 Annelies Demeyer1,2
Annelies Demeyer1,2 Ioannis Skordos1,2
Ioannis Skordos1,2 Yasmine Driege1,2Marja Kreike1,2Tino Hochepied1,2Mathijs Baens3
Yasmine Driege1,2Marja Kreike1,2Tino Hochepied1,2Mathijs Baens3 Jens Staal1,2*†
Jens Staal1,2*† Rudi Beyaert1,2*†
Rudi Beyaert1,2*†- 1VIB Center for Inflammation Research, Ghent, Belgium
- 2Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
- 3Center for Innovation and Stimulation of Drug Discovery (CISTIM), Leuven, Belgium
MALT1 is a central signaling component in innate and adaptive immunity by regulating NF-κB and other key signaling pathways in different cell types. Activities of MALT1 are mediated by its scaffold and protease functions. Because of its role in lymphocyte activation and proliferation, inhibition of MALT1 proteolytic activity is of high interest for therapeutic targeting in autoimmunity and certain lymphomas. However, recent studies showing that Malt1 protease-dead knock-in (Malt1-PD) mice suffer from autoimmune disease have somewhat tempered the initial enthusiasm. Although it has been proposed that an imbalance between immune suppressive regulatory T cells (Tregs) and activated effector CD4+ T cells plays a key role in the autoimmune phenotype of Malt1-PD mice, the specific contribution of MALT1 proteolytic activity in T cells remains unclear. Using T cell-conditional Malt1 protease-dead knock-in (Malt1-PDT) mice, we here demonstrate that MALT1 has a T cell-intrinsic role in regulating the homeostasis and function of thymic and peripheral T cells. T cell-specific ablation of MALT1 proteolytic activity phenocopies mice in which MALT1 proteolytic activity has been genetically inactivated in all cell types. The Malt1-PDT mice have a reduced number of Tregs in the thymus and periphery, although the effect in the periphery is less pronounced compared to full-body Malt1-PD mice, indicating that also other cell types may promote Treg induction in a MALT1 protease-dependent manner. Despite the difference in peripheral Treg number, both T cell-specific and full-body Malt1-PD mice develop ataxia and multi-organ inflammation to a similar extent. Furthermore, reconstitution of the full-body Malt1-PD mice with T cell-specific expression of wild-type human MALT1 eliminated all signs of autoimmunity. Together, these findings establish an important T cell-intrinsic role of MALT1 proteolytic activity in the suppression of autoimmune responses.
Introduction
MALT1 (PCASP1 or paracaspase-1) is ubiquitously expressed in multiple cell types, where it is involved in signaling leading to pro-inflammatory gene expression. The best known role of MALT1 is the activation of NF-κB signaling in response to T cell and B cell antigen receptor triggering, which is critical for the adaptive immune system. At the border between the innate and adaptive immune system, MALT1 acts downstream of several innate immune receptors on antigen-presenting dendritic cells (DC), which in turn regulate cytokine production that can influence the polarization of the adaptive immune system. MALT1 is also critical for NF-κB induction, but not for degranulation responses, downstream of several ITAM-coupled natural killer cell (NK) receptors and the Fcε receptor on mast cells (1). MALT1-deficient mice and patients show primary immunodeficiency (PID) via defects in T and B cells, in particular marginal zone (MZ) B cell development and antigen-induced T cell proliferation and activation, which leads to immunodeficiency and failure to respond to vaccination (2–8). A MALT1-deficient patient suffering from recurrent infections was cured by transplantation of mostly T cells (8). This indicates a major role for T cells as a determinant of the compromised immunity phenotypes caused by MALT1 deficiency. T cells are highly dependent on antigen receptor signaling, which requires MALT1. In contrast, B cells can be alternatively activated by innate receptors in a MALT1-independent manner. This might explain the lower phenotypic impact of MALT1 deficiency in B cells (2, 6, 7, 9). MALT1 signaling in non-lymphoid cells is also critical for optimal immunity, as illustrated by PID patients susceptible to fungal infections due to a CARD9 mutant that fails to recruit downstream BCL10 and MALT1 (10). Apart from its role in immune signaling through loss-of-function, aberrant activation or expression of MALT1 has been associated to different forms of B cell cancer, such as ABC-DLBCL (11–14), low-grade MALT lymphomas (15–19), and a complex type of PID called B cell expansion with NF-κB and T cell anergy (BENTA) (20, 21).
MALT1 belongs to the type 1 paracaspase family, an ancient protein family that originated before the last common ancestor of planulozoa (bilaterians and cnidarians) (22, 23). The type 1 paracaspases consist of an amino-terminal death domain, immunoglobulin domains and a caspase-like domain. Upon activation, MALT1 acts as a scaffold and recruits critical downstream signaling components such as TRAF6 and TAK1 in order to activate NF-κB and JNK/AP-1 dependent transcription (2, 3, 24, 25). The caspase-like domain has received plenty of research attention after it was established that paracaspases have protease activity (26, 27). Both the scaffold function leading to NF-κB and the protease activity of MALT1 are of critical importance, since they have been functionally conserved at least as far back as the last common ancestor of the three type 1 paracaspase paralogs in jawed vertebrates (22, 23). An ever growing list of paracaspase protease substrates—which at this moment includes auto-processing (28–30), adaptor proteins (27), transcription factors (31), deubiquitinases (26, 32), ubiquitin ligases (33–35) and specific RNA degrading enzymes (36–38)—illustrate that MALT1 protease activity removes inhibitory proteins and fine-tunes the type of responses that are induced (1, 39). There is only a single member of the paracaspase family in mammals (22). This, as well as the signal fine-tuning by an enzymatic activity, makes MALT1 protease activity a very attractive therapeutic target in humans for the treatment of autoimmunity and some cancers. At this moment, several independent studies have described active site or allosteric MALT1 protease inhibitors (11–14, 40–45), and mouse studies have already indicated the therapeutic value of some of these inhibitors in certain types of cancer (13, 14, 43, 46) and inflammatory diseases (40). Recently, four independent papers reported unexpected autoimmune phenotypes such as multi-organ inflammation, including the stomach, and ataxia in full-body Malt1-PD mice (4–6, 47), which could indicate a risk when inhibiting MALT1 protease activity for therapeutic reasons. While at first sight counter-intuitive, autoimmune or autoinflammatory diseases (AID) are in fact common in patients suffering from PID (48). The general conclusion from the different Malt1-PD mouse studies was that the disease was caused by an imbalance between effector CD4+ T cells and Tregs (4, 5). Much of the disease could be mitigated by deletion of IFN-γ in the Malt1-PD mice, indicating that IFN-γ is a primary driver of disease (5). However, another report also suggested a contribution from immune-regulatory IL-10 producing B-cells (Bregs) (47). Moreover, MALT1 also regulates the activation of DC cells, NK cells and mast cells (1), indicating the possibility that MALT1 activity in some of these cells may also regulate inflammatory T and B cell responses associated with autoimmunity in Malt1-PD mice. Given the complexity of MALT1 function in the development of lymphoid cells and activation of several immune cells, the study of cell-intrinsic functions of MALT1 requires Malt1 conditional knock-in (KI) mice. In order to better predict safety risks upon pharmacological MALT1 protease inhibition and get further insight into the role of T cells in the autoimmune pathology of Malt1-PD mice, we employed conditional KI mice lacking MALT1 proteolytic activity specifically in T cells (Malt1-PDT), and compared their phenotype with mice lacking MALT1 proteolytic activity in all cell types (Malt1-PD). To verify that only MALT1 protease deficiency in T cells can cause the disease, we also reconstituted Malt1-PD mice with transgenic T cell-specific expression of human wild-type (WT) MALT1. Taken together, we show that T cell-specific inactivation of MALT1 proteolytic activity phenocopies inactivation of MALT1 in all cell types, resulting in a disruption in T cell immune homeostasis and development of autoimmunity, indicating an important T cell-intrinsic role of MALT1 proteolytic activity in keeping normal immune homeostasis.
