 Nick P. Goplen1
Nick P. Goplen1 Su Huang1
Su Huang1 Bibo Zhu1
Bibo Zhu1 In Su Cheon1Young Min Son1Zheng Wang1Chaofan Li1
In Su Cheon1Young Min Son1Zheng Wang1Chaofan Li1 Qigang Dai1Li Jiang1Jie Sun1,2*
Qigang Dai1Li Jiang1Jie Sun1,2*- 1Thoracic Diseases Research Unit, Division of Pulmonary and Critical Care Medicine, Department of Medicine, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
- 2Department of Immunology, Mayo Clinic College of Medicine and Science, Rochester, MN, United States
Tissue resident memory CD8 T cells (TRM) serve as potent local sentinels and contribute significantly to protective immunity against intracellular mucosal pathogens. While the molecular and transcriptional underpinnings of TRM differentiation are emerging, how TRM establishment is regulated by other leukocytes in vivo is largely unclear. Here, we observed that expression of PPAR-γ in the myeloid compartment was a negative regulator of CD8 TRM establishment following influenza virus infection. Interestingly, myeloid deficiency of PPAR-γ resulted in selective impairment of the tissue-resident alveolar macrophage (AM) compartment during primary influenza infection, suggesting that AM are likely negative regulators of CD8 TRM differentiation. Indeed, influenza-specific CD8 TRM cell numbers were increased following early, but not late ablation of AM using the CD169-DTR model. Importantly, these findings were specific to the parenchyma of infected tissue as circulating memory T cell frequencies in lung and TCM and TEM in spleen were largely unaltered following macrophage ablation. Further, the magnitude of the effector response could not explain these observations. These data indicate local regulation of pulmonary TRM differentiation is alveolar macrophage dependent. These, findings could aid in vaccine design aimed at increasing TRM density to enhance protective immunity, or deflating their numbers in conditions where they cause overt or veiled chronic pathologies.
Introduction
Residual CD8 T cells from primary responses form a long-lived immunological memory barrier poised at multiple anatomical sites (1–5). CD8 resident memory T cells (TRM) precluded from the circulatory system migrating within either non-lymphoid or secondary draining lymphoid organs, represent nearly one-third of the total CD8 memory T cell pool following primary responses (6–9). Compared to circulating conventional memory T cells [i.e., effector memory (TEM) and central memory (TCM)], TRM offer rapid local protection from pathogens (10, 11). This protective immuno-surveillance is highly dependent on TRM density which can be greatly enhanced through in situ self-renewal, replenishment from circulating memory T cells, and de novo T cell differentiation following a secondary exposure (6–9, 12). Yet, little is known about the local cellular immune-networks that locally mediate differentiation and thereby regulate initial TRM density in the lung and elsewhere.
CD8 TRM begin their differentiation in secondary lymphoid organs in the context of TCR, co-stimulatory, and cytokine receptor signaling derived from sufficiently activated dendritic cells (13–17). Exogenous uptake of viruses or infected cells by DCs followed by cross-presentation of viral peptide to CD8 T cells in secondary lymphoid organs markedly enhances TRM differentiation (18–23). Following priming, TRM cells derive from the memory-precursor effector cell (MPEC) pool (17, 24). These early memory precursors (CD127+KLRG-1Lo, including ex-KLRG-1 MPECs) are not just precursors to TRM, but also TCM (17, 24–27).
Remarkably, circulating memory CD8 T cells receive all the required cues provided by professional antigen presenting cells for appreciable clonal expansion and full functional differentiation in vivo within the first 3 days following an acute inflammatory infection (14, 17, 28–31). In contrast, TRM commitment windows occur within 7–14 days and appear to be influenced by much later factors in the context of an inflamed tissue environment commensurate with exposure to TGF-β (27, 32–35). Additional TCR and CD28 signaling and cytokines such as IL-7, IL-15, IL-12, IL-18, IL-21, Type I interferons, and TNFa as well as interactions with stroma and extracellular matrix may be further epitope, tissue, or pathogen-specific requirements for TRM differentiation and or maintenance (24, 36–46). Hence, CD8 TRM undergo a second stage of differentiation at the site of infection and though context-dependent, exhibit distinct differentiation and maintenance requirements relative to their circulatory memory counterparts programmed early after activation (14, 24, 32, 46).
The cellular networks involved in this extra stage of differentiation from naive to MPEC CD8 T cell, to that which establishes the transcriptional program required for TRM residency (43), are just now being worked out and the focus of this study. In a model of intestinal Yersinia pseudotuberculosis infection, inflammatory macrophages derived from bone-marrow monocytes (CCR2-dependent migration) accumulate and positively regulate the differentiation of CD103− TRM at the site of inflammation via provision of signal 3 cytokines (IL-12 and type I IFNs) that dampen CD103 expression (40, 47). Therefore, inflammatory cytokines provided by bone marrow-derived macrophages can endow heterogenous TRM sentinel programming in the gut. Similarly, in vaccinia virus infection, inflammatory monocytes (Ly6chi, CCR2-dependent) were responsible for long-term maintenance of a subset of pulmonary TRM without affecting clonal expansion or contraction (48). Further, a network involving CD4 TRM in the female reproductive tract (FRT) forms positive-feedback loops with local macrophages that promote low-level constitutive IFNγ production from the CD4 TRM resulting in organization of local macrophage-mediated immune-clusters outside of secondary lymphoid tissue (49). Once residency is established, function of CD8 TRM in the FRT depend on CD301b+ cDC2 to provide TCR signals in the lamina propria (50). Thus, a limited number of studies have implicated macrophages as positive regulators of CD4 and CD8 TRM establishment, but the origins of the tissue-resident macrophages in question are not clear in all cases.
In this study, we investigated a genetic model in which the alveolar macrophage compartment has a known functional deficit in influenza responses and found this was associated with increased local CD8 memory T cell density. A model in which alveolar macrophages were ablated prior to infection, but not during T cell contraction, also exhibited enhanced flu-specific resident memory T cell density compared to controls. In contrast, no major alterations in the circulating TEM or TCM influenza responses were observed following alveolar macrophage ablation. This work suggests that manipulation of the alveolar macrophage compartment may be an attractive target for modulating TRM density for therapeutics or vaccine design.
Materials and Methods
Mice and Infection
Lyz2-cre, and PPAR-γfl/fl (C57BL/6) were purchased from the Jackson (Harbor, ME) Laboratory and bred in house. C57BL/6 CD169-DTR (51, 52) were received from Professor Tanaka (Tokyo, Japan). In all cases, wild-type control mice were transgene or Cre-negative littermates. All mice were housed in a specific pathogen-free environment and used under conditions fully reviewed and approved by the internal animal care and use committee (IACUC, approval #A00002035) guidelines at the Mayo Clinic (Rochester, MN). For influenza virus infection, influenza A/PR8/1934 strain (~120 pfu/mouse) was diluted in FBS-free DMEM media (Corning) on ice and inoculated in anesthetized mice through intranasal route (35 μl) as described before (53). This low dose was chosen because higher, easily tolerable doses in wt animals leads to death in macrophage-depleted animals (54). Where applicable, mice were re-challenged on day 53 with influenza A/X-31 strain (1.2 × 105 pfu/mouse) with daily treatment of FTY720 (25 μg/mouse) starting 1 day before re-challenge.
Cell Depletions
For depletion of CD169-positive cells at time-points indicated in text and figure legends, CD169-DTR mice and DTR-littermate controls were injected with diphtheria toxin (Sigma, DTx, 300 ng/mouse) every 3 days from the time indicated. Data were analyzed at day 3, 10, or 22 post-infection (DTx treatment started day minus 1 or day 10 post-infection) to assess CTL response or depletion efficiency, or at day 42 or 53 to analyze effects of macrophage depletion on cellular makeup of the lung environment following intravital labeling of white blood cells in the circulation as described below.
