 Carine El Sissy1
Carine El Sissy1 Jérémie Rosain1
Jérémie Rosain1 Paula Vieira-Martins1
Paula Vieira-Martins1 Pauline Bordereau1
Pauline Bordereau1 Aurélia Gruber1Magali Devriese1Loïc de Pontual4Muhamed-Kheir Taha5Claire Fieschi6,7Capucine Picard2,3
Aurélia Gruber1Magali Devriese1Loïc de Pontual4Muhamed-Kheir Taha5Claire Fieschi6,7Capucine Picard2,3 Véronique Frémeaux-Bacchi1,8*
Véronique Frémeaux-Bacchi1,8*- 1Assistance Publique – Hôpitaux de Paris (AP-HP), Laboratoire d'Immunologie, Hôpital Européen Georges-Pompidou, Paris, France
- 2Paris University, INSERM UMR1163, Imagine Institute, Paris, France
- 3Study Center for Primary Immunodeficiencies (AP-HP), Hôpital Necker-Enfants maladies Hospital, Paris, France
- 4Pediatrics Department, Jean Verdier Hospital, Assistance Publique des Hôpitaux de Paris, Paris 13 University, Bondy, France
- 5Invasive Bacterial Infection and National Reference Center for Meningococci, Pasteur Institut, Paris, France
- 6Department of Clinical Immunology, Hôpital Saint-Louis, Assistance Publique – Hôpitaux de Paris (AP-HP), Paris, France
- 7Inserm U1126, Centre Hayem, Hôpital Saint-Louis, Paris, France
- 8Centre de Recherche des Cordeliers, INSERM, Sorbonne Université, USPC, Université Paris Descartes, Complement and Diseases Team, Paris, France
The complement system is crucial for defense against pathogens and the removal of dying cells or immune complexes. Thus, clinical indications for possible complete complement deficiencies include, among others, recurrent mild or serious bacterial infections as well as autoimmune diseases (AID). The diagnostic approach includes functional activity measurements of the classical (CH50) and alternative pathway (AP50) and the determination of the C3 and C4 levels, followed by the quantitative analysis of individual components or regulators. When biochemical analysis reveals the causal abnormality of the complement deficiency (CD), molecular mechanisms remains frequently undetermined. Here, using direct sequencing analysis of the coding region we report the pathogenic variants spectrum that underlie the total or subtotal complement deficiency in 212 patients. We identified 107 different hemizygous, homozygous, or compound heterozygous pathogenic variants in 14 complement genes [C1Qβ (n = 1), C1r (n = 3), C1s (n = 2), C2 (n = 12), C3 (n = 5), C5 (n = 12), C6 (n = 9), C7 (n = 17), C8 β (n = 7), C9 (n = 3), CFH (n = 7), CFI (n = 18), CFP (n = 10), CFD (n = 2)]. Molecular analysis identified 17 recurrent pathogenic variants in 6 genes (C2, CFH, C5, C6, C7, and C8). More than half of the pathogenic variants identified in unrelated patients were also found in healthy controls from the same geographic area. Our study confirms the strong association of meningococcal infections with terminal pathway deficiency and highlights the risk of pneumococcal and auto-immune diseases in the classical and alternative pathways. Results from this large genetic investigation provide evidence of a restricted number of molecular mechanisms leading to complement deficiency and describe the clinical potential adverse events of anti-complement therapy.
Key Points
– Restricted number of molecular mechanisms leading to complement deficiency.
– meningococcal or pneumococcal infections and auto-immune diseases are adverse events of anti-complement therapy.
Introduction
The complement system is a key component of innate immunity and contributes to the elimination of pathogens, dying host cells and abnormal molecular structures (1, 2). Therefore, complement deficiencies disorders impair the immune system's ability to defend the body against foreign or abnormal cells that invade or attack it (3–5). Severe or multiple infections—mainly meningococcal infections—or severe autoimmune diseases (AID) in particular with childhood onset are the main features associated with complement deficiency (CD) (6–10). The complement is composed of a complex proteolytic cascade of more than 40 soluble and membranous proteins that interact in a various range of function. Inactive circulating complement proteins can be rapidly activated in a cascade fashion by three different activation pathways leading to a common terminal pathway. The classical pathway (CP) that involves the C1qrs complex, C2, and C4 is initiated by the binding of C1q to immune complexes. The activation of the lectin pathway (LP) is mediated by five pattern recognition molecules [as mannose binding lectin (MBL), ficolins, and collectins] that bind carbohydrate on pathogen surface and the MBL-associated serine proteases (MASP) (11). In contrast, the alternative pathway (AP) patrols for pathogen invasion with the constantly generation of the fluid phase C3 convertase complex C3(H2O)Bb formed with the spontaneous hydrolysis of C3, Factor B (FB), and Factor D (FD) that cleaves C3 at low level. Healthy host cells are protected against complement attacks by many regulation factors such as Factor H (FH) and Factor I (FI) in the plasma, CD46, and CD55 on cells and are thus resistant to damage of the low-grade activation (12). On pathogens that lack specific regulators of complement, C3b interacts with FB to form a surface-bound AP C3 convertase (C3bBb). Properdin (FP), a key positive regulator of the AP is required for an efficient C3b feedback loop and amplified AP activation (3). These three different pathways lead to the formation of C3 convertases (alternative and classical C3 convertases) which cleave C3 in the bioactive opsonin C3b and C3a following the rapid formation of the C5 convertase which cleaves C5 into C5a and C5b and induces the first step of the membrane attack complex (MAC) formation. The MAC assembly requires the sequential and irreversible association of complement components C5b, C6, C7, C8 (C8β and C8αγ), and C9. Therefore, the activation of the complement system induces opsonization of C3b and degraded fragments (iC3b/C3dg) of C3 on bacterial surface promoting phagocytosis and the capacity to kill pathogens by the MAC (13). The anaphylatoxins, C3a, and C5a are constantly released during complement activation and induce an inflammatory response (1).
CD are inherited as autosomal recessive traits, except for properdin deficiency that is X-linked recessive and C1 inhibitor deficiency that is inherited as an autosomal dominant pattern. If the MBL deficiency is common, CD are generally rare, with an estimated prevalence of 5% of all primary immunodeficiencies (7). Published data of CD are limited to single case reports (14–19), small case series (20, 21), and few multicenter studies (9, 22, 23), evaluating clinical manifestations. Here, we report the immunological, genetic, and clinical features of the largest worldwide cohort ever described of 212 patients with total or sub-total complement deficiencies.
