 Samuel Cern Cher Chiang
Samuel Cern Cher Chiang Jack J. Bleesing
Jack J. Bleesing Rebecca A. Marsh
Rebecca A. Marsh- 1Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 2Department of Pediatrics, University of Cincinnati, Cincinnati, OH, United States
Advances in flow cytometry have led to greatly improved primary immunodeficiency (PID) diagnostics. This is due to the fact that patient blood cells in suspension do not require further processing for analysis by flow cytometry, and many PIDs lead to alterations in leukocyte numbers, phenotype, and function. A large portion of current PID assays can be classified as “phenotyping” assays, where absolute numbers, frequencies, and markers are investigated using specific antibodies. Inherent drawbacks of antibody technology are the main limitation to this type of testing. On the other hand, “functional” assays measure cellular responses to certain stimuli. While these latter assays are powerful tools that can be used to detect defects in entire pathways and distinguish variants of significance, it requires samples with robust viability and also skilled processing. In this review, we concentrate on hemophagocytic lymphohistiocytosis (HLH), describing the principles and accuracies of flow cytometric assays that have been proven to assist in the screening diagnosis of primary HLH.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) can be described as a systemic hyperinflammatory syndrome. It is most often thought to be caused by an inability to clear an inciting infectious or other immunologic trigger. This leads to pathologic immune activation and a positive feedback loop of ever increasing cytokine secretion and cellular cytotoxicity that ultimately results in self harm (1, 2). HLH can be classified as “primary” or “secondary” depending on whether it occurs as a result of an inborn error leading to a dysfunctional immune system like perforin deficiency, or occurs in settings such as infection, malignancy, rheumatologic, or other disease without a known underlying inherited defect in the immune system (3–5). Primary HLH can be caused by mutations in a number of genes which affect cytotoxic lymphocyte granule-mediated cytotoxicity including PRF1, UNC13D, STX11, STXBP2, RAB27A (Griscelli Syndrome), AP3B1 (Hermansky-Pudlak syndrome type 2), and LYST (Chediak-Higashi Syndrome). Primary HLH can also include other genetic diseases such as XIAP deficiency, which is characterized by inflammasome dysregulation, and SAP deficiency which has a complicated mechanism of disease, though these diseases are usually classified as X-linked lymphoproliferative diseases (XLP) type 1 and type 2, respectively. Regardless, the classification of HLH into primary or secondary groups is sometimes difficult due to the varied phenotype presented and delays or limitations in obtaining genetic results. This has necessitated the development of faster diagnostic screening assays. Many excellent reviews exist on the subject of primary HLH and cytotoxic lymphocyte function, and the reader would be wise to refer to them for a deeper understanding on the subject (1, 6–10). In this review, we will focus on summarizing the laboratory assays currently used to screen for genetic abnormalities in primary HLH linked genes and explore their accuracy. We will also briefly discuss possible pitfalls and future directions in diagnosing diseases typically associated with HLH.
Perforin Deficiency
NK cells and cytotoxic T lymphocytes are often grouped together as cytotoxic lymphocytes. Their primary role is to kill virus infected or malignant cells (11, 12). Perforin, the pore forming protein, is encoded by the gene PRF1 and is a key player in this process as well as the archetypical example of primary HLH (13). PRF1 is also historically the first primary HLH gene to be identified and is often referred to as familial hemophagocytic lymphohistiocytosis type 2 (FHL2) (14). Perforin is stored within cytotoxic granules. Once secreted from cytotoxic lymphocyte granules, perforin oligomerizes on the surface of target cells to create pores which allow the penetration of contents such as granzymes into the target. Perforin is easily stained for intracellularly in NK cells using a conjugated monoclonal antibody. Perforin has been shown to be absent or highly reduced in persons with biallelic mutations for PRF1 gene. Staining can be performed using fresh whole blood or peripheral blood mononuclear cell (PBMC). First, the various lymphocyte lineages are extracellularly stained followed by cell fixation and permeabilization. Intracellular perforin is then stained for and the cells finally analyzed on a flow cytometer (15). To note, while freshly isolated NK cells contain perforin and are routinely used for perforin analysis, only a minority of cytotoxic T cells in “healthy” individuals express perforin. Perforin expression in resting bulk CD8+ cells thus varies greatly between individuals. To overcome this, bona fide effector T cells can be gated using CD57 if evaluation of perforin in resting T cells is desired (16, 17). This can greatly help in individuals with poor NK cell counts.
The diagnostic accuracy of perforin expression in NK cells for detecting biallelic PRF1 mutations has recently been published and is highly accurate with sensitivity of 96.6% and specificity of 89.5% for an overall area under the curve (AUC) of 0.971 (Table 1) (18, 20). These and other reports have also shown that PRF1 mutation carriers (a mutation in only one allele) often have clearly reduced perforin expression arguing for an allele dependent perforin expression (19, 26, 27).
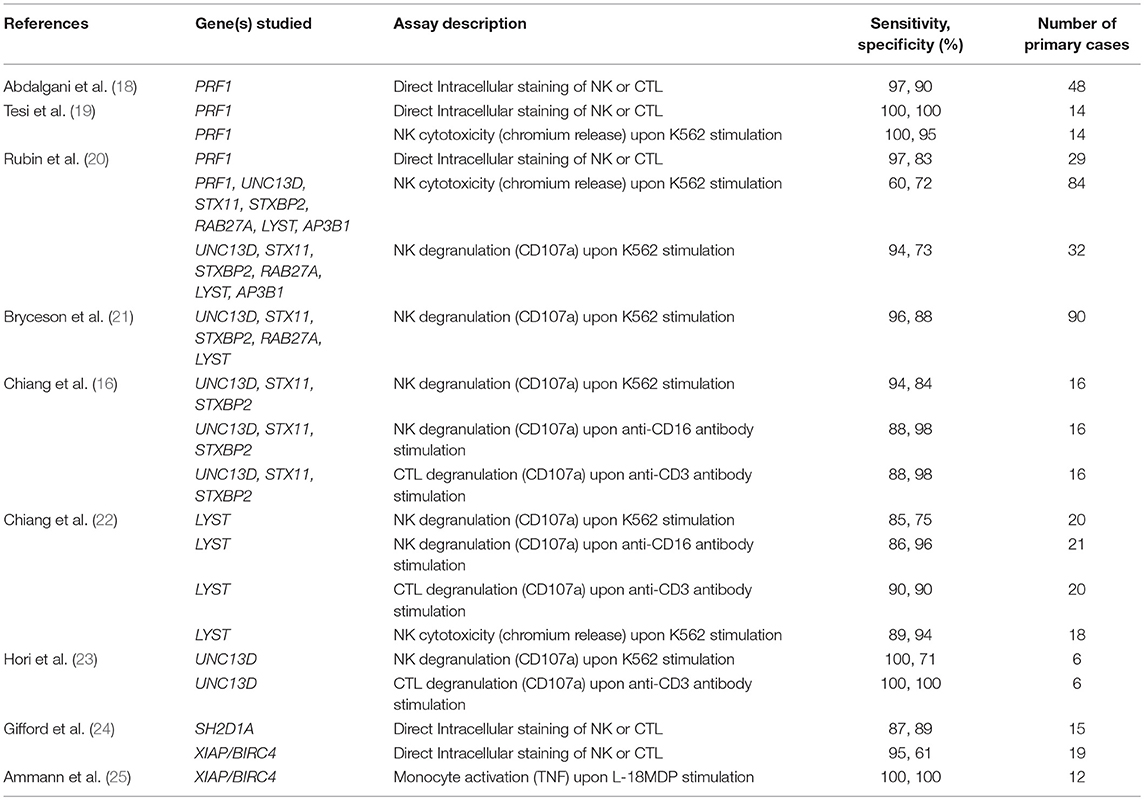
Table 1. Sensitivity and specificity results for the diagnosis of primary HLH and related diseases extrapolated from various studies using a range of immunological assays.
