 Barbara Seliger
Barbara Seliger- Institute of Medical Immunology, Martin Luther University Halle-Wittenberg, Halle (Saale), Germany
During the last decade, the dynamics of the cellular crosstalk have highlighted the significance of the host vs. tumor interaction. This resulted in the development of novel immunotherapeutic strategies in order to modulate/inhibit the mechanisms leading to escape of tumor cells from immune surveillance. Different monoclonal antibodies directed against immune checkpoints, e.g., the T lymphocyte antigen 4 and the programmed cell death protein 1/ programmed cell death ligand 1 have been successfully implemented for the treatment of cancer. Despite their broad activity in many solid and hematologic tumor types, only 20–40% of patients demonstrated a durable treatment response. This might be due to an impaired T cell tumor interaction mediated by immune escape mechanisms of tumor and immune cells as well as alterations in the composition of the tumor microenvironment, peripheral blood, and microbiome. These different factors dynamically regulate different steps of the cancer immune process thereby negatively interfering with the T cell –mediated anti-tumoral immune responses. Therefore, this review will summarize the current knowledge of the different players involved in inhibiting tumor immunogenicity and mounting resistance to checkpoint inhibitors with focus on the role of tumor T cell interaction. A better insight of this process might lead to the development of strategies to revert these inhibitory processes and represent the rational for the design of novel immunotherapies and combinations in order to improve their efficacy.
Introduction
It has been generally accepted that the development and progression of tumors is a result of an altered crosstalk between the tumor and the host immune system (1–3). The immune system not only suppresses tumor growth by destroying tumor cells or inhibiting their outgrowth, but also promotes tumor progression by either selecting for tumor escape variants or by establishing conditions within the tumor microenvironment (TME) and periphery that facilitate tumor outgrowth, which has been classified as a hallmark of cancer (4). These include an increased frequency of immune suppressive cells, metabolites, cytokines and soluble factors, hypoxia and acidic pH (5, 6). Further changes of the TME during neoplastic transformation are a selective ablation of immune effector cells and deletion or neutralization of cytokines, like interferon (IFN)-γ (7). Despite interferon (IFN)-γ exert pro-tumorigenic effects under certain circumstances dependent on the cellular and molecular context (8, 9), it represents a key mediator of immunosurveillance produced by natural killer (NK) cells and T cells known to promote cytotoxic activity of macrophages and enhance the expression of immune modulatory molecules on tumor cells (7). This results in the release of tumor associated antigens (TAA) for cross presentation by dendritic cells (DCs), which uptake and process these antigens into peptides then presented via the major histocompatibility complex (MHC) class I and class II molecules to CD8+ and CD4+ T cells, respectively. However, elimination of transformed cells can be incomplete due to a decreased tumor immunogenicity (10). This results first in an equilibrium state characterized by a balance between proliferation and killing of tumor cells by CD8+ T cells thereby maintaining the tumor at a subclinical stage, followed by the generation of tumor cells, which are resistant to immune rejection due to constant selective pressure of the immune system (2). These immune escape mechanisms are associated with the loss or downregulation of TAA and/or HLA class I surface molecules or aberrantly expression of the non-classical HLA-G and HLA-E antigens as well as co-inhibitory molecules (Table 1). This might be at least partially mediated by the induction of oncogenic pathways (11, 12) and changes in the tumor cell metabolism (13, 14).
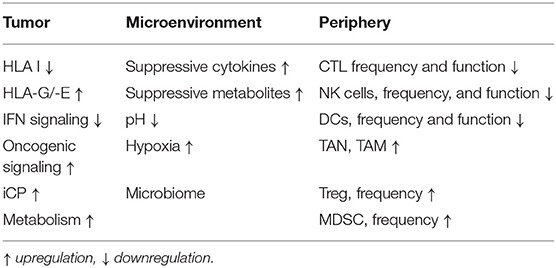
Table 1. Immune escape mechanisms.
However, interventions, such as chemotherapy, radiotherapy (RT), physico-chemical, and thermal ablation can promote the release of TAA and might overcome the dominant immune suppressive pathways leading to an increased immunogenicity (15–18). Therefore, the combination of immunotherapies with other strategies offers novel opportunities to recover immune activity and increase their efficacy, which result in a better patients' outcome. Indeed, this approach is currently investigated in a number of experimental models and clinical trials (19).
Players involved in mounting anti-tumor immune responses include in particular cells of the adaptive immune system, which protect and/or control tumor outgrowth and the interaction of the host against viral/pathogen infections and neoplastic transformation. The therapeutic potential of host-vs.-tumor activity has been analyzed by various groups and is based on CD4+ and CD8+ T cell responses, which are part of the cancer immune cycle and significantly influence the clinical outcome of patients (20, 21). It is well-known that the initial antigen-mediated activation of T cells is modulated by the engagement co-stimulatory signals with its ligands on antigen-presenting cells (APC). Under physiologic conditions, immune checkpoint pathways avoid auto-immunity by inducing inhibitory pathways important for maintaining self-tolerance thereby regulating the type and magnitude of T cell responses required to mount a proper anti-tumoral activity. During the last decade, a number of different inhibitory T cell and non-T cell iCP pathways have been well-characterized (Table 2) (31). The prototype is the cytotoxic T lymphocyte antigen 4 (CTLA-4; CD152), which competes with CD28 for the ligands CD80 and CD86, and antagonizes the T cell receptor (TCR) signaling (32–34). In addition, the interaction of the programmed cell death protein 1 (PD-1; CD279) with its ligands the programmed cell death 1 ligand 1 (PD-L1; CD274/B7-H1) and/or PD-L2 (CD273/B7-DC), negatively interferes with TCR signaling (35–38). Thus, immune checkpoints (iCPs) have either a stimulatory or inhibitory potential, the latter acting as “breaks” on the immune response. Recently, there exists evidence that inhibitory iCPs could be targeted by immune check point inhibitors (iCPIs) leading to an increased anti-tumoral response and patients' survival (39).
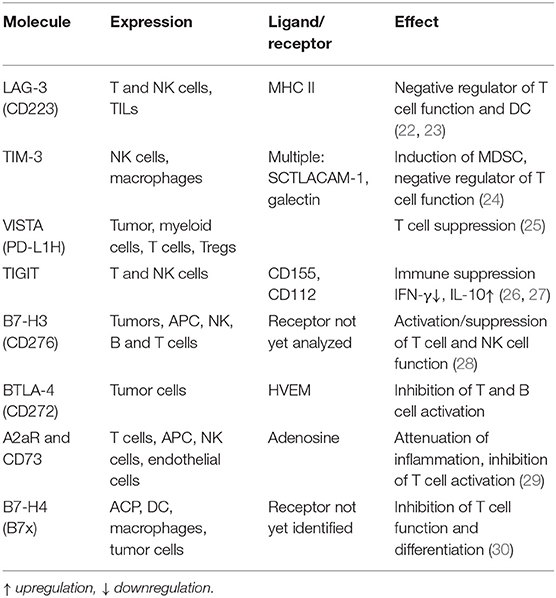
Table 2. T cell associated inhibitory immune checkpoint pathways.
