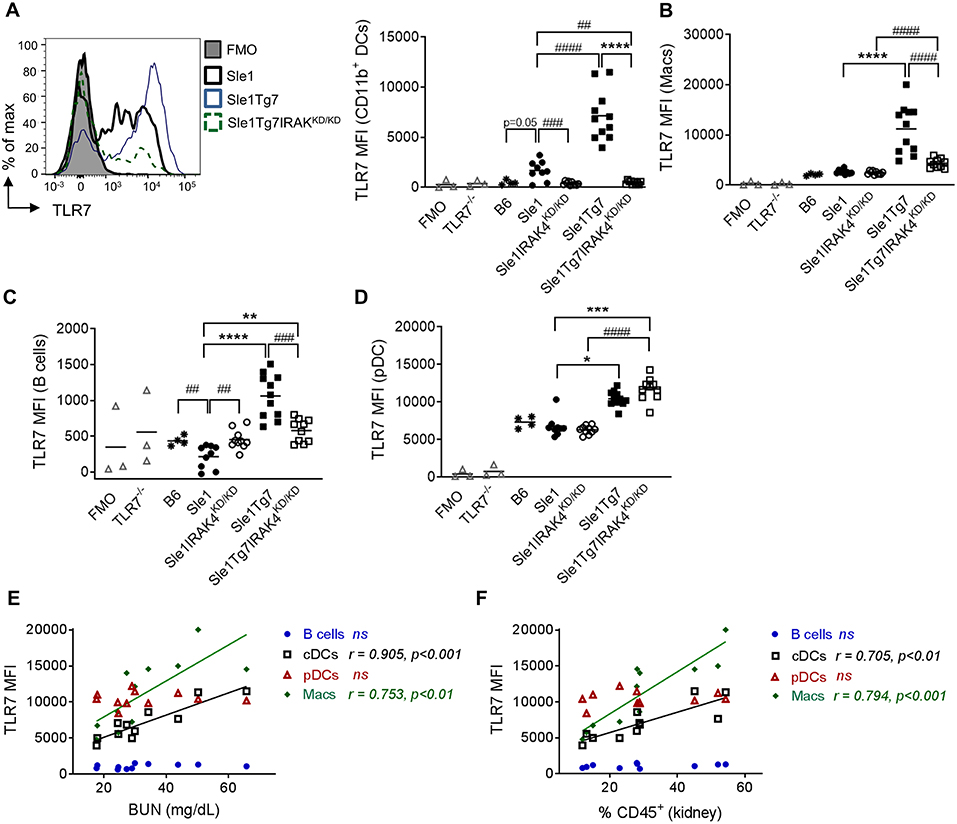
Figure 6. Expression of leukocytes in the kidney is modulated by disease and Tg7 transgene. Leukocytes in the kidney from 6 to 7 months old B6, Sle1, Sle1IRAK4KD/KD, Sle1Tg7, and Sle1Tg7IRAK4KD/KD female mice were analyzed for intracellular TLR7 expression using flow cytometry. TLR7−/− mice and fluorescent minus one (FMO) controls were used to determine specificity. (A) Representative overlay plots of intracellular TLR7 expression in CD11b+ DCs showing increases due to the transgenic and severe disease factors with cumulative data across all strains. Cumulative intracellular TLR7 protein expression in (B) CD11b+CD11c+F4/80+ macrophages (Macs), (C) B cells, (D) pDCs, (E,F) Correlation of TLR7 expression on B cells, CD11b+DCs (cDCs), pDCs, and macrophages (Macs) from Sle1Tg7 mice with (H) BUN and (I) percent of CD45 infiltration in kidney. Data from 9 to 11 mice per group is shown. Parametric data were assessed by 1-way ANOVA (with Bonferroni's multiple comparisons test) and non-parametric data with Kruskal-Wallis (with Dunn's multiple comparisons test) **P < 0.01, ***P < 0.001, ****P < 0.0001. Additional comparisons between two groups were made with a Student's t test or Mann-Whitney test for parametric and non-parametric data, respectively, indicated by # (##P < 0.01, ###P < 0.001, ####P < 0.0001). Correlations were performed by calculating Pearson's correlation coefficients (r) and their significance assessed by a two-tailed t-test.