The A91V alteration in PRF1 is unique. Having a high prevalence of 0.22 to 3.9% depending on the population studied, it has been assumed to be less pathologic (Figure 1) (28–31). However, in vitro studies have shown that A91V leads to reduced perforin function (32, 33). Individuals with A91V in both compound heterozygous and homozygous state can be identified by laboratory assays and show low to no residual protein expression, and such results may be indiscriminable from other pathologic PRF1 mutations (30, 34, 35).
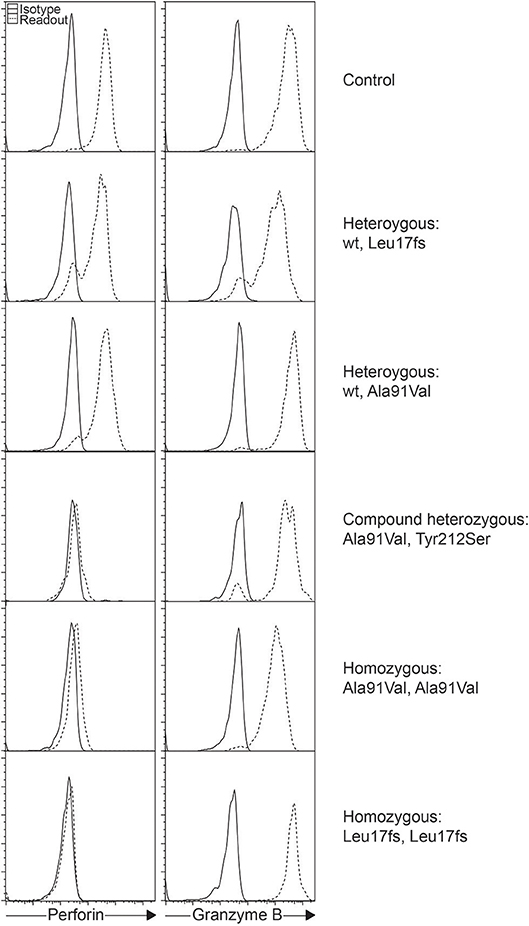
Figure 1. Intracellular staining of perforin and granzyme B in different individuals. Histograms represent gated NK cells showing varying levels of perforin expression as a consequence of the different PRF1 variants as shown.
The lack of perforin leads to an inability to kill target cells. This functional defect can be detected by lowered chromium release using the radioactive chromium cytotoxicity assay (36). Because the chromium release assay shows suboptimal accuracy, many have turned to screening for primary HLH diseases with perforin staining coupled with the degranulation/exocytosis/CD107a assay in place of or in addition to chromium release NK cell function testing. The CD107a assay examines if cytotoxic lymphocytes (NK cells and CTL) can release secretory lysosomes as described below, but this assay does not report if target cells are killed. Samples from patients with perforin deficiency will not show any degranulation abnormalities but is nonetheless often run to confirm normal degranulation. Typical perforin deficiency can thus be confidently diagnosed based on the lack of perforin staining, deficient NK cell cytotoxicity, but normal degranulation.
Secretory Granule Exocytosis Deficiency
Autosomal recessive mutations in UNC13D, STX11, or STXBP2 have been linked to primary HLH disease. These encode the proteins Munc13-4, syntaxin-11, or Munc18-2, and as diseases are known as FHL3, FHL4, or FHL5, respectively. The proteins encoded are crucial for perforin-containing secretory lysosome exocytosis, a process more commonly referred to as degranulation. Defects in RAB27A, LYST, and AP3B1, leading to Griscelli syndrome type 2 (GS2), Chediak-Higashi syndrome (CHS), and Hermansky-Pudlak syndrome type 2 (HPS2), respectively, also cause defective degranulation. These latter patients often manifest with HLH and usually, but not always, occulocutaneous albinism (22, 37–42). Together, these 6 genes can be grouped for diagnostic screening as they show a similar cellular phenotype of failed secretory lysosome content release and failure to kill target cells.
At this juncture, it is important to differentiate between the terms “NK cell degranulation” and “NK cell function,” as they are often thought to be one and the same. The NK degranulation assay, also known as CD107a or NK exocytosis assay, evaluates if CD107a containing secretory lysosomes are able to release their content and thus deposit CD107a on the external cell membrane where it is measured as a surrogate for degranulation (Figure 2). Under the microscope, CD107a and perforin often co-localize and so it is assumed that when granules bearing CD107a are externalized, perforin would also most likely be released at the immune synapse (43, 44). In the case of perforin deficiency, the CD107a assay is not useful as a screening tool because secretory lysosomes without perforin are still released and CD107a still expressed on the cell membrane. The CD107a assay is also unable to detect whether granules are headed toward the immune synapse where the target cell is being engaged. When stimulating NK cells in vitro with anti-CD16 antibody, the release of secretory lysosomes are non-polarized which would not be efficient for target cell elimination (43). The CD107a assay has been found useful for the diagnosis of FHL3-5, GS2, CHS, and HPS2, and possibly ORAI1, STIM1, and HPS10 (45–48), because in all these cases, secretory lysosomes are unable reach the cell membrane or fail to fuse with the cell membrane leading to the absence of surface CD107a after relevant stimulation. But, in cases of preserved detection of CD107a upregulation, additional testing to evaluate NK cell killing may be needed, as lysosome degranulation does not necessarily equate to the death of target cells.
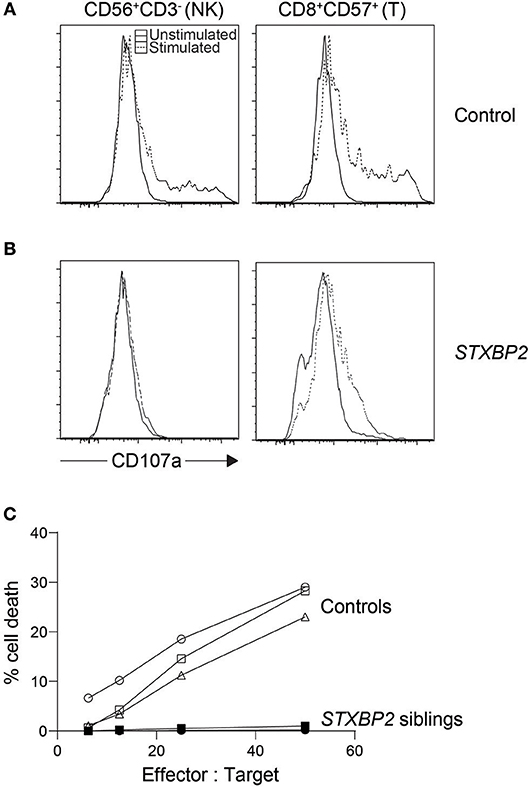
Figure 2. Cytotoxic lymphocyte evaluation of an STXBP2 patient. We performed NK cytotoxicity as well as NK and T cell degranulation using fresh PBMC from a case with homozygous c.1430C>T (p.Pro477Leu) mutations. While (A) control NK cells and CD8+CD57+ T cells degranulated as expected when stimulated, respectively with K562 or anti-CD3 antibody, (B) the patient's cytotoxic lymphocytes did not. (C) NK cytotoxicity was also evaluated via 51Cr release and found deficient. In addition, we included cytotoxicity data from a sibling carrying the same homozygous mutation.
