 Tytti Heinonen
Tytti Heinonen Eleonora Ciarlo
Eleonora Ciarlo Didier Le Roy
Didier Le Roy Thierry Roger
Thierry Roger- Infectious Diseases Service, Department of Medicine, Lausanne University Hospital and University of Lausanne, Epalinges, Switzerland
The sirtuins SIRT3 and SIRT5 are the main mitochondrial lysine deacetylase and desuccinylase, respectively. SIRT3 and SIRT5 regulate metabolism and redox homeostasis and have been involved in age-associated metabolic, neurologic and oncologic diseases. We have previously shown that single deficiency in either SIRT3 or SIRT5 had no impact on host defenses in a large panel of preclinical models of sepsis. However, SIRT3 and SIRT5 may compensate each other considering that they share subcellular location and targets. Here, we generated a SIRT3/5 double knockout mouse line. SIRT3/5 deficient mice multiplied and developed without abnormalities. Hematopoiesis and immune cell development were largely unaffected in SIRT3/5 deficient mice. Whole blood, macrophages and neutrophils from SIRT3/5 deficient mice displayed enhanced inflammatory and bactericidal responses. In agreement, SIRT3/5 deficient mice showed somewhat improved resistance to Listeria monocytogenes infection. Overall, the double deficiency in SIRT3 and SIRT5 has rather subtle impacts on immune cell development and anti-microbial host defenses unseen in single deficient mice, indicating a certain degree of overlap between SIRT3 and SIRT5. These data support the assumption that therapies directed against mitochondrial sirtuins, at least SIRT3 and SIRT5, should not impair antibacterial host defenses.
Introduction
The innate immune system plays a central role in host defenses. Innate immune cells among which monocytes/macrophages, granulocytes and dendritic cells (DCs) sense microbial and danger associated molecular patterns (MAMPs/DAMPs) through pattern recognition receptors (PRRs) expressed at the cell surface, in the cytoplasm and in endosomes. The best characterized PRRs belong to the families of Toll-like receptors (TLRs), C-type lectin receptors (CLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and cytosolic DNA sensors (1, 2). The binding of MAMPs/DAMPs to PRRs activates intracellular signaling cascades that induce the production of effector molecules involved in inflammation and host defense mechanisms, as well as the resolution of inflammation and tissue repair (3, 4). Immune cells are plastic and adapt their metabolism and responsiveness to their environment to execute their biological functions (5, 6).
Sirtuins belong to the family of so-called histone deacetylases (HDACs) that target lysine posttranscriptional modifications. Classical HDACs (HDAC1-11) are Zn2+-dependent, while sirtuins are NAD+-dependent lysine deacetylases. Sirtuins are homologs to yeast Sir2 that gained tremendous attention when it was shown to be activated upon caloric restriction and to increase lifespan (7). The mammalian genome encodes for seven sirtuins that target proteins by removing acetyl functional groups, but also acyl, glutaryl, malonyl, and succinyl groups as demonstrated lately (8). The list of targets of sirtuins has increased dramatically over the years, and high throughput proteomics analyses pinpointed to thousands of substrates for sirtuins. Accordingly, sirtuins are involved in the regulation of many biological and pathological processes and in the development of metabolic, neurodegenerative, cardiovascular, and oncologic diseases (9, 10).
SIRT3 and SIRT5 are mainly localized in the mitochondrial matrix, where SIRT3 is the main deacetylase (11) and SIRT5 is the main desuccinylase (12, 13). Of note, SIRT5 also catalyzes lysine demalonylation and deglutarylation (13, 14). SIRT3 promotes glucose and fatty acid metabolism, urea cycle and the activity of the electron transport chain. During caloric restriction, SIRT3 regulates mitochondrial acetylome and multiple metabolic pathways in the liver (15, 16). SIRT3 protects from oxidative stress by activating the reactive oxygen species (ROS) detoxifying enzyme superoxide dismutase 2 (SOD2) and the redox controlling enzyme isocitrate dehydrogenase 2 (IDH2) (17, 18). Similar to SIRT3, SIRT5 activates enzymes involved in ROS detoxification (i.e., SOD1, IDH1, and IDH2), promotes mitochondrial functions and integrity and regulates the urea cycle and other metabolic pathways (14, 19–25). The genetic ablation of SIRT3 or SIRT5 in mice has been associated with increased susceptibility to age-associated diseases including insulin resistance, obesity, neurodegeneration, cardiac dysfunction and fibrosis, while contrasting context-dependent effects have been reported for tumorigenesis (10, 26–30). Deficiencies in SIRT3 or SIRT5 have also been reported to promote colitis, acute lung injury and ischemia reperfusion injury (10, 31–36). Overall, targeting the activity of sirtuins and particularly mitochondrial sirtuins is viewed as an attractive therapeutic strategy to tackle the development of age-related disorders (10, 28–30). Considering that inflammation is an essential component of innate immune defenses, we analyzed the impact of SIRT3 and SIRT5 deficiencies on the response of mice subjected to a broad panel of preclinical models of bacterial and fungal sepsis (37, 38). Neither SIRT3 nor SIRT5 was critical to fight against infections. Additionally, SIRT3−/− mice were not particularly susceptible to cecal ligation and puncture (CLP), a stringent model of sepsis (39, 40). Hence, SIRT3 and SIRT5 appear to have a more prominent influence on chronic metabolic and inflammation-related disorders than on infectious diseases characterized by acute inflammatory reactions.