Our analysis of renal macrophages and B cells showed increases in TLR7 as a result of severe disease (Figures 6B,C). Additionally, given the fact that IRAK4 kinase perturbation did not reduce TLR7 expression in renal Sle1 macrophages, the increase in TLR7 in Sle1Tg7IRAK4KD/KD compared to Sle1IRAK4KD/KD may be attributed to the Tg7-BAC itself. However, since these mice have an increase in RNA-containing ICs, which provide a positive feedback loop of TLR7-Myddosome signaling, these assumptions may not be so simple.
In contrast to our findings in the spleen, TLR7 expression in kidney pDCs was dependent solely on the BAC-Tg7, since the elimination of IRAK kinase activity and disease had no effect (Figure 6D). The mixed cell populations, CD11b+Gr1+, CD11c+MHCII− and CD11c−MHCII− also showed similar gene and disease dependent changes (Figures S5A–C).
Additionally, we observed that TLR7 expression in renal CD11b+cDCs and macrophages, but not in pDCs and B cells, correlated with serum BUN and the total leukocyte infiltration (Figures 6E,F).
Discussion
This report provides several advancements in our understanding of the immunological events occurring in autoimmunity using murine models of SLE. We conclusively demonstrated that the kinase domain of IRAK4 is necessary for all autoimmune traits in mild Sle1 prone mice and in the TLR7-directed severe disease exhibited by the Sle1Tg7 model. Aside from Myddosome signaling, IRAK4 also plays a structural, kinase-independent role, which can mediate TAK1-independent NF-κB activation (37, 41). Our data and the work of others has effectively eliminated this as a major factor in the development of autoimmune traits (42, 45). Additionally, in this study, we comprehensively analyzed TLR7 protein expression across leukocyte subsets infiltrating the kidney, and in the spleen. We determined that TLR7 is regulated by the disease severity and IRAK4 kinase activity in a leukocyte and tissue specific manner. Importantly, we show that increased TLR7 protein in renal cDCs and macrophages correlates with measures of inflammation and kidney pathogenesis, pointing to their crucial role in the disease process.
Previous work has shown that IRAK4 phosphorylation is essential for the aggressive systemic autoimmunity and autoinflammation exhibited by the ABIN1 model (42). These mice develop key lupus phenotypes including ANA production and GN, however they also develop inflammation in tissues not commonly associated with clinical disease or exhibited by other lupus-prone murine models, such as liver and gut pathologies. Although many phenotypes can be attributed to increased TNFα signaling in the ABIN1 model, it seems unlikely that this is the mechanism of the lupus-like phenotypes (44). Congruent to this, we have shown that TNFα levels, even at the site of the diseased kidney, are not affected by systemic increases in TLR7 or the severe autoimmune environment (25). The ABIN1 studies also show that, despite the upregulation of type I IFNs, elimination of IFNAR1 only partially reduced the development of GN, and did not affect other phenotypes (42). Therefore, the cytokines and critical cells mediating the pathologies remain elusive. The widespread hyperactivation of the TLR family, together with BCR signaling suggests a more complicated mechanism of disease in this model. We and others have suggested a stepwise progression of the development of polygenic associated lupus (54, 55). The initial steps in disease development confer a loss of tolerance, the generation of autoreactive T and B cells and autoantibody development. The progression to severe disease requires an additional immune alteration, which in many cases has been attributed to innate dysregulation. Given the retention of autoimmune phenotypes following backcrossing to the B6 background by Cohen and colleagues, it is clear that this single mutation of ABIN1 mediates dysregulation of both immune tolerance and innate activation (42, 44). Certainly, the loss of MyD88, and disrupted IRAK1/4 signaling points to the essential nature of the TLR and IL1R families for the mechanism of aggressive disease.