Methods
Patients
Patients addressed for complement testing often present, amongst others, recurrent microbial infections, unexplained inflammation, auto immune disease (AID), or renal diseases. Patients included in this study came from different French departments of medicine. Complement deficient patients (273 index cases and 48 asymptomatic related individuals) diagnosed between 1988 and 2018 were reported in this study including 75 previously published patients (9, 15, 20, 24–26). Clinical data were available for 84% of the cohort (n = 230) (Table 1). Genetic screening was possible for 212 patients.
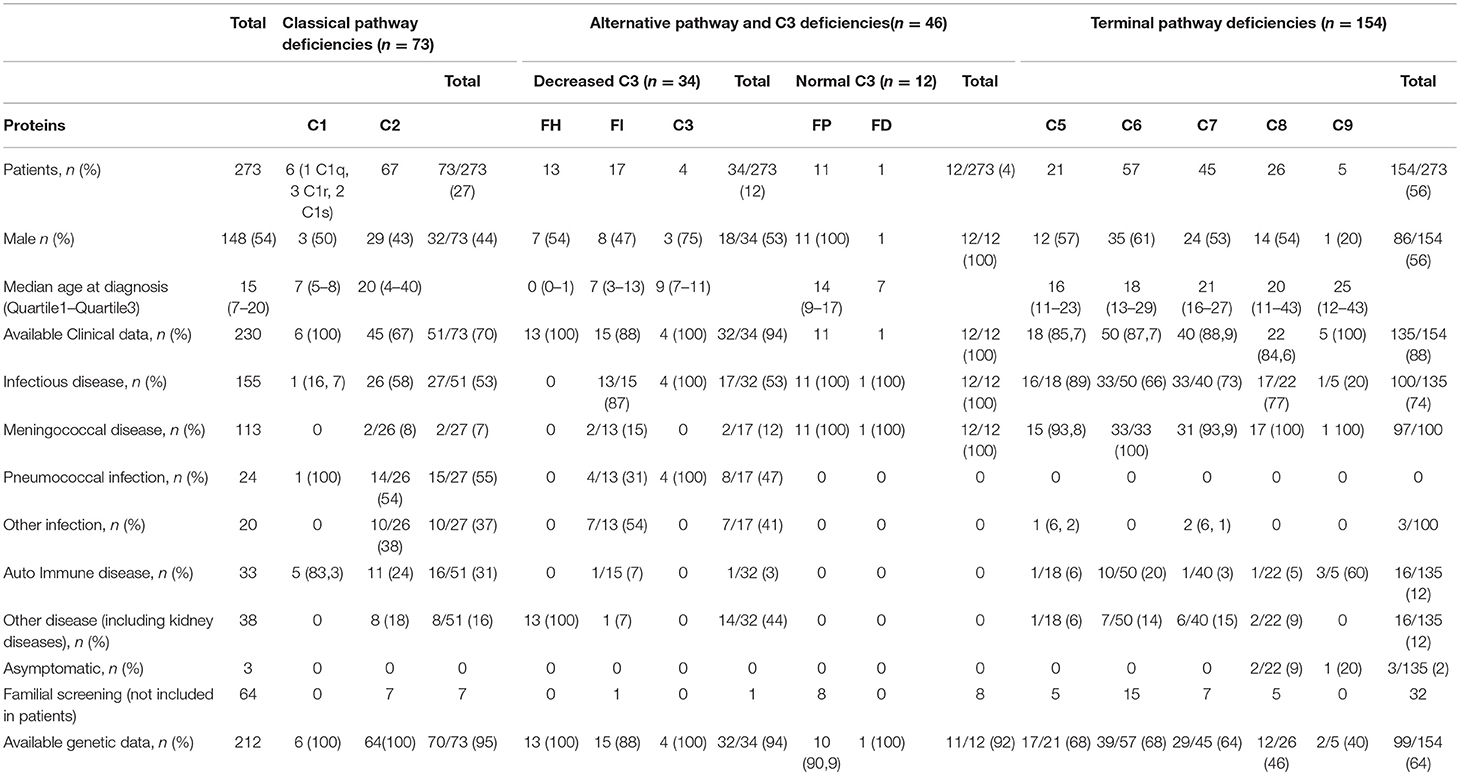
Table 1. Clinical characteristics of patients with complement deficiency.
Complement Assays
A functional complement activity assay of the classical (CH50) and alternative pathway (AP50) hemolytic activities were determined according to standard procedures (27). CH50 and AP50 are based on lysis of Ab-sensitized sheep and rabbit erythrocytes, respectively. To get complexion of the divalent cations calcium and magnesium needed for the activation of the classical pathway, and thus to avoid in vitro complement activation, blood was drawn directly into EDTA-containing tubes. The addition of Veronal-buffered saline (1.8 mM sodium barbiturate, 3.1 mM barbituric acid, and 0.15 M NaCl, pH 7.4, containing 0.75 mM Ca2+ and 2.5 mM Mg2+) restores the hemolytic function of EDTA-plasma samples. According to CH50 and AP50 results, further assays are performed to determine the missing complement component. The quantification of individual complement factors by ELISA or hemolytic assays may uncover quantitative or functional defects in protein. In routine diagnosis, C3 and C4 plasma levels are measured by nephelometry (Siemens, Inc., Newark, DE). Levels of classical complement components (C1q, C1r, C1s), of alternative pathway (FH, FI, FP, FD, FB) and of terminal pathway components (C5, C6, C7, C8, C9) were measured by home-made enzyme-linked immunosorbent assay (ELISA) [antibodies are described in Supplemental Table 1 and in (15, 24, 28)]. Hemolytic FH and FI assays were used to diagnose functional FH and FI deficiencies, respectively. CH50, AP50, and ELISA assays were performed on plasma from EDTA whole blood (plasma EDTA). Results were expressed as a percentage of the mean result obtained on a reference plasma EDTA pool obtained from 190 healthy donors.