Tissue Processing, Cellular Isolation, and Data Analysis
Animals were injected intravenously with 3 μg of CD45 or CD8β antibody labeled with various fluorochromes. Two minutes post-injection, animals were euthanized with an overdose of ketamine/xylazine. Following euthanasia, spleens were removed and BAL samples were taken by repeated gentle instillation and removal of PBS via the trachea as previously reported (55). The right ventrical was gently perfused with PBS (10 mL). Lungs were instilled with 1 mL of digestion buffer [90%DMEM 10% PBS + Calcium and Magnesium with 180 U/mL Type 2 Collagenase (Worthington) and 15 μg/mL DNase (Sigma) additives]. Tissue was processed on a gentleMACS tissue disrupter (Miltenyi) for 40 min at 37°C followed by hypotonic lysis of red blood cells in ammonium-chloride-potassium buffer and filtering through 70 μm mesh. Fc-gamma receptors were blocked with anti CD16/32 (2.4G2). Cell surfaces were immuno-stained with following cocktails of fluorochrome-conjugated Abs (Biolegend) Siglec-F (E50-2440), CD11c (N418), CD11b (M1/70), merTK (2B10c42), CD64 (X54-4/7.1), Ly6G (1A8), I-A/I-E (M5/114.15.2), Ly6C (HK1.4), immuno-staining was performed at 4°C for 30 min. Cells were washed twice with FACS buffer (PBS, 2 mM EDTA, 2% FBS, 0.09% Sodium Azide), prior to fixation and ran on an Attune NxT auto sampler (Life Technologies). FCS files for myeloid stains were analyzed with FlowJo 10.2 (Tree Star) and processed in a similar way to previous reports (56) as shown in Supplementary Figure 1. Following intra vital labeling, single cell lung or spleen suspensions were immuno-stained with antibodies against: CD8α (53-6.7) CD69 (H1.2F3), CD44 (IM7), CD11a (M17/4), CD103 (2E7), PD-1 (29F.1A12), and NP366−374-Db and PA224−233-Db tetramers (NIH Tetramer Core Facility) on ice for 45–60 min. Following exclusion of doublets and gating on total CD8 T cells, residency was determined by increased CD69 expression on CD8α+ cells protected from circulatory labeling per Supplementary Figure 1. Nearly 100% of cells in the lung defined this way express hi levels of CD44 at the time-points investigated. CD103 was ignored in analyses because in the lung 6 weeks after infection, CD103 expression is TRM epitope-specific (46). Central and Effector memory CD8 T cells (TCM and TEM, respectively) were differentiated by CD62L expression (TCM = Hi, TEM = Lo) on splenic CD8 T cells at least 42 days post-infection that were Db-NP or -PA-tetramer+ CD44Hi CD127+.
Statistical Analysis
Quantitative data are presented as mean ± Standard of Deviation. Unpaired two-tailed Student's t-test (two-tailed, unequal variance) were used to determine statistical significance with Prism software (Graphpad). We considered α < 0.05 as significant in all statistical tests and denoted within figures as a *.
Results
Myeloid PPAR-γ Deficiency Increases CD8 TRM Establishment
Peroxisome proliferator-activated receptor gamma (PPAR-γ) is a member of the nuclear hormone receptor superfamily. It is a lipid sensing transcription factor that regulates lipid uptake and glucose metabolism (57). Because of the vital importance of the myeloid compartment to mucosal respiratory infections (54), we investigated a model from mice with a conditional PPAR-γ deletion known to have prolonged recovery from influenza infection in multiple contexts, including obesity (54, 58, 59). In this model, Lyz2-driven Cre expression in PPAR-γfl/fl animals present with transgene penetrance in macrophages, neutrophils and circulating monocytes; however, only alveolar macrophages expressed significant amounts of PPAR-γ protein. We and others previously found that PPAR-γ in alveolar macrophages suppressed inflammation and accelerated recovery following influenza or RSV infections without affecting viral clearance (54, 59). This genetic model exhibits enhanced acute morbidity and prolonged recovery from influenza infection measured by changes in body weight (Figure 1A).
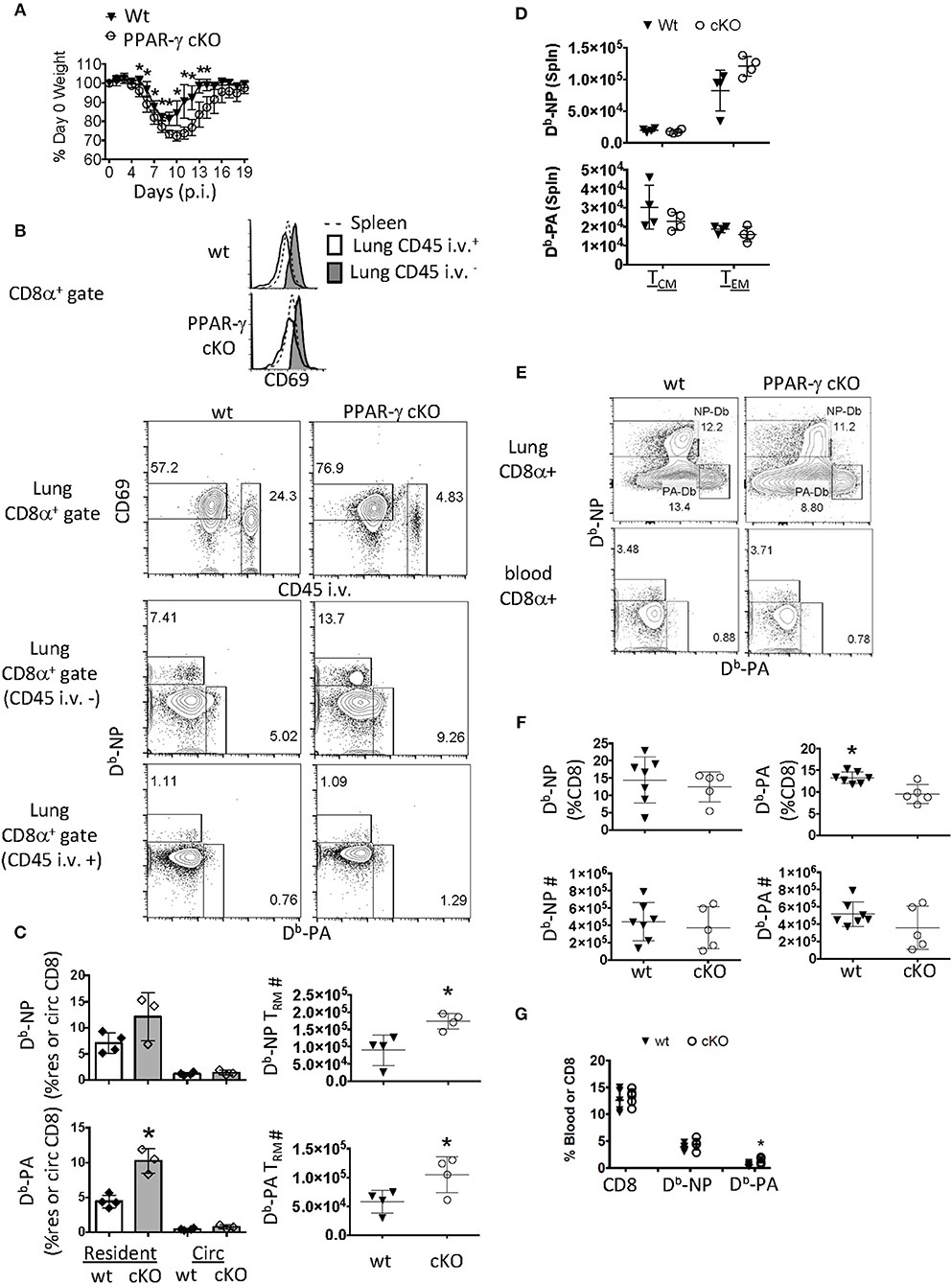
Figure 1. PPAR-γ (Lyz2-Cre cKO) mice exhibit enhanced flu-specific TRM response. Wt C57BL/6 and PPAR-γfl/fl x Lyz2-Cre mice were infected with PR8 influenza virus. (A) Percentage of initial body weight was monitored from day 0 to 19. (B) CD8 T cells were assayed by flow cytometry day 60 post-infection. From lung digests and spleens following intravenous injection of CD45 Ab (CD45 i.v.) to label circulating white blood cells. The lung-resident (CD45 i.v.–, filled), lung circulating (CD45 i.v.+, open) and splenic (dotted) CD8 T cell compartments were assessed for CD69 expression (top panel). Dot plots of CD69 and CD45 i.v. staining in whole lung CD8 T cells (second panel) and Db-NP and PA specific CD8 T cells in each of the lung compartments (bottom two panels). (C) Frequencies (left) or numbers (right) of influenza-specific Db-NP and Db-PA tetramer+ cells were quantitated by flow cytometry. Resident memory T cells were defined as (CD8α+ CD69+ tetramer + CD45− i.v.–) and circulating (Circ) memory T cells were defined as (CD8α+ tetramer+ CD45+ i.v). (D) Numbers of influenza-specific Db-NP (top) and Db-PA tetramer+ cells (bottom) in the spleens were measured by flow cytometry. Central (TCM) and effector (TEM) memory T cells from the spleen were defined as CD8α+ CD44Hi CD127+tetramer+ and were, respectively, CD62LHi/Lo. (E–G) Lung and blood effector T cells were quantified at 10 days post infection. (E) Representative FACS-plot of Db-NP or Db-PA tetramer staining in CD8 T cells of the whole lung or blood. (F) Frequencies (upper panel) and numbers (lower panel) of influenza-specific Db-NP and Db-PA tetramer+ CD8 T cells in lungs. (G) Frequencies of Db-NP and Db-PA tetramer+ CD8 T cells in blood. *p < 0.05 for cKO compared to wt. Data are representative of three experimental replicates, except (D,G).