In our studies we were able to analyse the contribution of IRAK4-mediated signaling at different stages of disease. We and others have used B6.Sle1 mice as an example of step 1 in the pathway to severe disease, given their mild phenotypes and that they rarely develop GN in the absence of an additional susceptibility locus (24, 47, 51, 56). The sle1 susceptibility locus on chromosome 1 is shared across most spontaneous murine model systems (54). Previous work by Wakeland et al. has shown that the NZM2410-derived sle1 phenotypes are conferred by polymorphisms in the SLAMF members, Ly108, CD82, CD244, CD229, and CD319 (57, 58). Our data shows that IRAK4 phosphorylation is required for the break in tolerance to nuclear antigens and ANA production in Sle1 mice. In addition, all mild autoimmune traits exhibited by these mice appear to be prevented. The reductions in splenic B and T cell activation and expansion are akin to the B6 phenotypes, suggesting that MyD88/IRAK pathway disruption is targeting spontaneous autoimmune mechanisms, rather that core developmental processes. Our data also suggests that while the MyD88/IRAK pathway is critical for these Sle1-conferred phenotypes, changes in TLR7 at the protein level do not play a significant role. In addition, it is likely that the loss in tolerance mediated by the SLAMF polymorphisms in Sle1 mice develops with the assistance of other TLR/IL1R family members, given the promiscuous nature of the Myddosome and IRAK4 kinase activity.
To examine severe disease, we used our low copy TLR7 BAC transgenic on the Sle1 background, Sle1Tg7. These mice develop similar phenotypes to the BXSB strain without the male dependency for GN. Therefore, cohorts of the same sex or comparisons between sexes can be analyzed, allowing for sex-independent or sex-dependent immunological pathway analysis. Furthermore, Sle1 mice have a female sex bias similar to clinical disease, most likely attributed to the incomplete X inactivation, or the XX complex itself (59, 60).
An additional advantage of using B6.Sle1/Sle1Tg7 mice was the retention of the same non-autoimmune background (B6), eliminating influences of other loci. In these studies, we were able to confirm that the elimination of proteinuria and splenomegaly seen in BXSBYaa.IRAK4KD/KD mice was not due to the dilution of susceptibility loci present in the BXSB background by the introduction of the B6 non-autoimmune genome (45). However, in contrast to the data from Murphy et al. our heterozygous Sle1Tg7IRAK4WT/KD mice developed severe disease in a similar manner to Sle1Tg7IRAK4KD/IRAK4KD homozygotes (data not shown). The increased disease in the BXSB F2 heterozygous highlights additional loci which contribute to disease and are not present in the B6 background of Sle1Tg7 mice. Similar to the findings of the homozygous BXSBYaa.IRAK4KD/KD male mice shown in the above study, we saw a prevention in the expansion of B cells and plasma cells, with IRAK4 kinase elimination. The splenomegaly by most spontaneous severe lupus models is also characterized by increases in germinal center B cells and T follicular helper cells, together with T cell activation (25, 26, 51). We were also able to show that these changes were prevented with IRAK4 kinase perturbation. A more in-depth analysis of the CD11c+ lineage excluded neutrophils and revealed that CD11b+ cDCs and their putative precursors, CD11c+MHC− cells, comprised the majority of this expansion. Our splenic analysis also showed that in the majority of leukocytes, TLR7 was upregulated not only through the genomic contribution of the Tg7-BAC, but also by the intense autoimmune environment observed in the Sle1Tg7 mice.