Genomic DNA Sequencing
Genetic testing was performed on 212/273 patients (77.7%). Genomic DNA was extracted and purified from whole blood using standard procedures. Screening of type 1 C2 deficiency was performed by polymerase chain reaction (PCR) and gel migration on agarose as previously described (29). Amplifications by PCR (GoTaq DNA Polymerase, Promega, USA) of all exons and flanking splices sites of all the complement components were performed using specific forward and reverse primers (available on request). PCR products were purified (Multiscreen plates, Millipore, USA) and sequenced by the Big Dye terminator cycle sequencing methods using the same forward and reverse primers that for amplification (QiaQuick PCR Purification Kit, Qiagen SA, Hilden, Germany). Sequence analyses were then conducted (Applied Biosystems, Waltham, Massachusetts, USA) and read using Sequencher 5.0 (Gene Codes Corporation, USA). The interpretation of mutations was performed by comparing sequences of patients with the reference sequences exons using Alamut 2.3 (Interactive Biosoftware, France).
Ethics
This study was carried out in accordance with the recommendations of French Ethics Committee (“Comité de Protection des Personnes Ile de France 5,” reference a-11–15, number 1922081) with written informed consent from all subjects.
Results
Screening Strategy
CH50 and AP50 were used to screen deficiency in the complement pathways (30). Classical pathway deficiency (CPD) was suggested by a decreased CH50 activity (below 10% of normal values) with normal AP50 activity. Terminal pathway deficiency (TPD) was defined by undetectable CH50 and AP50 activities (below 10% of normal values). The addition of normal plasma (8 μL) to the complement deficient plasma restored its ability to sustain CH50 activity, whereas the addition of plasma depleted of the deficient component did not. Patients with total TPD had undetectable CH50 and AP50, patients with sub-total deficiencies (SD) in C5, C6, or C7 had a low but detectable CH50 activity. Normal CH50 activity and an undetectable AP50 activity with a normal C3 level suggest a deficiency in one of the alternative pathway components (i.e., FB, FD, or FP). Significantly isolated reduction of C3 can reveal C3, FI or FH deficiencies (C3D, CFID, and CFHD). Quantitative or functional measurements of individual complement components followed by the screening of the all coding regions of the identified genes lead to the final diagnosis.
We diagnosed 273 patients with CD including 73 (27%) patients with CPD, 46 (17%) with AP deficiency (APD), and 154 (56%) with TPD. Patients with APD were divided into patients with low plasma C3 level (C3, Factor I and Factor H deficiencies, n = 34) or normal C3 level (Properdin and Factor D deficiencies, n = 12).
Applying sequencing analysis we identified 107 pathogenic variants in 14 complement genes (Supplemental Table 2 and Supplemental Figures 1–3).
Patients' Characteristics
The median age at diagnosis was 15 (Q1–Q3:7–20). A total of 148 patients were male (148/273, 54.2%). Clinical data were available for 230 patients (230/273, 84.2%). Patients suffered from infectious diseases (n = 155/230, 67.3%), auto-immune disease (n = 33, 14.3%), kidney disease [atypical Hemolytic Uremic Syndrome (aHUS) or C3 Glomerulopathy] (n = 14, 6%). The type of infections was mostly meningococcal (n = 113, 72.9%) or pneumococcal (n = 24, 15.5%) (Table 1).
Genetic and Clinical Characteristic of CPD
We identified 73 patients (73/273; 27%) with an undetectable CH50 activity associated with a low level of one of the components of the C1qrs complex (C1q: n = 1, C1r: n = 3, C1s: n = 2) or C2 protein (n = 67). Clinical data and molecular characterization of the genetic defects were available in 51 (51/73, 69.8%) and 73 (100%) patients, respectively.
Median age of diagnosis was 26 years; the sex ratio (M/F) was 0.44. A total of 27 and 16 patients were diagnosed after infections (n = 27/51; 52.9%) including Streptococcus pneumoniae in 53.8% or with Systemic Lupus Erythematosus (SLE) (16/51; 31.4%), respectively.
C2 deficiency (C2D) was confirmed in 67 patients from 67 kindred. Median age of diagnosis was 20 years; the sex ratio (M/F) was 0.43. Half of C2D patients (26/45, 57.8%) presented with recurrent or severe infections mostly due to S. pneumoniae (n = 14, 53.8%). Two patients (7.7%) presented with meningococcal meningitis. Other clinical manifestations in C2 deficient patients included AID (n = 11, 24.4%), abnormalities in serum immunoglobulin concentrations and various clinical manifestations such as renal, pulmonary or cutaneous manifestations (Table 1).
Genetic analysis revealed that 54 patients carried the homozygous 28 base-pairs deletion (Del28pb) in the C2 gene. Compound heterozygosity (heterozygous Del28pb combined with another rare variant) or two heterozygous rare variants was identified in 9 and 1 patients, respectively. The 12 molecular defects are small deletion or insertion (n = 3), non-sense variant (n = 2), canonical splice sites (n = 2) and missense variant (n = 5) (Table 2, Supplemental Figure 1).
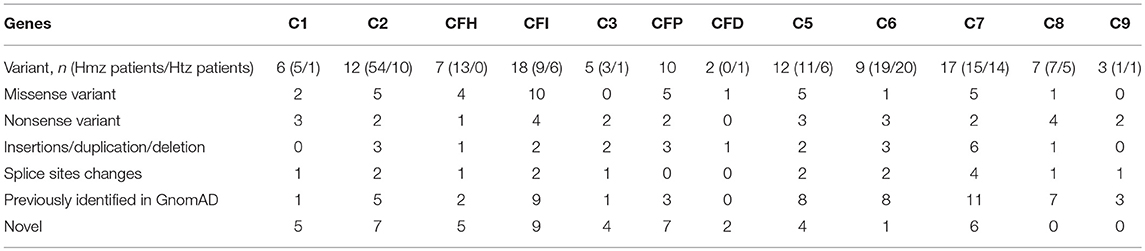
Table 2. Variants spectrum associated with quantitative and functional deficiency.
Six unrelated patients were diagnosed with a complete deficiency of one of the C1qrs complex component. Five patients presented pediatric onset of AID and one had recurrent ear, nose and throat infections. The median age at diagnosis was 7 years (Q1–Q3: 5–8), with a sex ratio (M/F) of 2 (Table 1). Six very rare genetic changes with minor allele frequency <0.0001% were identified in the coding exons of the C1q (n = 1), C1r (n = 3), and C1s (n = 2). Five patients had homozygous variants (1 in C1QB, 2 in C1R, and 2 in C1s) and one C1r deficient patient carried two C1R heterozygous variants (Table 2, Supplemental Figure 1).