Results
T Cell-Specific Loss of MALT1 Proteolytic Activity Is Sufficient to Cause Ataxia and Weight Retardation to the Same Extent as Full-Body Loss of MALT1 Proteolytic Activity
To characterize the contribution of hematopoietic cells in the autoimmune phenotype of Malt1-PD mice, all previous studies have relied on crosses with other knock-out (KO) lines (e.g., IFN-γ KO), bone marrow transplants and injection of WT Tregs (4–6, 47). However, none of these experiments allow an undeniable conclusion on the specific role of T cells in the different autoimmune responses observed in Malt1-PD mice. In order to further characterize this phenotype, we generated T cell-specific Malt1-PDT mice and compared their phenotype with that of full-body Malt1-PD mice that we also generated. We used the F1 offspring from the Malt1FL/FL CD4-Cre X Malt1PD/+ cross, where the Malt1PD/FL CD4-Cre offspring represent Malt1-PDT mice. The remaining F1 offspring (Malt1PD/FL, Malt1+/FL, and Malt+/FL CD4-Cre) all behaved as WT and were consequently all used as phenotypic controls (Supplemental Figures 1A–C). To obtain a positive phenotypic control, Malt1PD/+ were crossed with Malt1−LacZ/−LacZ KO mice, where Malt1PD/−LacZ F1 offspring represent full-body Malt1-PD mice and Malt1+/−LacZ offspring represent healthy controls with the same gene dosage as our Malt1-PDT mice (Supplemental Figures 1A–C). Just like in the full-body Malt1-PD mice, the T cell-specific Malt1-PDT mice displayed a weight retardation, starting from approximately 9 weeks of age, and ataxia, starting on average at 11–12 weeks of age (Figures 1A–D). This is also similar to what was previously observed for other full-body Malt1-PD mice in several independent studies (4–6, 47). A WT mouse will stretch its hind legs when suspended by its tail to maintain its balance (Figure 1A). A mouse suffering from ataxia will flail around with its hind legs at an early stage of disease and start clutching its hind paws (and sometimes also front paws) at a more advanced stage (Figures 1A,B). We scored a mouse as having ataxia at the moment the hind paws were completely clutched while hanging by the tail. Although time of onset was not different (Figures 1C,D), we noticed that the overall symptoms were more severe in females than in males, which is similar to what has been previously observed in an independent Malt1-PD mouse line (47). This observation is also consistent with the sexual dimorphism in autoimmune diseases seen in humans (49, 50). Since the T cell-specific Malt1-PDT mice show as severe disease development as the full-body Malt1-PD mice, we can conclude that absence of MALT1 protease activity in T cells only is sufficient to induce ataxia and weight loss to a similar extent as induced by its absence in all body cells.
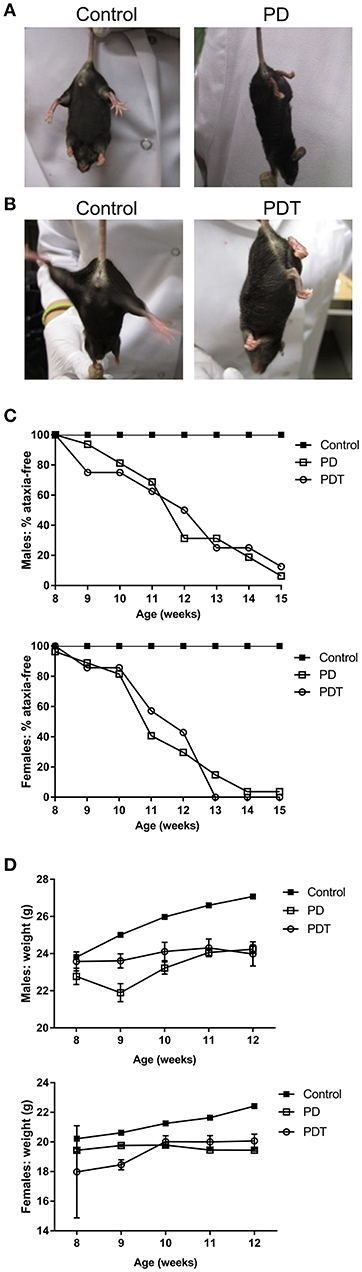
Figure 1. Malt1-PD and Malt1-PDT mice display ataxia and weight retardation. (A,B) Representative pictures of Malt1-PD and -PDT mice suffering from ataxia, characterized by leg clutching. (C) Ataxia onset in male and female control, Malt1-PD and -PDT mice monitored from week 8 to 15 (male mice: control n = 41, Malt1-PD n = 16, and Malt1-PDT n = 8; female mice: control n = 37, Malt1-PD n = 26 and Malt1-PDT n = 7). (D) Weight retardation in male and female Malt1-PD and -PDT mice monitored from week 8-12 until termination. Values after week 12 are excluded due to a too low number of surviving Malt1-PDT mice, causing very broad 95% confidence intervals. Male mice: week 8: control n = 41, Malt1-PD n = 9 and Malt1-PDT n = 6; week 9: control n = 49, Malt1-PD n = 8 and Malt1-PDT n = 8; week 10: control n = 50, Malt1-PD n = 11 and Malt1-PDT n = 7; week 11: control n = 61, Malt1-PD n = 16 and Malt1-PDT n = 8; week 12: control n = 59, Malt1-PD n = 16 and Malt1-PDT n = 7 and female mice: week 8: control n = 37, Malt1-PD n = 10 and Malt1-PDT n = 3; week 9: control n = 49, Malt1-PD n = 18 and Malt1-PDT n = 6; week 10: control n = 50, Malt1-PD n = 23 and Malt1-PDT n = 5; week 11: control n = 56, Malt1-PD n = 27 and Malt1-PDT n = 6; week 12: control n = 54, Malt1-PD n = 26 and Malt1-PDT n = 6.
T Cell-Specific Loss of MALT1 Proteolytic Activity Causes Multi-Organ Inflammation
After birth, mice were checked regularly and no external signs of suffering could be observed before the development of ataxia. However, upon sacrifice we noticed that the stomach of Malt1-PD and -PDT mice was more rigid and vascularised (Figures 2A,B). By means of H&E staining we also observed severe gastritis, plus immune infiltration in lacrimal and salivary glands indicating that the mice already had developed inflammation in other organs (Figures 2C–H). In order to minimize the suffering of mice, we euthanized them as soon as possible after the observation of ataxia. To assess whether disease development in Malt1-PD and -PDT mice was due to the same mechanism, we evaluated cytokine production by CD44+ CD4+ T cells in cervical lymph nodes (cLN) of mice already suffering from ataxia. We decided to analyze cLN as they are located close to known inflamed organs. Both Malt1-PD and -PDT mice have a slight reduction in total CD4+ T cells and an increase in CD4+ T cells that express CD44, so-called effector CD4+ T cells compared to WT control mice (Figures 3A,B). We also observed a strong upregulation of IFN-γ-producing CD44+CD4+ T cells in Malt1-PD and -PDT mice, indicating a T cell intrinsic role for MALT1 protease activity in restricting Th1 responses (Figure 3C). We further assessed serum cytokine levels in Malt1-PD and -PDT mice. For IL-2 and TNF there was a significant, but mild increase in the Malt1-PD compared to control mice, which was no longer detectable in Malt1-PDT mice. No increase in IL-4, IL-6, IL-17 and IFN-γ could be detected in serum of Malt1-PD and -PDT mice (Figure 3D). Therefore, we can conclude that both Malt1-PD and -PDT mice do not suffer from systemic inflammation, but instead suffer from local Th1-associated multi-organ inflammation.
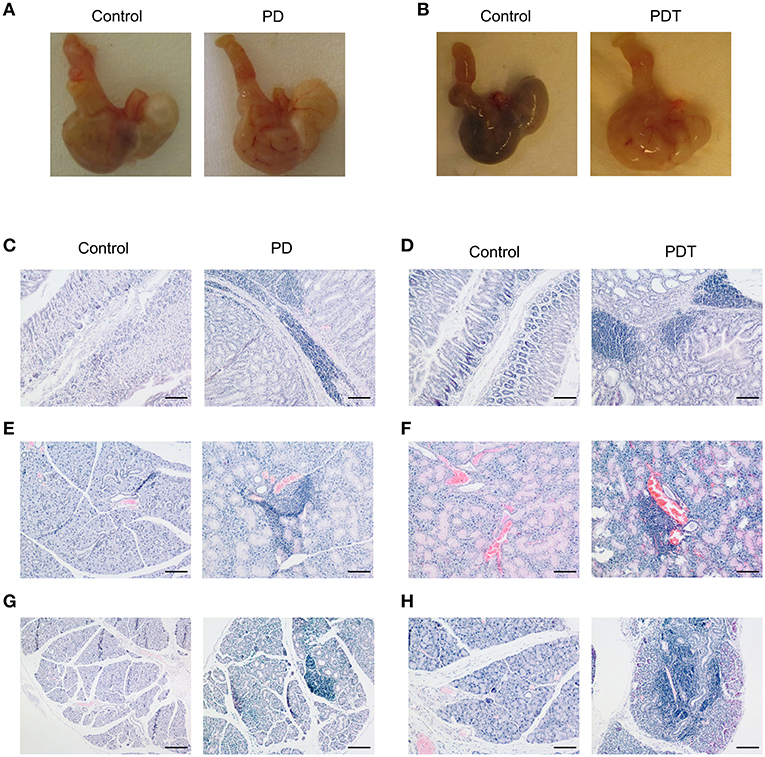
Figure 2. Multi-organ inflammation in Malt1-PD and Malt1-PDT mice. (A,B) Representative pictures of stomach of Malt1-PD and -PDT mice with increased vascularization compared to their corresponding controls. (C,D) H&E staining showing immune infiltration in the submucosa of the stomach of Malt1-PD (C) and Malt1-PDT (D) mice. Scale bar 100 μm. (E,F) H&E staining showing immune infiltration in the salivary gland of Malt1-PD (E) and Malt1-PDT (F) mice. Scale bar 100 μm. (G,H) H&E staining showing immune infiltration in the lacrimal gland of Malt1-PD (G) and Malt1-PDT (H) mice. Scale bar 100 μm.