As such, the often crowned “gold standard” chromium release assay still holds relevance since described in the 1960s (49, 50). In this assay, K562 cells (ATCC, CCL-243) first preloaded with radioactive chromium-51 will be killed by NK cells and the extent to which the stored chromium is freed is taken to represent the percentage of K562 killed (51–53). No published data exists exploring the accuracies of NK cytotoxicity assay in diagnosing each subtype of primary HLH, possibly due to sample number limitations. Only one recent study attempted to systematically quantify the accuracy of the chromium release NK cell function assay when used in the clinical lab setting for diagnosing PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, and AP3B1 mutations, and found it lacking with a sensitivity of 60% and specificity of 72% (Table 1) (20).
The low accuracy of this assay, often used during acute phase HLH, may be partly blamed on the assay's dependency on the NK cell percentage in the sample. HLH patients normally experience large expansions of CD8 T cells, and stressed blood samples from these patients often leave large numbers of RBC and cell debris in the peripheral blood mononuclear cell (PBMC) suspension after ficoll. This leads to an artificially low NK cell percentage which is often unaccounted for, giving an impression of reduced NK function when in fact it is due to the overwhelming number of other cells in the mix. Because the assay is sensitive as such, care must be taken when interpreting poor NK cytotoxicity results especially during acute HLH as it could indicate poor sample quality rather than dysfunctional NK cells. While this assay has many limitations, the result distinctly demonstrates whether or not target cells are finally killed (Figure 2) (54). Numerous flow-, colorimetric-, and imaging-based cytotoxicity assays have been touted as possible chromium release assay replacements but no large cohort of primary HLH cases has been validated on any of these platforms (55–59). Pending such reports, the chromium release assay is still the only published clinical standard for NK functional studies.
Therefore, we currently rely on the CD107a NK cell degranulation assay for the screening diagnosis of primary HLH related to mutations in UNC13D, STX11, STXBP2, RAB27A, LYST, and AP3B1. The most commonly used NK degranulation assay tests rested PBMC stimulated with the myelogenous leukemia cell line K562 (21). After co-incubation for several hours, the percentage of NK cells bearing surface CD107a or the fluorescence intensity of CD107a positive NK cells is then evaluated. Persons with a defect in secretory lysosome transport or membrane fusion will show greatly reduced surface CD107a levels (Figure 2). A pan European study found 97% of FHL3-5 and 85% of GS2 and CHS cases had abnormal percentage of NK cell degranulation (<5% CD107a+ NK cells) to give an overall sensitivity of 96% and specificity of 88% in diagnosing a genetic degranulation disorder (Table 1) (21). A follow-up study on a North American cohort evaluated CD107a mean channel fluorescence (MCF) of NK cells instead of percentage of degranulating cells (20). It found 93.8% of patients with biallelic mutations in an HLH-associated degranulation gene with lowered CD107a MCF but only 60.4% of individuals without biallelic mutations in relevant genes with normal CD107a levels, giving an overall area under the curve of 0.86. More recently, a cohort of 21 CHS cases has likewise confirmed the CD107a assay is able to accurately identify primary defects in NK degranulation (22). In the first two studies, a sizable portion of controls were found to have lowered NK degranulation. This could be due to technical issues, stress during blood sample transport, medications leading to reduced lymphocyte reaction, or epigenetic changes resulting in NK cells with a particularly skewed functional response (60–63). So while better than the chromium release assay, the NK-K562 degranulation assay, like all diagnostic assays, is not perfect.
To overcome the shortcomings stemming from an overreliance on any single test, NK degranulation can also be evaluated through other means, for example, via stimulation using PMA, activating antibodies such as anti-CD16 targeting the Fc receptor, or activation of synergistic NK receptors (16, 64, 65). Preliminary data has found Fc stimulation induced degranulation returns 88% sensitivity and 98% specificity in a cohort of 16 FHL3-5 (Table 1) (16). We can thus infer that both NK cell natural cytotoxicity and antibody-dependent cellular cytotoxicity are defective in classical primary HLH. This is an important point to note as immunodeficiencies could affect only one specific pathway. For instance, a certain CD16 (FcγRIIIA) mutation was found to impair natural NK cytotoxicity but Fc specific function was intact (66). Current standard clinical tests limited to only K562 stimulation would be insufficient for detecting abnormalities in such cases.
Cytotoxic T lymphocytes have also been found defective in degranulation in the context of primary HLH due to mutations in the genes required for normal degranulation. Previously, T cell blasts had to be grown up over weeks in order to sufficiently stimulate perforin production in T cells and generate enough cell numbers for experimentation (21). More recently, it was noticed that specific populations of T cells, namely CD3+CD8+CD57+ contain perforin and granzymes ex vivo without prior need for stimulation (17). This population of bone fide effector cells, by virtue of perforin expression, was found to efficiently degranulate upon anti-CD3 antibody stimulation. Crucial to our context, when tested on primary HLH samples, CD3+CD8+CD57+ T cell degranulation was defective to a similar level as in NK cells (16). A small confirmatory study found high sensitivity with a cohort of biallelic pathogenic UNC13D variants (23). With multiple ways to induce degranulation on multiple cell types, we could speculate on possible undiscovered immunodeficiencies that affect only NK cells or T cells and detectable only with a combination of various degranulation assays.
Like perforin, it is possible to directly detect Munc13-4, syntaxin11, Munc18-2, and Rab27a with antibodies (67–69). However, this is usually performed with western blot. One exception is Munc13-4 detection in platelets with flow cytometry (70, 71). Although this assay has been found to be highly accurate for predicting UNC13D mutations, the antibody used is polyclonal and not commercially available.
Taken together, when primary HLH is suspected, performing the triad of perforin staining, NK and/or T cell degranulation, and NK cytotoxicity will give a more complete evaluation of cytotoxic cell activity and improve HLH diagnosis. While all the assays are individually accurate, we suggest moving toward a “multiplexing” of degranulation assays in the future to increase confidence in diagnosis, provide security should any one cell population be poorly represented, and pave the way for detecting degranulation deficiencies in specific pathways or cell types. Additionally, validating a radioactivity-free killing assay that accounts for effector cell counts would be highly useful for true assessment of cytotoxic lymphocyte function.
X-linked Diseases
The genes SH2D1A and XIAP/BIRC4 encode the proteins SAP and XIAP, respectively. Deficiencies in these proteins lead to X-linked lymphoproliferative disease type (XLP) 1 and 2 (72, 73). As their names imply, both genes are X-linked and often manifest HLH with Epstein-Barr virus (EBV) infection (74–76) but beyond that, XLP1 and XLP2 have quite different phenotypes and share little functional or structural similarities (77).