SIRT3 and SIRT5 share subcellular location and targets, so they might compensate each other in single knockout mice. To bypass this hurdle, we generated a SIRT3/5 deficient mouse line. SIRT3/5−/− mice were fertile and developed without apparent abnormalities. In vitro and in vivo investigations revealed somewhat enhanced inflammatory and bactericidal responses of whole blood, macrophages, and neutrophils and a moderate improved resistance to Listeria monocytogenes in the double knockouts. Altogether SIRT3 and SIRT5 have subtle, redundant roles during antimicrobial host defenses. Overall, therapies directed against mitochondrial sirtuins should not dramatically impact on antimicrobial host defenses.
Materials and Methods
Key Resources
See Supplementary Information.
Ethics Statement
Animal experiments were approved by the Service des Affaires Vétérinaires, Direction Générale de l'Agriculture, de la Viticulture et des Affaires Vétérinaires (DGAV), état de Vaud (Epalinges, Switzerland; authorizations 876.9 and 877.9) and performed according to Swiss and ARRIVE guidelines (http://www.nc3rs.org.uk/arrive-guidelines).
Mice
C57BL/6J mice were from Charles River Laboratories (Saint-Germain-sur-l'Arbresle, France). SIRT3−/− and SIRT5−/− C57BL/6J mice were described (41, 42) and obtained from Prof. Johan Auwerx, Laboratory for Integrative and Systems Physiology, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland. SIRT3−/− males were crossed with SIRT5−/− females. Thirty-two SIRT3/5+/− females were crossed with 16 SIRT3/5+/− males. Among the 205 F2 mice, 4 males and 8 females were double knockout mice and used to establish the SIRT3/5−/− mouse line. All mice used in this study were 7–14-week old, housed under specific pathogen-free conditions and exempt of mouse hepatitis virus and murine norovirus. For genotyping purposes, DNA was extracted and analyzed by PCR using the Mouse Direct PCR Kit (Bimake, Houston, TX) and primers pairs described in Supplementary Information.
Cells and Reagents
Bone marrow (BM) cells were cultured 7 days in IMDM or RPMI 1640 supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin (Invitrogen, San Diego, CA), 10% heat inactivated fetal bovine serum (Biochrom GmbH, Berlin, DE) and 50 U/ml macrophage colony-stimulating factor (ImmunoTools, Friesoythe, Germany) or 30% L929 cell supernatant to generate BM-derived macrophages (BMDMs) (43, 44). Cells were seeded in half-area 96-well plates (2.5 × 104 cells/well), 96-well plates (2 × 105 cells/well) and 6-well plates (3 × 106 cells/well) without growth factors. Neutrophils were isolated from the bone marrow using the Neutrophil isolation kit (Miltenyi, Bergisch Gladbach, Germany) and plated in 96-well plates (105 cells/well). Salmonella minnesota ultra pure lipopolysaccharide (LPS) was from List Biologicals Laboratories (Campbell, CA), Pam3CSK4 from EMC microcollections GmBH (Tübingen, Germany), and CpG ODN 1826 (CpG) and poly(I:C) from Invivogen (San Diego, CA). Monosodium urate (MSU) crystals were prepared as described (45). Listeria monocytogenes 10403 s was grown in brain heart infusion broth (BD Biosciences, Erembodegem, Belgium). Bacteria were washed with 0.9% NaCl and adjusted at 1010 cfu/ml. When required, bacteria were heat-inactivated for 2 h at 70°C.