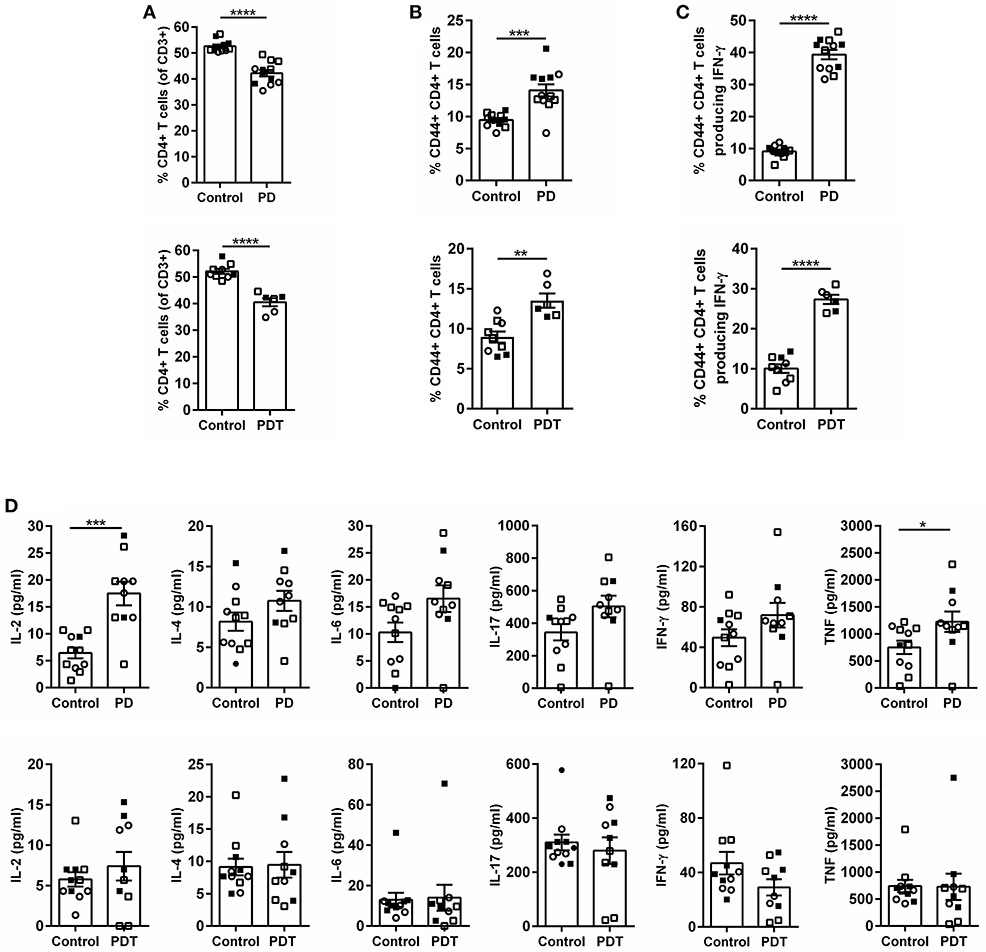
Figure 3. Inflammation in Malt1-PD and Malt1-PDT mice is local and not systemic. (A) CD4+ T cells in Malt1-PD (top panel) and Malt1-PDT (bottom panel) mice. (B) CD44+CD4+ T cells in Malt1-PD (top panel) and Malt1-PDT (bottom panel) mice. (C) CD44+CD4+ T cells producing IFN-γ in Malt1-PD (top panel) and Malt1-PDT (bottom panel) mice. For (A–C) lymphocytes from cLN were stimulated for 4 h with PMA/ionomycin and brefeldin A to determine the different cell populations by flow cytometry. Malt1-PD mice: n = 11, corresponding control mice: n = 12; Malt1-PDT mice: n = 6, corresponding control mice: n = 9. (D) Serum levels of IL-2, IL-4, IL-6, IL-17, IFN-γ, and TNF in Malt1-PD (top panels) and Malt1-PDT (bottom panels) mice. Data represent three separate serum collections: collection 1 = filled squares, collection 2 = open squares and collection 3 = open circles. Malt1-PD mice: n = 10, corresponding control mice: n = 11 and Malt1-PDT mice: n = 11, corresponding control mice: n = 10. The mean ± SEM is indicated on the graphs. The statistical significance between groups was calculated with an unpaired 2 tailed Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
A T Cell-Intrinsic Role for MALT1 Proteolytic Activity Is Critical for Thymic nTreg Development
The best known Tregs are Foxp3+CD25+CD4+ T cells (51), which have two distinct developmental origins. Some develop in the thymus at a young age—the so-called natural Tregs (nTregs). Others mature in the periphery from naïve conventional T cells during extended exposure to antigen or under inflammatory conditions—the so-called induced Tregs (iTregs). Both populations are genetically distinct and have non-redundant functions (52, 53). MALT1 has been shown to be specifically required for thymic Treg development, while induced peripheral Treg formation in aged mice is not inhibited by MALT1 deficiency (4, 5, 54). The ability to induce Treg formation in Malt1-KO mice is also consistent with previous in vitro differentiation studies using a high dose of anti-CD3 to stimulate the TCR (55). This might indicate a threshold effect which is influenced by MALT1. Therefore, we investigated the role of MALT1 proteolytic activity in thymic Treg development in young healthy (ataxia-free) Malt1-PD and -PDT mice. A clear defect in thymic Treg development could be observed, since Foxp3+CD25+CD4+ T cells were almost absent in the thymus of young Malt1-PD and -PDT mice (Figures 4A,B). Furthermore, thymic Treg development could be rescued upon transgenic expression of human WT MALT1 in cells targeted by CD4-Cre (Figures 4D,E). This clearly indicates a T cell-intrinsic role for MALT1 protease activity in nTreg development.
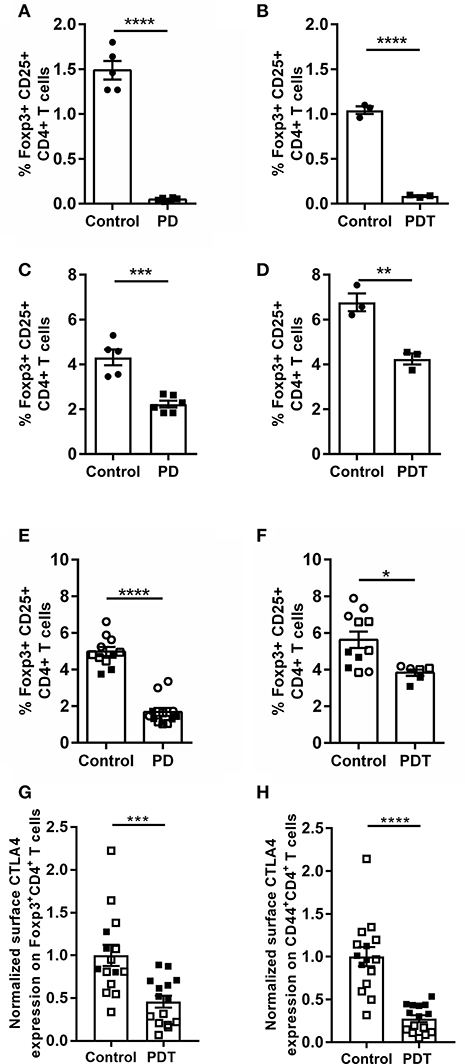
Figure 4. Reduced Treg frequency and reduced surface CTLA-4 expression on Tregs and effector CD4+ T cells in Malt1-PD and Malt1-PDT mice. (A,B) Treg frequency (Foxp3+CD25+CD4+CD8− T cells) in thymus of young Malt1-PD (n = 6) (A) and Malt-PDT (n = 3) (B) mice and their corresponding controls (n = 5 and n = 3, respectively). (C,D) Treg frequency in cLN of young Malt1-PD (n = 6) (C) and Malt1-PDT (n = 3) (D) mice and their corresponding controls (n = 5 and n = 3, respectively). (E,F) Treg frequency in Malt1-PD (n = 11) (E) and Malt1-PDT (n = 6) (F) mice suffering from ataxia and their corresponding controls (n = 12 and n = 9, respectively). Lymphocytes were stimulated for 4 h with PMA/ionomycin and the data represent three individual experiments: experiment 1 = filled squares, experiment 2 = open squares and experiment 3 = open circles. (G,H) Normalized CTLA-4 expression on the surface of Tregs (G) and CD44+CD4+ T cells (H) from young disease free Malt1-PDT mice (n = 15) and their corresponding controls (n = 15). The individual percentages of Foxp3+CD4+ T cells or CD44+CD4+ T cells that express CTLA-4 on their surface is normalized against the average percentage of the corresponding control mice of each individual experiment. Lymphocytes were stimulated for 4 h with PMA/ionomycin and data represent two individual experiments: experiment 1 = filled squares, experiment 2 = open squares. For (A–H): all data were obtained via flow cytometry. The mean ± SEM is indicated on the graphs. The statistical significance between groups was calculated with an unpaired 2 tailed Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Tregs are present in the cLN of young Malt1-PD and -PDT mice, albeit at lower levels than in control mice (Figures 4C,D). The higher Treg numbers found in cLN of young Malt1-PDT mice could be a secondary effect from inflammation-induced formation of iTregs or peripheral proliferation of the few remaining nTregs. The nearly absent levels of Tregs in thymus, but detectable levels of Tregs in the periphery is a common feature of Malt1-PD and -PDT mice, as well as Malt1-KO mice (4, 5, 47, 54, 56).