Similar to perforin, SAP and XIAP monoclonal antibodies exist and have been validated clinically for direct intracellular protein detection (Figures 3, 4) (78–80). However, care must be taken when reading such reports as certain pathologic variants have been found to preserve antibody binding leading to false negative (false normal) results (81–83). Also, while the absence of binding can be equated with the absence of that protein and thus strongly suggests a defect, the binding of an antibody to its antigen says nothing about the function of the protein bound. As such, patients expressing normal SAP and XIAP levels, or for that matter all direct antibody phenotyping tests, should still be sequenced if clinically suspicious. Bimodal staining patterns are also useful in identifying female carriers as well as estimating the level of chimerism for transplant monitoring (24, 79). For XIAP, there has been reports of non-random X inactivation in some female carriers. Lymphocytes bearing the wild-type allele have been seen selected in some while others show the opposite, skewing toward the defective X chromosome at risk for disease manifestations (73, 84, 85). Direct screening of SAP returns 87% sensitivity and 89% specificity for the prediction of pathologic mutations in SH2D1A while direct screening of XIAP gives 95% sensitivity and 61% specificity (Table 1) (24, 86).
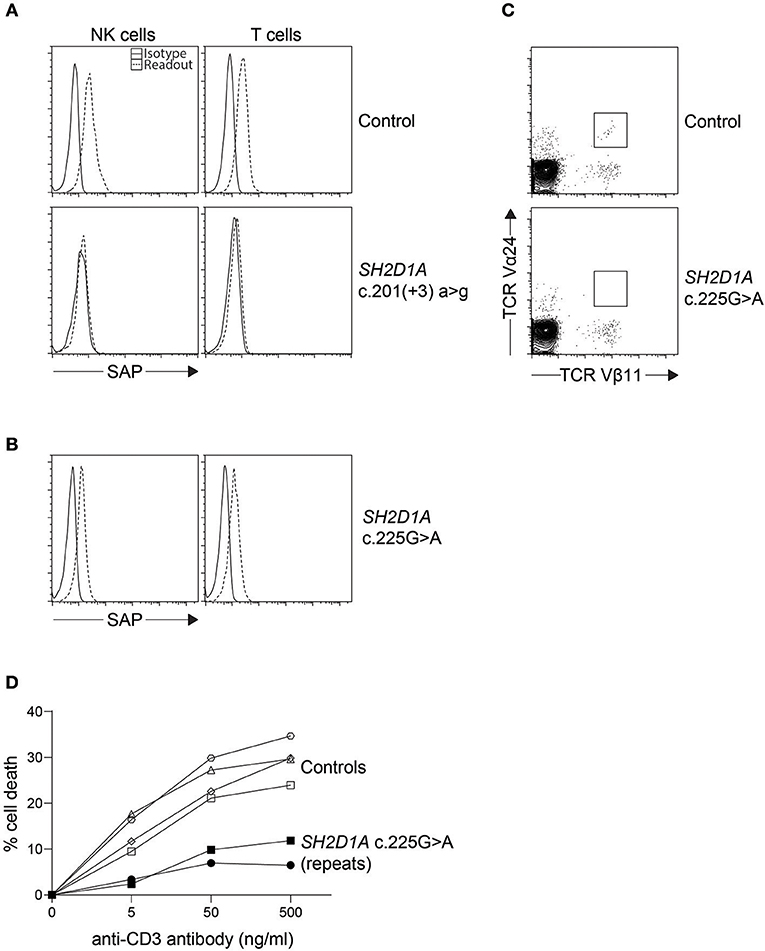
Figure 3. Indirect diagnosis of XLP1. (A) While most SH2D1A mutations result in absent or lowly expressing SAP protein levels, we found (B) a clinically suspicious patient with c.125G > A (p.Cys42Tyr) missense mutation with only a slight reduction in SAP protein expression by flow cytometry. The patient was thus further evaluated for (C) iNKT numbers on bulk CD3+ cells and (D) restimulation-induced cell death (RICD) via anti-CD3 antibody repeated on two occasions. The low iNKT counts and reduced cell death upon TCR restimulation provided evidence that the missense SH2D1A variant found was indeed pathological.
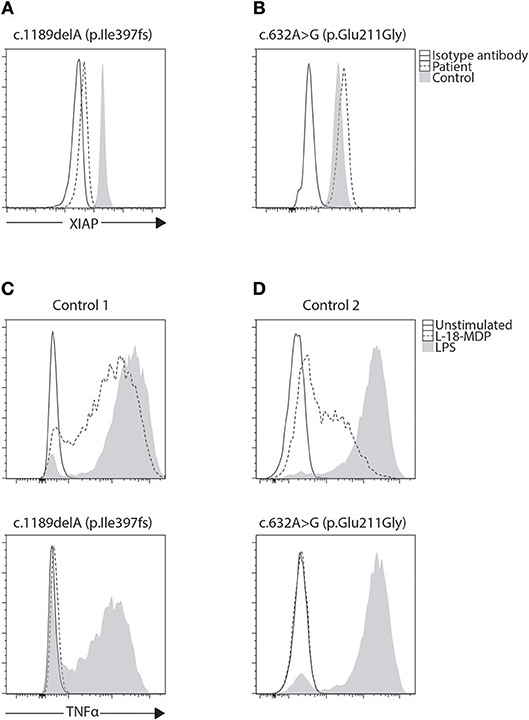
Figure 4. Phenotyping and functional evaluation for XLP2. (A) While a majority of BIRC4 mutations present with absent or lowly expressing XIAP protein levels, we found (B) a patient with c.632A>G (p.Glu211Gly) variant of uncertain significance (VUCS) with only a slight reduction in XIAP protein expression by flow cytometry. Functional evaluation of XIAP can be done through stimulation of NOD2 with L18-MDP. This signaling pathway requires XIAP for TNF transcription through NF-κB. (C,D) When examined, both these cases show equally defective TNF production regardless of XIAP expression revealing the VUCS is in fact a damaging mutation. LPS acts as a positive control that signals through TLR4 demonstrating preserved cellular function in patient cells.
It has been demonstrated that both SAP and XIAP are required for the development of normal invariant NKT (iNKT) cells and for normal T cell restimulation-induced cell death (RICD) (73, 76, 87, 88). As such, iNKT quantification and RICD assays can be performed for cases where direct staining is inconclusive, or if further supporting data is desired (Figure 3). A more sophisticated cytotoxic assay looking at inhibitory 2B4 signaling in NK cells has also been reported to discriminate functional SAP deficiency (89). Likewise, a functional test exists where XIAP function is investigated downstream of NOD2 stimulation on monocytes. Following stimulation with L18-MDP, TNF is normally produced by CD14 positive cells. However, patients with pathologic mutations in XIAP, even where XIAP protein staining was found normal or in patients with milder clinical phenotype, all had equally defective TNF production and could easily be discriminated (Figure 4) (25). A cutoff of 10% TNF-producing monocytes perfectly distinguished 12 XIAP patients from 29 healthy controls and 6 female carriers (Table 1). Subsequent reports demonstrated the assay's usefulness in diagnosing inflammatory bowel disease (IBD) cases with novel XIAP mutations (90, 91). By performing phenotyping as well as functional assays side by side, it is hoped that future cases might be more accurately identified.
Other Primary Immunodeficienices
A host of patients with other diseases such as ALPS, CGD, CVID, and SCID, as well as variants in genes including BTK, CARMIL2, CD27, ITK, LRBA, MAGT1, NEMO, PIK3CD, RAG2, WAS, NLR genes, and STAT genes, have been implicated with possible HLH (92–94). The assays described so far including NK cell degranulation and cytotoxicity will be of little diagnostic use here except to rule out defective secretory lysosome transport. For some genes, there exist flow cytometric assays that can assist with diagnosis. For example T, B, and NK specific subset phenotyping panels can pick up ALPS (increased double negative T cells), X-linked agammaglobulinemia due to mutations in BTK (low B cell counts or BTK expression), mutations in CD27 (absent surface expression of CD27), mutations in MAGT1 (lowered NKG2D expression), and a variety of SCID disorders (very low B, T, and/or NK counts, reduced recent thymic emigrants and CD45RA expression) (95). The neutrophil oxidative burst assay is an excellent assay for the diagnosis of CGD (96). WAS can be accurately diagnosed through direct staining of intracellular WAS protein (97). Multiple excellent reviews exist for PID diagnostics (98, 99).