Given this phenotype, we surmised that the flu infection was more severe in cKO mice and that the local CD8 memory T cell response may reflect this (60). Sixty days following infection, we noticed a selective upregulation of CD69 in the parenchymal CD8 T cell compartment of wt and cKO mice compared to CD8 T cells circulating through the lung or spleen (Figure 1B, top panel). The gating scheme used for resident and circulating flu-specific T cells throughout the manuscript can be seen in Supplementary Figure 1A. We observed an increase in frequency and number of lung-resident CD69+ CD8 T cells precluded from circulation from cKO vs. wt animals 60 days post-infection (p.i.) (Figure 1B, second and third panels). Flu-specific polyclonal CD8 TRM cells against Db-restricted NP366−374 and PA224−233 epitopes increased 2-fold each in cKO animals relative to wt controls. This difference was not seen in memory T cells circulating through the lung (Figure 1B, bottom panel and Figure 1C). These changes in local TRM density could not be explained by differences in circulating memory T cells as no changes were observed in the magnitude of TEM or TCM responses in the spleen (Figure 1D). An increase in TRM establishment might readily be explained if cKO animals had a larger effector T cell pool early in the immune response (61). However, in this regard, we found that although cKO and wt littermates had differences in CD8 TRM cell frequency, CD8 effector T cell numbers in whole lungs were unchanged 10 days post-infection (Figures 1E,F). There was a slight increase in frequency of Db-PA CD8 T cells from cKO animals in the peripheral blood (Figures 1E,G), however, as noted, this did not carry over into differences in the number of circulatory TEM or TCM cells. Therefore, Lyz2-driven PPAR-γ deficiency led to enhanced TRM establishment without majorly affecting influenza-specific effector or circulatory memory T cell responses.
Myeloid PPAR-γ Deficiency Impairs AM Compartment Following Influenza Infection
Since the macrophage compartment has been shown to influence TRM number (40, 48), we examined two populations of lung macrophages including fetal-derived alveolar (CD64+ CD11bint Siglec Fhi) and adult monocyte-derived macrophages (CD64+ CD11bhi Siglec FLo) prior to and after influenza infection by flow cytometry in whole lung and airway lumen (Figures 2A–C). Gating strategy for myeloid cells in lung compartments is displayed in Supplementary Figure 1B. The pulmonary myeloid compartment showed a kinetic decrease of tissue-resident alveolar macrophage, but not monocyte-derived macrophages or circulating monocyte numbers 4, 10, 15, and even 30 days post-infection in cKO vs. wt lungs and bronchial alveolar lavage fluid (BAL). Together, the data indicated that PPAR-γ in the myeloid compartment is important for AM response kinetics following influenza infection and has moderate effects on the magnitude of other myeloid cell responses in the lung.
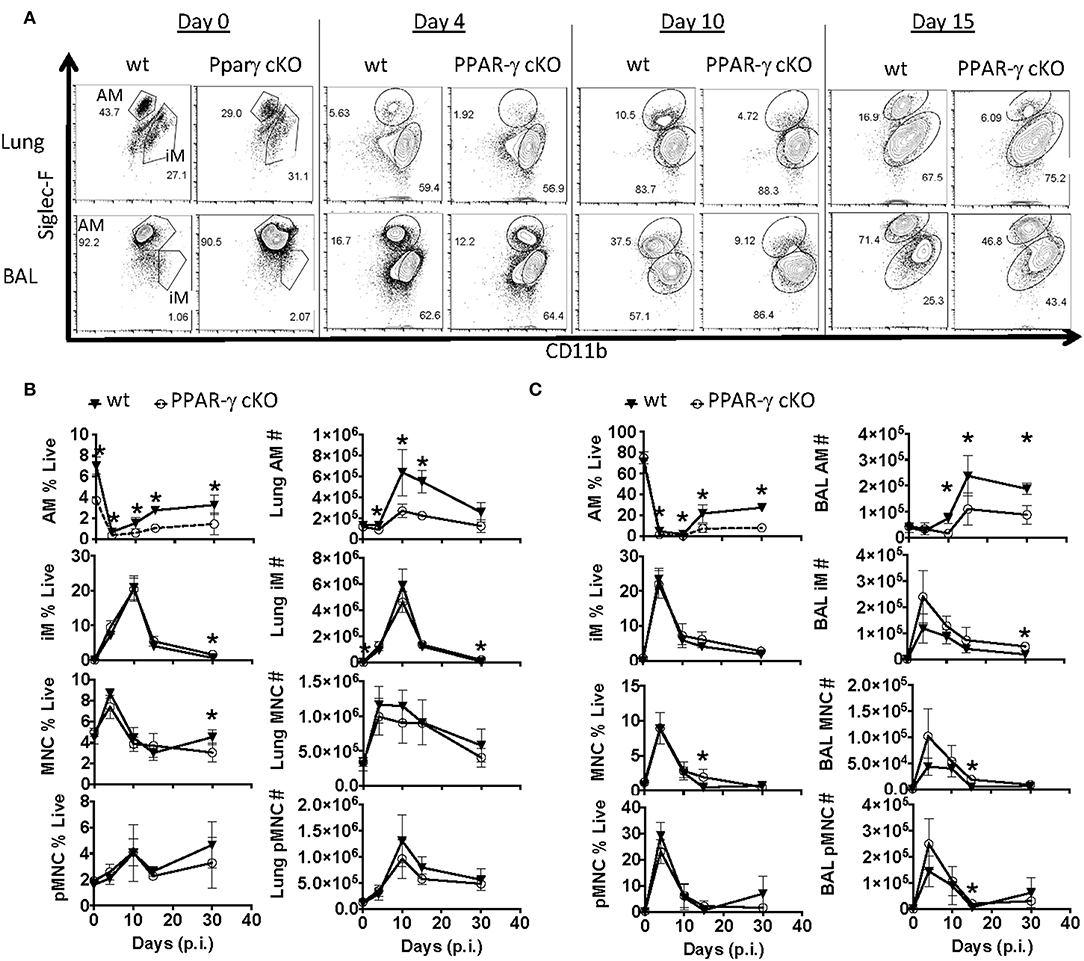
Figure 2. PPAR-γ cKO mice exhibit abnormal alveolar macrophage kinetics following influenza challenge. Wt C57BL/6 and PPAR-γf (cKO) mice were infected with influenza PR8 virus. Alveolar macrophage (CD64+ Siglec-FHi CD11bint), inflammatory macrophage (CD64+ Siglec-FLo CD11bHi), monocyte (CD64− Siglec-F− Ly6G− CD11bHi) and neutrophil (Ly6GHi CD11bHi) frequencies and numbers were determined by flow cytometry. (A) Representative FACS plots of alveolar macrophages (AM) and inflammatory macrophages (iM) within lung (top panel) or BAL fluid (bottom panel). (B,C) Frequencies (left) and numbers (right) of alveolar macrophages (AM), inflammatory macrophages (iM), monocytes (MNC), and neutrophils (pMNC) in (B) lung and (C) BAL at indicated days post-infection (p.i.). *p < 0.05 for cKO compared to wt. Data are compiled from 2 to 3 experimental replicates.