In addition to a decrease in Tregs, also a decrease in the amount of Bregs was suggested to contribute to the autoimmune disease phenotype in Malt1-PD mice (47). Bregs can originate from many different B cell lineages, where marginal zone (MZ) B cells serve as an important source in the spleen (57). MZ B cells are disrupted in Malt1-KO and Malt1-PD mice. We therefore investigated MZ B cell development in T cell-specific Malt1-KO and Malt1-PDT mice compared to WT and full-body Malt1-PD and -KO mice. In contrast to full body Malt1-PD and -KO mice, MZ B cell development is completely normal in both T cell-specific Malt1-KO and Malt1-PDT mice (Supplemental Figures 2A–D). Furthermore, in contrast to Malt1-PD mice, Bregs were not absent in the Malt1-PDT mice (Figure 5). Together, these findings argue against a contribution from a decrease in IL-10 producing Bregs to the autoimmune phenotype.
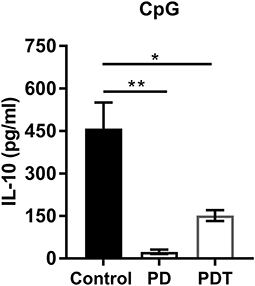
Figure 5. Bregs are almost absent in Malt1-PD mice, but are present in Malt1-PDT mice. IL-10 secretion by control (n = 4), Malt1-PD (n = 3), and Malt1-PDT (n = 3) splenic B cells after overnight stimulation with TLR9 ligand CpG. The mean ± SEM is indicated on the graphs. The statistical significance between groups was calculated with an unpaired 2 tailed Student's t-test: *p < 0.05 and **p < 0.01.
Loss of MALT1 Proteolytic Activity Reduces Expression of the Treg Functionality Marker CTLA-4
Previously Jaworski et al. (4) and Bornancin et al. (47) reported a reduction of peripheral Tregs in Malt1-PD mice. We could confirm this in cLN from young disease-free Malt1-PD mice and in mice already suffering from ataxia. Notably, at the time of sacrifice, when the mice had developed ataxia, the number of peripheral Tregs in Malt1-PDT mice is higher than in Malt1-PD mice (Figures 4E,F), indicating that also MALT1 protease-dependent signals from non-T cells may promote peripheral Treg induction. Nevertheless, both Malt1-PD and -PDT mice developed equally severe disease symptoms, indicating that disease development in the Malt1-PD and -PDT cannot be solely explained by reduced peripheral iTreg levels. Therefore, we assessed the functionality of Tregs by means of their surface expression of CTLA-4, which binds CD80 and CD86 proteins found on the surface of antigen presenting cells. These proteins provide, by binding to CD28 on the T cells, the costimulatory signal needed for optimal T cell activation. By expressing CTLA-4, Tregs can downregulate CD80 and CD86 expression on antigen presenting cells, leading to reduced T cell proliferation, survival and IL-2 production (58). We have previously observed, via flow cytometry on splenocytes, that the percentage of Foxp3+CD4+ T cells that express CTLA-4 on their surface is reduced in Malt1-KO mice compared to WT mice (unpublished data). To assess the effect of MALT1 proteolytic activity on the percentage of Tregs (Foxp3+CD4+ T cells) with surface CTLA-4 expression, we isolated cells from cLN of young disease-free Malt1-PDT mice and analyzed CTLA-4 surface expression by flow cytometry. This revealed that the percentage of surface CTLA-4 expressing Tregs was also reduced in Malt1-PDT mice compared to control mice (Figure 4G). The reduced expression of CTLA-4 is not the result of a generalized defect in effector Treg differentiation, since expression of other Treg effector markers is not affected (Supplemental Figures 3A–D). CTLA-4 can also be expressed on effector CD4+ T cells (CD44+CD4+ T cells), where reduced expression is associated with elevated tissue infiltration and suppression of autoreactivity (59, 60). As for the Tregs, also here the percentage of CTLA-4 surface-positive CD44+CD4+ T cells was found to be reduced in young disease-free Malt1-PDT mice (Figure 4H). Together, these results clearly demonstrate that MALT1 proteolytic activity in T cells regulates CTLA-4 surface expression in Tregs and CD44+CD4+ T cells, indicating that MALT1 proteolytic activity influences Treg functionality in a T cell-intrinsic manner, since even a moderate decrease in CTLA-4 expression can lead to autoimmunity in humans (61). The reduced CTLA-4 expression could thus be an important factor regulating autoimmunity in the Malt1-PDT mice.
T Cell-Specific Transgenic Human MALT1 Reconstitution Rescues MALT1 Protease-Deficient Mice From Autoimmune Disease
Since both full-body Malt1PD/− and T cell-specific Malt1PD/FL CD4-Cre mice equally develop an inflammatory phenotype, we can conclude that the loss of MALT1 proteolytic activity in cells targeted by CD4-Cre is sufficient to induce a full autoimmune response. To verify that MALT1 enzymatic activity in other cells not targeted by CD4-Cre may still contribute to the development of autoimmune disease, we generated Malt1PD/− Rosa26LSL-MALT1-WT CD4-Cre mice in which all cells endogenously express protease-dead MALT1, and where human WT MALT1 is specifically expressed as a transgene in cells that have experienced CD4-Cre activity. These mice did no longer show gastritis and immune infiltration in lacrimal and salivary glands as observed in Malt-PD mice (Figures 6A,B), nor did they show an increase in IFN-γ producing CD4+ T cells (Figure 6C). Moreover, transgenic expression of WT MALT1 in cells targeted by CD4-Cre in a Malt1-PD background suppressed the strong drop in the amount of Tregs in the thymus and cLN of Malt1-PD mice (Figures 6D,E). The complete rescue of the autoimmune phenotype in full-body Malt1-PD mice by T cell-specific WT MALT1 expression further demonstrates that only T cell MALT1 protease activity is critical to suppress the autoimmune phenotype.
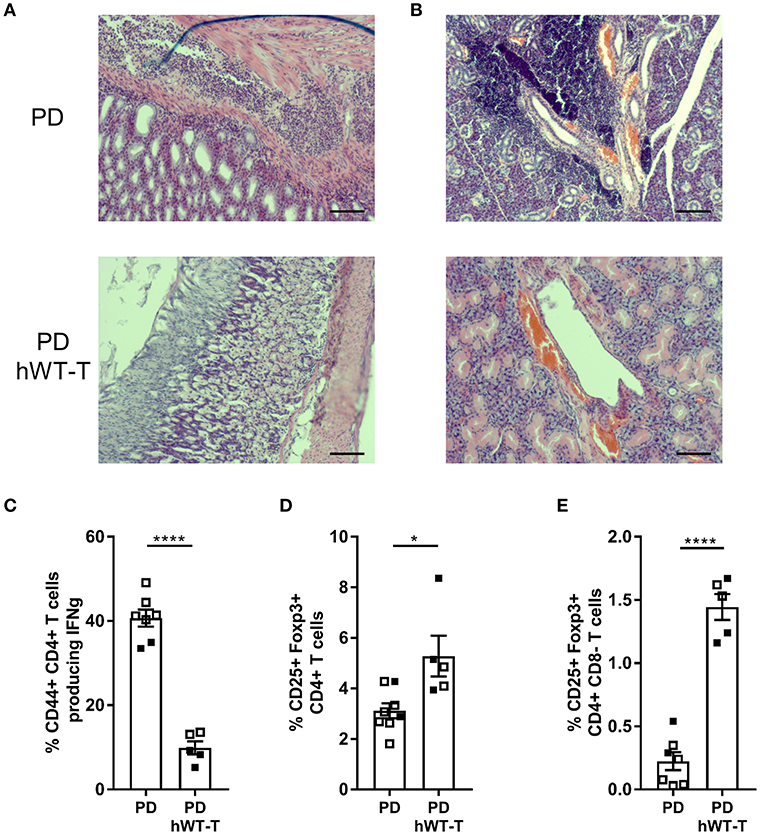
Figure 6. Expression of hMALT1 in T cells of Malt1-PD mice rescues the disease phenotype. (A,B) H&E staining of stomach (A) and salivary gland (B) of Malt1-PD (PD) and Malt1-PD mice expressing wild type human MALT1 in T cells (Malt1PD/- Rosa26LSL-MALT1-WT CD4-Cre = PD hWT-T) mice. (C) Percentage of CD44+CD4+ T cells producing IFN-γ in PD and PD hWT-T mice. (D) Treg frequency (Foxp3+CD25+CD4+CD8− T cells) in thymus of PD and PD hWT-T mice. (E) Treg frequency in cLN of PD and PD hWT-T mice. For (C–E): all data were obtained via flow cytometry and for (E), lymphocytes were stimulated for 4 h with PMA/ionomycin. Data represent two individual experiments: experiment 1 = open squares and experiment 2 = filled squares. Malt1-PD mice: total n = 7 and Malt1-PD hWT-T mice: total n = 5, age 13–18 weeks. The mean ± SEM is indicated on the graphs. The statistical significance between groups was calculated with an unpaired 2 tailed Student's t-test: *p < 0.05 and ****p < 0.0001.