Genetic and Clinical Characteristic of APD With Low C3 Level
Among the AP deficient subjects, 34 (12.5% of the whole cohort) suffered from FI (n = 17), FH (n = 13), or C3 (n = 4) deficiency.
Among the 17 patients with FI deficiency, 9 were females (52.9%) and the median age at diagnosis was 7 years. Clinical and molecular mechanisms were available in 15 patients. Thirteen patients (13/15; 86.7%) presented with meningococcal disease (n = 2), S. pneumoniae infection (n = 4) and recurrent respiratory infections (n = 7). The FI level was within the normal range (n = 1), below the normal range (n = 2) and undetectable (n = 13). In all patients' plasma FH was below the normal ranges.
Patients carry two copies of the same rare variant (n = 9) or two rare variants (n = 6) (Table 2, Figure 1, Supplemental Figure 2). The molecular basis was missense (n = 10), nonsense (n = 4) variants, insertions/duplication (n = 2) or splice sites changes (n = 2) (Supplemental Figure 2). Nine out the 17 variants were referenced with a minor allele frequency below 0.01 and 8 were not reported. All were predicted to be pathogenic using Polyphen-2 (data not shown). Two variants (p.Ile340Thr; p.Asp524Val) had no effect on FI synthesis but caused a defect of FI activity (not effective in inactivating membrane-bound C3b) (31, 32). Thirteen unrelated patients were diagnosed with a complete FH deficiency. All patients had low C3 levels secondary to alternative pathway consumption (Min–Max: 40–234 mg/L). Mean CH50 activity of these patients was 33% (<10 to 79%). FH level was below 0.1% of normal values in 4 cases and comprised between 2 and 12% of normal values in 8 cases. One patient had a normal FH level (data not shown).
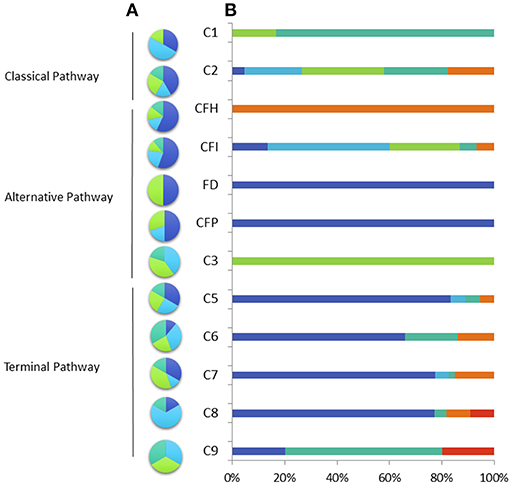
Figure 1. Percentage of variant type (A) nonsense (light blue), missense (dark blue), deletion or insertion (green), splice site mutation (blue green) and clinical phenotype (B) in the French cohort of patients with total and subtotal deficiency. Invasive meningococcal disease (dark blue), other infection (light blue), auto-immune disease (blue green), renal disease (orange), pneumococcal infection (green) other affection (red).
The age at diagnosis was below 1 year in 10 out the 13 cases. All patients had a renal disease, 10 patients presented with atypical aHUS (76.9%) and 3 with C3 glomerulopathy (C3G) (23.1%). All patients presented variants at the homozygous state (Table 2, Supplemental Figure 2). Half of the cases carry the small deletion (c.3693_3696delATAG p.*1232Ilefs*38) and were from North African descends.
Four unrelated patients were found to have a complete deficiency in C3 (Table 1). All presented with S. pneumoniae infections (Table 1, Figure 1). Three patients presented with homozygous variants and one was compound heterozygous (Supplemental Figure 2).
Genetic and Clinical Characteristic of APD With Normal C3 Level
Among the AP deficient subjects, 12 (4.4% of the whole cohort) presented FP deficiency (n = 11) or FD deficiency (n = 1). All these patients presented with Neisseria meningitidis infections.
X-linked FP deficiency was identified in 11 male patients from 11 unrelated families including 9 cases of FP type 1 deficiency (FP levels below 0.3 μg/ml) and 2 cases of FP type II deficiency (FP level between 2 and 5 μg/ml). Median age of diagnosis was 14 years, ranging from 1 to 23 years. Eight asymptomatic family members were diagnosed when the familial segregation was performed. The screening of the molecular genetic was performed in 10 out the 11 pedigrees. The genetic changes were missense variants (n = 5) non-sense variant (n = 2), or small deletion resulting in frameshift (n = 3) (Table 2, Supplemental Figure 2).
We here report the fifth patient with a complete FD deficiency (17, 33–35) (Supplemental Figure 4A). The patient presented at 3 years old a purpura fulminans successfully treated by cefotaxime. Diagnosis of N. meningitidis infection was confirmed by PCR on cerebrospinal fluid which identified a group B strain from clonal complex 41/44 (B:41/44). Assessment of the complement system was performed at the age of 7 years old. CH50 activity was normal but he had undetectable AP50 activity (<10%). FP and FB levels were normal. Addition of purified FD (CompTech) at the physiological concentration of 2 μg/ml restored the AP50 activity (Supplemental Figure 4B). Genetic study of CFD identified one heterozygous frameshift mutation (c.677–678delinsTTCT) and one heterozygous missense rare variant (c.653T>C, p.Leu218Pro) (Supplemental Figure 4C). Each allele was found in one parent, allowing to conclude that the alleles are on different chromosomes. The Leu218 residue is buried in a beta-strand (Supplemental Figure 4D) and is located between the p.Ser208 directly involved in the serine-protease activity of FD and the self-inhibitory loop of FD (p.Val221 to p.Val227, Supplemental Figure 4E).
Genetic and Clinical Characteristic of TPD
A total of 139 patients were diagnosed with complete TPD: C5D (n = 20, 14%), C6D (n = 52, 36.8%), C7D (n = 37, 26.2%), C8D (n = 26, 18.4 %), C9D (n = 5; 3.5%), and 15 with SD: C5SD (n = 1/15, 7%), C6SD (n = 4, 27%), C7SD (n = 10, 66%). Patients with available clinical data (n = 135) presented with N. meningitidis infections in 72% (n = 97/135) or with AI diseases in 12% (n = 16/135). The screening of the genetic abnormalities was performed on 99 patients (99/154 = 64%). A total of 53 and 47 patients carried homozygous variants (53/99 = 53%) or two heterozygous (47/99 = 47%) variants.