Early AM Depletion Minimally Affects Effector CD8 T Cell Response
Since we have narrowed down the influenza response defect in these mice to alveolar macrophages (59) while observing their frequency changes correlated with TRM establishment, we suspected alveolar macrophages may be playing a heretofore unappreciated role in CD8 TRM differentiation and or maintenance. To assess whether alveolar macrophages were important for TRM establishment, we utilized a transgenic model (CD169-DTR) (51, 52, 62). Parabiosis experiments demonstrate the vast majority of these CD169-expressing cells are tissue-resident macrophages in gut and kidney models (51, 52). With regards to the lung, we reasoned we were primarily depleting resident alveolar macrophages in a well-characterized model (51, 52, 63, 64). To verify this assumption, we monitored ablation efficiency by Immuno-staining myeloid cells from the naïve lungs of CD169-DTR and littermate control animals treated once with Diptheria toxin (DTx). We then examined CD169 expression in CD64 positive macrophages and CD64 negative cells in both the circulation and parenchyma 3 days later (Figure 3A). Higher CD169 expression was evident in the CD64+ vs. negative compartment. Importantly, DTx treatment did not cause changes in the low levels of CD169 expression in CD64− cells, suggesting DTx treatment did not majorly affect the CD64− compartment (Figure 3B). The vast majority (93.1%) of CD169+ cells in lungs were resident alveolar macrophages that were largely depleted following DTx treatment (Figures 3B,C). Simultaneously, there was an enrichment of monocyte derived macrophages (CD11bhi SiglecFLo CD64+), Ly6CLo circulating monocytes, and circulating neutrophils (Figure 3C). To understand the model in the context of influenza infection, we administered DTx 1 day prior to infection and characterized lungs 3 days later (Figure 3D). Alveolar macrophages were clearly decreased from the pulmonary compartment in contrast to inflammatory macrophages. As previously reported (63), this was also accompanied by an increase in circulating monocyte and neutrophil numbers as seen in the uninfected animals (Figure 3D). Therefore, within the lung, DTx treatment in CD169-DTR mice rather selectively depleted Siglec Fhi alveolar macrophages (CD64+ CD11chi CD11bint), while leading to numeric increases in other inflammatory myeloid populations in the presence or absence of infection. This population has been shown to be a CCR2-independent resident population from an embryonic origin that self-maintains through homeostatic proliferation even after depletion by DTx (CD169-DTR mice) or respiratory virus challenge (62).
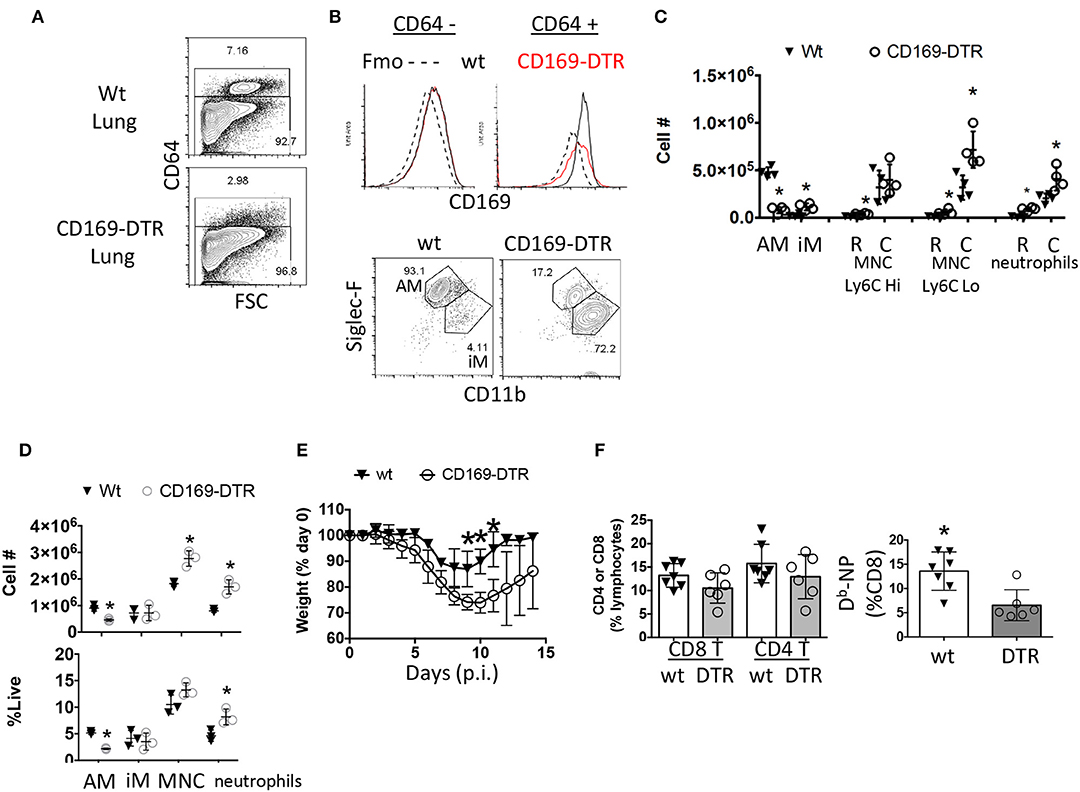
Figure 3. Early ablation of CD169+ cells leads to enhanced morbidity without enhancing circulating effector CD8 T cell responses. (A–C) Naïve Wt littermates or CD169-DTR mice were treated with DTx then euthanized 4 days later. (A) CD64 positive and negative compartments were assessed by flow cytometry. (B) CD169 expression in CD64 positive and negative cells from the lung were measured by flow cytometry and compared to a fluorescent-minus-once (Fmo) control for CD169 antibody specificity (top). CD64+ cells were classified as AM (Siglec-FHi CD11bint) or iM (Siglec-FLo CD11bHi) (B, bottom). (C) Lung resident (R, CD45− i.v.) and circulating (C, CD45+ i.v.) alveolar macrophages (AM) inflammatory macrophages (iM), monocytes (MNC), and neutrophils were quantitated by flow cytometry. (D–F) Wt littermates or CD169-DTR mice were treated with DTx then infected with influenza PR8. (D) Lung myeloid cell populations were quantified by flow cytometry at day 3 post-infection. (E) Percentage of initial body weight was measured daily for 14 days post-infection (p.i.). (F) Percentage of CD4 and CD8 T cells (left) or % of Db-NP-specific CD8 T cells from blood PBMCs 10 days post-infection. *p < 0.05 for CD169-DTR compared to wt. Data are representative of 2–3 experimental replicates except (A–C).
Macrophage numbers recover by 7 days post DTx treatment in CD169-DTR BALB/c mice leading us to choose every third day to administer DTx (51, 52). As has been previously reported (63), absence of CD169+ cells prior to infection led to prolonged recovery of weight loss following influenza infection (Figure 3E). We found a small, but noticeable decrease in Flu-specific Db-NP effector cell frequency in the blood in the CD169-DTR model (Figure 3F). Thus, depletion of alveolar macrophages mimicked morbidity in the conditional PPAR-γ knockout animals with impaired macrophage function and did not enhance the magnitude of the CD8 T cell response in the circulation.
Early AM Depletion Promotes TRM Development
In contrast to the comparable effector CD8 T cell responses, 6 weeks post-infection, the depletion of alveolar macrophages pre-infection (Figure 4A) resulted in significant increases in flu-specific (NP and PA Db-restricted epitopes) CD8 TRM frequencies (Figure 4B). However, AM depletion did not alter memory CD8 T cells against PA and NP epitopes circulating through the lung or in the spleen 42 days after infection with the exception of a minor increase in PA-specific CD8 T cell frequency in the lung vasculature (Figures 4B–E). We wondered if this increased TRM density could enhance protective immunity. To test this, we withdrew DTx treatment 42 days after infection and rechallenged the mice at day 53 to allow the reconstitution of the AM compartment. We observed increased lung CD8 TRM, but not circulating CD8 memory T cells or splenic TCM and TEM numbers against both NP and PA epitopes prior to the re-challenge in this model (Figures 4F,G). AM numbers were equivalent between CD169-DTR and littermate controls (Figure 4H). We also observed an increased number of inflammatory macrophages at day 53 with no changes in neutrophils or monocytes either in the parenchyma or circulation (Figure 4I).