A second group of primary immunodeficiency genes demonstrate defective NK cell activity without pronounced HLH. However, before suggesting that NK degranulation and cytotoxicity assays could be used in helping with the diagnosis of these PIDs, larger cohorts of patients must be collected for evaluation to confirm and explore cytotoxic lymphocytes further including: whether or not both NK and CTL are affected, if both degranulation and cytotoxicity are defective, and if the majority of mutations in that gene share the same phenotype. Genes in this group include AP3D1, CTSC, FERMT3, GATA2, IRF8, MYH9, ORAI1, and STIM1 (45, 47, 48, 100–107). From this list, we know that not all persons for whom NK cell function is defective should be labeled primary HLH. Moreover, a thorough evaluation is hampered as many of publications lack NK degranulation or cytotoxicity data, something we hope future endeavors will address. These genes are thus currently not grouped together with the “classical” primary HLH family because clinical HLH is not usually the outstanding feature. Most are also very rare leading to difficulty in performing large cohort evaluations of cytotoxic lymphocyte activity.
The Future of HLH Diagnostics
The HLH field has come some ways since the HLH-2004 criteria were established (108). A European cohort of cases with clinical HLH and PID other than defects in cytotoxicity found 63 cases, 80% of which were CGD and CID (109). Across the Atlantic, another HLH cohort was comprised of only 19% primary HLH disorders, with 58% of patients having other PIDs including genes associated with inflammasome function (92). We reason the high percentage of “non-classical-HLH” cases is a reflection of improved HLH awareness within the community and should be looked upon positively. These and other studies looking into specific sensitivities of various HLH-2004 criteria have found them wanting (110–112). The concern often cited is the inability to distinguish between primary HLH, secondary HLH, and other PIDs. A simple solution that can easily be adopted today is increased screening. As can be concluded from Table 1, many subtypes of primary HLH can be diagnosed with good accuracy. As such, the fulfillment of HLH criteria should act as an actionable gateway to seriously consider PID by performing various laboratory tests as discussed. This in tandem with advanced sequencing should more often than not provide conclusive diagnosis for all the common primary HLH cases. As previously mentioned, we believe the field of HLH diagnostics will move toward a “multiplexing” of screening assays to more quickly screen for multiple defects simultaneously.
The evaluation of gene expression signatures is an exciting development that could help untangle some of the primary vs. secondary HLH questions going forward. Unique interferon-stimulated gene signatures have been found in systemic lupus erythematosus differentiating it from rheumatoid arthritis and control samples (113, 114). Other studies successfully used the interferon score to identify various Mendelian Type-I IFN-mediated autoinflammatory diseases (115, 116). Preliminary work to define a HLH signature has also been performed with favorable results (117, 118). While research on this area is in its infancy today, we postulate a future where specific gene expression fingerprints from tens or hundreds of genes would be elucidated for the various shades of HLH. We could then quickly and accurately segregate HLH into several subcategories as well as deduce their disease status. The signatures could not only act as a “precision” diagnostic tool but also afford us a deeper cellular mechanistic understanding on the pathobiology of various closely related diseases, and thus opportunities for “precision” therapeutics. We are excited to see what the future holds in terms of HLH diagnostics.
Author Contributions
RM initiated the manuscript which SC wrote and JB edited.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Kathryn Quinn, Carrie Gifford, Joyce Collett, and the entire staff of the Diagnostic Immunology Laboratory.
References
1. Janka GE, Lehmberg K. Hemophagocytic syndromes–an update. Blood Rev. (2014) 28:135–42. doi: 10.1016/j.blre.2014.03.002
2. Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. (2015) 212:307–17. doi: 10.1084/jem.20140964
3. Bracaglia C, Prencipe G, De Benedetti F. Macrophage activation syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J. (2017) 15:5. doi: 10.1186/s12969-016-0130-4
4. Machowicz R, Janka G, Wiktor-Jedrzejczak W. Similar but not the same: differential diagnosis of HLH and sepsis. Crit Rev Oncol Hematol. (2017) 114:1–12. doi: 10.1016/j.critrevonc.2017.03.023
5. Wang H, Xiong L, Tang W, Zhou Y, Li F. A systematic review of malignancy-associated hemophagocytic lymphohistiocytosis that needs more attentions. Oncotarget. (2017) 8:59977–85. doi: 10.18632/oncotarget.19230
6. Ham H, Billadeau DD. Human immunodeficiency syndromes affecting human natural killer cell cytolytic activity. Front Immunol. (2014) 5:2. doi: 10.3389/fimmu.2014.00002
7. Marsh RA, Haddad E. How i treat primary haemophagocytic lymphohistiocytosis. Br J Haematol. (2018) 182:185–99. doi: 10.1111/bjh.15274
8. Marsh RA, Jordan MB, Talano JA, Nichols KE, Kumar A, Naqvi A, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: a review of the published experience. Pediatr Blood Cancer. (2017) 2017:64. doi: 10.1002/pbc.26308
9. Risma KA, Marsh RA. Hemophagocytic lymphohistiocytosis: clinical presentations and diagnosis. J Allergy Clin Immunol Pract. (2019) 7:824–32. doi: 10.1016/j.jaip.2018.11.050
10. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. (2017) 49:20–6. doi: 10.1016/j.coi.2017.08.004
11. Ljunggren HG, Karre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
12. Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol. (2019) 105:1319–29. doi: 10.1002/JLB.MR0718-269R
13. Voskoboinik I, Trapani JA. Perforinopathy: a spectrum of human immune disease caused by defective perforin delivery or function. Front Immunol. (2013) 4:441. doi: 10.3389/fimmu.2013.00441
14. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
15. Chiang S, Bryceson Y. Measurement of NK cell phenotype and activity in humans. In: Detrick B, Schmitz J, Hamilton R, editors. Manual of Molecular and Clinical Laboratory Immunology, Eighth Edition. Washington, DC: ASM Press (2016). p. 300–9.
16. Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood. (2013) 121:1345–56. doi: 10.1182/blood-2012-07-442558
17. Chattopadhyay PK, Betts MR, Price DA, Gostick E, Horton H, Roederer M, et al. The cytolytic enzymes granyzme A, granzyme B, and perforin: expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J Leukoc Biol. (2009) 85:88–97. doi: 10.1189/jlb.0208107
18. Abdalgani M, Filipovich AH, Choo S, Zhang K, Gifford C, Villanueva J, et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood. (2015) 126:1858–60. doi: 10.1182/blood-2015-06-648659
19. Tesi B, Chiang SC, El-Ghoneimy D, Hussein AA, Langenskiold C, Wali R, et al. Spectrum of atypical clinical presentations in patients with biallelic PRF1 missense mutations. Pediatr Blood Cancer. (2015) 62:2094–100. doi: 10.1002/pbc.25646
20. Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. (2017) 129:2993–9. doi: 10.1182/blood-2016-12-753830
21. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. (2012) 119:2754–63. doi: 10.1182/blood-2011-08-374199
22. Chiang SCC, Wood SM, Tesi B, Akar HH, Al-Herz W, Ammann S, et al. Differences in granule morphology yet equally impaired exocytosis among cytotoxic T Cells and NK cells from chediak-higashi syndrome patients. Front Immunol. (2017) 8:426. doi: 10.3389/fimmu.2017.00426
23. Hori M, Yasumi T, Shimodera S, Shibata H, Hiejima E, Oda H, et al. A CD57(+) CTL Degranulation Assay Effectively Identifies Familial Hemophagocytic Lymphohistiocytosis Type 3 Patients. J Clin Immunol. (2017) 37:92–9. doi: 10.1007/s10875-016-0357-3
24. Gifford CE, Weingartner E, Villanueva J, Johnson J, Zhang K, Filipovich AH, et al. Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin Cytom. (2014) 86:263–71. doi: 10.1002/cyto.b.21166
25. Ammann S, Elling R, Gyrd-Hansen M, Duckers G, Bredius R, Burns SO, et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol. (2014) 176:394–400. doi: 10.1111/cei.12306
26. Hussein AA, Hamadah T, Qandeel M, Sughayer M, Amarin R, Mansour A, et al. Hematopoietic stem cell transplantation of an adolescent with neurological manifestations of homozygous missense PRF1 mutation. Pediatr Blood Cancer. (2014) 61:2313–5. doi: 10.1002/pbc.25166
27. Kim JY, Shin JH, Sung SI, Kim JK, Jung JM, Ahn SY, et al. A novel PRF1 gene mutation in a fatal neonate case with type 2 familial hemophagocytic lymphohistiocytosis. Korean J Pediatr. (2014) 57:50–3. doi: 10.3345/kjp.2014.57.1.50
28. Martinez-Pomar N, Lanio N, Romo N, Lopez-Botet M, Matamoros N. Functional impact of A91V mutation of the PRF1 perforin gene. Hum Immunol. (2013) 74:14–7. doi: 10.1016/j.humimm.2012.10.011
29. Santoro A, Cannella S, Trizzino A, Lo Nigro L, Corsello G, Arico M. A single amino acid change A91V in perforin: a novel, frequent predisposing factor to childhood acute lymphoblastic leukemia? Haematologica. (2005) 90:697–8.
30. Tesi B, Lagerstedt-Robinson K, Chiang SC, Ben Bdira E, Abboud M, Belen B, et al. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med. (2015) 7:130. doi: 10.1186/s13073-015-0244-1
31. Zur Stadt U, Beutel K, Weber B, Kabisch H, Schneppenheim R, Janka G. A91V is a polymorphism in the perforin gene not causative of an FHLH phenotype. Blood. (2004) 104:1909; author reply 10. doi: 10.1182/blood-2004-02-0733
32. Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, et al. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood. (2007) 110:1184–90. doi: 10.1182/blood-2007-02-072850
33. Voskoboinik I, Thia MC, Trapani JA. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood. (2005) 105:4700–6. doi: 10.1182/blood-2004-12-4935
34. Mancebo E, Allende LM, Guzman M, Paz-Artal E, Gil J, Urrea-Moreno R, et al. Familial hemophagocytic lymphohistiocytosis in an adult patient homozygous for A91V in the perforin gene, with tuberculosis infection. Haematologica. (2006) 91:1257–60.
35. Sanchez IP, Leal-Esteban LC, Alvarez-Alvarez JA, Perez-Romero CA, Orrego JC, Serna ML, et al. Analyses of the PRF1 gene in individuals with hemophagocytic lymphohystiocytosis reveal the common haplotype R54C/A91V in Colombian unrelated families associated with late onset disease. J Clin Immunol. (2012) 32:670–80. doi: 10.1007/s10875-012-9680-5
36. Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. (2008) 45:15–21. doi: 10.1136/jmg.2007.052670
37. Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle EM, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. (2006) 108:81–7. doi: 10.1182/blood-2005-11-4413
38. Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. (2006) 107:4857–64. doi: 10.1182/blood-2005-11-4398
39. Wood SM, Meeths M, Chiang SC, Bechensteen AG, Boelens JJ, Heilmann C, et al. Different NK cell-activating receptors preferentially recruit Rab27a or Munc13-4 to perforin-containing granules for cytotoxicity. Blood. (2009) 114:4117–27. doi: 10.1182/blood-2009-06-225359
40. Netter P, Chan SK, Banerjee PP, Monaco-Shawver L, Noroski LM, Hanson IC, et al. A novel Rab27a mutation binds melanophilin, but not Munc13-4, causing immunodeficiency without albinism. J Allergy Clin Immunol. (2016) 138:599–601 e3. doi: 10.1016/j.jaci.2015.12.1337
41. Tesi B, Rascon J, Chiang SCC, Burnyte B, Lofstedt A, Fasth A, et al. A RAB27A 5' untranslated region structural variant associated with late-onset hemophagocytic lymphohistiocytosis and normal pigmentation. J Allergy Clin Immunol. (2018) 142:317–21 e8. doi: 10.1016/j.jaci.2018.02.031
42. Dell'Acqua F, Saettini F, Castelli I, Badolato R, Notarangelo LD, Rizzari C. Hermansky-Pudlak syndrome type II and lethal hemophagocytic lymphohistiocytosis: Case description and review of the literature. J Allergy Clin Immunol Pract. (2019). doi: 10.1016/j.jaip.2019.04.001. [Epub ahead of print].
43. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. (2005) 202:1001–12. doi: 10.1084/jem.20051143
44. Krzewski K, Gil-Krzewska A, Nguyen V, Peruzzi G, Coligan JE. LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood. (2013) 121:4672–83. doi: 10.1182/blood-2012-08-453738
45. Ammann S, Schulz A, Krageloh-Mann I, Dieckmann NM, Niethammer K, Fuchs S, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. (2016) 127:997–1006. doi: 10.1182/blood-2015-09-671636
46. Fuchs S, Rensing-Ehl A, Speckmann C, Bengsch B, Schmitt-Graeff A, Bondzio I, et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol. (2012) 188:1523–33. doi: 10.4049/jimmunol.1102507
47. Klemann C, Ammann S, Heizmann M, Fuchs S, Bode SF, Heeg M, et al. Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J Allergy Clin Immunol. (2017) 140:1721–4. doi: 10.1016/j.jaci.2017.05.039
48. Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U.S.A. (2011) 108:3324–9. doi: 10.1073/pnas.1013285108
49. Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. (1968) 14:181–96.