Pathogenic variants were identified in the coding exons or flanking splice sites of the C5 (n = 11), C6 (n = 9), C7 (n = 17), C8 b chain (n = 7), and C9 (n = 3) (Figure 1, Supplemental Figure 3). The pathogenic variants were missense (n = 11), nonsense (n = 14), small deletions (n = 12) or located in splice sites (n = 10). We identified 15 recurrent mutations accounted for 78% of the pathogenic alleles (153/197 alleles) (Table 3). The variant allele frequencies ranged from 0.0008 to 0.23% and are variable according to the geographical origin. Four of these variants: (c.960_962delCAA p.Asn320del in C5 gene; c.1879delG p.Asp627Thrfs*4 in C6 gene; c.1135G>C p.Gly379Arg in the C7 gene, and c.1282C>T p.Arg428* in C8B gene) accounted for 41% of the molecular defects (80/197).
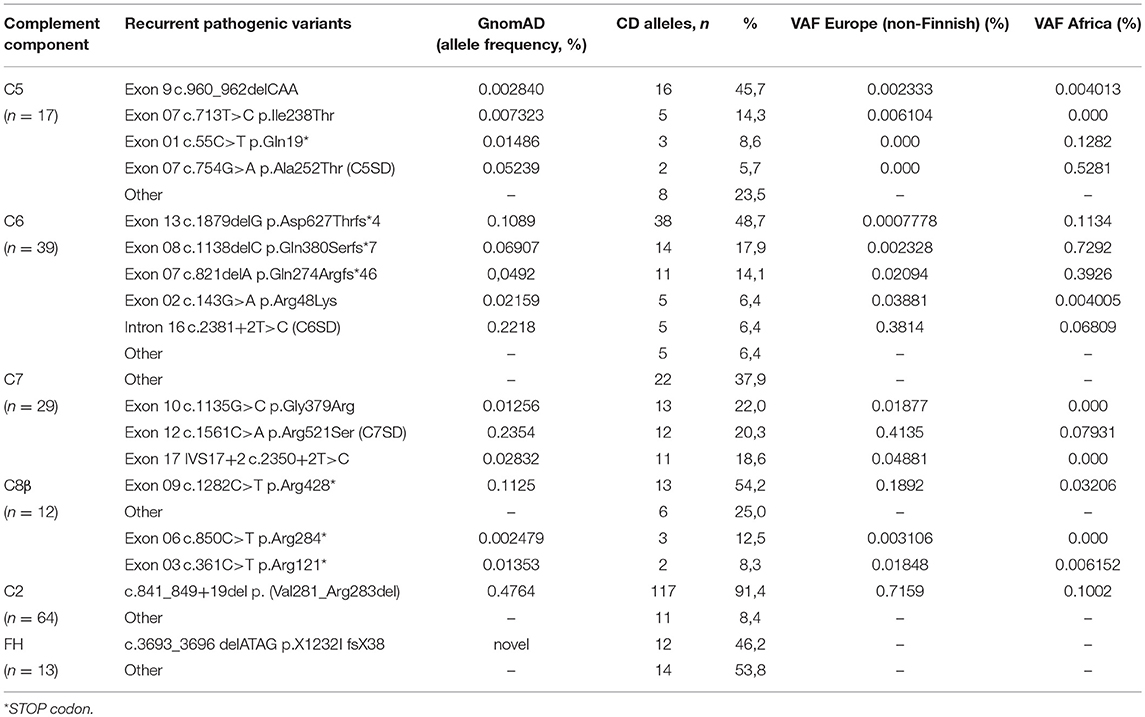
Table 3. Recurrent Pathogenic rare variant identified in the cohort.
SD is defined by the presence of low but detectable and functional residual complement components. TP subtotal deficiency of C5 (C5 level: 2%), C6 (C6 level: 2%), and C7 (C7 level ranging from 0.1 to 1%) were found in 15 patients. The molecular alterations of the SD were the C5 missense variant p.Ala252Thr, the C6 splice site c.2381+2T>C variant and the C7 missense variant p.Arg521Ser. Compound heterozygosity (combined SD and D alleles) or two SD variants were identified in 11 (C6SD/C6D, n = 3; C7SD/C7D, n = 8) and 4 patients (C5SD/C5SD, n = 1; C6SD/C6SD, n = 1; C7SD/C7SD, n = 2) respectively. The C5SD patient who carry the homozygous p.Ala252Thr variant presented with a normal AP50 hemolytic activity.
Discussion
We here report the spectrum of complement genes pathogenic variants in the largest worldwide cohort of patients with subtotal or total complement deficiency.
Except for FP, we posited autosomal recessive transmission of the abnormal allele in these families and sought homozygous or compound heterozygous variants within the same gene. Variant frequency in the general population is a key criterion used in the clinical interpretation of sequence variants. As strong disease-predisposing alleles are likely to be deleterious, we focused on rare variants with a minor allele frequency below 0.1%. In our study a total of 48 variants (48/107, 45%) were not found in the Genome Aggregation Database (gnomAD) that has the ability to identified ultra-rare variant. However, 9 variants (9/48, 19%) not detected in gnomAD, have been already reported in patients with CD from unrelated families, including 5 variants (5/10, 50%) identified in FP deficient patients suggesting a common ancestral origin for these groups of families or a mutational hotspot (36–38).