Our work also provides a detailed analysis of the immunological events occurring not only in the spleen, but also in the diseased kidney. Approximately 50% of SLE patients develop GN and obvious challenges restrict comprehensive understanding of the inflammatory events occurring. Given the burgeoning data on the role of TLR7 in SLE pathogenesis, analysis at the protein level within the tissue may prove to be invaluable. Overall IRAK4 kinase ablation prevented both mild and severe kidney disease in the Sle1 and Sle1Tg7 models, respectively, together with the associated T cell and DC infiltrate. Our examination of TLR7 protein levels using flow cytometry showed that the autoimmune inflammatory environment was able to augment TLR7 expression, with the exception of renal infiltrating pDCs. Moreover, renal CD11b+cDC TLR7 expression was associated with key disease parameters, including BUN levels and the renal leukocyte infiltrate. This may suggest that the development of a soluble TLR7 assay as a urine biomarker could prove to be clinically relevant. However, given the endosomal location of TLR7, it is unclear as to whether this is possible and therefore warrants further investigation.
Previous data from a number of groups has shown that TLR7 plays a critical role in the initial stages of autoimmunity and development of ANAs (18–20). Interestingly, elimination of TLR7 in murine SLE results in similar phenotypes to our IRAK4 kinase deficient Sle1 mice, including a decreased GC formation, splenic TFH frequencies and serum autoantibody titers (18, 19, 61, 62). In addition, elimination of the downstream transcription factor, IRF5 in MRLlpr lupus prone mice results in a similar ablation of all autoimmune phenotypes (63). In patient genome wide association studies (GWAS), IRF5 polymorphisms have also associated with disease development (33–36). Congruent to these findings, the elimination of IRAK4 signaling prevents TLR-induced IKKβ phosphorylation and the subsequent translocation of the transcription factor IRF5 to the nucleus (40). This prevents the TLR induced (and Myd88 dependent) cytokine production. At the same time, the Myddosome complex becomes more stable and NFβB nuclear translocation remains intact (40, 64). Taken together these findings indicate that the autoimmunity observed across multiple model systems is dependent on a TLR7, MyD88-IRAK4 kinase, and IRF5 dependent signaling cascade, which is independent of NFkB nuclear translocation and cytokine production. Given that the region encompassing Sle1 on chromosome is a common susceptibility locus, additionally identified in NZB/W and in MRLlpr mice, and previous work showing TLR7 and MyD88 elimination preventing autoimmune phenotypes, we propose that IRAK kinase perturbations in these polygenic murine models would result in eliminating autoimmunity. Further studies into the signaling, nuclear translocation and cytokine production would shed further light on these mechanisms, particularly given the bias toward TLR7 of our model system.
We and others have previously shown that IFNα and TLR7 ligands, such as RNA-containing viruses and TLR7 agonist R848 can increase TLR7 mRNA expression (51, 65). Since RNA-containing immune complexes also act as TLR7 ligands (66), they may increase TLR7 expression in lupus models and clinical disease through a positive feedback loop. Several studies support this hypothesis: SLE serum increases TLR7 expression (7) and SLE patients with an anti-RNA-associated antibody profile have preferentially higher TLR7 expression (67). TLR7 ligand hyper-responsiveness has been identified as the main driver of lupus-like pathology in pristane-induced mouse models of SLE (48, 68). Our studies in TLR9-deficient mice have also attributed the increase in TLR7 protein expression to an anti-RNA antibody profile and severe disease (47). In the present study we have shown that disease can directly contribute to the increase of TLR7 expression in various cell types. Eliminating downstream TLR7 (and other TLR) signaling through IRAK4 by using IRAK4 kinase deficient mice prevents the development of disease and TLR7 upregulation beyond the effects of the BAC. Blocking signal transduction and preventing TLR7 upregulation by inhibiting IRAK4 thus represents a possible therapeutic target for SLE treatment.