50. Wigzell H. Quantitative titrations of mouse H-2 antibodies using Cr-51-labelled target cells. Transplantation. (1965) 3:423–31. doi: 10.1097/00007890-196505000-00011
51. Bryant J, Day R, Whiteside TL, Herberman RB. Calculation of lytic units for the expression of cell-mediated cytotoxicity. J Immunol Methods. (1992) 146:91–103. doi: 10.1016/0022-1759(92)90052-U
52. Pross HF, Maroun JA. The standardization of NK cell assays for use in studies of biological response modifiers. J Immunol Methods. (1984) 68:235–49. doi: 10.1016/0022-1759(84)90154-6
53. Whiteside TL, Bryant J, Day R, Herberman RB. Natural killer cytotoxicity in the diagnosis of immune dysfunction: criteria for a reproducible assay. J Clin Lab Anal. (1990) 4:102–14. doi: 10.1002/jcla.1860040207
54. Pollock RE, Zimmerman SO, Fuchshuber P, Lotzova E. Lytic units reconsidered: pitfalls in calculation and usage. J Clin Lab Anal. (1990) 4:274–82. doi: 10.1002/jcla.1860040408
55. Fassy J, Tsalkitzi K, Goncalves-Maia M, Braud VM. A Real-time cytotoxicity assay as an alternative to the standard chromium-51 release assay for measurement of human NK and T Cell cytotoxic activity. Curr Protoc Immunol. (2017) 118:7 42 1–7:12. doi: 10.1002/cpim.28
56. He L, Hakimi J, Salha D, Miron I, Dunn P, Radvanyi L. A sensitive flow cytometry-based cytotoxic T-lymphocyte assay through detection of cleaved caspase 3 in target cells. J Immunol Methods. (2005) 304:43–59. doi: 10.1016/j.jim.2005.06.005
57. Piriou L, Chilmonczyk S, Genetet N, Albina E. Design of a flow cytometric assay for the determination of natural killer and cytotoxic T-lymphocyte activity in human and in different animal species. Cytometry. (2000) 41:289–97. doi: 10.1002/1097-0320(20001201)41:4<289::AID-CYTO7>3.0.CO;2-5
58. Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev Vaccines. (2010) 9:601–16. doi: 10.1586/erv.10.49
59. van der Haar Avila I, Marmol P, Cany J, Kiessling R, Pico de Coana Y. Evaluating antibody-dependent cell-mediated cytotoxicity by flow cytometry. Methods Mol Biol. (2019) 1913:181–94. doi: 10.1007/978-1-4939-8979-9_13
60. Meehan AC, Mifsud NA, Nguyen TH, Levvey BJ, Snell GI, Kotsimbos TC, et al. Impact of commonly used transplant immunosuppressive drugs on human NK cell function is dependent upon stimulation condition. PLoS ONE. (2013) 8:e60144. doi: 10.1371/journal.pone.0060144
61. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. (2015) 42:443–56. doi: 10.1016/j.immuni.2015.02.008
62. Theorell J, Gustavsson AL, Tesi B, Sigmundsson K, Ljunggren HG, Lundback T, et al. Immunomodulatory activity of commonly used drugs on Fc-receptor-mediated human natural killer cell activation. Cancer Immunol Immunother. (2014) 63:627–41. doi: 10.1007/s00262-014-1539-6
63. Wai LE, Fujiki M, Takeda S, Martinez OM, Krams SM. Rapamycin, but not cyclosporine or FK506, alters natural killer cell function. Transplantation. (2008) 85:145–9. doi: 10.1097/01.tp.0000296817.28053.7b
64. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
65. Cruikshank M, Anoop P, Nikolajeva O, Rao A, Rao K, Gilmour K, et al. Screening assays for primary haemophagocytic lymphohistiocytosis in children presenting with suspected macrophage activation syndrome. Pediatr Rheumatol Online J. (2015) 12 Suppl 1:48. doi: 10.1186/s12969-015-0043-7
66. Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. (2012) 122:3769–80. doi: 10.1172/JCI64837
67. Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. (2000) 25:173–6. doi: 10.1038/76024
68. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. (2005) 14:827–34. doi: 10.1093/hmg/ddi076
69. Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. (2009) 119:3765–73. doi: 10.1172/JCI40732
70. Murata Y, Yasumi T, Shirakawa R, Izawa K, Sakai H, Abe J, et al. Rapid diagnosis of FHL3 by flow cytometric detection of intraplatelet Munc13-4 protein. Blood. (2011) 118:1225–30. doi: 10.1182/blood-2011-01-329540
71. Shibata H, Yasumi T, Shimodera S, Hiejima E, Izawa K, Kawai T, et al. Human CTL-based functional analysis shows the reliability of a munc13-4 protein expression assay for FHL3 diagnosis. Blood. (2018) 131:2016–25. doi: 10.1182/blood-2017-10-812503
72. Purtilo DT, Cassel CK, Yang JP, Harper R. X-linked recessive progressive combined variable immunodeficiency (Duncan's disease). Lancet. (1975) 1:935–40. doi: 10.1016/s0140-6736(75)92004-8
73. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. (2006) 444:110–4. doi: 10.1038/nature05257
74. Marsh RA. Epstein-Barr Virus and Hemophagocytic Lymphohistiocytosis. Front Immunol. (2017) 8:1902. doi: 10.3389/fimmu.2017.01902
75. Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, et al. X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatr Res. (1995) 38:471–8. doi: 10.1203/00006450-199510000-00001
76. Snow AL, Pandiyan P, Zheng L, Krummey SM, Lenardo MJ. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol Rev. (2010) 236:68–82. doi: 10.1111/j.1600-065X.2010.00917.x
77. Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood. (2010) 116:3398–408. doi: 10.1182/blood-2010-03-275909
78. Kanegane H, Yang X, Zhao M, Yamato K, Inoue M, Hamamoto K, et al. Clinical features and outcome of X-linked lymphoproliferative syndrome type 1 (SAP deficiency) in Japan identified by the combination of flow cytometric assay and genetic analysis. Pediatr Allergy Immunol. (2012) 23:488–93. doi: 10.1111/j.1399-3038.2012.01282.x
79. Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. (2010) 362:1–9. doi: 10.1016/j.jim.2010.08.010
80. Zhao M, Kanegane H, Kobayashi C, Nakazawa Y, Ishii E, Kasai M, et al. Early and rapid detection of X-linked lymphoproliferative syndrome with SH2D1A mutations by flow cytometry. Cytometry B Clin Cytom. (2011) 80:8–13. doi: 10.1002/cyto.b.20552
81. Erdos M, Uzvolgyi E, Nemes Z, Torok O, Rakoczi E, Went-Sumegi N, et al. Characterization of a new disease-causing mutation of SH2D1A in a family with X-linked lymphoproliferative disease. Hum Mutat. (2005) 25:506. doi: 10.1002/humu.9339
82. Morra M, Simarro-Grande M, Martin M, Chen AS, Lanyi A, Silander O, et al. Characterization of SH2D1A missense mutations identified in X-linked lymphoproliferative disease patients. J Biol Chem. (2001) 276:36809–16. doi: 10.1074/jbc.M101305200
83. Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol. (2013) 149:133–41. doi: 10.1016/j.clim.2013.07.004
84. Yang X, Hoshino A, Taga T, Kunitsu T, Ikeda Y, Yasumi T, et al. A female patient with incomplete hemophagocytic lymphohistiocytosis caused by a heterozygous XIAP mutation associated with non-random X-chromosome inactivation skewed towards the wild-type XIAP allele. J Clin Immunol. (2015) 35:244–8. doi: 10.1007/s10875-015-0144-6
85. Holle JR, Marsh RA, Holdcroft AM, Davies SM, Wang L, Zhang K, et al. Hemophagocytic lymphohistiocytosis in a female patient due to a heterozygous XIAP mutation and skewed X chromosome inactivation. Pediatr Blood Cancer. (2015) 62:1288–90. doi: 10.1002/pbc.25483