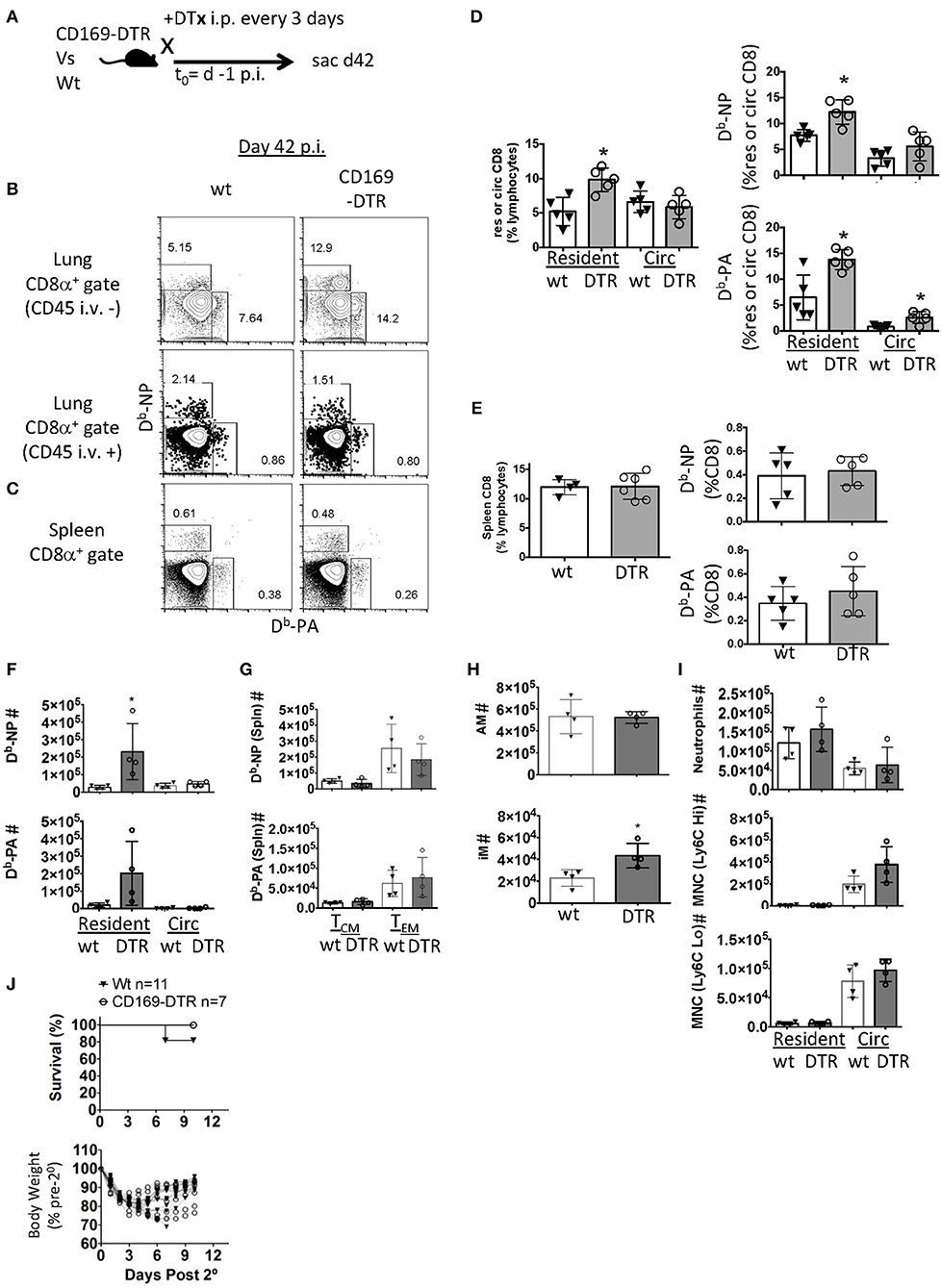
Figure 4. Early alveolar macrophage ablation enhances flu-specific TRM establishment. (A) Wt C57BL/6 or CD169-DTR mice were infected with influenza PR8. (A) Schematic of experimental design showing that mice were treated with DTx on day minus 1 and every third day, until day 42 p.i. (B) Representative FACS plots of lung resident (CD45 i.v.–) or circulating (CD45 i.v.+) Db-NP or Db-PA memory T cells within CD8 T cell compartment in Wt or CD169-DTR mice. (C) Representative FACS plots of splenic Db-NP or Db-PA memory T cells within the CD8 T cell compartment in Wt or CD169 DTR mice. (D) Frequencies of CD8 T cells in lung resident or circulating lymphocytes (left). Frequencies of influenza-specific Db-NP and Db-PA tetramer+ memory T cells in lung resident or circulating (Circ) CD8 T cells (D, right). (E) Frequency of CD8 T cells in splenic lymphocytes (left). Frequencies of influenza-specific Db-NP and Db-PA tetramer+ memory T cells in splenic CD8 T cells (E, right). (F–J) C57BL/6 or CD169-DTR mice were infected with influenza PR8. Mice were treated with DTx on day minus 1 and every third day until day 42 p.i. mice were sacrificed or rechallenged with influenza X31 at day 53 pi. (F) Numbers of lung resident (CD45 i.v.–) or circulating (CD45 i.v.+) Db-NP or Db-PA memory T cells at day 53 p.i. (G) Numbers of splenic Db-NP or Db-PA memory T cells at day 53 p.i. (H) Alveolar macrophages (AM, top) or inflammatory macrophages (IM, bottom) were quantitated in the lungs. (I) Neutrophils, Ly6CHi or Ly6CLo monocytes in the parenchyma (CD45 i.v.–) or circulation (CD45 i.v.+). (J) Mice were treated with FTY720 at day 52 p.i. and re-infected with X-31 on day 53. FTY720 treatment was maintained throughout the challenge phase. Hose survival (top) and weight loss and recovery of individual mice (bottom) were measured to Wt C57. (B–E) Antigen-specific lung resident, circulating (Circ) and splenic Db-NP and -PA CD8 T cell frequencies were measured. DTx treatment was stopped on day 42 and (F) flu-specific lung-circulating, –resident, and (G) splenic central and effector memory cells were quantitated on day 53. (H,I) Myeloid cells were quantitated in the parenchyma and circulation. (J) Mice were treated with FTY720 at day 52 p.i. and re-infected with X-31 on day 53. FTY720 treatment was maintained and survival (top) and weights (bottom) were measured to day 10 following X-31 infection (days post 2°). *p < 0.05 for CD169-DTR compared to wt. Data are representative of three experimental replicates except (F–J) which are single experiments.
To investigate TRM contribution to secondary immunity in this model, PR8-immune mice were treated with S1P1R antagonist (FTY720) to prevent lymphocytes access to the circulation. Mice were then challenged with a high dose of a heterosubtypic influenza A strain (H3N2, X-31). Two of the Wt mice died following high does of X-31 (n = 11) while no CD169-DTR mice were lost (n = 7), however this was not a statistically significant finding. We also observed no significant differences in changes in weight loss or recovery (Figure 4J). Together the data suggested alveolar macrophage depletion caused enhanced establishment of TRM despite a blunted effector pool, although whether this enhanced TRM presence is associated with increased immune protection requires further studies.
Late AM Depletion Does Not Enhance TRM Responses
The preceding experiments did not indicate when alveolar macrophages might be playing a role to limit TRM establishment or maintenance. To address whether alveolar macrophages are needed early vs. late in the effector response to influence local T cell immunity, we next depleted alveolar macrophages 10 days post-infection and maintained the DTx regimen until d42 post-infection (Figure 5A). Following 12 days of this regimen, AM were depleted 100-fold in the BAL and 20-fold in the lung (Figure 5B). This was associated with a skewing of circulating monocyte frequencies, but not numbers (Figure 5B). In contrast to the effects of early alveolar macrophage depletion, loss of CD169+ cells during T cell contraction did not significantly change the establishment of Flu-specific TRM, lung-circulating, or splenic memory CD8 T cells (Figures 5C–G). Since these results, with respect to local resident memory CD8 T cells, conflict with models ablating CCR2-dependent bone marrow-derived mucosal macrophages (40), it is therefore plausible that embryonic-derived resident macrophages and inflammatory macrophages have distinct functions in the differentiation and or maintenance of CD8 TRM (Figure 5H).