General Strategies of Tumors to Facilitate Tumor Suppression by Analyzing the Composition and Frequency of Immune Cells in the Tumor Microenvironment (TME) and Peripheral Blood (PB)
The impaired anti-tumoral immune response represents an important hallmark of solid tumors and hematopoietic malignancies and involves many distinct mechanisms at the tumor site, in the tumor microenvironment (TME) and in the peripheral blood (21). The generation of an inflammatory and immune suppressive milieu in the TME induces tumor escape mechanisms, such as downregulation of classical HLA class I antigens and an upregulation of HLA-G and -E as well as iCPs including e.g., PD-L1 in the TME and CTLA-4 in the lymphoid tissues leading to evasion of adaptive immune responses (40–42). Furthermore, an upregulation of other immune inhibitory molecules like PD-1, T cell immunoglobulin and mucin domain-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), 2B4, and T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif (TIGIT) have been reported, which is accompanied by a reduced IFN-γ and TFN-α secretion of T cells (43–45). An altered TME is further characterized by an altered cellular composition and activity of tumor infiltrating immune cells. Next to a reduced frequency and activity of immune effector cells, such as CD8+ T cells, NK cells, an increased frequency of immune suppressive cells, such as tumor associated neutrophils (TANs), myeloid-derived suppressor cells (MDSC), tumor associated macrophages (TAM), tumor associated fibroblasts (TAF), regulatory T cells (Treg), and stroma cells leading to a complex interaction network of heterogeneous immune and non-immune cell populations with overlapping and opposite functions (46). In addition, soluble factors, like the transcriptional growth factor (TGF)-β, interleukin (IL)-10, the vascular endothelial growth factor (VEGF)A, chemokines as well as metabolites, e.g., arginase, hypoxia, and low pH, have been identified to be responsible for the establishment of an immune suppressive TME (47, 48). Furthermore, a reduced frequency and impaired function of effector cells and capacity of dendritic cells to present antigen as well as an increased number of immune suppressive cells were also found in peripheral blood. Both an immune suppressive TME and a reduced immune function of PB are associated with a poor patients' outcome (49–52). There exists evidence that functional T cell responses could be missed by analyzing only PBL (53) and no TIL. Therefore, it is essential to determine the composition, organization and function of the TME and PB of individual patients as well as the tumor itself to predict potential anti-tumoral effects of antigen specific T cells, since this has been shown to have prognostic relevance and therapeutic implications (47, 54).
Immune Checkpoint Inhibitors and Patients' Response
Novel immunotherapeutic approaches have recently revolutionized the treatment of solid and hematopoietic tumors. The clinical success of monoclonal antibodies (mAb) directed against CTLA-4 and the PD-1/PD-L1 pathway was a breakthrough achievement (55–57). The anti-CTLA-4 mAb Ipilimumab was the first iCPI approved by the Food and Drug Administration (FDA) (58, 59) followed by the approval of the anti-PD-1 mAbs Pembrolizumab and Nivolumab in 2014 or 2016, respectively. The anti-PD-L1 mAbs Durvalumab, Atezolizumab, and Avelumab were FDA approved in 2017 after promising results in non-small cell lung cancer (NSCLC), urothelial carcinoma and Merkel cell carcinoma (60–63).
Despite the rapid progress of approvals for iCPI, the accumulated experience demonstrated that approximately only one-third of patients had a durable response upon single iCPI treatment. Thus, the majority of patients do not benefit from iCPI alone, which might be due to primary, adaptive and acquired resistance mechanisms (64). Therefore, a number of clinical trials using iCPIs across all tumor types using different combinations, e.g., chemotherapy, iCPIs, chimeric antigen receptor, hypermethylating agents, CDK4 inhibitors, RT and targeted therapies, are currently conducted. Some of these combinations have achieved response rates over 50% (57, 65, 66). Regarding the combination of RT with iCPI it is noteworthy that RT could not only stimulate immune responses, but could also exert immune suppressive effects (67, 68). In this context, scheduling of iCPI therapy is important for the therapeutic outcome in combination with RT (69, 70), which has been shown to shape the T cell receptor repertoire of TIL (71). However, there is still an urgent need to explore biomarkers to predict response to these treatments and to identify combinations of agents to improve treatment efficacy, overall survival (OS) of patients and mitigate toxicities of these treatment options.
Immune Modulatory Molecules and their Relevance for T Cell Responses and Patients' Outcome
Expression of Classical and Non-classical HLA Class I Antigens
This topic has been reviewed and discussed by various authors (72–75). HLA class I surface expression is frequently downregulated or lost in solid and hematopoietic tumors. These abnormalities have functional relevance, since they impair T cell recognition of tumors. Furthermore, HLA class I alterations have been associated with a worse patients' outcome and a reduced overall survival (OS) and play a role in the resistance to iCPI therapy. The underlying molecular mechanisms of impaired HLA class I surface expression are diverse and often associated with deficiencies in the expression of components of the antigen processing machinery (APM) and IFN pathways as recently summarized (72, 76). This could be due to either structural alterations or deregulation at the transcriptional, epigenetic or posttranscriptional level of these molecules (77). Furthermore, HLA class I expression has been associated with an increased density of tumor infiltrating lymphocytes (TIL) and an increased anti-tumoral T cell response (78).
Next to the impaired expression of HLA class I antigens, a frequent overexpression of HLA-G and/or –E was found in tumors of distinct origin, but not in adjacent normal tissues or in healthy controls. This was accompanied by a reduced T cell and NK cell recognition and a bad patients' prognosis (79–83). Soluble HLA-G levels (sHLA-G) were also frequently detected and inversely correlate to numbers of activated T cells suggesting that sHLA-G promotes tumor immune escape through activation of immune responses (84).
Expression of Immune Checkpoints: Challenges and Pitfalls
Next to alterations of HLA class I antigens, high expression levels of co-inhibitory checkpoints, such as e.g., PD-L1 and B7-H4, in various tumor entities are often associated with the clinical outcome of cancer patients (85–88). An altered expression pattern of PD-L1 was found in primary and metastatic bladder tumors suggesting a dynamic nature of the TME (89). The capacity of the PD-L1 mediated immune suppression was inversely proportional to the antigenicity of the tumor (90). Since PD-L1 expression on both tumor and host's immune cells could lead to escape from immune surveillance, PD-L1 expression has been suggested as a biomarker for prediction of prognosis and response to iCPI (91). Its expression correlates with the adaptive immune resistance in several tumor types, including melanoma, NSCLC, Merkel cell carcinoma, breast cancer, mismatch-repair deficient tumors, and Hodgkin's lymphoma (91–93). However, PD-L1 expression does not reliably predict response to iCPI. In melanoma, tumor PD-L1 expression showed a significant correlation with response to five out of eight iCPI studies treating patients with anti-PD-1 mAb, while it did not predict response to anti-CTLA-4 therapy (94). Furthermore, some patients negative for PD-L1 expression can have a response to iCPI (92). In NSCLC, no association of PD-L1 expression with response has been reported with Nivolumab, while PD-L1 expression on at least 50% of NSCLC lesions almost doubled the response rate to Pembrolizumab from 19 to about 45% (95). In contrast to pre-treatment biopsies, tumor biopsies in early treatment phase obtained from metastatic melanoma patients treated sequentially receiving CTLA-4 and PD-1 iCPI showed high PD-1 and PD-L1 expression levels in responders (96). In NSCLC cells, the PD-L1 genomic locus amplification correlated with PD-L1 expression and anti-tumor responses (97, 98). Despite a significant heterogeneity was observed, higher levels of CTLA-4 and PD-L2 expression were found in melanoma patients, who benefit from CTLA-4 antibodies (99, 100). In contrast, PD-L1, PD-L2, and CTLA-4 expression did not correlate to anti-PD-1-responsiveness of melanoma patients (101). In addition to the discrepant results on the role of PD-L1 expression for prognosis and iCPI response, there exist some limitations regarding the analysis of the PD-L1 expression, including membranous vs. cytoplasmic expression, expression by multiple cell types in the TME, focal expression in tumor samples, changes in the expression during disease progression, upon radiation, chemotherapy, and epigenetic drugs and in particular the variability of laboratory techniques and anti-PD-L1 antibodies employed for immunohistochemistry (IHC) (102).