86. Marsh RA, Villanueva J, Zhang K, Snow AL, Su HC, Madden L, et al. A rapid flow cytometric screening test for X-linked lymphoproliferative disease due to XIAP deficiency. Cytometry B Clin Cytom. (2009) 765):334–44. doi: 10.1002/cyto.b.20473
87. Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med. (2005) 11:340–5. doi: 10.1038/nm1189
88. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med. (2005) 201:695–701. doi: 10.1084/jem.20042432
89. Meazza R, Tuberosa C, Cetica V, Falco M, Parolini S, Grieve S, et al. Diagnosing XLP1 in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. (2014) 134:1381–7 e7. doi: 10.1016/j.jaci.2014.04.043
90. Cifaldi C, Chiriaco M, Di Matteo G, Di Cesare S, Alessia S, De Angelis P, et al. Novel X-linked inhibitor of apoptosis mutation in very early-onset inflammatory bowel disease child successfully treated with HLA-haploidentical hemapoietic stem cells transplant after removal of alphabeta(+) T and B cells. Front Immunol. (2017) 8:1893. doi: 10.3389/fimmu.2017.01893
91. Girardelli M, Arrigo S, Barabino A, Loganes C, Morreale G, Crovella S, et al. The diagnostic challenge of very early-onset enterocolitis in an infant with XIAP deficiency. BMC Pediatr. (2015) 15:208. doi: 10.1186/s12887-015-0522-5
92. Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. (2018) 132:89–100. doi: 10.1182/blood-2017-11-814244
93. Mukda E, Trachoo O, Pasomsub E, Tiyasirichokchai R, Iemwimangsa N, Sosothikul D, et al. Exome sequencing for simultaneous mutation screening in children with hemophagocytic lymphohistiocytosis. Int J Hematol. (2017) 106:282–90. doi: 10.1007/s12185-017-2223-3
94. Pasic S, Micic D, Kuzmanovic M. Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott-Aldrich syndrome. Acta Paediatr. (2003) 92:859–61. doi: 10.1111/j.1651-2227.2003.tb02548.x
95. Abraham RS, Aubert G. Flow cytometry, a versatile tool for diagnosis and monitoring of primary immunodeficiencies. Clin Vaccine Immunol. (2016) 23:254–71. doi: 10.1128/CVI.00001-16
96. Epling CL, Stites DP, McHugh TM, Chong HO, Blackwood LL, Wara DW. Neutrophil function screening in patients with chronic granulomatous disease by a flow cytometric method. Cytometry. (1992) 13:615–20. doi: 10.1002/cyto.990130609
97. Chiang SCC, Vergamini SM, Husami A, Neumeier L, Quinn K, Ellerhorst T, et al. Screening for Wiskott-Aldrich syndrome by flow cytometry. J Allergy Clin Immunol. (2018) 142:333–5 e8. doi: 10.1016/j.jaci.2018.04.017
98. Boldt A, Bitar M, Sack U. Flow Cytometric Evaluation of Primary Immunodeficiencies. Clin Lab Med. (2017) 37:895–913. doi: 10.1016/j.cll.2017.07.013
99. Kanegane H, Hoshino A, Okano T, Yasumi T, Wada T, Takada H, et al. Flow cytometry-based diagnosis of primary immunodeficiency diseases. Allergol Int. (2018) 67:43–54. doi: 10.1016/j.alit.2017.06.003
100. Gruda R, Brown AC, Grabovsky V, Mizrahi S, Gur C, Feigelson SW, et al. Loss of kindlin-3 alters the threshold for NK cell activation in human leukocyte adhesion deficiency-III. Blood. (2012) 120:3915–24. doi: 10.1182/blood-2012-02-410795
101. Mace EM, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest. (2017) 127:306–20. doi: 10.1172/JCI86276
102. Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. (2004) 173:7277–81. doi: 10.4049/jimmunol.173.12.7277
103. Schaballie H, Rodriguez R, Martin E, Moens L, Frans G, Lenoir C, et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J Allergy Clin Immunol. (2015) 136:816–9 e4. doi: 10.1016/j.jaci.2015.03.009
104. Andzelm MM, Chen X, Krzewski K, Orange JS, Strominger JL. Myosin IIA is required for cytolytic granule exocytosis in human NK cells. J Exp Med. (2007) 204:2285–91. doi: 10.1084/jem.20071143
105. Sanborn KB, Mace EM, Rak GD, Difeo A, Martignetti JA, Pecci A, et al. Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood. (2011) 118:5862–71. doi: 10.1182/blood-2011-03-344846
106. Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. (2013) 121:2669–77. doi: 10.1182/blood-2012-09-453969
107. Meade JL, de Wynter EA, Brett P, Sharif SM, Woods CG, Markham AF, et al. A family with Papillon-Lefevre syndrome reveals a requirement for cathepsin C in granzyme B activation and NK cell cytolytic activity. Blood. (2006) 107:3665–8. doi: 10.1182/blood-2005-03-1140
108. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
109. Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. (2015) 100:978–88. doi: 10.3324/haematol.2014.121608
110. Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2008) 51:402–4. doi: 10.1002/pbc.21564
111. Otrock ZK, Hock KG, Riley SB, de Witte T, Eby CS, Scott MG. Elevated serum ferritin is not specific for hemophagocytic lymphohistiocytosis. Ann Hematol. (2017) 96:1667–72. doi: 10.1007/s00277-017-3072-0
112. Schram AM, Campigotto F, Mullally A, Fogerty A, Massarotti E, Neuberg D, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood. (2015) 125:1548–52. doi: 10.1182/blood-2014-10-602607
113. El-Sherbiny YM, Psarras A, Md Yusof MY, Hensor EMA, Tooze R, Doody G, et al. A novel two-score system for interferon status segregates autoimmune diseases and correlates with clinical features. Sci Rep. (2018) 8:5793. doi: 10.1038/s41598-018-24198-1
114. Zhang R, Yang X, Wang J, Han L, Yang A, Zhang J, et al. Identification of potential biomarkers for differential diagnosis between rheumatoid arthritis and osteoarthritis via integrative genomewide gene expression profiling analysis. Mol Med Rep. (2019) 19:30–40. doi: 10.3892/mmr.2018.9677
115. Kim H, de Jesus AA, Brooks SR, Liu Y, Huang Y, VanTries R, et al. Development of a validated interferon score using nanostring technology. J Interferon Cytokine Res. (2018) 38:171–85. doi: 10.1089/jir.2017.0127
116. Rice GI, Melki I, Fremond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. (2017) 37:123–32. doi: 10.1007/s10875-016-0359-1
117. Sumegi J, Nestheide S, Aronow B, Fletcher D, Keddache M, Villanueva J, et al. MicroRNA activation signature in patients with hemophagocytic lymphohistiocytosis and reversibility with disease-specific therapy. J Allergy Clin Immunol. (2016) 137:309–12. doi: 10.1016/j.jaci.2015.06.006
Keywords: flow cytometry, HLH, hemophagocytic lymphohistiocytosis, primary immunodeficiencies, clinical diagnostics, diagnostic accuracy, clinical laboratory tests, XLP
Citation: Chiang SCC, Bleesing JJ and Marsh RA (2019) Current Flow Cytometric Assays for the Screening and Diagnosis of Primary HLH. Front. Immunol. 10:1740. doi: 10.3389/fimmu.2019.01740
Received: 17 April 2019; Accepted: 10 July 2019;
Published: 23 July 2019.
Edited by:
Roshini Sarah Abraham, Nationwide Children's Hospital, United StatesReviewed by:
Kai Lehmberg, University Medical Center Hamburg-Eppendorf, GermanyHirokazu Kanegane, Tokyo Medical and Dental University, Japan
Copyright © 2019 Chiang, Bleesing and Marsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Cern Cher Chiang, c2FtLmNoaWFuZ0BjY2htYy5vcmc=; Rebecca A. Marsh, cmViZWNjYS5tYXJzaEBjY2htYy5vcmc=