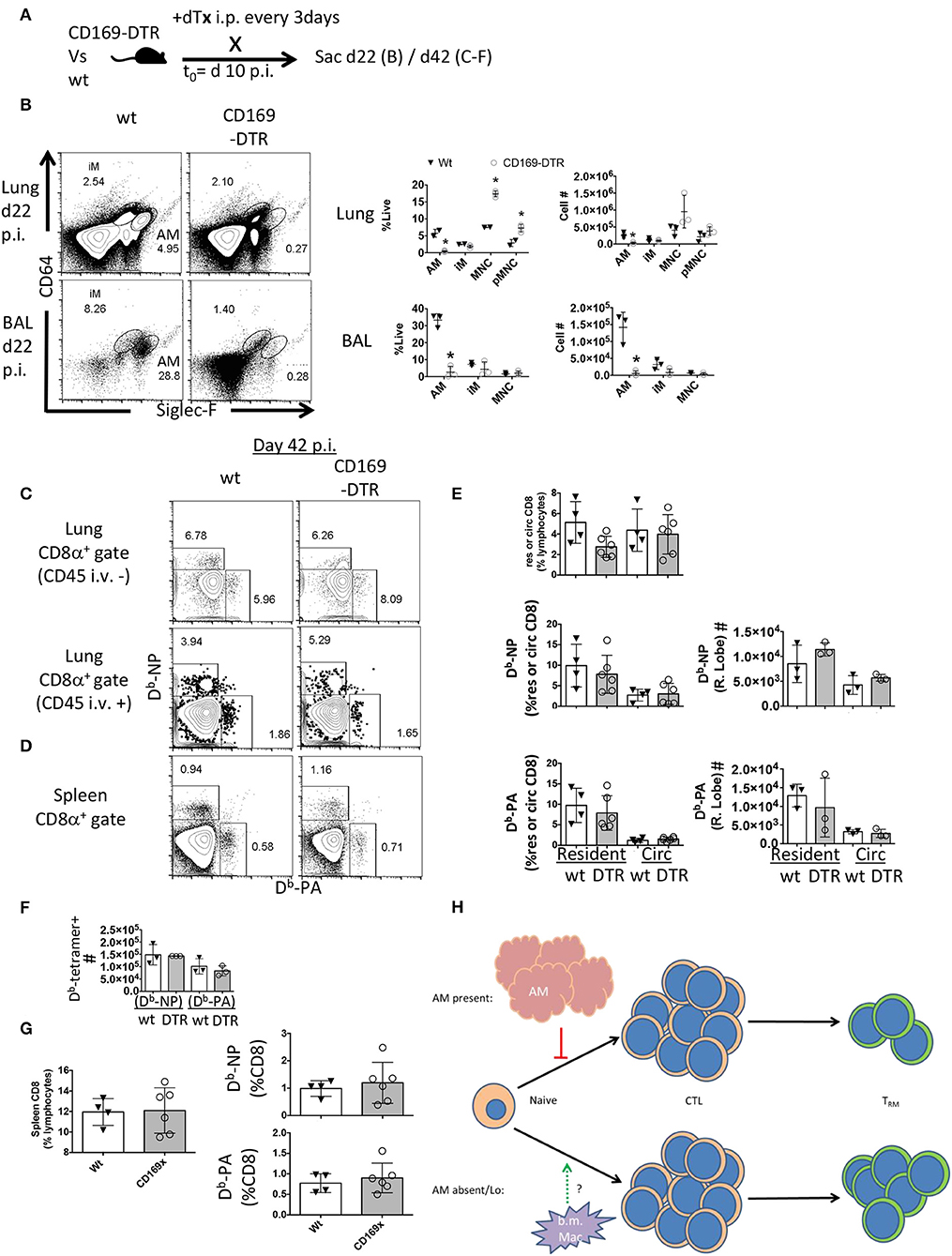
Figure 5. Late CD169+ ablation does not affect TRM establishment. Wt C57BL/6 and CD169-DTR mice were infected with influenza. (A) Experimental schematic showing DTx regimen began day 10 post-influenza PR8 infection. (B) Representative FACS plots of alveolar macrophages (AM) and inflammatory macrophages (iM) in Wt or CD169 DTR mice (left). Frequency (middle) and cell number (right) of AM, iM, and monocytes (MNC) 22 days post influenza infection in the Lung (top) and BAL fluid (bottom). (C) Representative FACS plots of influenza-specific Db-NP and Db-PA tetramer+ memory resident (CD45 i.v.–) and circulating (CD45 i.v.+) CD8 T cells in the lung 42 days post-infection. (D) Representative FACS plots of Db-NP and Db-PA tetramer+ memory. (E) Frequencies of lung CD8 T cells in lung lymphocyte populations in the parenchyma (resident) or circulation (Circ) (top) or of Db-tetramer positive memory T cells in the CD8 compartments (left) with Numbers of same (right). (F) Numbers of splenic Db-NP or Db-PA memory CD8 T cells at day 42 p.i. (G) Frequencies of CD8 T cells from lymphocytes in the spleen (left) or Db-NP or Db-PA memory T cells in the CD8 compartment at day 42 p.i. (right). (H) Model indicating the early, but not late presence of resident alveolar macrophages (AM) regulates the CD8 resident memory T cell compartment size without affecting magnitude of effector (CTL) response. Data are representative of three experimental replicates, except (F) which is a single experiment. *p < 0.05 for CD169-DTR compared to wt.
Discussion
Our findings demonstrate TRM differentiation is regulated early by tissue-resident alveolar macrophages largely independent of the magnitudes of either the effector CTL response or circulating memory T cell pools. This study therefore suggests alveolar macrophages regulate an intercellular immune-network unique to the inflamed tissue and, under certain circumstances, might influence local secondary immunity by governing mucosal TRM density. Conversely, previous studies have revealed that CCR2-dependent monocytes and macrophages are important in the development of CD8 TRM following infection (40, 47). Thus, it is possible that bone-marrow and embryonic-derived macrophages exhibit opposing roles for TRM differentiation (40, 62). Alternatively, macrophage subsets could differentially influence local adaptive immune responses dependent on location or nature of the pathogen-specific response. Though we did not narrow observations to macrophage-intrinsic vs. extrinsic in this study, mechanistically, we find a role for PPAR-γ in the myeloid compartment in limiting TRM density, potentially contributing to blunted secondary and or heterosubtypic immunity. We have independently found that PPAR-γ from alveolar macrophages serves to limit immune response-induced long-term damage following influenza infection demarking this pathway in macrophages as a signaling axis for immunity and pathology (unpublished).
Though we did not find a role for enhanced immunity in mice with early macrophage depletion under the tested conditions, this model likely abolishes any innate memory features that AM develop from viral infections in an effector CD8 T cell-help dependent manner (65, 66). Trained immunity of local innate cells contribute to subsequent responses long after a primary infection. Since we only tested one viral dose in the secondary challenge model, more investigation is required to determine whether protective immunity is enhanced when AM are absent early from a primary response and whether this is dependent on changes in TRM density.
CD8 T cell Immune responses to influenza infection primarily involve the lung and lung-draining mediastinal lymph nodes (67). Peripheral macrophages have not been reported to be involved in CD8 T cell responses to influenza (63). Given that macrophages are not playing a role in peripheral memory T cell differentiation, in contrast to the local infection site in this study, we think it likely that increases in CD8 TRM in our model are from depleting embryonic-derived alveolar macrophages (62). There is also a transient influx of neutrophils into lungs of diphtheria toxin treated CD169-DTR animals compared to wt while not affecting DCs in lung draining lymph nodes (63, 64). Notably, acute neutrophil depletion, at a time when we observed AM playing a role, has shown no relevance to TRM differentiation or maintenance in influenza models despite delaying CTL recruitment (68, 69). Further, macrophages in secondary lymphoid tissue aid in transferring antigen to cross-priming DCs (70). Therefore, we would expect opposite results with regards to long-term TRM establishment if non-local macrophages were playing a major role in that regard (19). We find it more plausible that the loss of embryonic-derived alveolar macrophages released homeostatic controls gearing the lung environment to further favor TRM differentiation.