Somatic Mutations and Neoantigen Load
Increasing evidence demonstrated that mutations lead to the generation of neoantigens, which are presented by HLA class I molecules and can be recognized by CD8+ cytotoxic T cells (CTL) (77). Thus, the tumor mutational burden (TMB) might be correlated with the level of response to T cell based immunotherapies. Indeed, a systemic review of melanoma patients showed that responses to iCPIs correlated with TMB, neoantigen load, and immune-related gene expression (103, 104). Microsatellite instable (MSI) colorectal carcinoma (CRC) has large mutational burdens, higher immune cell infiltration and higher response rates to PD-1 blockade (105). However, a high TMB does not always predict responders to iCPI therapy, which might be due to neoantigen heterogeneity and an extremely diverse array of somatic mutations (106, 107). It is noteworthy that T cell epitopes have a similarity to bacterial and viral antigens suggesting a cross reactivity of T cells to intestinal bacterial and viral antigens, which can also modulate the iCPI therapy (108). In addition, PD-L1 expression can be controlled by driver mutations and oncogenic signaling (109–112).
Immune Profiling Signatures and iCPI
Genetic and immune heterogeneity was found in melanoma responding to immunotherapy. Mutanome and individual gene-based expression analysis demonstrated mesenchymal and T cell suppressive inflammatory or angiogenic tumor phenotypes, which were associated with innate anti-PD-1 resistance (113). Genes, which were higher expressed in non-responding pre-treatment tumors, include molecules involved in epithelial to mesenchymal transition (EMT), immune suppression and chemotaxis of monocytes and macrophages (114). Interestingly, a dormant TIL phenotype characterized by an elevated TMB and intra-tumoral CD3 signal, elevated TILs with low activation and proliferation was associated with a favorable response to iCPI (115). The IFN signature is correlated with an improved prognosis and iCPI response or resistance to iCPIs (116). Furthermore, T cell diversification reflects antigen selection in the blood of patients on iCPI treatment (117). Recently, a 15-gene pre-treatment classifier model was identified to predict response to anti-CTLA-4 treatment (118).
Role of Immune Cell Subpopulations for Tumor Immunity and the Effect of iCPI
Tumor-Infiltrating Cytotoxic Lymphocytes (CTL) and iCPI
The success of checkpoint blockade depends on the presence of TIL, particularly of CD8+ CTL, in the TME. These CTL are located at the invasive tumor margin and intratumorally, and are negatively regulated by the PD-1/PD-L1-mediated adaptive immune resistance (119, 120). In metastatic melanoma, the presence of CTL at the tumor margin predicted better response to iCPI. Colon cancers with MSI are highly infiltrated with T cells, particularly with CTL, relative to microsatellite-stable (MSS) colon cancers (121–123). Members of the CCL and CXCL chemokine families have been associated with T cell recruitment to melanoma metastases (124, 125). Higher levels of CCL2, CXCL4, and CXCL12 have been noted in tumors responding to iCPI therapy (126).
So far, it is not clear whether CD8+ effector memory cells might explain the durable response observed in many patients. Interestingly, brisk CTL infiltrates at time of progression in patients on iCPI treatment were observed suggesting an impaired activity of effector immune cells in the TME leading to therapeutic resistance. Despite CD8+ T cell responses against tumor cells are well-understood, information about the role of CD4+ T cell immunity in cancer is limited. Tumor specific CD4+ T cells have a broad activity beyond the provision of helper signals to CD8+ T cells (127, 128). CD4+ T cells exhibit anti-tumor effects and Th1 T cells are involved in the killing of tumor cells by secretion of cytokines that activate death receptors on tumor cells and induce epitopes spreading. Furthermore, CD4+ Th1 T cells can activate DC functions. The secretion of IL-4 from CD4+ T cells could establish long-term memory immune responses and further recruit eosinophils and macrophages.
Tumor-Infiltrating Regulatory T Cells (Tregs)
Tumor-infiltrating CD4+ Tregs were frequently detected in the TME and suppress CTL activity leading to reduced anti-tumor T cell responses (129). They promote tumor growth by iCP expression (CTLA-4, PD-1 and others) as well as production of IL-10 and TGF-β. An increased frequency of Tregs correlates with disease progression and metastasis in both experimental models and humans (130). CTLA-4 blockade expands the Treg frequency and high levels of soluble CD25 interleukin-2 receptor chair-alpha (IL2Rα) has been correlated with resistance to anti-CTLA-4 therapy (131). This was confirmed by Treg depletion potentiating the iCPI therapy (132). It has been suggested that early recruitment of Tregs to the TME inhibits an effective tumor response and lack of response to iCPI. PD-1 blockade with Nivolumab attenuated the activated T cell phenotypes during the course of therapy, promoted CTL proliferation and resistance to Treg-mediated suppression by down-regulating the intracellular expression of FoxP3, while Tregs increased during disease progression (133). An increased ratio of CTL to Treg in tumor tissues has been associated with response to CTLA-4 and PD-1 blockade.
Natural Killer Cells as Players for Innate Immune Responses
Natural killer (NK) cells are effector cells of the innate immune system and important players in mounting innate anti-tumoral immune responses by their ability to directly target and eliminate viral infections as well as neoplastic transformed cells (134). Under pathological conditions and during inflammation, the NK cell activation depend on the balance between inhibitory as well as activating signals, which determine the NK cell mediated cytotoxicity. In addition, NK cells are involved in other immune regulatory processes and could modulate adaptive immune responses, since they share characteristics with adaptive lymphocytes (134). They could also interact with mast cells and effect tumorgenesis due to the production of pro-angiogenic factors and thus play an important role alone or in combination with mast cells in the regulation of angiogenesis (135). There is increasing evidence that NK cells are involved in regulating metastatic dissemination. NK cells are often shown to reduce metastatic efficacy of tumor cell lines in vivo, while low NK cell activity is correlated with advanced disease and metastasis formation (136, 137). Furthermore, the presence of tumor-infiltrating NK cells is a positive prognostic marker for multiple tumors (138–140).