Discussion
MALT1 proteolytic activity plays a key role in both immune cells and non-immune cells (62, 63), and mice expressing a catalytically inactive MALT1 mutant (Malt1-PD mice) were previously shown to suffer from severe autoimmunity (4–6, 47). Conditional genetic models are essential to determine the critical cell type responsible for disease development in Malt1-PD mice. We studied the T cell-intrinsic functions of MALT1 proteolytic activity in the regulation of immune and autoimmune responses by comparing Malt1-PD mice with Malt1-PDT mice that specifically express catalytically inactive MALT1 in T cells. We report that both lines develop similar disease symptoms and autoimmunity, indicating that MALT1 proteolytic activity suppresses autoimmunity in a T cell-intrinsic manner. Autoimmune disease in Malt1-PD mice was previously explained by a disrupted balance between effector CD4+ T cells and the number of Tregs (4, 5, 47). Both Malt1-PD and Malt1-PDT mice have almost no nTregs in their thymus, indicating a T cell-intrinsic role of MALT1 protease activity in thymic Treg development. nTregs inhibit autoreactive T cells to become mature Th1 cells (53, 64). If induced peripheral Tregs cannot compensate for the lack of thymus-derived nTregs, this may explain the Th1-dependent autoimmune phenotype of Malt1-PD and Malt1-PDT mice. Therefore, it might be interesting to investigate whether inducible genetic inactivation of MALT1 proteolytic activity in adult animals results in a similar phenotype. Moreover, less well-known Foxp3-negative regulatory T cell populations (e.g., Tr1 and Th3 cells) (52) may also play a role. Of note, the reduction of Foxp3+ Tregs is less pronounced in Malt1-PDT mice compared to Malt1-PD mice, suggesting that also MALT1 protease-dependent signals from cells not targeted by CD4-Cre contribute to the decrease in Tregs. One non-T cell population that could influence Treg numbers and which is different between Malt1-PD and Malt1-PDT mice are the Breg cells (65, 66).
As Malt1-PD and Malt1-PDT mice develop equally severe disease symptoms, disease development cannot be solely explained by reduced peripheral Treg levels. Therefore, we also assessed the Treg surface expression of CTLA-4, which is a known Treg functionality marker and whose deficiency partially leads to similar disease symptoms as those seen in Malt1-PD and -PDT mice (64, 67–70). In addition, in vivo models for colitis and diabetes demonstrate the importance of CTLA-4 for the Treg inhibitory function (69). It is believed that the inhibitory effect of Tregs occurs via CTLA-4 mediated trans-endocytosis of CD80 and CD86 proteins from antigen presenting cells (71), which impairs CD28 co-stimulation that is required for optimal T cell activation (58, 71). We found that the number of Tregs that expressed CTLA-4 on their surface was much lower in Malt1-PDT mice compared to WT controls. We therefore propose that not the reduced number of Treg per se, but rather impaired Treg functionality is responsible for autoimmune disease development in Malt1-PDT mice. Of note, also the number of effector CD4+ T cells expressing CTLA-4 on their surface was found to be reduced in Malt1-PDT mice compared to controls. CTLA-4 on effector CD4+ T cells can also inhibit T cell responses in a cell extrinsic way, but their suppressive capacities are modest compared to Tregs (72, 73). Nevertheless, reduced CTLA-4 expression on effector CD4+ T cells might also contribute to the disease phenotype observed in our Malt1-PD and -PDT mice.
CTLA-4 expression on T cells depends on mTORC1 activation downstream of TCR stimulation (74). Of interest, mTORC1 activation was recently shown to require MALT1 protease activity via regulation of the glutamine import protein ASCT2 (75, 76). Reduced CTLA-4 expression on Tregs and effector CD4+ T cells from Malt1-PDT mice might therefore reflect reduced mTORC1 activation. Moreover, since CTLA-4 mRNA is directly targeted for degradation by the MALT1 substrates Regnase-1, Roquin-1, and Roquin-2 (37), reduced CTLA-4 expression on Tregs and effector CD4+ T cells from Malt1-PDT mice may also reflect its post-transcriptional regulation by MALT1. Based on our results, we propose that future risk assessments of MALT1 protease inhibition related to autoimmunity could focus on the effect of MALT1 inhibitors on Treg differentiation, maintenance and functionality. It should be mentioned that there is an important difference between genetic and pharmacological approaches to assess the role of MALT1 proteolytic activity in immune homeostasis. In case of genetic inactivation, MALT1 function is disrupted already in the germline (in case of full-body KI mice) or is removed during early T cell differentiation (in case of conditional CD4-Cre KI mice). In contrast, upon pharmacological inhibition with for example small compound inhibitors of MALT1, Tregs are MALT1 protease proficient before the treatment is started, which may result in a different outcome. While the current paper was written, however, several studies using Malt1FL/PD Foxp3-CreTg/+ mice that specifically express catalytically inactive MALT1 in Tregs indicated that post-development loss of MALT1 proteolytic activity in Tregs leads to severe autoimmune disease (77–79), which might raise concerns regarding the safety of pharmacological inhibition of MALT1 protease activity. Based on our results on CTLA-4 and the above mentioned other studies, it would be highly interesting to see whether Foxp3-Cre conditional KO of Regnase-1 and/or Roquin-1 and Roquin-2 can rescue the autoimmune disease in Malt1-PD mice. Noteworthy, a MALT1 protease inhibitor will dampen both effector T cells and Tregs, which can still lead to a different immune homeostasis phenotype compared to the autoimmune phenotype upon Treg-specific loss of MALT1 proteolytic activity. Importantly, there is already indirect evidence indicating that therapeutic targeting of MALT1 protease activity in adult mice with small compound inhibitors is safe. Firstly, extended treatment of mice with the MALT1 protease inhibitor mepazine did not reduce Treg numbers (40). As a counterexample, long-term treatment of psychiatric patients with chlorpromazine, another MALT1 inhibitor, is associated with the induction of autoimmunity markers and occasionally results in a lupus-like disease (80, 81). It is however important to stress that both these phenothiazine compounds also have other MALT1-independent activities that can be responsible for the observed effects (82). Another argument in favor of safety for MALT1 inhibitor treatment in adults is the fact that MALT1 seems to be specifically required for the development of nTregs, which develop at a young age in the thymus (54). A third compelling argument is that only a small amount of functional (WT) Tregs is enough to suppress the autoimmune phenotype of Malt1-PD mice (5). Since therapeutic inhibition of enzymatic activity is never complete and can be adjusted by dosage and treatment regimes, it might be that the remaining functional Tregs are enough to prevent autoimmunity. Nevertheless, a major future challenge will be to generate and characterize inducible genetic models to emulate the effects of long-term inhibitor treatment in adults.
Materials and Methods
Mouse Rosa26LSL-hMALT1-WT Transgenesis
Cloning of RMCE-Targeting Constructs
Plasmids of the cloned genes were deposited in the BCCM/GeneCorner plasmid collection along with detailed descriptions of cloning strategy and plasmid sequence (http://www.genecorner.ugent.be/). Human MALT1 (LMBP 9104, LMBP 9105) was cloned into pENTR221 together with a genotype-specific barcode sequence downstream of the stop codon for genotyping. The ENTR clones were integrated in the pRMCE_DV1 (LMBP 8870) destination vector to generate RMCE targeting vectors for MALT1 (LMBP 9111, LMBP 9112).
RMCE Targeting
RMCE compatible mouse ES-cells [G4 ROSALUC (83)] were cultured in standard ES-cell medium containing 500 ml KO-DMEM (Thermo Fisher Scientific), 15% serum (Hyclone), 100 μM Non-essential amino acids (Thermo Fisher Scientific), 1x Glutamax (Thermo Fisher Scientific), 100 μM β-mercaptothanol (Thermo Fisher Scientific) and 2,000 units LIF/ml (VIB Protein Core). ES-cells were co-transfected with 2.5 μg of a FlpE expressing plasmid (84) and 2.5 μg of targeting vector using lipofectamin 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. G418 selection (200 μg/ml) was applied 48 h after transfection. After 7–10 days, individual G418 resistant, RMCE targeted ES cell clones were picked and further expanded. Correct targeting events were confirmed by Southern blot analysis (5′ probe, EcoRI, wt = 15,5 kb/mutant = 4.5 kb/wtRosaluc allele = 6 kb; EGFP internal probe, KpnI, single integration = 11 kb) and by PCR; the targeted allele generates a band of 560 bp (primers: FW 5′ AAA GTC GCT CTG AGT TGT TAT 3′ and REV 5′ GCG GCC TCG ACT CTA CGA TA 3′). Correctly targeted ES-cells were aggregated with outbred Swiss morula, which were then implanted into pseudopregnant Swiss mice.