We do not anticipate that effector T cells are interacting directly with AM given that retained TRM are eventually found in the interstitial tissue and alveolar macrophages are confined within the smallest of airways. Yet, we do not discount this possibility for memory T cells found within alveoli (44, 60, 71, 72). Rather, together with others', our data indicates an abrupt loss of resident macrophage function or number results in this niche temporarily being replaced by monocyte-derived macrophages that migrate into tissues and positively influence TRM differentiation and or maintenance (40, 48). More thorough studies addressing the leukocyte kinetics, direct links, and model disparities are needed. Our data do not refute a model whereby embryonic-derived alveolar macrophages limit known niches for TRM by limiting the damage or disrepair of local inflamed tissue following respiratory viral clearance (60, 73–75).
Surprisingly, late ablation of alveolar macrophages (DTx treatment at day 10), did not influence TRM differentiation nor was their continual depletion necessary when ablation was initiated prior to infection (Figure 4F). Collectively, this suggests alveolar macrophages existing prior to pathogen encounter play an early role in imposing their limits on TRM programming. The direct or indirect mechanisms by which early TRM differentiation is regulated by alveolar macrophages is currently being pursued. Ultimately, altering TRM density through manipulation of the local macrophage pool may circumvent patient-specific roadblocks, such as an individual's HLA alleles or T cell repertoires, which restrict off-the-shelf immunotherapies.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The studies involving animals were fully reviewed and approved by the internal animal care and use committee (IACUC, approval #A00002035) guidelines at the Mayo Clinic (Rochester, MN).
Author Contributions
Experiments were designed by NG, SH, and JS. NG and SH performed and analyzed the experiments. NG and JS prepared the manuscript. All the other authors provided substantial insight that drove experimental design.
Funding
This study was funded in part by the US National Institutes of Health RO1s AI112844, AG047156, and HL126647 (JS), T32 AG049672 (NG), Mayo Clinic Kogod Aging Center Pilot grant and Mayo Clinic Center for Biomedical Discovery discretionary fund to JS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank NIH tetramer core facility for providing tetramers for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02332/full#supplementary-material
Supplementary Figure 1. Gating scheme for circulating and parenchymal myeloid cells and TRM. 53 days post-infection per DTx regimen in Figures 4F–I, lung digests (Wt shown) were performed after intravenous labeling of circulating leukocytes with anti CD45. (A) Following lymphocyte gating in lung samples and exclusion of doublets, viable (ZomB dye Lo) CD8 T cells were gated and compartmentalized into CD69+CD45 i.v.– (resident) or CD45 i.v.+ (circulating) populations (left) and Db-NP and Db-PA tetramer positive memory CD8 T cells in each compartment (right). (B) Myeloid cells from the same experiment were examined after excluding doublets from total lung cells and gating on live cells (ZomB dye Lo). Alveolar (AM) and inflammatory (iM) macrophages were segmented by Siglec-F and CD11b expression in cells expressing CD64 (top panel). Ly6GHi cells were examined for CD11b expression and classified as neutrophils (pMNC) and separated into resident vs. circulating on basis of CD45 i.v. staining (–resident, + circulating; middle right panel). Monocytes (MNC) were CD11bHi CD64− cells excluded for DCs (CD11cHi MHCIIHi), neutrophils (Ly6GHi+ CD11bHi), and eosinophils (CD64− Siglec-F−). Monocytes were compartmentalized by Ly6C expression and whether they had access to the circulation (CD45 i.v.+) or not (CD45 i.v.–).
References
1. Jungi TW. Immunological memory to Listeria monocytogenes in rodents: evidence for protective T lymphocytes outside the recirculating lymphocyte pool. J Reticuloendothel Soc. (1980) 28:405–17.
2. Jungi TW, Jungi R. Immunological memory to Listeria monocytogenes in rodents. IV Studies on origin and fate of tissue-positioned T memory cells. Immunology. (1981) 44:789–98.
3. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. (2013) 31:137–61. doi: 10.1146/annurev-immunol-032712-095954
4. Carbone FR. Tissue-resident memory T cells and fixed immune surveillance in nonlymphoid organs. J Immunol. (2015) 195:17–22. doi: 10.4049/jimmunol.1500515
5. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. (2011) 477:216–9. doi: 10.1038/nature10339
6. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. (2014) 346:98–101. doi: 10.1126/science.1254536
7. Schenkel JM, Fraser KA, Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol. (2014) 192:2961–4. doi: 10.4049/jimmunol.1400003
8. Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. (2018) 19:173–82. doi: 10.1038/s41590-017-0029-3
9. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. (2018) 19:183–91. doi: 10.1038/s41590-017-0027-5
10. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science. (2014) 346:101–5. doi: 10.1126/science.1254803
11. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. (2009) 10:524–30. doi: 10.1038/ni.1718
12. Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting edge: tissue-resident memory T cells generated by multiple immunizations or localized deposition provide enhanced immunity. J Immunol. (2017) 198:2233–7. doi: 10.4049/jimmunol.1601367
13. Janeway CA Jr. A trip through my life with an immunological theme. Annu Rev Immunol. (2002) 20:1–28. doi: 10.1146/annurev.immunol.20.080801.102422
14. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. (2009) 183:1695–704. doi: 10.4049/jimmunol.0900592
15. Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature. (2011) 471:629–32. doi: 10.1038/nature09863
16. Akue AD, Lee JY, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol. (2012) 188:2516–23. doi: 10.4049/jimmunol.1102213
17. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1+ effector CD8+ T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity. (2018) 48:716–29 e718. doi: 10.1016/j.immuni.2018.03.015
18. Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci USA. (2006) 103:10729–34. doi: 10.1073/pnas.0601956103
19. Iborra S, Martinez-Lopez M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, et al. Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity. (2016) 45:847–60. doi: 10.1016/j.immuni.2016.08.019
20. Enamorado M, Khouili SC, Iborra S, Sancho D. Genealogy, dendritic cell priming, and differentiation of tissue-resident memory CD8+ T cells. Front Immunol. (2018) 9:1751. doi: 10.3389/fimmu.2018.01751
21. Wakim LM, Smith J, Caminschi I, Lahoud MH, Villadangos JA. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol. (2015) 8:1060–71. doi: 10.1038/mi.2014.133
22. Waithman J, Zanker D, Xiao K, Oveissi S, Wylie B, Ng R, et al. Resident CD8+ and migratory CD103+ dendritic cells control CD8 T cell immunity during acute influenza infection. PLoS ONE. (2013) 8:e66136. doi: 10.1371/journal.pone.0066136
23. Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, et al. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat Commun. (2014) 5:4674. doi: 10.1038/ncomms5674
24. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
25. O'Sullivan JA, Zloza A, Kohlhapp FJ, Moore TV, Lacek AT, Dulin NO, et al. Priming with very low-affinity peptide ligands gives rise to CD8+ T-cell effectors with enhanced function but with greater susceptibility to transforming growth factor (TGF)β-mediated suppression. Cancer Immunol Immunother. (2011) 60:1543–51. doi: 10.1007/s00262-011-1043-1
26. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity. (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
27. Ma C, Zhang N. Transforming growth factor-beta signaling is constantly shaping memory T-cell population. Proc Natl Acad Sci USA. (2015) 112:11013–7. doi: 10.1073/pnas.1510119112
28. Prlic M, Hernandez-Hoyos G, Bevan MJ. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J Exp Med. (2006) 203:2135–43. doi: 10.1084/jem.20060928
29. van Blijswijk J, Schraml BU, Reis e Sousa C. Advantages and limitations of mouse models to deplete dendritic cells. Eur J Immunol. (2013) 43:22–6. doi: 10.1002/eji.201243022