Tumor-Infiltrating Regulatory Myeloid Cells
Tumor-infiltrating myeloid cells comprise MDSCs, tumor-associated granulocytes, TAMs and DCs, generate and promote both immunogenic and tolerogenic responses (141–143). MDSCs are heterogeneous immune-suppressive immature myeloid cells that can be divided into a polymorphonuclear subset and a monocytic subset. They support tumor growth, epithelial to mesemchymal transition (EMT) and predict poor prognosis of patients, but their role in tumorgenesis has still to be defined (144, 145). MDSCs exert their effects by producing immune suppressive factors, like arginine 1 (Arg-1) expression, nitric oxide (NO), cyclooxygenase-2 (COX-2), reactive oxygen species (ROS), and activate Treg via CD40–CD40L interactions (146–148). In melanoma, elevated levels of CXCL17 were found, which recruits MDSCs and predicts non-responders to iCPI (149).
Tumor-associated neutrophils (TANs) and TAMs have been classified as an anti-tumor (type 1) or pro-tumor (type 2) phenotype. Pro-tumor effects of TANs include dampening of CTL response, increased angiogenesis, and modulation of cellular trafficking. Type 1 TAMs (M1) produce immune stimulatory cytokines, like IL-6, IL-12 and CXCL9, that promote recruitment of CTLs, while type 2 TAMs (M2) exhibit an immune suppressive signature and support tumor growth by release of angiogenic factors, like IL-10 and CCL22, matrix remodeling mediated by proteases, and by inhibition of CTL and DC activity (150–153). In addition, TAMs promote Tregs by inducing the skewing of blood-derived CD4+ T cells toward an immunosuppressive phenotype due to their decreased production of effector cytokines, increased IL-10 production and enhanced expression of the co-inhibitory molecules PD-1 and TIM-3 (154, 155). However, the interaction between TA-specific CD4+ Th1 cells and TAMs might shift the intra-tumoral M1/M2 ratio toward an M1 phenotype (155). PD-L1 expression of monocytes and TAMs promote immune evasion and correlate with disease progression in hepatocellular carcinoma. This might be mediated by a hypoxia inducible factor 1α induced increased expression of the receptor TREM1 in TMAs resulting in immune suppression mediated by Treg recruitment, which was associated with disease progression as well as resistance to anti-PD-L1 treatment (156). Fc-gamma receptors (FcγRs) expressed by M2 TAMs facilitate anti-tumor response to CTLA-4 inhibition through Treg depletion (157, 158). Tumor-infiltrating eosinophils promote infiltration of CTLs by polarization of TAMs and normalization of the tumor vasculature, and predict a better prognosis in colon cancer (159).
The heterogenic family of DCs, including classical (cDCs) and plasmacytoid DCs (pDCs), are antigen-presenting cells (APC) that prime and regulate CTL responses. Anti-viral immune responses rely heavily on pDC-derived type I IFNs, while pDCs in tumors exert immunosuppressive activities. In contrast, tumor-infiltrating cDC increase T cell activation in lung cancer and melanoma patients forming tertiary lymphoid clusters, which are associated with better outcomes (160, 161). Tertiary lymphoid clusters also correlated with improved survival in pancreatic cancer (162). The rare subgroup of CD103(+) (integrin αE)+ DCs are strong stimulators of CTL and dependent on different transcription factors, like IRF8, Zbtb46, and Batf3. These CD103 cDCs (Batf3-cDC, cDC1) are also associated with CTL and increased OS for patients with breast, head and neck or lung cancer (163). In lung adenocarcinoma murine models, immunogenic chemotherapy (oxaliplatin-cyclophosphamide) has been reported to up-regulate toll-like receptor 4 (TLR-4) on tumor-infiltrating Batf3-cDCs, which leads to the recruitment of CTLs and sensitization to iCPIs (164).
Gut Microbiome, Immune Cell Interaction—iCPI Therapy
T cells as members of the adaptive immune system are involved in gut homeostasis, inflammation, and carcinogenesis (165). Recently, association between microbiota profiles, cancer susceptibility, and responsiveness to cancer therapy has been suggested (166–168). Indeed, microbiota could modify the immune response and influence the response to chemotherapy and immunotherapy (169, 170). Emerging evidence has suggested that the cross-talk between the gut microbiome and immune cells plays a role in determining responses to iCPI therapy. Indeed, the composition of the gut microbiome has been associated with response to iCPI in pre-clinical models as well as in patients. For example, in murine melanoma, commensal Bifidobacterium has been reported to promote the efficacy of anti-PD-L1 therapy by augmenting the function of DCs leading to CTL priming and infiltration (171). Recent studies in melanoma, lung, and kidney cancer patients have demonstrated an association of commensal gut microbiome with response to iCPI (172). Baseline gut microbiota enriched with Faecalibacterium and other Firmicutes is associated with a better response (173). In melanoma patients responding to iCPI more abundant species included Bifidobacterium, Collinsella, Enterococcus, Clostridiales, Rominococcus and Faecalibacterium, while low levels of Akkermansia muciniphila were observed in epithelial cancers not responding to iCPI (174). Patients with a favorable gut microbiome had increased expression of cytolytic T cell markers and APM components, and an increased ratio of CD8+ CTLs to FoxP3+CD4+ Tregs. Furthermore, metagenomic studies revealed functional differences in gut bacteria in responders. These are characterized by an enrichment of anabolic pathways and an enhanced systemic and antitumor immunity in responding patients with a favorable gut microbiome as well as in germ-free mice receiving fecal transplants from responding patients (172). Thus, the modulation of the components in the gut microbiome can augment anti-tumor immunotherapy. However, there exist several challenges including optimal composition of the gut microbiome and the therapeutic strategy to achieve that composition.
Resistance to Checkpoint Inhibitors
Abnormalities of the HLA class I antigen and IFN signaling pathways often correlate with the development of resistances to various kinds of immunotherapies including iCPI treatment and adoptive cell therapy (ACT) (64, 175–178). These could be categorized into intrinsic and acquired immune resistance (177, 179, 180) and are associated with an altered tumor T cell interaction. An increased knowledge of these processes might lead to the reprogramming of the immunologically “cold” TME characterized by a low immune cell infiltration and low TCR diversity (181, 182) and an increased T cell function (183–185). Combining the modulation of the immune cell repertoire and the reduction of immune suppressive metabolites and cytokines of the TME and enhancement of T cell tumor interaction with iCPI and/or vaccinations or even targeted therapies are currently tested in diverse clinical trials (186, 187).
Conclusions
There is strong evidence of an emerging role of T cell tumor interactions for the outcome of patients in general and regarding the efficacy of immunotherapies including iCPIs. The pathways involved in the regulation of the interaction between tumor and T cells are broad and highly dynamic. Tumors developed a plethora of adaptions leading to escape from counter-regulations of the immune system. This is mediated by ineffective T cell responses due to low tumor immunogenicity and the suppressive influence of the TME. The use of iCPI showed that the manipulation of inhibitory signaling pathways creates anti-tumoral immune responses. However, the efficacy of iCPIs is still limited. Thus, a better understanding of these processes might lead to the development of innovative therapies in order to reactivate T cell responses.
Author Contributions
BS designed the project and wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to acknowledge Maria Heise for excellent secretarial help.