Interestingly, we showed that half of the pathogenic variants identified in our cohort (59/107, 55%) were previously identified in healthy control but were overrepresented in CD patients. Deleterious variants in complement genes are rare, but the incidence varies considerably within populations. C7 deficiency caused by the pathogenic variant p.Gly379Arg is reported in 1% of the Israeli Moroccan Jewish population (39). C6 deficiency has a high prevalence in western Cape South Africans and in African Americans (40, 41) and the C5 variant p.Ala252Thr leading to subtotal C5 deficiency is prevalent in sub–Saharan Africa (22). In our study, we identified 15 recurrent deleterious variants in genes coding for the lytic complex. The pathogenic variant (c-960–962delCAA) found in half of the patients with C5 deficiency in our cohort has been identified in North-African populations, suggesting that this pathogenic allele may have a higher incidence within this restrictive geographic area (21, 42). Three changes of the C7 gene (p.Gly379Arg, p.Arg521Ser, and 5′ splice donor site of intron 17) with a significantly higher frequency of the variant allele frequency in a European rather than in an African population account for 65.5% of the C7 molecular defects identified in our cohort (24, 39). Approximately 1% of Western European healthy individuals, exhibit the pathogenic allele in C2 gene caused by a 28-base pair genomic deletion. This deletion causes skipping of exon 6 during RNA splicing, resulting in generation of a premature termination codon (29). Our study showed that 90% of the patients with complete C2D carried this molecular defect. Altogether, we showed that half of the CD are explained by a restricted number of molecular abnormalities.
We identified 3 overexpressed variants in the C6 gene (63/78 C6D alleles) reported with a higher frequency in healthy donor from the Black African communities with an estimated frequency ranging from 0.1 to 1% when compared to the European population (41). Many countries outside Africa have been a common destination for African immigrants over the years. All these “at risk variants” can be spread to other countries and might have arisen spontaneously in one allele in an individual many generations ago. The higher risk of invasive meningococcal disease (IMD) in patients with TPD have been extensively described (43). These patients usually develop IMD with bacterial isolates belonging to unusual phenotypes and genotypes (9). The high frequency of deficient alleles in individuals of African descent leading to IMD could impact medical care and genetic counseling in the general population. Indeed, a selective screening of the 3 most frequent pathogenic variants could be a reasonable option to identify C6–deficient individuals requiring long term protection. All these observations highly suggest that the identified variants are pathogenic and shows the growing trend to identifying pathogenic variants previously restricted to single, ethnically isolated regions in many different ethnic groups worldwide.
The majority of variants was non-sense (n = 28) or small deletion/insertion/duplication (n = 23) variants that possibly impaired transcription or translation. We identified 4 missense mutations leading to SD in C5 (p.Ala252Thr) (26), C7 (p.Arg521Ser) (20), and in FP (p.Glu244Lys and p.Arg102Trp (26). Interestingly, we identified 38 missense variants highly suggestive to be responsible for CD. Variants can either prevent synthesis of a protein resulting in quantitative deficiency or can lead to impaired protein synthesis resulting in functional deficiency. Three missense variants in CFH gene affect conserved cysteine residues characteristic of SCR modules and therefore block the normal secretion of the protein as previously demonstrated and explain the undetectable FH in patient's plasma (44). Three rare genetic variants in CFI (p.Val152Met, p.Gly162Asp, p.Ala258Thr) have been found in patients with age-macular degeneration and in aHUS and low serum FI antigenic levels (45). Our data confirm that these 3 variants impair the secretion and lead to FID. An original finding in this study is the identification of 4 functional deficient variants, one in C1QB (p.Gly90Ser) (25), one in CFH (p.Arg53Cys) (46), and two in CFI (p.Ile340Thr, p.Asp524Val) (47, 48). The structural and functional consequences of the rare missense variant remain unexplored in more than half of the newly identified variant. Bioinformatics prediction using structure function maps is useful to determine the likely consequences of the variants. All missense variants identified in the deficient patients are classified probably damaging by Polyphen-2 (Supplemental Table 3). Altogether, except in few cases, CD are secondary to quantitative complement deficiency assessed by the lack of detectable protein level. Further studies using recombinant proteins expression system are needed to confirm these hypotheses.
Recently, an increasing number of drugs targeting several complement proteins are being evaluated (49) and clinical data collected from studies could help predict the side effects of these complement-targeted drugs. Our study confirmed that only a restricted set of pyogenic bacteria, N. meningitidis and S. pneumoniae appear to be prominent in patients with CD. We identified 24 patients with documented pneumococcal infection, among whom 58.3% (n = 14) presented C2D, 16.7% (n = 4) complete C3D and 20.8% (n = 5) CFID, none presented with TPD. For the 2005–2015 period, the incidence rate of IMD in France ranged from 0.78 and 1.23 per 100.000 inhabitant with 5,772 cases of IMD that were reported, of which 4,090 (71%) of cases among <1 to 24 years old (mean of 409 cases per year for this population) (50). During our study period, the number of patients with TP deficiency has ranged from 2 to 5 cases per year. If considering the median age of complement deficiency diagnosis, around 1% of the patients that presented with IMD had a TP deficiency confirming the high risk of meningococcal infection in individuals without functional lytic complexes. Interestingly patients with TP, FD, and FP deficiencies are associated with IMD but C3D and FID patients suffered of pneumococcal infections. Although frequently asymptomatic, we confirmed that homozygous C2D are associated with severe infections and AID (23). Surprisingly half of the patients with C6D were found to have an AID but no IMD. As our series of patients is small and the analysis is based only on index cases, we cannot exclude a fortuitous association. Therefore, AID could develop as a side effect of treatment with new drugs targeting the C6 and should be monitored.
Current complement analysis goes far beyond the determination of CH50, C3, C4, and AP50 (30, 51). Abnormal test results were followed up by quantitative immunochemical measurements of individual complement components following by the screening of the all coding regions of the identified genes. MBL deficiency has been associated, although not conclusively, with increased susceptibility to several infections (52). However, alleles that confer MBL deficiency are common with a frequency up to 30%, which suggests a redundant role of the lectin pathway in host defense (53). Therefore, we do not perform the screening of MBL deficiency in the systematic exploration of the immunity in clinical practice.
Despite phenotypic differences, simple and routine exploration can reveal all CD, but the genetic characterization confirms the diagnosis. The presence of pathogenic variants frequent in the African population provides an opportunity to identified highly susceptible individuals for IMD. Our work supports the recommendation to systemically explore patients with IMD with atypical meningococcal isolates for CD and particularly for TPD. Our data provide strong support that complement inhibition confers high susceptibility to Neisseria and pneumococcal infections but also to AID and strategies to mitigate this risk are mandatory.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
Conception and design: VF-B. CE, JR, PV-M, PB, AG, and MD carried out the complement assessment and the sequencing analysis. Provision of study materials and patients: LdP, CF, CP, and M-KT. Data collection and assembly: VF-B and CE. Data analysis and interpretation: VF-B, CE, JR, M-KT, CF, and CP. Manuscript: VF-B and CE. Final approval of manuscript: all authors.