Mice
In order to generate conditional Malt1-KO mice, mice were generated from the EUCOMM Malt1tm1a(EUCOMM)Hmgu/+ ES cells (85), which were back-crossed to a germ line Flp deleter (86) to generate Malt1FL/+ mice. These mice were further crossed to the CD4-Cre (87) line and back-crossed to generate Flp-negative T cell-specific Malt1-KO (Malt1FL/FL CD4-Cre) (88–91). The Malt1tm1a(EUCOMM)Hmgu/+ mice were also back-crossed with a germline Cre-line and back crossed to generate Cre-negative co-isogenic Malt1−LacZ/−LacZ full-body KO mice. Malt1PD/+ (the mouse MALT1 C472A mutation that disrupts protease activity) mice were generated in a C57/BL6 background by CRISPR/Cas9 (92) by Cyagen Biosciences Inc. Malt1-PD mice were always maintained as heterozygotes (Malt1PD/+) in breeding to avoid suffering from the autoimmune phenotype. Rosa26LSL-MALT1-WT transgenic mice were crossed with CD4-Cre and subsequently back-crossed with full-body Malt1 KO mice, and later back-crossed to Malt1PD/+ mice to generate rescued Malt1PD/− Rosa26LSL−MALT1−WT CD4-Cre and Malt1PD/PD Rosa26LSL−MALT1−WT CD4-Cre mice where all cells except T cells are MALT1 protease deficient. The transgenes were confirmed by western blot (Supplemental Figure 4) for specific expression of MALT1 only in presence of CD4-Cre using a human-specific rabbit monoclonal anti-MALT1 antibody (EP603Y, Abcam), anti-mouse MALT1 (sc-28246, Santa Cruz) and anti-Cre (69050-3, Merck Millipore). Mice used in these experiments were kept at room temperature, fed ad libitum in an SPF facility (IRC, VIB-UGent). All experiments were done according to the UGent ethical guidelines with the local ethical committee approval number: EC 2015-031.
Genotyping
The Malt1 Flox-allele or KO allele from Eucomm was monitored with the primers MALTcKO-F (GTTTCTCAGGTCTTTAGTTCATGTC), CoMLT-3-R (TATACTCTACATCTCCATGGT), MALTcKO-R (TTGTTTTGCAGATCTCTGCC), and MLT-LacZ-F (TCGCTACCATTACCAGTTGGT) resulting in 280 bp (WT), 400 bp (FL), 345 (KO), or 514 bp (KO-LacZ) PCR products. SNP-specific primers were designed as described in Staal et al. (93), with the allele-specific nucleotides close to the 3′ end of the primer. The Malt1 C472A KI was monitored with the primers F-MALT-KICA-GT (CCCACTCCCAGGATTGTTATATT), R-MALT1-KICA-GT (TGCTCTAGATCCACAGGTGTGGTT), KI-MALT-CA-F (AATGTGTTCCTGTTGGATATGGCCAG), and KI-MALT-WT-R (GAGACATTTTACCTTTTCCGACAC) resulting in 461 bp (allele-independent), 304 bp (PD) and 201 bp (WT) PCR fragments. The Rosa26LSL-MALT1-WT transgenes were genotyped with the primers RMCE-MALT-F (GCAGCTACTTGGTATCAAAGGATCT), RMCE-MALT-R (TTTCCACAACTATCCATGTCGC), generating an allele-independent 896 bp PCR fragment, and the allele-specific primer RMCE-MALT-WT-R (CCAATTTCTGAAAAATAGTGTCCAAC) generating an allele specific band of 537 bp. Flp was detected with the primers Flp-F (TTAGTTCAGCAGCACATGATG) and Flp-R (GGAGGATTTGATATTCACCTG), resulting in a 370 bp PCR fragment. Cre was detected with the primers Cre-F (TGCCACGACCAAGTGACAGCAATG) and Cre-R (AGAGACGGAAATCCATCGCTCG) producing a 374 bp PCR fragment. All genotypings were done using the GoTaq Green Hot Start (Promega) master mix, with a typical PCR program: 5 min 95°C denaturation, 35–40 cycles [30 s 95°C|30 s 52–60°C|60 s 72°C] and 10 min 72°C final elongation.
Flow Cytometry
Data were obtained with a LSRII flow cytometer (BD Biosciences) and FlowJo Software (Treestar, Inc., Ashland, Ore) was used for data analysis.
Detection of Tregs
Cells from thymus and cLN were surfaced stained with Fixable Viability Dye eFluor506 (eBioscience), anti-CD16/CD32 Fc block (clone 2.4G2; BD Biosciences), anti-CD3-eFluor450 (clone 17A2; eBioscience), anti-CD4-FITC (clone GK1.5; BD Biosciences or eBioscience), anti-CD25-PercPcy5.5 (clone PC61; BD Biosciences) for 20 min. Next, cells were permeabilized for 30 min, followed by 30 min of intracellular staining for anti-Foxp3-PE (clone FJK-16s; eBioscience). For the intracellular staining, the Foxp3 buffer set (eBioscience) was used and all incubation steps were done on ice.
Analysis of Effector Markers on Tregs
Cells from cLN cultured in complete medium (RPMI 1640 medium supplemented with 10% FCS, sodium pyruvate, L-glutamine, antibiotics and β-mercaptoethanol) were stimulated with PMA (50 ng/ml) and ionomycin (1 μg/ml) for 4 h at 37°C. The cells were stained as mentioned above, but anti-CD44-APC-efluor780 (clone IM7; eBioscience) and anti-CTLA-4 PE-eFluor610 (clone UC10-4B9; eBioscience) were also included in the surface staining. For the other Treg effector markers, non-stimulated cells from cLN were stained as mentioned above, but anti-CD44-AlexaFluor700 (clone IM7; BD Pharmingen), anti-CD62L-eFluor450 (clone MEL-14; eBioscience) and anti-KLRG1-APC (clone 2F1; eBioscience) or anti-TNFR2-BV421 (clone TR75-89; BD Pharmingen) and anti-ICOS-PE-Cy5 (clone 7E.17G9; eBioscience) were also included in the surface stain.
Analysis of Cytokines by Intracellular Cytokine Staining
Cells from cLN were cultured in complete medium and stimulated with PMA (50 ng/ml), ionomycin (500 ng/ml) and brefeldin A (1 μg/ml) for 4–5 h at 37°C. Stimulated cells were washed, surface stained with anti-CD16/CD32, Fixable Viability Dye eFluor 506, anti-CD3-eFluor450, anti-CD4-FITC, anti-CD44-APCeFluor780, for 20 min. Next, cells were fixed and permeabilized for 30 min using the Foxp3 buffer set, followed by intracellular staining with anti-IFNγ-PE-Cy7 (clone XMG1.2; BD Pharmingen) for 30 min.
Detection of MZ B Cells
Spleen cells were surface stained with Fixable Viability Dye eFluor506 (eBioscience), anti-CD16/CD32 Fc block (clone 2.4G2; BD Biosciences), anti-CD3-PE-Cy5 (145-2C11; Tonbo Biosciences), anti-CD45R/B220 (clone RA3-6B2; BD Biosciences), anti-CD21/CD35-eFluor450 (clone 4E3; eBioscience), and anti-CD23-PE-Cy7 (clone B3B4,eBioscience) for 20 min.
Cytokine Analysis
Peripheral blood samples were collected for serum preparation. The levels of IL-2 (171-G5003M), IL-4 (171-G5005M), IL-6 (171-G5007M), IL-17 (171-G5013M), IFN-γ (171-G5017M), and TNF (171-G5023M) were determined by Bio-Plex (Biorad) according to the manufacturer's conditions. B cells were purified from spleen with the MagniSort Mouse B cell Enrichment Kit (eBioscience), cultured in complete medium (RPMI 1640 medium supplemented with 10% FCS, sodium pyruvate, L-glutamine, antibiotics and β-mercaptoethanol) and stimulated overnight with 20 μM CpG1826 (Invivogen) as described (47). The level of IL-10 (171G5009M) in medium was determined by Bio-Plex (Biorad) according to manufacturer's conditions.
Histology
Stomach, lacrimal gland and salivary gland were fixed with 4% paraformaldehyde and imbedded in paraffin. Sections (5 μm) were stained with haematoxylin and eosin. Images (100× magnification) were acquired with a BX51 discussion microscope (Olympus) with PixeLink camera under 100× magnification.
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
All experiments were done according to the UGent ethical guidelines with the local ethical committee approval number: EC 2015-031.
Author Contributions
AD and JS designed the experiments. AD performed all experiments, except for the weighing and ataxia scoring done by JS, YD, and AD. IS did experiments for MZ B cells. YD and MK assisted with genotyping of mice. The Malt1PD/+ C472A PD mice were generated by MB. TH generated the Rosa26LSL−hMALT1−WT transgenic mice and the Malt1tm1a(EUCOMM)Hmgu/+ mice from EUCOMM ES cells. AD, JS, MB, and RB contributed to the scientific discussion. AD, JS, and RB wrote the manuscript.
Funding
Research in the authors' lab was supported by grants from the Fund for Scientific Research Flanders (FWO), Belgian Foundation Against Cancer; Ghent University Concerted Research Actions (GOA), the VIB Grand Challenges Program (VIB-GC01-C01). AD was supported by a predoctoral fellowship from the IWT. JS was supported by a postdoctoral fellowship from the FWO.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Part of the work was done during a research collaboration between VIB, CD3, and AstraZeneca on MALT1 inhibitors (94).
Acknowledgments
Ulf Sivars and colleagues (Respiratory, Inflammation and Autoimmunity Diseases (RIA) iMed Bioscience, AstraZeneca R&D, Gothenburg, Sweden) for scientific discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01898/full#supplementary-material
References
1. Demeyer A, Staal J, Beyaert R. Targeting MALT1 proteolytic activity in immunity, inflammation and disease: good or bad? Trends Mol Med. (2016) 22:135–50. doi: 10.1016/j.molmed.2015.12.004
2. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. (2003) 19:749–58.
3. Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. (2003) 302:1581–4. doi: 10.1126/science.1090769
4. Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, et al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. (2014) 33:2765–81. doi: 10.15252/embj.201488987
5. Gewies A, Gorka O, Bergmann H, Pechloff K, Petermann F, Jeltsch KM, et al. Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep. (2014) 9:1292–305. doi: 10.1016/j.celrep.2014.10.044
6. Yu JW, Hoffman S, Beal AM, Dykon A, Ringenberg MA, Hughes AC, et al. MALT1 protease activity is required for innate and adaptive immune responses. PLoS ONE. (2015) 10:e0127083. doi: 10.1371/journal.pone.0127083
7. McKinnon ML, Rozmus J, Fung S-Y, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol. (2014) 133:1458–62, 1462.e1–7. doi: 10.1016/j.jaci.2013.10.045
8. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol. (2015) 35:135–46. doi: 10.1007/s10875-014-0125-1
9. Hua Z, Hou B. TLR signaling in B-cell development and activation. Cell Mol Immunol. (2013) 10:103–6. doi: 10.1038/cmi.2012.61
10. De Bruyne M, Hoste L, Bogaert DJ, Van den Bossche L, Tavernier SJ, Parthoens E, et al. A CARD9 founder mutation disrupts NF-κB signaling by inhibiting BCL10 and MALT1 recruitment and signalosome formation. Front Immunol. (2018) 9:2366. doi: 10.3389/fimmu.2018.02366
11. Ferch U, Kloo B, Gewies A, Pfänder V, Düwel M, Peschel C, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. (2009) 206:2313–2320. doi: 10.1084/jem.20091167
12. Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. (2009) 106:19946–51. doi: 10.1073/pnas.0907511106
13. Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. (2012) 22:812–24. doi: 10.1016/j.ccr.2012.11.003
14. Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. (2012) 22:825–37. doi: 10.1016/j.ccr.2012.11.002
15. Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H, et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene. (1999) 18:5785–94.
16. Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. (1999) 93:3601–9.
17. Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, et al. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. (2001) 276:19012–9. doi: 10.1074/jbc.M009984200
18. Streubel B, Lamprecht A, Dierlamm J, Cerroni L, Stolte M, Ott G, et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood. (2003) 101:2335–9. doi: 10.1182/blood-2002-09-2963
19. Hu S, Du M-Q, Park S-M, Alcivar A, Qu L, Gupta S, et al. cIAP2 is a ubiquitin protein ligase for BCL10 and is dysregulated in mucosa-associated lymphoid tissue lymphomas. J Clin Invest. (2006) 116:174–81. doi: 10.1172/JCI25641
20. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol. (2015) 136:819–21e1. doi: 10.1016/j.jaci.2015.03.008
21. Desjardins M, Arjunaraja S, Stinson JR, Dorjbal B, Sundaresan J, Niemela J, et al. A unique heterozygous CARD11 mutation combines pathogenic features of both gain- and loss-of-function patients in a four-generation family. Front Immunol. (2018) 9:2944. doi: 10.3389/fimmu.2018.02944
22. Hulpiau P, Driege Y, Staal J, Beyaert R. MALT1 is not alone after all: identification of novel paracaspases. Cell Mol Life Sci CMLS. (2016) 73:1103–16. doi: 10.1007/s00018-015-2041-9
23. Staal J, Driege Y, Haegman M, Borghi A, Hulpiau P, Lievens L, et al. Ancient origin of the CARD-coiled coil/Bcl10/MALT1-like paracaspase signaling complex indicates unknown critical functions. Front Immunol. (2018) 9:1136. doi: 10.3389/fimmu.2018.01136
24. Sun L, Deng L, Ea C-K, Xia Z-P, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. (2004) 14:289–301. doi: 10.1016/S1097-2765(04)00236-9
25. Lork M, Staal J, Beyaert R. Ubiquitination and phosphorylation of the CARD11-BCL10-MALT1 signalosome in T cells. Cell Immunol. (2018) 340:103877. doi: 10.1016/j.cellimm.2018.11.001
26. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. (2008) 9:263–71. doi: 10.1038/ni1561
27. Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol. (2008) 9:272–81. doi: 10.1038/ni1568
28. Baens M, Bonsignore L, Somers R, Vanderheydt C, Weeks SD, Gunnarsson J, et al. MALT1 auto-proteolysis is essential for NF-κB-dependent gene transcription in activated lymphocytes. PLoS ONE. (2014) 9:e103774. doi: 10.1371/journal.pone.0103774
29. Ginster S, Bardet M, Unterreiner A, Malinverni C, Renner F, Lam S, et al. Two antagonistic MALT1 auto-cleavage mechanisms reveal a role for TRAF6 to unleash MALT1 activation. PLoS ONE. (2017) 12:e0169026. doi: 10.1371/journal.pone.0169026
30. Baens M, Stirparo R, Lampi Y, Verbeke D, Vandepoel R, Cools J, et al. Malt1 self-cleavage is critical for regulatory T cell homeostasis and anti-tumor immunity in mice. Eur J Immunol. (2018) 48:1728–38. doi: 10.1002/eji.201847597
31. Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton J-E, Guzzardi M, et al. Malt1-dependent RelB cleavage promotes canonical NF-κB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA. (2011) 108:14596–601. doi: 10.1073/pnas.1105020108
32. Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Damme PV, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. (2011) 30:1742–52. doi: 10.1038/emboj.2011.85
33. Elton L, Carpentier I, Staal J, Driege Y, Haegman M, Beyaert R. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-κB signaling. FEBS J. (2016) 283:403–12. doi: 10.1111/febs.13597
34. Klein T, Fung S-Y, Renner F, Blank MA, Dufour A, Kang S, et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat Commun. (2015) 6:8777. doi: 10.1038/ncomms9777
35. Douanne T, Gavard J, Bidère N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J Cell Sci. (2016) 129:1775–80. doi: 10.1242/jcs.185025
36. Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-induced cleavage of regnase-1 in CD4+ helper T cells regulates immune activation. Cell. (2013) 153:1036–49. doi: 10.1016/j.cell.2013.04.034
37. Jeltsch KM, Hu D, Brenner S, Zöller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. (2014) 15:1079–89. doi: 10.1038/ni.3008
38. Yamasoba D, Sato K, Ichinose T, Imamura T, Koepke L, Joas S, et al. N4BP1 restricts HIV-1 and its inactivation by MALT1 promotes viral reactivation. Nat Microbiol. (2019) 2019:3. doi: 10.1038/s41564-019-0460-3
39. Afonina IS, Elton L, Carpentier I, Beyaert R. MALT1–a universal soldier: multiple strategies to ensure NF-κB activation and target gene expression. FEBS J. (2015) 282:3286–97. doi: 10.1111/febs.13325
40. Mc Guire C, Elton L, Wieghofer P, Staal J, Voet S, Demeyer A, et al. Pharmacological inhibition of MALT1 protease activity protects mice in a mouse model of multiple sclerosis. J Neuroinflamm. (2014) 11:124. doi: 10.1186/1742-2094-11-124
41. Lim SM, Jeong Y, Lee S, Im H, Tae HS, Kim BG, et al. Identification of β-lapachone analogs as novel MALT1 inhibitors to treat an aggressive subtype of diffuse large B-cell lymphoma. J Med Chem. (2015) 58:8491–502. doi: 10.1021/acs.jmedchem.5b01415
42. Bardet M, Unterreiner A, Malinverni C, Lafossas F, Vedrine C, Boesch D, et al. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol Cell Biol. (2018) 96:81–99. doi: 10.1111/imcb.1018
43. Fontán L, Qiao Q, Hatcher JM, Casalena G, Us I, Teater M, et al. Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J Clin Invest. (2018) 128:4397–412. doi: 10.1172/JCI99436
44. Scott DA, Hatcher JM, Liu H, Fu M, Du G, Fontán L, et al. Quinoline and thiazolopyridine allosteric inhibitors of MALT1. Bioorg Med Chem Lett. (2019) 29:1694–8. doi: 10.1016/j.bmcl.2019.05.040
45. Tran TD, Wilson BAP, Henrich CJ, Staudt LM, Krumpe LRH, Smith EA, et al. Secondary metabolites from the fungus Dictyosporium sp. and their MALT1 inhibitory activities. J Nat Prod. (2019) 82:154–62. doi: 10.1021/acs.jnatprod.8b00871
46. Jacobs KA, André-Grégoire G, Maghe C, Li Y, Thys A, Harford-Wright E, et al. Control of the endo-lysosome homeostasis by the paracaspase MALT1 regulates glioma cell survival. bioRxiv. (2019) 2019:582221. doi: 10.1101/582221
47. Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil-Allaoui I, et al. Deficiency of MALT1 paracaspase activity results in unbalanced regulatory and effector T and B cell responses leading to multiorgan inflammation. J Immunol. (2015) 194:3723–34. doi: 10.4049/jimmunol.1402254
48. Carneiro-Sampaio M, Coutinho A. Early-onset autoimmune disease as a manifestation of primary immunodeficiency. Front Immunol. (2015) 6:185. doi: 10.3389/fimmu.2015.00185
49. Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X Chromosome dose and sex bias in autoimmune diseases: increased 47,XXX in systemic lupus erythematosus and Sjögren's syndrome. Arthritis Rheumatol. (2015) 68:1290–300. doi: 10.1002/art.39560
50. Ramien C, Taenzer A, Lupu A, Heckmann N, Engler JB, Patas K, et al. Sex effects on inflammatory and neurodegenerative processes in multiple sclerosis. Neurosci Biobehav Rev. (2016) 67:137–46. doi: 10.1016/j.neubiorev.2015.12.015
51. Peterson RA. Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol Pathol. (2012) 40:186–204. doi: 10.1177/0192623311430693
52. Lehtimäki S, Lahesmaa R. Regulatory T cells control immune responses through their non-redundant tissue specific features. Front Immunol. (2013) 4:294. doi: 10.3389/fimmu.2013.00294
53. Lin X, Chen M, Liu Y, Guo Z, He X, Brand D, Zheng SG. Advances in distinguishing natural from induced Foxp3+ regulatory T cells. Int J Clin Exp Pathol. (2013) 6:116–23.