RNA Analyses
RNA was extracted (RNeasy kit) and reverse transcribed (QuantiTect reverse transcription kit) (Qiagen, Hilden, Germany). PCRs were performed in triplicate with 1.25 μl cDNA, 1.25 μl H2O, 0.62 μl primers [Supplementary Information and (46)] and 3.12 μl KAPA SYBR Green Fast (Kapa Biosystems, Wilmington, MA) using a QuantStudio™ 12K Flex system (Life Technologies, Carlsbad, CA). Gene expression was normalized to actin expression.
Western Blot Analyses
Total and nuclear proteins were extracted, submitted to PAGE and transferred onto membranes as described (47, 48). Membranes were incubated with primary and secondary HRP-coupled antibodies and revealed by chemiluminescence [Supplementary Information and (49)]. Images were recorded with a Fusion Fx system (Vilber Lourmat, Collégien, France). Full length blots are presented in Supplementary Figure S1.
Flow Cytometry
Single cell suspensions from thymus, spleen and BM were incubated with 2.4 G2 to block Fc receptors and stained with antibodies described in Supplementary Information (50). For hematopoietic stem cells (HSC) and progenitor cells, lineage cocktail contained antibodies directed against CD45R (B220), CD3e, CD11b, CD19, Ly6C/G, Ter119/Ly-76. Data were acquired using an Attune Nxt flow cytometer (ThermoFisher, Waltham, MA) and analyzed using FlowJo version 10.2 (FlowJo LLC, Ashland, OR). Gating strategies are presented in Supplementary Figure S2.
ROS Measurement
BMDMs were plated in half-area black 96-well plates in RPMI without phenol red (Invitrogen). Cells were incubated for 10 min at 37°C with 5 μM MitoSOX (Thermofisher). Stimuli were added and fluorescence (Ex510, Em580) recorded using a Synergy plate reader (BioTek, Winooski, VT). Neutrophils in HBSS without calcium and magnesium (ThermoFisher) were incubated for 1 h with 100 nM PMA (Enzo Life Sciences, Farmingdale, NY) and 5 μM MitoSOX during the last 10 min of incubation. ROS were measured by flow cytometry.
Cytokine Measurement
Cytokines were quantified by ELISA (Supplementary Information) or Luminex using a custom ProCarta kit (ENA-78/CXCL5, G-CSF, IFNγ, IL-1α, IL-1β, IL-3, IL-6, IL-10, IL-12p40, IL-17A, IL-18, IP-10/CXCL10, KC/CXCL1, MCP-1/CCL2, MIP-1α/CCL3, MIP-2/CXCL2, TNF) (Invitrogen, Carlsbad, CA) and a bioplex 200 system (Bio-Rad, Hercules, CA) (51).
Metabolic Activity
The metabolic activity of BMDMs was measured using the XF Cell Mito Stress, Glycolysis Stress and Mito Fuel Flex Test Kits on a 96-well format Seahorse XFe® system (Agilent Technologies, Santa Clara, CA) (46).
Neutrophil Killing and NETosis Assays
Neutrophils were incubated with live L. monocytogenes for 1 h in RPMI medium. Serial dilutions of reaction mixtures were plated on blood agar plates (BD Biosciences). Twenty-four hours later, colonies were enumerated. To measure NETosis, neutrophils were incubated for 3 h with 100 nM PMA and 5 μM of the cell impermeable dye Sytox green. Fluorescence (Em504, Ex523) was recorded using the Synergy plate reader.
In vivo Models
Listeriosis was induced by challenging intravenously (i.v.) age and sex-matched mice with a low (7.3 × 103 cfu) or a high (0.9–1 × 105 cfu) inoculum of L. monocytogenes. Blood and organs were collected 1–3 days post-infection to quantify bacteria and cytokines and analyze cell populations. A model of endotoxemia was developed by challenging mice intraperitoneally (i.p.) with 10 mg/kg LPS. Body weight loss, severity score and survival were registered at least twice daily (52, 53) by 2–3 operators. The severity score was graded from 0 to 4 based on the mobility, the posture, the appearance and the weight loss of mice (detailed criteria were approved by the Service des Affaires Vétérinaires, DGAV, and are available upon request). Mice were sacrificed when they met a severity score of 4. A mice found dead was assigned a score of 5.
Statistical Analyses
Groups were compared by variance analysis followed by two-tailed unpaired Student's t-test or a Mann-Whitney test when appropriate. Survival was analyzed using the Kaplan-Meier method. P < 0.05 was used to indicate statistical significance. Analyses were performed using PRISM 8.0.1 (GraphPad Software, San Diego, CA).