30. van Blijswijk J, Schraml BU, Rogers NC, Whitney PG, Zelenay S, Acton SE, Reis e Sousa C. Altered lymph node composition in diphtheria toxin receptor-based mouse models to ablate dendritic cells. J Immunol. (2015) 194:307–15. doi: 10.4049/jimmunol.1401999
31. Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. (2002) 17:211–20. doi: 10.1016/S1074-7613(02)00365-5
32. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
33. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. (2013) 39:687–96. doi: 10.1016/j.immuni.2013.08.019
34. Hu Y, Lee YT, Kaech SM, Garvy B, Cauley LS. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J Immunol. (2015) 194:2407–14. doi: 10.4049/jimmunol.1402369
35. Yu CI, Becker C, Wang Y, Marches F, Helft J, Leboeuf M, et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity. (2013) 38:818–30. doi: 10.1016/j.immuni.2013.03.004
36. Kohlmeier JE, Miller SC, Woodland DL. Cutting edge: antigen is not required for the activation and maintenance of virus-specific memory CD8+ T cells in the lung airways. J Immunol. (2007) 178:4721–5. doi: 10.4049/jimmunol.178.8.4721
37. Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, et al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med. (2015) 21:1272–9. doi: 10.1038/nm.3962
38. Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol. (2016) 17:414–21. doi: 10.1038/ni.3396
39. Tian Y, Cox MA, Kahan SM, Ingram JT, Bakshi RK, Zajac AJ. A Context-Dependent Role for IL-21 in Modulating the Differentiation, Distribution, and Abundance of Effector and Memory CD8 T Cell Subsets. J Immunol. (2016) 196:2153–66. doi: 10.4049/jimmunol.1401236
40. Bergsbaken T, Bevan MJ, Fink PJ. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8+ Tissue-Resident Memory T Cells. Cell Rep. (2017) 19:114–24. doi: 10.1016/j.celrep.2017.03.031
41. Pavelko KD, Bell MP, Harrington SM, Dong H. B7-H1 influences the accumulation of virus-specific tissue resident memory T cells in the central nervous system. Front Immunol. (2017) 8:1532. doi: 10.3389/fimmu.2017.01532
42. Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol. (2017) 2:aag2031. doi: 10.1126/sciimmunol.aag2031
43. Behr FM, Chuwonpad A, Stark R, van Gisbergen K. Armed and ready: transcriptional regulation of tissue-resident memory CD8 T cells. Front Immunol. (2018) 9:1770. doi: 10.3389/fimmu.2018.01770
44. McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol. (2018) 11:1071–8. doi: 10.1038/s41385-018-0003-x
45. Topham DJ, Reilly EC. Tissue-resident memory CD8+ T cells: from phenotype to function. Front Immunol. (2018) 9:515. doi: 10.3389/fimmu.2018.00515
46. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol. (2019) 4:aaw1217. doi: 10.1126/sciimmunol.aaw1217
47. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol. (2015) 16:406–14. doi: 10.1038/ni.3108
48. Desai P, Tahiliani V, Stanfield J, Abboud G, Salek-Ardakani S. Inflammatory monocytes contribute to the persistence of CXCR3hi CX3CR1lo circulating and lung-resident memory CD8 + T cells following respiratory virus infection. Immunol Cell Biol. (2018) 96:370–8. doi: 10.1111/imcb.12006
49. Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. (2014) 346:93–8. doi: 10.1126/science.1257530
50. Shin H, Kumamoto Y, Gopinath S, Iwasaki A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat Commun. (2016) 7:13346. doi: 10.1038/ncomms13346
51. Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, et al. Intestinal CD169+ macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat Commun. (2015) 6:7802. doi: 10.1038/ncomms8802
52. Karasawa K, Asano K, Moriyama S, Ushiki M, Monya M, Iida M, et al. Vascular-resident CD169-positive monocytes and macrophages control neutrophil accumulation in the kidney with ischemia-reperfusion injury. J Am Soc Nephrol. (2015) 26:896–906. doi: 10.1681/ASN.2014020195
53. Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. (2009) 15:277–84. doi: 10.1038/nm.1929
54. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. (2014) 10:e1004053. doi: 10.1371/journal.ppat.1004053
55. Yao S, Jiang L, Moser EK, Jewett LB, Wright J, Du J, et al. Control of pathogenic effector T-cell activities in situ by PD-L1 expression on respiratory inflammatory dendritic cells during respiratory syncytial virus infection. Mucosal Immunol. (2015) 8:746–59. doi: 10.1038/mi.2014.106
56. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
57. Nobs SP, Kopf M. PPAR-gamma in innate and adaptive lung immunity. J Leukoc Biol. (2018) 104:737–41. doi: 10.1002/JLB.3MR0118-034R
58. Huang S, Jiang L, Cheon IS, Sun J. Targeting peroxisome proliferator-activated receptor-gamma decreases host mortality after influenza infection in obese mice. Viral Immunol. (2019) 32:161–9. doi: 10.1089/vim.2019.0016
59. Huang S, Zhu B, Cheon IS, Goplen NP, Jiang L, Zhang R, et al. PPAR-gamma in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J Virol. (2019) 93:e00030–19. doi: 10.1128/JVI.00030-19
60. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. (2016) 213:3057–73. doi: 10.1084/jem.20160938
61. Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature. (1994) 369:652–4. doi: 10.1038/369652a0
62. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
63. Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, et al. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol. (2014) 44:2003–12. doi: 10.1002/eji.201344359
64. Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, et al. Tissue-resident CD169+ macrophages form a crucial front line against plasmodium infection. Cell Rep. (2016) 16:1749–61. doi: 10.1016/j.celrep.2016.07.010
65. Netea MG, Joosten LAB. Trained immunity and local innate immune memory in the lung. Cell. (2018) 175:1463–5. doi: 10.1016/j.cell.2018.11.007
66. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. (2018) 175:1634–50 e1617. doi: 10.1016/j.cell.2018.09.042
67. Turner DL, Bickham KL, Farber DL, Lefrancois L. Splenic priming of virus-specific CD8 T cells following influenza virus infection. J Virol. (2013) 87:4496–506. doi: 10.1128/JVI.03413-12
68. Lim K, Hyun YM, Lambert-Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science. (2015) 349:aaa4352. doi: 10.1126/science.aaa4352
69. Reilly EC, Lambert-Emo K, Topham DJ. The effects of acute neutrophil depletion on resolution of acute influenza infection, establishment of tissue resident memory (trm), and heterosubtypic immunity. PLoS ONE. (2016) 11:e0164247. doi: 10.1371/journal.pone.0164247
70. Backer R, Schwandt T, Greuter M, Oosting M, Jungerkes F, Tuting T, et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci USA. (2010) 107:216–21. doi: 10.1073/pnas.0909541107
71. Richter M, Ray SJ, Chapman TJ, Austin SJ, Rebhahn J, Mosmann TR, et al. Collagen distribution and expression of collagen-binding alpha1beta1 (VLA-1) and alpha2beta1 (VLA-2) integrins on CD4 and CD8 T cells during influenza infection. J Immunol. (2007) 178:4506–16. doi: 10.4049/jimmunol.178.7.4506
72. Gilchuk P, Hill TM, Guy C, McMaster SR, Boyd KL, Rabacal WA, et al. A distinct lung-interstitium-resident memory CD8+ T cell subset confers enhanced protection to lower respiratory tract infection. Cell Rep. (2016) 16:1800–9. doi: 10.1016/j.celrep.2016.07.037
73. Takamura S. Persistence in temporary lung niches: a survival strategy of lung-resident memory CD8+ T cells. Viral Immunol. (2017) 30:438–50. doi: 10.1089/vim.2017.0016
74. Keeler SP, Agapov EV, Hinojosa ME, Letvin AN, Wu K, Holtzman MJ. Influenza A virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J Immunol. (2018) 201:2354–68. doi: 10.4049/jimmunol.1800671
Keywords: tissue-resident memory, alveolar macrophage, CD8 T cell differentiation, influenza, PPAR-γ, CD69, CD169
Citation: Goplen NP, Huang S, Zhu B, Cheon IS, Son YM, Wang Z, Li C, Dai Q, Jiang L and Sun J (2019) Tissue-Resident Macrophages Limit Pulmonary CD8 Resident Memory T Cell Establishment. Front. Immunol. 10:2332. doi: 10.3389/fimmu.2019.02332
Received: 05 April 2019; Accepted: 16 September 2019;
Published: 10 October 2019.
Edited by:
Vladimir Badovinac, The University of Iowa, United StatesReviewed by:
Martin J. Richer, McGill University, CanadaSara Hamilton, University of Minnesota Twin Cities, United States
Copyright © 2019 Goplen, Huang, Zhu, Cheon, Son, Wang, Li, Dai, Jiang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Sun, c3VuLmppZUBtYXlvLmVkdQ==