54. Brüstle A, Brenner D, Knobbe-Thomsen CB, Cox M, Lang PA, Lang KS, et al. MALT1 is an intrinsic regulator of regulatory T cells. Cell Death Differ. (2015) 24:1214–23. doi: 10.1038/cdd.2015.104
55. Brüstle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. (2012) 122:4698–709. doi: 10.1172/JCI63528
56. Mc Guire C, Wieghofer P, Elton L, Muylaert D, Prinz M, Beyaert R, et al. Paracaspase MALT1 deficiency protects mice from autoimmune-mediated demyelination. J Immunol. (2013) 190:2896–903. doi: 10.4049/jimmunol.1201351
57. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
58. Hou TZ, Qureshi OS, Wang CJ, Baker J, Young SP, Walker LSK, et al. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J Immunol. (2015) 194:2148–59. doi: 10.4049/jimmunol.1401876
59. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci USA. (2010) 107:1524–8. doi: 10.1073/pnas.0910341107
60. Klocke K, Holmdahl R, Wing K. CTLA-4 expressed by FOXP3+ regulatory T cells prevents inflammatory tissue attack and not T-cell priming in arthritis. Immunology. (2017) 152:125–37. doi: 10.1111/imm.12754
61. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
62. Juilland M, Thome M. Holding all the CARDs: how MALT1 controls CARMA/CARD-dependent signaling. Front Immunol. (2018) 9:1927. doi: 10.3389/fimmu.2018.01927
63. Ruland J, Hartjes L. CARD-BCL-10-MALT1 signalling in protective and pathological immunity. Nat Rev Immunol. (2018) 19:118–34. doi: 10.1038/s41577-018-0087-2
64. DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM. CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J Immunol. (2005) 175:7135–42. doi: 10.4049/jimmunol.175.11.7135
65. Wang R-X, Yu C-R, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. (2014) 20:633–41. doi: 10.1038/nm.3554
66. Chien C-H, Chiang B-L. Regulatory T cells induced by B cells: a novel subpopulation of regulatory T cells. J Biomed Sci. (2017) 24:86. doi: 10.1186/s12929-017-0391-3
67. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8.
68. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7.
69. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
70. Walker LSK. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
71. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
72. Corse E, Allison JP. Cutting edge: CTLA-4 on effector T cells inhibits in trans. J Immunol. (2012) 189:1123–7. doi: 10.4049/jimmunol.1200695
73. Wang CJ, Kenefeck R, Wardzinski L, Attridge K, Manzotti C, Schmidt EM, et al. Cutting edge: cell-extrinsic immune regulation by CTLA-4 expressed on conventional T cells. J Immunol. (2012) 189:1118–22. doi: 10.4049/jimmunol.1200972
74. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
75. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
76. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, Shiva S, Kane LP. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal. (2014) 7:ra55. doi: 10.1126/scisignal.2005169
77. Cheng L, Deng N, Yang N, Zhao X, Lin X. Malt1 protease is critical in maintaining function of regulatory T cells and may be a therapeutic target for antitumor immunity. J Immunol. (2019) 2019:ji1801614. doi: 10.4049/jimmunol.1801614
78. Rosenbaum M, Gewies A, Pechloff K, Heuser C, Engleitner T, Gehring T, et al. Bcl10-controlled Malt1 paracaspase activity is key for the immune suppressive function of regulatory T cells. Nat Commun. (2019) 10:2352. doi: 10.1038/s41467-019-10203-2
79. Di Pilato M, Kim EY, Cadilha BL, Prüßmann JN, Nasrallah MN, Seruggia D, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. (2019) 570:112–6. doi: 10.1038/s41586-019-1215-2
80. Zucker S, Zarrabi HM, Schubach WH, Varma A, Derman R, Lysik RM, et al. Chlorpromazine-induced immunopathy: progressive increase in serum IgM. Medicine. (1990) 69:92–100.
81. Abbes W, Imen B, Hanene A, Mouna S, Kamilia K, Zouheir B, et al. Chlorpromazine-induced lupus with circulating anticoagulant. A case report. Eur Psychiatry. (2017) 41:S748–9. doi: 10.1016/j.eurpsy.2017.01.1389
82. Meloni L, Verstrepen L, Kreike M, Staal J, Driege Y, Afonina IS, et al. Mepazine inhibits RANK-induced osteoclastogenesis independent of its MALT1 inhibitory function. Molecules. (2018) 23:E3144. doi: 10.3390/molecules23123144
83. Haenebalcke L, Goossens S, Naessens M, Kruse N, Farhang Ghahremani M, Bartunkova S, et al. Efficient ROSA26-based conditional and/or inducible transgenesis using RMCE-compatible F1 hybrid mouse embryonic stem cells. Stem Cell Rev. (2013) 9:774–85. doi: 10.1007/s12015-013-9458-z
84. Schaft J, Ashery-Padan R, van der Hoeven F, Gruss P, Stewart AF. Efficient FLP recombination in mouse ES cells and oocytes. Genes. (2001) 31:6–10. doi: 10.1002/gene.1076
85. Brown SDM, Moore MW. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome. (2012) 23:632–40. doi: 10.1007/s00335-012-9427-x
86. Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. (2000) 25:139–40. doi: 10.1038/75973
87. Wolfer A, Bakker T, Wilson A, Nicolas M, Ioannidis V, Littman DR, et al. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat Immunol. (2001) 2:235–41. doi: 10.1038/85294
88. Drennan MB, Govindarajan S, Verheugen E, Coquet JM, Staal J, McGuire C, et al. NKT sublineage specification and survival requires the ubiquitin-modifying enzyme TNFAIP3/A20. J Exp Med. (2016) 213:1973–81. doi: 10.1084/jem.20151065
89. Kip E, Staal J, Verstrepen L, Tima HG, Terryn S, Romano M, et al. MALT1 controls attenuated rabies virus by inducing early inflammation and T cell activation in the brain. J Virol. (2018) 92:e02029–17. doi: 10.1128/JVI.02029-17
90. Kip E, Staal J, Tima HG, Verstrepen L, Romano M, Lemeire K, Suin V, et al. Inhibition of MALT1 decreases neuroinflammation and pathogenicity of virulent rabies virus in mice. J Virol. (2018) 2018:18. doi: 10.1128/JVI.00720-18
91. Gilis E, Gaublomme D, Staal J, Venken K, Dhaenens M, Lambrecht S, et al. MALT1-deletion in T-cells protects against the development of auto-immune arthritis, but causes spontaneous osteoporosis. Arthritis Rheumatol. (2019) 2019:e41029. doi: 10.1002/art.41029
92. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. (2013) 153:910–8. doi: 10.1016/j.cell.2013.04.025
93. Staal J, Kaliff M, Bohman S, Dixelius C. Transgressive segregation reveals two Arabidopsis TIR-NB-LRR resistance genes effective against Leptosphaeria maculans, causal agent of blackleg disease. Plant J. (2006) 46:218–30. doi: 10.1111/j.1365-313x.2006.02688.x
Keywords: autoimmunity, inflammation, Treg, Breg, CTLA-4, paracaspase, protease, lymphocyte
Citation: Demeyer A, Skordos I, Driege Y, Kreike M, Hochepied T, Baens M, Staal J and Beyaert R (2019) MALT1 Proteolytic Activity Suppresses Autoimmunity in a T Cell Intrinsic Manner. Front. Immunol. 10:1898. doi: 10.3389/fimmu.2019.01898
Received: 10 May 2019; Accepted: 26 July 2019;
Published: 14 August 2019.
Edited by:
Laurence Morel, University of Florida, United StatesReviewed by:
Ranjeny Thomas, University of Queensland, AustraliaMargot Thome, Université de Lausanne, Switzerland
Copyright © 2019 Demeyer, Skordos, Driege, Kreike, Hochepied, Baens, Staal and Beyaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Staal, amVucy5zdGFhbEBpcmMudmliLXVnZW50LmJl; Rudi Beyaert, cnVkaS5iZXlhZXJ0QGlyYy52aWItdWdlbnQuYmU=
†These authors share last authorship