Results
SIRT3/5 Deficiency Has No Dramatic Impact on Mouse Development and Macrophage Metabolism
We generated a SIRT3/5 double knockout mouse line (SIRT3/5−/−, see Materials and Methods) to study the interaction between SIRT3 and SIRT5. Genomic-DNA based PCR genotyping (Figure 1A) and Western blotting analyses (Figure 1B) confirmed the truncation of the Sirt3 and Sirt5 genes and the absence of SIRT3 and SIRT5 protein expression in SIRT3/5−/− mice. Fecundity and development were normal. The size (Figure 1C) and the female/male sex ratio (Figure 1D) of the litters as well as the weight of adult female and male mice (Figure 1E) were like those of SIRT3/5+/+, SIRT3−/−, and SIRT5−/− mouse lines. Autopsy did not reveal gross abnormalities in SIRT3/5−/− mice.
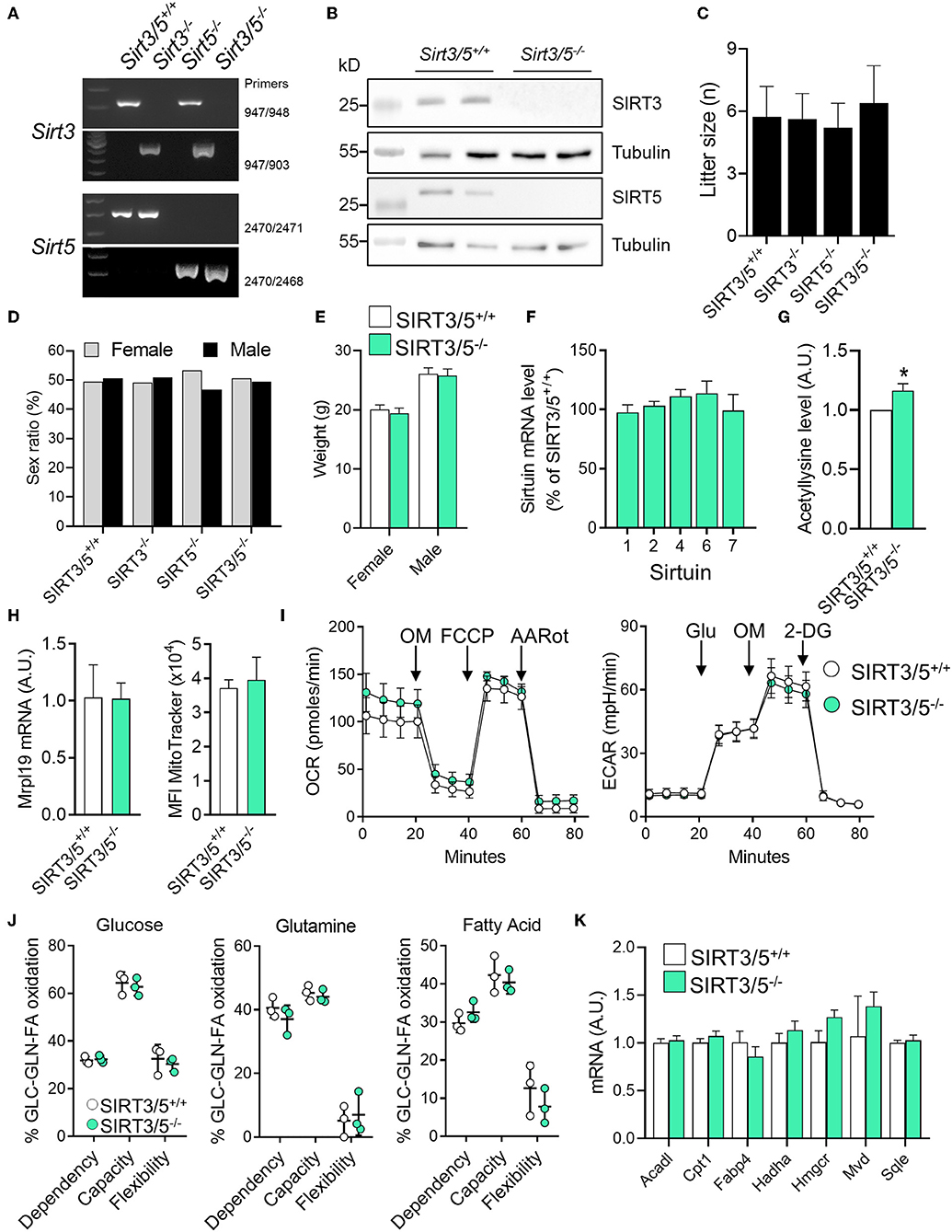
Figure 1. SIRT3/5 deficiency has no evident impact on mouse development and macrophage metabolism. (A) Genotyping of SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mice by PCR. Genomic DNA was amplified by PCR using the primer pairs indicated on the right and reaction mixtures were electrophoresed through agarose gels. (B) SIRT3, SIRT5, and tubulin expression in the liver of SIRT3/5+/+ and SIRT3/5−/− mice measured by Western blotting (Full blots are presented in Supplementary Figure S1). (C,D) Size (C) and sex ratio (in percentage, D) of litters from SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mouse lines. (E) Weight of adult SIRT3/5+/+ and SIRT3/5−/− female and male mice. (F) Sirtuin mRNA expression levels in SIRT3/5−/− BMDMs, expressed relative to the mRNA levels in SIRT3/5+/+ BMDMs set at 100%. Data are means ± SD from one experiment performed with three mice analyzed in triplicate. (G) Acetyllysine levels in total protein extracts from SIRT3/5+/+ and SIRT3/5−/− BMDMs were measured by Western blotting and imaging. Values were normalized to those obtained using SIRT3/5+/+ BMDMs set at 1. Data are means ± SD from one experiment performed with three mice. P = 0.009. (H) Mrpl19 mRNA expression levels assessed by RT-PCR and median fluorescence intensity (MFI) of MitoTracker measured by flow cytometry in SIRT3/5+/+ and SIRT3/5−/− BMDMs. Data are means ± SD from four mice aged 10–12 weeks. (I) Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse technology. OM, oligomycin; FCCP, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; AARot, rotenone+Antimycin