Abbreviations
AML, acute myeloid leukemia; APC, antigen-presenting cells; Arg-1, arginine 1; cDC, classical dendritic cell; COX-2, cyclooxygenase−2; CRC, colorectal carcinoma; CTL, cytotoxic lymphocytes; CTLA-4, T lymphocyte antigen 4; DC, dendritic cell; ECM, extracellular matrix; EMT, epithelial to mesenchymal transition; FcγR, Fc-gamma receptor; FDA, Food and Drug Administration; FOXP3, forkhead box P3; gzmb, granzyme B; HLA, human leukocyte antigen; HNSCC, head and neck squamous cell cancer; ICOS, inducible T cell costimulatory; iCPI, immune check point inhibitors; iCP, immune checkpoint; IFN, interferon; IL, interleukin; IL-2Rα, interleukin-2 receptor chain-alpha; LAG-3, lymphocyte activation gene-3; M1, type 1 TAM; M2, type 2 TAM; mAb, monoclonal antibody; MDSC, myeloid-derived-suppressor cell; MHC, major histocompatibility complex; MSI, microsatellite-instable; MSS, microsatellite-stable; NK, natural killer; NO, nitric oxide; NSCLC, non-small cell lung cancer; OS, overall survival; PD-1, programmed cell death protein 1; pDC, plasmacytoid dendritic cell; PD-L1, programmed cell death 1 ligand 1; RCC, renal cell cancer; ROS, reactive oxygen species; RT, radiation therapy; SCLC, small cell lung carcinoma; TAA, tumor associated antigen; TAF, tumor associated fibroblasts; TAM, tumor-associated macrophage; TAN, tumor-associated neutrophil; TCR, T cell receptor; Teff, effector T cell; Tex, exhausted CD8+ T cell; TGF-β, transforming growth factor-beta; TIGIT, T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif; TIL, tumor infiltrating lymphocyte; TIM-3, T cell immunoglobulin and mucin domain containing protein-3; TLR-4, toll-like receptor 4; TMB, tumor mutational burden; TME, tumor microenvironment; Treg, regulatory T cell.
References
1. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
2. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
3. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
6. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. (2017) 8:248. doi: 10.3389/fimmu.2017.00248
7. Lin CF, Lin CM, Lee KY, Wu SY, Feng PH, Chen KY, et al. Escape from IFN-γ-dependent immunosurveillance in tumorigenesis. J Biomed Sci. (2017) 24:10. doi: 10.1186/s12929-017-0317-0
8. Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. (2011) 17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482
9. Kursunel MA, Esendagli G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. (2016) 31:73–81. doi: 10.1016/j.cytogfr.2016.07.005
10. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
11. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236
12. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. (2013) 3:1355–63. doi: 10.1158/1535-7163.TARG-13-B290
13. VanderMolen KM, Little JG, Sica VP, El-Elimat T, Raja HA, Oberlies NH, et al. Safety assessment of mushrooms in dietary supplements by combining analytical data with in silico toxicology evaluation. Food Chem Toxicol. (2017) 103:133–47. doi: 10.1016/j.fct.2017.03.005
14. Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: emerging concepts and therapeutic opportunities. Biochim Biophys Acta Rev Cancer. (2017) 1868:212–20. doi: 10.1016/j.bbcan.2017.04.002
15. Martinez-Zubiaurre I, Chalmers AJ, Hellevik T. Radiation-induced transformation of immunoregulatory networks in the tumor stroma. Front Immunol. (2018) 9:1679. doi: 10.3389/fimmu.2018.01679
16. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. (2017) 14:247–58. doi: 10.1038/nrclinonc.2016.183
17. Keisari Y. Tumor abolition and antitumor immunostimulation by physico-chemical tumor ablation. Front Biosci. (2017) 22:310–47. doi: 10.2741/4487
18. Haen SP, Pereira PL, Salih HR, Rammensee HG, Gouttefangeas C. More than just tumor destruction: immunomodulation by thermal ablation of cancer. Clin Dev Immunol. (2011) 2011:160250. doi: 10.1155/2011/160250
19. Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. (2016) 2:286–94. doi: 10.1016/j.trecan.2016.05.002
20. Reiser J, Banerjee A. Effector, memory, and dysfunctional CD8+ T cell fates in the antitumor immune response. J Immunol Res. (2016) 2016:8941260. doi: 10.1155/2016/8941260
21. Ostroumov D, Fekete-Drimusz N, Saborowski M, Kühnel F, Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci. (2018) 75:689–713. doi: 10.1007/s00018-017-2686-7
22. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
23. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. (2018) 9:176–89. doi: 10.18632/genesandcancer.180
24. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. (2017) 276:97–111. doi: 10.1111/imr.12520
25. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VIST. Immunol Rev A. (2017) 276:66–79. doi: 10.1111/imr.12525
26. Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. (2018) 67:1659–67. doi: 10.1007/s00262-018-2246-5
27. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. (2017) 38:20–8. doi: 10.1016/j.it.2016.10.002
28. Ni L, Dong C. New B7 family checkpoints in human cancers. Mol Cancer Ther. (2017) 16:1203–11. doi: 10.1158/1535-7163.MCT-16-0761
29. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
30. Podojil JR, Miller SD. Potential targeting of B7-H4 for the treatment of cancer. Immunol Rev. (2017) 276:40–51. doi: 10.1111/imr.12530
31. Aihara K, Higuchi T, Hirobe M. Increasing 5-lipoxygenase inhibitory activities by oxidative conversion of O-methoxyphenols to catechols using a Cu2+-ascorbic acid-O2 system. Chem Pharm Bull. (1990) 38:842–4. doi: 10.1248/cpb.38.842
32. Fallarino F, Fields PE, Gajewski TF. B7–1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J Exp Med. (1998) 188:205–10. doi: 10.1084/jem.188.1.205
33. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7–1 (CD80) and B7–2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. (1994) 1:793–801. doi: 10.1016/S1074-7613(94)80021-9
34. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
35. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. (1999) 5:1365–9. doi: 10.1038/70932
36. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
37. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
38. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. (2001) 193:839–46. doi: 10.1084/jem.193.7.839
39. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
40. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
41. Menter T, Tzankov A. Mechanisms of immune evasion and immune modulation by lymphoma cells. Front Oncol. (2018) 8:54. doi: 10.3389/fonc.2018.00054
42. Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. (2017) 14:203–20. doi: 10.1038/nrclinonc.2016.168
43. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. (2013) 190:4899–909. doi: 10.4049/jimmunol.1300271
44. Alspach E, Lussier DM, Schreiber RD. Interferon gamma and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol. (2019) 11. doi: 10.1101/cshperspect.a028480
45. Shimizu K, Iyoda T, Okada M, Yamasaki S, Fujii SI. Immune suppression and reversal of the suppressive tumor microenvironment. Int Immunol. (2018) 30:445–54. doi: 10.1093/intimm/dxy042
46. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
47. Tuccitto A, Shahaj E, Vergani E, Ferro S, Huber V, Rodolfo M, et al. Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch. (2018) 474:407–20. doi: 10.1007/s00428-018-2477-z
48. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol. (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
49. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. (2018) 391:2128–39. doi: 10.1016/S0140-6736(18)30789-X
50. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. J Pathol. (2014) 232:199–209. doi: 10.1002/path.4287
51. Gnjatic S, Bronte V, Brunet LR, Butler MO, Disis ML, Galon J, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer. (2017) 5:44. doi: 10.1186/s40425-017-0243-4
52. Taube JM, Galon J, Sholl LM, Rodig SJ, Cottrell TR, Giraldo NA, et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod Pathol. (2018) 31:214–34. doi: 10.1038/modpathol.2017.156
53. Tjin EP, Konijnenberg D, Krebbers G, Mallo H, Drijfhout JW, Franken KL, et al. T-cell immune function in tumor, skin, and peripheral blood of advanced stage melanoma patients: implications for immunotherapy. Clin Cancer Res. (2011) 17:5736–47. doi: 10.1158/1078-0432.CCR-11-0230
54. Petitprez F, Sun CM, Lacroix L, Sautès-Fridman C, de Reyniès A, Fridman WH. Quantitative analyses of the tumor microenvironment composition and orientation in the era of precision medicine. Front Oncol. (2018) 8:390. doi: 10.3389/fonc.2018.00390
55. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
56. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
57. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. (2018) 52(Pt 2):39–52. doi: 10.1016/j.semcancer.2017.10.001
59. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
60. Kim ES. Avelumab: first global approval. Drugs. (2017) 77:929–37. doi: 10.1007/s40265-017-0749-6
61. Rosell R, Karachaliou N. Avelumab in non-small-cell lung cancer. Lancet Oncol. (2018) 19:1423–4. doi: 10.1016/S1470-2045(18)30683-1
62. Bommareddy PK, Kaufman HL. Avelumab and other recent advances in Merkel cell carcinoma. Future Oncol. (2017) 13:2771–83. doi: 10.2217/fon-2017-0305
63. Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, Aljumaily R, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. (2018) 19:51–64. doi: 10.1016/S1470-2045(17)30900-2
64. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
65. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. (2016) 13:273–90. doi: 10.1038/nrclinonc.2016.25
66. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
67. Wennerberg E, Vanpouille-Box C, Bornstein S, Yamazaki T, Demaria S, Galluzzi L. Immune recognition of irradiated cancer cells. Immunol Rev. (2017) 280:220–30. doi: 10.1111/imr.12568
68. Frey B, Rückert M, Deloch L, Rühle PF, Derer A, Fietkau R, et al. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol Rev. (2017) 280:231–48. doi: 10.1111/imr.12572
69. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. (2014) 74:5458–68. doi: 10.1158/0008-5472.CAN-14-1258
70. Escorcia FE, Postow MA, Barker CA. Radiotherapy and immune checkpoint blockade for melanoma: a promising combinatorial strategy in need of further investigation. Cancer J. (2017) 23:32–9. doi: 10.1097/PPO.0000000000000236
71. Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res. (2018) 6:139–50. doi: 10.1158/2326-6066.CIR-17-0134
72. Garrido F, Perea F, Bernal M, Sánchez-Palencia A, Aptsiauri N, Ruiz-Cabello F. The escape of cancer from T cell-mediated immune surveillance: HLA class I loss and tumor tissue architecture. Vaccines. (2017) 5:7. doi: 10.3390/vaccines5010007
73. Cai L, Michelakos T, Yamada T, Fan S, Wang X, Schwab JH, et al. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol Immunother. (2018) 67:999–1009. doi: 10.1007/s00262-018-2131-2
74. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. (2017) 171:1259–71.e11. doi: 10.1016/j.cell.2017.10.001
75. Ferns DM, Heeren AM, Samuels S, Bleeker MCG, de Gruijl TD, Kenter GG, et al. Classical and non-classical HLA class I aberrations in primary cervical squamous- and adenocarcinomas and paired lymph node metastases. J Immunother Cancer. (2016) 4:78. doi: 10.1186/s40425-016-0184-3
76. Aptsiauri N, Ruiz-Cabello F, Garrido F. The transition from HLA-I positive to HLA-I negative primary tumors: the road to escape from T-cell responses. Curr Opin Immunol. (2018) 51:123–32. doi: 10.1016/j.coi.2018.03.006
77. Seliger B. Molecular mechanisms of HLA class I-mediated immune evasion of human tumors and their role in resistance to immunotherapies. HLA. (2016) 88:213–20. doi: 10.1111/tan.12898
78. Perea F, Sánchez-Palencia A, Gómez-Morales M, Bernal M, Concha Á, García MM, et al. HLA class I loss and PD-L1 expression in lung cancer: impact on T-cell infiltration and immune escape. Oncotarget. (2018) 9:4120–33. doi: 10.18632/oncotarget.23469
79. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: an immune checkpoint molecule. Adv Immunol. (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001
80. Morandi F, Rizzo R, Fainardi E, Rouas-Freiss N, Pistoia V. Recent advances in our understanding of HLA-G biology: lessons from a wide spectrum of human diseases. J Immunol Res. (2016) 2016:4326495. doi: 10.1155/2016/4326495
81. Seliger B. The non-classical antigens of HLA-G and HLA-E as diagnostic and prognostic biomarkers and as therapeutic targets in transplantation and tumors. Clin Transpl. (2013) 465–72.
82. Morandi F, Pistoia V. Interactions between HLA-G and HLA-E in physiological and pathological conditions. Front Immunol. (2014) 5:394. doi: 10.3389/fimmu.2014.00394
83. Swets M, König MH, Zaalberg A, Dekker-Ensink NG, Gelderblom H, van de Velde CJ, et al. HLA-G and classical HLA class I expression in primary colorectal cancer and associated liver metastases. Hum Immunol. (2016) 77:773–9. doi: 10.1016/j.humimm.2016.03.001
84. Xu YF, Lu Y, Cheng H, Jiang J, Xu J, Long J, et al. High expression of human leukocyte antigen-g is associated with a poor prognosis in patients with PDAC. Curr Mol Med. (2015) 15:360–7. doi: 10.2174/1566524015666150401102218
85. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. (2016) 9:5023–39. doi: 10.2147/OTT.S105862
86. Shen L, Qian Y, Wu W, Weng T, Wang FXC, Hong B, et al. B7-H4 is a prognostic biomarker for poor survival in patients with pancreatic cancer. Hum Pathol. (2017) 66:79–85. doi: 10.1016/j.humpath.2017.05.023
87. Zhang K, Lv S, Lin Z, Tang D. CdS:Mn quantum dot-functionalized g-C3N4 nanohybrids as signal-generation tags for photoelectrochemical immunoassay of prostate specific antigen coupling DNAzyme concatamer with enzymatic biocatalytic precipitation. Biosens Bioelectron. (2017) 95:34–40. doi: 10.1016/j.bios.2017.04.005
88. Wu L, Deng WW, Yu GT, Mao L, Bu LL, Ma SR, et al. B7-H4 expression indicates poor prognosis of oral squamous cell carcinoma. Cancer Immunol Immunother. (2016) 65:1035–45. doi: 10.1007/s00262-016-1867-9
89. Pichler R, Heidegger I, Fritz J, Danzl M, Sprung S, Zelger B, et al. PD-L1 expression in bladder cancer and metastasis and its influence on oncologic outcome after cystectomy. Oncotarget. (2017) 8:66849–64. doi: 10.18632/oncotarget.19913
90. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. (2017) 5:106–17. doi: 10.1158/2326-6066.CIR-16-0391
91. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
92. Ancevski Hunter K, Socinski MA, Villaruz LC. PD-L1 testing in guiding patient selection for PD-1/PD-L1 inhibitor therapy in lung cancer. Mol Diagn Ther. (2018) 22:1–10. doi: 10.1007/s40291-017-0308-6