Funding
This work was supported by grants from the Délégation Régionale à la Recherche Clinique, Assistance Publique – Hôpitaux de Paris (Programme Hospitalier de Recherche Clinique (AOM08198), by the EU FP7 Grant 2012-305608 (EURenOmics), the Fondation Du Rein (FRM, Prix 2012 FDR), the Association pour l'Information et la Recherche dans les maladies Rénales génétiques (AIRG France).
Conflict of Interest Statement
VF-B received fees from Alexion Pharmaceuticals for invited lectures and/or board membership. VF-B is the recipient of a research grant from Alexion Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to acknowledge physicians who participated to the study: Lubka Roumenina, Marie-Agnès Dragon-Durey, Lionel Galicier, Brigitte Bader Meunier, Nizar Mahlaoui, Despina Moshous, and Felipe Suarez. The authors thank J. Blouin and N. Poulain for their excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01936/full#supplementary-material
References
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
3. Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence. (2014) 5:98–126. doi: 10.4161/viru.26515
4. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
5. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
6. Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. (1991) 4:359–95. doi: 10.1128/CMR.4.3.359
7. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. (2014) 61:110–17. doi: 10.1016/j.molimm.2014.06.030
8. Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis. (2014) 59:244–51. doi: 10.1093/cid/ciu274
9. Rosain J, Hong E, Fieschi C, Vieira Martins P, El Sissy C, Deghmane AE, et al. Strains responsible for invasive meningococcal disease in patients with terminal complement pathway deficiencies. J Infect Dis. (2017) 215:1331–8. doi: 10.1093/infdis/jix143
10. Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. (2016) 7:55. doi: 10.3389/fimmu.2016.00055
11. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. (2016) 274:74–97. doi: 10.1111/imr.12468
12. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. (2016) 274:152–71. doi: 10.1111/imr.12475
13. Heesterbeek DA, Bardoel BW, Parsons ES, Bennett I, Ruyken M, Doorduijn DJ, et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. Embo J. (2019) 38:e99852. doi: 10.15252/embj.201899852
14. Westra D, Kurvers RA, van den Heuvel LP, Wurzner R, Hoppenreijs EP, van der Flier M, et al. Compound heterozygous mutations in the C6 gene of a child with recurrent infections. Mol Immunol. (2014) 58:201–5. doi: 10.1016/j.molimm.2013.11.023
15. Dragon-Durey MA, Quartier P, Fremeaux-Bacchi V, Blouin J, de Barace C, Prieur AM, et al. Molecular basis of a selective C1s deficiency associated with early onset multiple autoimmune diseases. J Immunol. (2001) 166:7612–16. doi: 10.4049/jimmunol.166.12.7612
16. Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. (2001) 369:1667–9. doi: 10.1056/NEJMc1306326
17. Sprong T, Roos D, Weemaes C, Neeleman C, Geesing CL, Mollnes TE, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. (2006) 107:4865–70. doi: 10.1182/blood-2005-07-2820
18. Helminen M, Seitsonen S, Jarva H, Meri S, Jarvela IE. A novel mutation W388X underlying properdin deficiency in a Finnish family. Scand J Immunol. (2012) 75:445–8. doi: 10.1111/j.1365-3083.2012.02674.x
19. Moya-Quiles MR, Bernardo-Pisa MV, Martinez P, Gimeno L, Bosch A, Salgado G, et al. Complement component C6 deficiency in a Spanish family: implications for clinical and molecular diagnosis. Gene. (2013) 521:204–6. doi: 10.1016/j.gene.2013.03.027
20. Rameix-Welti MA, Regnier CH, Bienaime F, Blouin J, Schifferli J, Fridman WH, et al. Hereditary complement C7 deficiency in nine families: subtotal C7 deficiency revisited. Eur J Immunol. (2007) 37:1377–85. doi: 10.1002/eji.200636812
21. Colobran R, Franco-Jarava C, Martin-Nalda A, Baena N, Gabau E, Padilla N, et al. Novel mutations causing C5 deficiency in Three North-African families. J Clin Immunol. (2016) 36:388–96. doi: 10.1007/s10875-016-0275-4
22. Franco-Jarava C, Comas D, Orren A, Hernandez-Gonzalez M, Colobran R. Complement factor 5 (C5) p.A252T mutation is prevalent in, but not restricted to, sub-Saharan Africa: implications for the susceptibility to meningococcal disease. Clin Exp Immunol. (2017) 189:226–31. doi: 10.1111/cei.12967
23. Jonsson G, Truedsson L, Sturfelt G, Oxelius VA, Braconier JH, Sjoholm AG. Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine. (2005) 84:23–34. doi: 10.1097/01.md.0000152371.22747.1e
24. Dragon-Durey MA, Fremeaux-Bacchi V, Blouin J, Barraud D, Fridman WH, Kazatchkine MD. Restricted genetic defects underlie human complement C6 deficiency. Clin Exp Immunol. (2003) 132:87–91. doi: 10.1046/j.1365-2249.2003.02099.x
25. Roumenina LT, Sene D, Radanova M, Blouin J, Halbwachs-Mecarelli L, Dragon-Durey MA, et al. Functional complement C1q abnormality leads to impaired immune complexes and apoptotic cell clearance. J Immunol. (2011) 187:4369–73. doi: 10.4049/jimmunol.1101749
26. Pedersen DV, Roumenina L, Jensen RK, Gadeberg TA, Marinozzi C, Picard C, et al. Functional and structural insight into properdin control of complement alternative pathway amplification. Embo J. (2017) 36:1084–99. doi: 10.15252/embj.201696173
28. Roumenina LT, Loirat C, Dragon-Durey MA, Halbwachs-Mecarelli L, Sautes-Fridman C, Fremeaux-Bacchi V. Alternative complement pathway assessment in patients with atypical HUS. J Immunol Methods. (2011) 365:8–26. doi: 10.1016/j.jim.2010.12.020
29. Johnson CA, Densen P, Hurford RK Jr, Colten HR, Wetsel RA. Type I human complement C2 deficiency. A 28-base pair gene deletion causes skipping of exon 6 during RNA splicing. J Biol Chem. (1992) 267:9347–53.