A; Glu, glucose; 2-DG, 2-deoxyglucose. Data are means ± SD from four mice aged 10–12 weeks analyzed in quadruplicate. (J) Mitochondrial fuel usage by SIRT3/5+/+ and SIRT3/5−/− BMDMs measured using the Seahorse technology. (K) Acadl, Cpt1, Fabp4, Hadha, Hmgcr, Mvd, and Sqle mRNA expression levels were quantified by RT-PCR. Gene expression levels were normalized to actin levels. A.U., arbitrary unit. Data are means ± SD from three mice aged 10–12 weeks analyzed in triplicate.
The dual deletion of SIRT3 and SIRT5 was not compensated by an increased expression of mRNA encoding for SIRT1, SIRT2, SIRT4, SIRT6, and SIRT7 in bone marrow derived macrophages (BMDMs) (Figure 1F). There was around 20% increase of total protein acetylation in SIRT3/5−/− BMDMs (Figure 1G). SIRT3/5 deficiency did not alter the mitochondrial mass, evaluated by measuring mitochondrial ribosomal protein L19 (Mrpl19) mRNA levels and the fluorescence intensity of the mitochondrial dye MitoTracker (Figure 1H). The oxygen consumption rate (OCR), which reflects mitochondrial respiration, was weakly increased in SIRT3/5−/− BMDMs (Figure 1I). The extracellular acidification rate (ECAR), a readout of the glycolytic activity, was not affected in resting and LPS stimulated SIRT3/5−/− BMDMs (Figure 1I and Supplementary Figure S3A). OCR and ECAR were similarly affected in resting SIRT3−/− BMDMs (Supplementary Figure S3B) and SIRT5−/− BMDMs (38). The dependency, capacity, and flexibility of BMDMs to oxidize the mitochondrial fuels glucose, glutamine and fatty acids were identical for SIRT3/5+/+ and SIRT3/5−/− BMDMs (Figure 1J). SIRT3/5+/+ and SIRT3/5−/− BMDMs expressed similar levels of a number of genes encoding for molecules involved in the fatty acid metabolism, i.e., Acadl (acyl-CoA dehydrogenase long chain), Cpt1 (carnitine palmitoyltransferase 1), Fabp4 (fatty acid binding protein 4), Hadha (hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha), Hmgcr (3-hydroxy-3-methylglutaryl-CoA reductase), Mvd (mevalonate diphosphate decarboxylase) and Sqle (squalene epoxidase) (Figure 1K). Finally, the OCR of SIRT3/5+/+ and SIRT3/5−/− BMDMs was not different before and after addition of etimoxir, an inhibitor of Cpt1 (88.0 vs. 85.5% inhibition in SIRT3/5+/+ vs. SIRT3/5−/− BMDMs; n = 6; P = 0.3). Hence, SIRT3/5 deficiency had no strong impact on basic metabolic parameters of BMDMs.