Over 90 patents for small molecule IRAK4 inhibitors have been filed from 2016 to 2018, primarily for autoimmune diseases and cancer (46, 69). In keeping with the data from lupus mice shown here and by others (42, 45), the IRAK4 inhibitor BMS-986126 suppressed the development of lupus in the NZB/NZW and MRLlpr models (46). Similarly, IRAK4 inhibitors, ND-2158 and ND-2110 attenuated TNFα production from TLR4/9-stimulated human PBMCs and autoimmune disease in collagen-induced arthritis and gout murine models (70). In separate studies relating to SLE pathogenesis, IRAK4 inhibitor, ND-2158 reduced cytokine production by human plasmacytoid dendritic cells and natural killer cells stimulated by RNA- ICs (71). However, in these studies did not find an increase in TNF or IFNα production in SLE PBMCs compared to controls, perhaps due to medication or leukocyte exhaustion. Given these findings and the broad role of IRAK4, it is unclear as to whether the toxicity of the side effects will prove too limiting for clinical use (72).
In summary, the findings in our study show a dependency on IRAK4 kinase activity for increased TLR7 protein levels, together with mild and severe autoimmune phenotypes of the Sle1 and Sle1Tg7 models, respectively. The regulation of TLR7 expression is not as simple as one expects, and depends on the pathogenic environment and the individual leukocyte involved. Further studies are required to further elucidate these factors, including cytokine induced nascent expression of TLR7 which may exacerbate BAC-induced TLR7 expression.
Taken together, our findings show the importance of increased signaling through TLR7 in SLE and targeting this pathway, may lead to a suppression of severe disease development.
Ethics Statement
This study was carried out in accordance with the recommendations of the Biological Resource Center of A*STAR. The protocol was approved by the IACUC committee, protocol ID 161176.
Author Contributions
TC, HKL, HYL, ST, OZ, WYO, HY, LB, LR, BA, TPT, and A-MF performed the experiments. TC, HKL, DM, LB, LR, TBT, and A-MF analysed the data. A-MF, TC, and HKL wrote the manuscript. A-MF, DMZ, JSM, and JEC designed the research studies. A-MF, DMZ, MFM, JSM, JEC, and LHKL provided reagents, mice and/or statistical support.
Funding
This study was funded through a joint research agreement by Merck & Co., Inc., Kenilworth, NJ, USA and A*STAR, and core funding from the A*STAR Research Entities; Singapore Immunology Network (SIgN) to A-MF and the Institute for Molecular and Cell Biology to JEC.
Conflict of Interest Statement
The authors declare that this study received funding through a joint innovation grant between Merck & Co., Inc. and A*STAR, Singapore. LB and LR are employees of Merck. MFM, JSM, and DMZ were employees of Merck at the time of the study. DM was an employee of Tessa at the time of the study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Katja Lakota, Ph.D and Tinka Svec, MSc from The University Medical Centre Ljubljana, Department of Rheumatology, for the HEp-2 slides scoring and subsequent discussion. We thank the Flow Cytometry cores at IMCB and SIgN at A*STAR.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01546/full#supplementary-material
References
1. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.40
2. Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. (2009) 10:373–9. doi: 10.1038/gene.2009.39
3. Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. (2009) 120:2012–24. doi: 10.1161/CIRCULATIONAHA.108.771170
4. Doedens JR, Jones WD, Hill K, Mason MJ, Gersuk VH, Mease PJ, et al. l. Blood-Borne RNA correlates with disease activity and IFN-stimulated gene expression in systemic lupus erythematosus. J Immunol. (2016) 197:2854–63. doi: 10.4049/jimmunol.1601142
5. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. (2002) 3:196–200. doi: 10.1038/ni758
6. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. (2004) 101:5598–603. doi: 10.1073/pnas.0400937101
7. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
8. Lood C, Arve S, Ledbetter J, Elkon KB. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J Exp Med. (2017) 214:2103–19. doi: 10.1084/jem.20161512
9. Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol. (2008) 152:482–7. doi: 10.1111/j.1365-2249.2008.03646.x
10. García-Ortiz H, Velázquez-Cruz R, Espinosa-Rosales F, Jiménez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis. (2010) 69:1861–5. doi: 10.1136/ard.2009.124313
11. Lee YH, Choi SJ, Ji JD, Song GG. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus. (2016) 25:593–601. doi: 10.1177/0961203315622823
12. Kawasaki A, Furukawa H, Kondo Y, Ito S, Hayashi T, Kusaoi M, et al. TLR7 single-nucleotide polymorphisms in the 3' untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther. (2011) 13:R41. doi: 10.1186/ar3277
13. Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci USA. (2010) 107:15838–43. doi: 10.1073/pnas.1001337107
14. Deng Y, Zhao J, Sakurai D, Kaufman KM, Edberg JC, Kimberly RP, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. (2013) 9:e1003336. doi: 10.1371/journal.pgen.1003336
15. Dillon SP, Kurien BT, Li S, Bruner GR, Kaufman KM, Harley JB, et al. Sex chromosome aneuploidies among men with systemic lupus erythematosus. J Autoimmun. (2012) 38(2–3):J129–34. doi: 10.1016/j.jaut.2011.10.004
16. Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X chromosome dose and sex bias in autoimmune diseases: increased prevalence of 47,XXX in systemic lupus erythematosus and sjogren's syndrome. Arthritis Rheumatol. (2016) 68:1290–1300. doi: 10.1002/art.39560
17. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol. (2018) 3:eaap8855. doi: 10.1126/sciimmunol.aap8855
18. Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. (2006) 25:429–40. doi: 10.1016/j.immuni.2006.07.014
19. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. (2010) 184:1840–8. doi: 10.4049/jimmunol.0902592
20. Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. (2010) 34:339–48. doi: 10.1016/j.jaut.2009.11.001
21. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. (2006) 312:1669–72. doi: 10.1126/science.1124978
22. Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. (2008) 38:1971–8. doi: 10.1002/eji.200838138
23. Santiago-Raber ML, Kikuchi S, Borel P, Uematsu S, Akira S, Kotzin BL, et al. Evidence for genes in addition to Tlr7 in the Yaa translocation linked with acceleration of systemic lupus erythematosus. J Immunol. (2008) 181:1556–62. doi: 10.4049/jimmunol.181.2.1556
24. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. (2006) 103:9970–5. doi: 10.1073/pnas.0603912103
25. Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol. (2012) 189:5786–96. doi: 10.4049/jimmunol.1202195
26. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. (2007) 27:801–10. doi: 10.1016/j.immuni.2007.09.009
27. Lee BL, Barton GM. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. (2014) 24:360–9. doi: 10.1016/j.tcb.2013.12.002
28. Mouchess ML, Arpaia N, Souza G, Barbalat R, Ewald SE, Lau L, et al. Transmembrane mutations in Toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity. (2011) 35:721–32. doi: 10.1016/j.immuni.2011.10.009
29. Li X. IRAK4 in TLR/IL-1R signaling: possible clinical applications. Eur J Immunol. (2008) 38:614–8. doi: 10.1002/eji.200838161
30. Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev. (2009) 227:248–63. doi: 10.1111/j.1600-065X.2008.00733.x
31. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
32. Fu Q, Zhao J, Qian X, Wong JL, Kaufman KM, Yu CY, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum. (2011) 63:749–54. doi: 10.1002/art.30193
33. Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. (2008) 40:204–10. doi: 10.1038/ng.81