30. Ricklin D, Barratt-Due A, Mollnes TE. Complement in clinical medicine: clinical trials, case reports and therapy monitoring. Mol Immunol. (2017) 89:10–21. doi: 10.1016/j.molimm.2017.05.013
31. Haerynck F, Stordeur P, Vandewalle J, Van Coster R, Bordon V, De Baets F, et al. Complete factor I deficiency due to dysfunctional factor I with recurrent aseptic meningo-encephalitis. J Clin Immunol. (2013) 33:1293–301. doi: 10.1007/s10875-013-9944-8
32. Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. (2010) 77:339–49. doi: 10.1038/ki.2009.472
33. Hiemstra PS, Langeler E, Compier B, Keepers Y, Leijh PC, van den Barselaar MT, et al. Complete and partial deficiencies of complement factor D in a Dutch family. J Clin Invest. (1989) 84:1957–61. doi: 10.1172/JCI114384
34. Biesma DH, Hannema AJ, van Velzen-Blad H, Mulder L, van Zwieten R, Kluijt I, et al. A family with complement factor D deficiency. J Clin Invest. (2001) 108:233–40. doi: 10.1172/JCI12023
35. Sng CCT, O'Byrne S, Prigozhin DM, Bauer MR, Harvey JC, Ruhle M, et al. A type III complement factor D deficiency: structural insights for inhibition of the alternative pathway. J Allergy Clin Immunol. (2018) 142:311–14 e316. doi: 10.1016/j.jaci.2018.01.048
36. Truedsson L, Westberg J, Fredrikson GN, Sjoholm AG, Kuijper EJ, Fijen CA, et al. Human properdin deficiency has a heterogeneous genetic background. Immunopharmacology. (1997) 38:203–6. doi: 10.1016/S0162-3109(97)00087-8
37. Fijen CA, van den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, et al. Properdin deficiency: molecular basis and disease association. Mol Immunol. (1999) 36:863–7. doi: 10.1016/S0161-5890(99)00107-8
38. van den Bogaard R, Fijen CA, Schipper MG, de Galan L, Kuijper EJ, Mannens MM. Molecular characterisation of 10 Dutch properdin type I deficient families: mutation analysis and X-inactivation studies. Eur J Hum Genet. (2000) 8:513–18. doi: 10.1038/sj.ejhg.5200496
39. Halle D, Elstein D, Geudalia D, Sasson A, Shinar E, Schlesinger M, et al. High prevalence of complement C7 deficiency among healthy blood donors of Moroccan Jewish ancestry. Am J Med Genet. (2001) 99:325–7. doi: 10.1002/ajmg.1183
40. Hobart MJ, Fernie BA, Fijen KA, Orren A. The molecular basis of C6 deficiency in the western Cape, South Africa. Hum Genet. (1998) 103:506–12. doi: 10.1007/s004390050858
41. Orren A, Owen EP, Henderson HE, van der Merwe L, Leisegang F, Stassen C, et al. Complete deficiency of the sixth complement component (C6Q0), susceptibility to Neisseria meningitidis infections and analysis of the frequencies of C6Q0 gene defects in South Africans. Clin Exp Immunol. (2012) 167:459–71. doi: 10.1111/j.1365-2249.2011.04525.x
42. Owen EP, Wurzner R, Leisegang F, Rizkallah P, Whitelaw A, Simpson J, et al. A complement C5 gene mutation, c.754G>A:p.A252T, is common in the Western Cape, South Africa and found to be homozygous in seven percent of Black African meningococcal disease cases. Mol Immunol. (2015) 64:170–6. doi: 10.1016/j.molimm.2014.11.010
43. Tedesco F, Nurnberger W, Perissutti S. Inherited deficiencies of the terminal complement components. Int Rev Immunol. (1993) 10:51–64. doi: 10.3109/08830189309051171
44. Ault BH, Schmidt BZ, Fowler NL, Kashtan CE, Ahmed AE, Vogt BA, et al. Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem. (1997) 272:25168–75. doi: 10.1074/jbc.272.40.25168
45. Kavanagh D, Yu Y, Schramm EC, Triebwasser M, Wagner EK, Raychaudhuri S, et al. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet. (2015) 24:3861–70. doi: 10.1093/hmg/ddv091
46. Merinero HM, Garcia SP, Garcia-Fernandez J, Arjona E, Tortajada A, Rodriguez de Cordoba S. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int. (2018) 93:470–81. doi: 10.1016/j.kint.2017.07.015
47. Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. (2004) 41:e84. doi: 10.1136/jmg.2004.019083
48. Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol. (2008) 45:95–105. doi: 10.1016/j.molimm.2007.05.004
49. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
50. Parent du Chatelet I, Deghmane AE, Antona D, Hong E, Fonteneau L, Taha MK, et al. Characteristics and changes in invasive meningococcal disease epidemiology in France, 2006-2015. J Infect. (2017) 74:564–74. doi: 10.1016/j.jinf.2017.02.011
51. Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of Serological Complement Biomarkers in Disease. Front Immunol. (2018) 9:2237. doi: 10.3389/fimmu.2018.02237
52. Casanova JL, Abel L. Human Mannose-binding lectin in immunity: friend, foe, or both? J Exp Med. (2004) 199:1295–9. doi: 10.1084/jem.20040537
Keywords: complement, genetic variants, meningococcal infections, deficiency, auto immune diseases
Citation: El Sissy C, Rosain J, Vieira-Martins P, Bordereau P, Gruber A, Devriese M, de Pontual L, Taha M-K, Fieschi C, Picard C and Frémeaux-Bacchi V (2019) Clinical and Genetic Spectrum of a Large Cohort With Total and Sub-total Complement Deficiencies. Front. Immunol. 10:1936. doi: 10.3389/fimmu.2019.01936
Received: 07 May 2019; Accepted: 30 July 2019;
Published: 08 August 2019.
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Kristina N. Ekdahl, Uppsala University, SwedenLourdes Isaac, University of São Paulo, Brazil
Copyright © 2019 El Sissy, Rosain, Vieira-Martins, Bordereau, Gruber, Devriese, de Pontual, Taha, Fieschi, Picard and Frémeaux-Bacchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Véronique Frémeaux-Bacchi, dmVyb25pcXVlLmZyZW1lYXV4LWJhY2NoaUBhcGhwLmZy