93. Liu D, Wang S, Bindeman W. Clinical applications of PD-L1 bioassays for cancer immunotherapy. J Hematol Oncol. (2017) 10:110. doi: 10.1186/s13045-017-0479-y
94. Brüggemann C, Kirchberger MC, Goldinger SM, Weide B, Konrad A, Erdmann M, et al. Predictive value of PD-L1 based on mRNA level in the treatment of stage IV melanoma with ipilimumab. J Cancer Res Clin Oncol. (2017) 143:1977–84. doi: 10.1007/s00432-017-2450-2
95. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
96. Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin Cancer Res. (2017) 23:5024–33. doi: 10.1158/1078-0432.CCR-16-0698
97. Inoue Y, Yoshimura K, Mori K, Kurabe N, Kahyo T, Mori H, et al. Clinical significance of PD-L1 and PD-L2 copy number gains in non-small-cell lung cancer. Oncotarget. (2016) 7:32113–28. doi: 10.18632/oncotarget.8528
98. Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. (2016) 11:62–71. doi: 10.1016/j.jtho.2015.09.010
99. Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H, et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci USA. (2016) 113: E7769–77. doi: 10.1073/pnas.1607836113
100. Obeid JM, Erdag G, Smolkin ME, Deacon DH, Patterson JW, Chen L, et al. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology. (2016) 5:e1235107. doi: 10.1080/2162402X.2016.1235107
101. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. (2014) 20:5064–74. doi: 10.1158/1078-0432.CCR-13-3271
102. Balar AV, Weber JS. PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunol Immunother. (2017) 66:551–64. doi: 10.1007/s00262-017-1954-6
103. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386
104. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. (2017) 9:eaah3560. doi: 10.1126/scitranslmed.aah3560
105. Chang L, Chang M, Chang HM, Chang F. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Appl Immunohistochem Mol Morphol. (2018) 26:e15–21. doi: 10.1097/PAI.0000000000000575
106. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. (2016) 17:e542–51. doi: 10.1016/S1470-2045(16)30406-5
107. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. (2017) 545:60–5. doi: 10.1038/nature22079
108. Sioud M. T-cell cross-reactivity may explain the large variation in how cancer patients respond to checkpoint inhibitors. Scand J Immunol. (2018) 87. doi: 10.1111/sji.12643
109. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. (2014) 20:3446–57. doi: 10.1158/1078-0432.CCR-13-2797
110. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 Expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. (2015) 21:4014–21. doi: 10.1158/1078-0432.CCR-15-0016
111. Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, Pereira C, Pros E, Romero OA, et al. MET-Oncogenic and JAK2-inactivating alterations are independent factors that affect regulation of PD-L1 expression in lung cancer. Clin Cancer Res. (2018) 24:4579–87. doi: 10.1158/1078-0432.CCR-18-0267
112. Li M, Liu F, Zhang F, Zhou W, Jiang X, Yang Y, et al. Genomic ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in gallbladder cancer: a whole-exome sequencing analysis. Gut. (2019) 68:1024–33. doi: 10.1136/gutjnl-2018-316039
113. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
114. Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. (2016) 22:3630–42. doi: 10.1158/1078-0432.CCR-15-1434
115. Gettinger SN, Choi J, Mani N, Sanmamed MF, Datar I, Sowell R, et al. A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun. (2018) 9:3196. doi: 10.1038/s41467-018-05032-8
116. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. (2016) 167:1540–54.e12. doi: 10.1016/j.cell.2016.11.022
117. Weis J, Schroder JM. The influence of fat tissue on neuroma formation. J Neurosurg. (1989) 71:588–93. doi: 10.3171/jns.1989.71.4.0588
118. Friedlander P, Wassmann K, Christenfeld AM, Fisher D, Kyi C, Kirkwood JM, et al. Whole-blood RNA transcript-based models can predict clinical response in two large independent clinical studies of patients with advanced melanoma treated with the checkpoint inhibitor, tremelimumab. J Immunother Cancer. (2017) 5:67. doi: 10.1186/s40425-017-0272-z
119. Inozume T, Yaguchi T, Ariyasu R, Togashi Y, Ohnuma T, Honobe A, et al. Analysis of the tumor reactivity of tumor-infiltrating lymphocytes in a metastatic melanoma lesion that lost MHC class I expression after anti-PD-1 therapy. J Invest Dermatol. (2019) 139:1490–6. doi: 10.1016/j.jid.2019.01.007
120. Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res. (2019) 79:1214–25. doi: 10.1158/0008-5472
121. Carethers JM, Murali B, Yang B, Doctolero RT, Tajima A, Basa R, et al. Influence of race on microsatellite instability and CD8+ T cell infiltration in colon cancer. PLoS ONE. (2014) 9:e100461. doi: 10.1371/journal.pone.0100461
122. Boissière-Michot F, Lazennec G, Frugier H, Jarlier M, Roca L, Duffour J, et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology. (2014) 3:e29256. doi: 10.4161/onci.29256
123. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. (2018) 8:730–49. doi: 10.1158/2326-6074.TUMIMM17-PR03
124. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
125. Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, et al. Oncogenic BRAF(V600E) governs regulatory T-cell recruitment during melanoma tumorigenesis. Cancer Res. (2018) 78:5038–49. doi: 10.1158/0008-5472.CAN-18-0365
126. Jamal R, Lapointe R, Cocolakis E, Thébault P, Kazemi S, Friedmann JE, et al. Peripheral and local predictive immune signatures identified in a phase II trial of ipilimumab with carboplatin/paclitaxel in unresectable stage III or stage IV melanoma. J Immunother Cancer. (2017) 5:83. doi: 10.1186/s40425-017-0290-x
127. Hanson HL, Kang SS, Norian LA, Matsui K, O'Mara LA, Allen PM. CD4-directed peptide vaccination augments an antitumor response, but efficacy is limited by the number of CD8+ T cell precursors. J Immunol. (2004) 172:4215–24. doi: 10.4049/jimmunol.172.7.4215
128. Bedoui S, Heath WR, Mueller SN. CD4+ T-cell help amplifies innate signals for primary CD8+ T-cell immunity. Immunol Rev. (2016) 272:52–64. doi: 10.1111/imr.12426
129. Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. (2009) 30:626–35. doi: 10.1016/j.immuni.2009.05.002
130. Nakamura R, Sakakibara M, Nagashima T, Sangai T, Arai M, Fujimori T, et al. Accumulation of regulatory T cells in sentinel lymph nodes is a prognostic predictor in patients with node-negative breast cancer. Eur J Cancer. (2009) 45:2123–31. doi: 10.1016/j.ejca.2009.03.024
131. Hannani D, Vétizou M, Enot D, Rusakiewicz S, Chaput N, Klatzmann D, et al. Anticancer immunotherapy by CTLA-4 blockade: obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. (2015) 25:208–24. doi: 10.1038/cr.2015.3
132. Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. (2017) 127:3472–83. doi: 10.1172/JCI90499
133. Yamaguchi K, Mishima K, Ohmura H, Hanamura F, Ito M, Nakano M, et al. Activation of central/effector memory T cells and T-helper 1 polarization in malignant melanoma patients treated with anti-programmed death-1 antibody. Cancer Sci. (2018) 109:3032–42. doi: 10.1111/cas.13758
134. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
135. Ribatti D, Tamma R, Crivellato E. Cross talk between natural killer cells and mast cells in tumor angiogenesis. Inflamm Res. (2019) 68:19–23. doi: 10.1007/s00011-018-1181-4
136. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. (2008) 27:5932–43. doi: 10.1038/onc.2008.267
137. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
138. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. (1997) 79:2320–8. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p
139. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. (2000) 88:577–83. doi: 10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
140. Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. (2002) 35:23–8. doi: 10.1016/S0169-5002(01)00292-6
141. Pyzer AR, Cole L, Rosenblatt J, Avigan DE. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int J Cancer. (2016) 139:1915–26. doi: 10.1002/ijc.30232
142. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
143. Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. (2016) 274:290–306. doi: 10.1111/imr.12464
144. Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. (2017) 8:14979. doi: 10.1038/ncomms14979
145. Sangaletti S, Tripodo C, Santangelo A, Castioni N, Portararo P, Gulino A, et al. Mesenchymal transition of high-grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep. (2016) 17:233–48. doi: 10.1016/j.celrep.2016.08.075
146. Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K, Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest. (2012) 41:635–57. doi: 10.3109/08820139.2012.695417
147. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. (2009) 69:1553–60. doi: 10.1158/0008-5472.CAN-08-1921
148. Gantt S, Gervassi A, Jaspan H, Horton H. The role of myeloid-derived suppressor cells in immune ontogeny. Front Immunol. (2014) 5:387. doi: 10.3389/fimmu.2014.00387
149. Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mabs and are dependent on CD8 T cells and crosspriming. Cancer Res. (2016) 76:5994–6005. doi: 10.1158/0008-5472.CAN-16-0549
150. Szebeni GJ, Vizler C, Nagy LI, Kitajka K, Puskas LG. Pro-tumoral inflammatory myeloid cells as emerging therapeutic targets. Int J Mol Sci. (2016) 17:E1958. doi: 10.3390/ijms17111958
151. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. (2014) 5:75. doi: 10.3389/fphys.2014.00075
152. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
153. Fuxe J, Karlsson MC. TGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol. (2012) 22:455–61. doi: 10.1016/j.semcancer.2012.05.004
154. Dannenmann SR, Thielicke J, Stöckli M, Matter C, von Boehmer L, Cecconi V, et al. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology. (2013) 2:e23562. doi: 10.4161/onci.23562
155. Eisel D, Das K, Dickes E, König R, Osen W, Eichmüller SB. Cognate interaction with CD4(+) T cells instructs tumor-associated macrophages to acquire M1-like phenotype. Front Immunol. (2019) 10:219. doi: 10.3389/fimmu.2019.00219
156. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking TREM-1(+) Tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-PD-L1 resistance in liver cancer. Hepatology. (2019) 70:198–214. doi: 10.1002/hep.30593
157. Grugan KD, McCabe FL, Kinder M, Greenplate AR, Harman BC, Ekert JE, et al. Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J Immunol. (2012) 189:5457–66. doi: 10.4049/jimmunol.1201889
158. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
159. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. (2015) 5:43–51. doi: 10.1158/2159-8290.CD-14-0863
160. Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. (2016) 7:407. doi: 10.3389/fimmu.2016.00407
161. Rodriguez AB, Peske JD, Engelhard VH. Identification and characterization of tertiary lymphoid structures in murine melanoma. Methods Mol Biol. (2018) 1845:241–57. doi: 10.1007/978-1-4939-8709-2_14
162. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
163. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
164. Xu A, Zhang L, Yuan J, Babikr F, Freywald A, Chibbar R, et al. TLR9 agonist enhances radiofrequency ablation-induced CTL responses, leading to the potent inhibition of primary tumor growth and lung metastasis. Cell Mol Immunol. (2018). doi: 10.1038/s41423-018-0184-y. [Epub ahead of print].
165. Yang Y, Xu C, Wu D, Wang Z, Wu P, Li L, et al. Gammadelta T cells: crosstalk between microbiota, chronic inflammation, and colorectal cancer. Front Immunol. (2018) 9:1483. doi: 10.3389/fimmu.2018.01483
166. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. (2012) 22:292–8. doi: 10.1101/gr.126573.111
167. Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. (2002) 2:28–37. doi: 10.1038/nrc703
168. Baker SR, Swanson NA. Clinical applications of tissue expansion in head and neck surgery. Laryngoscope. (1990) 100:313–9. doi: 10.1288/00005537-199003000-00020
169. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
170. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. (2013) 342:971–6. doi: 10.1126/science.1240537
171. Smits HH, van Beelen AJ, Hessle C, Westland R, de Jong E, Soeteman E, et al. Commensal Gram-negative bacteria prime human dendritic cells for enhanced IL-23 and IL-27 expression and enhanced Th1 development. Eur J Immunol. (2004) 34:1371–80. doi: 10.1002/eji.200324815
172. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
173. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
174. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
175. Andersen R, Westergaard MCW, Kjeldsen JW, Müller A, Pedersen NW, Hadrup SR, et al. T-cell responses in the microenvironment of primary renal cell carcinoma-implications for adoptive cell therapy. Cancer Immunol Res. (2018) 6:222–35. doi: 10.1158/2326-6066.CIR-17-0467
176. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
177. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
178. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. (2016) 16:121–6. doi: 10.1038/nrc.2016.2
179. Seliger B. Immune modulatory microRNAs as a novel mechanism to revert immune escape of tumors. Cytokine Growth Factor Rev. (2017) 36:49–56. doi: 10.1016/j.cytogfr.2017.07.001
180. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350:207–11. doi: 10.1126/science.aad0095
181. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
182. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. (2013) 210:1389–402. doi: 10.1084/jem.20130066
183. Hodi FS. Overcoming immunological tolerance to melanoma: targeting CTLA-4. Asia Pac J Clin Oncol. (2010) 6(Suppl 1):S16–23. doi: 10.1111/j.1743-7563.2010.01271.x
184. Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol. (2007) 179:7244–53. doi: 10.4049/jimmunol.179.11.7244
185. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. (2015) 5:915–9. doi: 10.1158/2159-8290.CD-15-0563
186. Puzanov I, Diab A, Abdallah K, Bingham CO, Brogdon C, Dadu R, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. (2017) 5:95. doi: 10.1186/s40425-017-0300-z
Keywords: T cells, tumor growth, tumor microenvironment, microbiome, inflammation, checkpoint inhibitors
Citation: Seliger B (2019) The Role of the Lymphocyte Functional Crosstalk and Regulation in the Context of Checkpoint Inhibitor Treatment—Review. Front. Immunol. 10:2043. doi: 10.3389/fimmu.2019.02043
Received: 15 March 2019; Accepted: 12 August 2019;
Published: 06 September 2019.
Edited by:
Anil Shanker, Meharry Medical College, United StatesReviewed by:
Udo S. Gaipl, University Hospital Erlangen, GermanyYona Keisari, Tel Aviv University, Israel
Copyright © 2019 Seliger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Seliger, YmFyYmFyYS5zZWxpZ2VyQHVrLWhhbGxlLmRl