34. Kelly JA, Kelley JM, Kaufman KM, Kilpatrick J, Bruner GR, Merrill JT, et al. Interferon regulatory factor-5 is genetically associated with systemic lupus erythematosus in African Americans. Genes Immun. (2008) 9:187–94. doi: 10.1038/gene.2008.4
35. Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthrit Rheum. (2008) 58:2481–7. doi: 10.1002/art.23613
36. Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet. (2008) 17:2868–76. doi: 10.1093/hmg/ddn184
37. Fraczek J, Kim TW, Xiao H, Yao J, Wen Q, Li Y, et al. The kinase activity of IL-1 receptor-associated kinase 4 is required for interleukin-1 receptor/toll-like receptor-induced TAK1-dependent NFkappaB activation. J Biol Chem. (2008) 283:31697–705. doi: 10.1074/jbc.M804779200
38. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol. (2014) 5:367. doi: 10.3389/fimmu.2014.00367
39. Zhu J, Mohan C. Toll-like receptor signaling pathways–therapeutic opportunities. Mediators Inflamm. (2010) 2010:781235. doi: 10.1155/2010/781235
40. Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R, et al. IRAK4 kinase activity controls Toll-like receptor induced inflammation through the transcription factor IRF5 in primary human monocytes. J Biol Chem. (2017) 292:18689–98. doi: 10.1074/jbc.M117.796912
41. Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem. (2004) 279:40653–8. doi: 10.1074/jbc.M402666200
42. Nanda SK, Lopez-Pelaez M, Arthur JS, Marchesi F, Cohen P, et al. Suppression of IRAK1 or IRAK4 catalytic activity, but not Type 1 IFN signaling, prevents lupus nephritis in mice expressing a Ubiquitin binding-defective mutant of ABIN1. J Immunol. (2016) 197:4266–73. doi: 10.4049/jimmunol.1600788
43. Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, et al.Oshima S, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. (2009) 457:906–9. doi: 10.1038/nature07575
44. Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med. (2011) 208:1215–28. doi: 10.1084/jem.20102177
45. Murphy M, Pattabiraman G, Manavalan TT, Medvedev AE. Deficiency in IRAK4 activity attenuates manifestations of murine Lupus. Eur J Immunol. (2017) 47:880–91. doi: 10.1002/eji.201646641
46. Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J Immunol. (2017) 198:1308–19. doi: 10.4049/jimmunol.1600583
47. Celhar T, Yasuga H, Lee HY, Zharkova O, Tripathi S, Thornhill SI, et al. TLR9 deficiency breaks tolerance to RNA-associated antigens and upregulates TLR7 protein in Sle1 mice. Arthritis Rheumatol. (2018) 70:1597–609. doi: 10.1002/art.40535
48. Bossaller L, Christ A, Pelka K, Nündel K, Chiang PI, Pang C, et al. TLR9 Deficiency leads to accelerated renal disease and myeloid lineage abnormalities in pristane-induced murine lupus. J Immunol. (2016) 197:1044–53. doi: 10.4049/jimmunol.1501943
49. Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, et al. Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immunol. (1997) 158:6019–28.
50. Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. (2004) 65:521–30. doi: 10.1111/j.1523-1755.2004.00443.x
51. Celhar T, Hopkins R, Thornhill SI, De Magalhaes R, Hwang SH, Lee HY, et al. RNA sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc Natl Acad Sci USA. (2015) 112:E6195–204. doi: 10.1073/pnas.1507052112
52. Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods. (1998) 212:187–92. doi: 10.1016/S0022-1759(98)00015-5
53. Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. (2008) 38:1948–60. doi: 10.1002/eji.200837925
54. Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. (2006) 92:1–69. doi: 10.1016/S0065-2776(06)92001-X
55. Lu R, Munroe ME, Guthridge JM, Bean KM, Fife DA, Chen H, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. (2016) 74:182–93. doi: 10.1016/j.jaut.2016.06.001
56. Zhu J, Liu X, Xie C, Yan M, Yu Y, Sobel ES, et al. T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J Clin Invest. (2005) 115:1869–78. doi: 10.1172/JCI23049
57. Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. (2004) 21:769–80. doi: 10.1016/j.immuni.2004.10.009
58. Wong EB, Soni C, Chan AY, Domeier PP, Shwetank, Abraham T, et al. B cell-intrinsic CD84 and Ly108 maintain germinal center B cell tolerance. J Immunol. (2015) 194:4130–43. doi: 10.4049/jimmunol.1403023
59. Smith-Bouvier DL, Divekar AA, Sasidhar M, Du S, Tiwari-Woodruff SK, King JK, et al. A role for sex chromosome complement in the female bias in autoimmune disease. J Exp Med. (2008) 205:1099–108. doi: 10.1084/jem.20070850
60. Souyris M, Mejía JE, Chaumeil J, Guéry JC. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin Immunopathol. (2019) 41:153–64. doi: 10.1007/s00281-018-0712-y
61. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. (2014) 192:4525–32. doi: 10.4049/jimmunol.1400098
62. Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol. (2014) 193:4400–14. doi: 10.4049/jimmunol.1401720
63. Tada Y, Kondo S, Aoki S, Koarada S, Inoue H, Suematsu R, et al. Interferon regulatory factor 5 is critical for the development of lupus in MRL/lpr mice. Arthritis Rheum. (2011) 63:738–48. doi: 10.1002/art.30183
64. De Nardo D, Balka KR, Cardona Gloria Y, Rao VR, Latz E, Masters SL. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J Biol Chem. (2018) 293:15195–207. doi: 10.1074/jbc.RA118.003314
65. Marshall JD, Heeke DS, Gesner ML, Livingston B, Van Nest G. Negative regulation of TLR9-mediated IFN-alpha induction by a small-molecule, synthetic TLR7 ligand. J Leukoc Biol. (2007) 82:497–508. doi: 10.1189/jlb.0906575
66. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. (2006) 6:823–35. doi: 10.1038/nri1957
67. Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol. (2013) 33:954–64. doi: 10.1007/s10875-013-9887-0
68. Han S, Zhuang H, Xu Y, Lee P, Li Y, Wilson JC, et al. Maintenance of autoantibody production in pristane-induced murine lupus. Arthritis Res Ther. (2015) 17:384. doi: 10.1186/s13075-015-0886-9
69. McElroy WT. Interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitors: an updated patent review (2016–2018). Expert Opin Ther Pat. (2019) 29:243–59. doi: 10.1080/13543776.2019.1597850
70. Kelly PN, Romero DL, Yang Y, Shaffer AL, Chaudhary D, Robinson S, et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. (2015) 212:2189–201. doi: 10.1084/jem.20151074
71. Hjorton K, Hagberg N, Israelsson E, Jinton L, Berggren O, Sandling JK, et al. Cytokine production by activated plasmacytoid dendritic cells and natural killer cells is suppressed by an IRAK4 inhibitor. Arthritis Res Ther. (2018) 20:238. doi: 10.1186/s13075-018-1702-0
Keywords: TLR7, IRAK4, anti-nuclear antibody, dendritic cells, SLE, lupus
Citation: Celhar T, Lu HK, Benso L, Rakhilina L, Lee HY, Tripathi S, Zharkova O, Ong WY, Yasuga H, Au B, Marlier D, Lim LHK, Thamboo TP, Mudgett JS, Mackey MF, Zaller DM, Connolly JE and Fairhurst A-M (2019) TLR7 Protein Expression in Mild and Severe Lupus-Prone Models Is Regulated in a Leukocyte, Genetic, and IRAK4 Dependent Manner. Front. Immunol. 10:1546. doi: 10.3389/fimmu.2019.01546
Received: 10 January 2019; Accepted: 20 June 2019;
Published: 10 July 2019.
Edited by:
Anne Davidson, Feinstein Institute for Medical Research, United StatesReviewed by:
Stefania Gallucci, Temple University, United StatesEdith Janssen, Janssen Research and Development, United States
Betsy J. Barnes, Feinstein Institute for Medical Research, United States
Copyright © 2019 Celhar, Lu, Benso, Rakhilina, Lee, Tripathi, Zharkova, Ong, Yasuga, Au, Marlier, Lim, Thamboo, Mudgett, Mackey, Zaller, Connolly and Fairhurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna-Marie Fairhurst, YW5uYW1hcmllQGltY2IuYS1zdGFyLmVkdS5zZw==
†These authors have contributed equally to this work
‡Present Address: John S. Mudgett, Genesis Biotechnology Group, Hamilton, NJ, United States
Matthew F. Mackey, GlaxoSmithKline, MA, United States
Dennis M. Zaller, Celgene, Bedford, MA, United States