SIRT3/5 Deficient Mice Have Minor Abnormalities of Leukocyte Development
The bone marrow is the main source of hematopoietic stem cells (HSC) and progenitors of immune cells during adulthood (54, 55). The number of CD45+ hematopoietic cells per leg (femur + tibia) was identical in SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mice (Figure 2A), as well as the composition of the HSC pool which is made of lineage negative, Sca1 positive, c-kit positive (LSK) cells, long-term (LT)-HSC, short-term (ST)-HSC, and multipotent progenitors (MPP) (Figure 2B). Accordingly, the percentage and the absolute number of T cells, B cells, neutrophilic granulocytes and monocytes were similar in SIRT3/5+/+ and SIRT3/5−/− mice, while some minor changes were observed in SIRT3−/− and SIRT5−/− mice (decreased CD3+ T cells and CD19+ B cells and increased monocytes) (Figure 2C and Table 1).
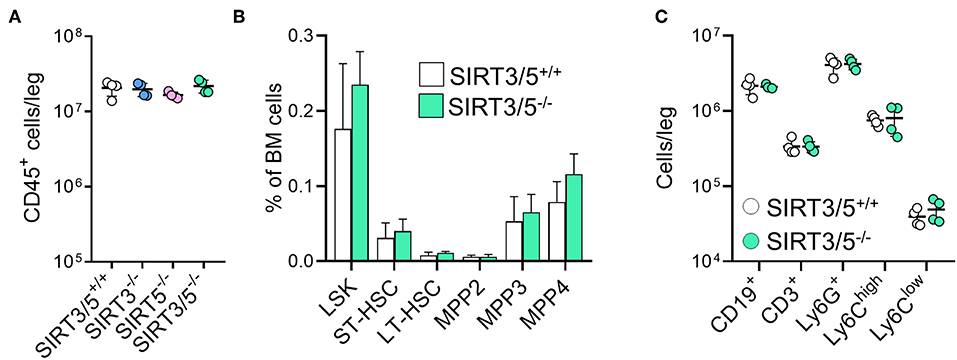
Figure 2. SIRT3/5 deficient mice have a normal number of hematopoietic stem cells and progenitors in the bone marrow. (A) Number of CD45+ bone marrow cells per leg of SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mice aged 10–14 weeks. (B) Percentage of lineage negative, Sca1 positive, c-kit positive (LSK) cells, long-term (LT)-hematopoietic stem cells (HSC), short-term (ST)-HSC, multipotent progenitors (MPP), MPP2, MMP3, and MPP4 among BM cells. Data are means ± SD from eight mice. (C) Number of CD19+, CD3+, Ly6G+, Ly6Chigh, or Ly6Clow cells per leg of SIRT3/5+/+ and SIRT3/5−/− mice.
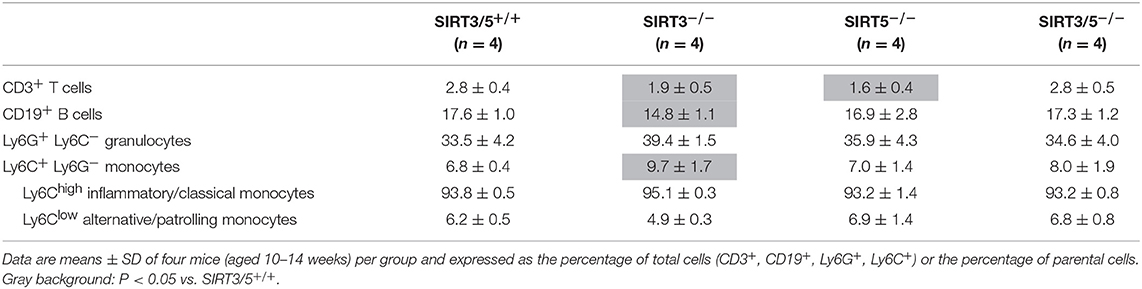
Table 1. Bone marrow leukocyte subsets.
SIRT3/5−/− mice, but not single knockout mice, showed a slight reduction of thymus cellularity when compared to SIRT3/5+/+ mice (SIRT3/5+/+ vs. SIRT3/5−/− mice: 12.7 ± 1.8 vs. 8.3 ± 1.2 million cells; P = 0.03). However, the proportion of CD4/CD8 single positive, double positive and double negative (DN1-DN4) thymocytes was comparable in all mouse lines (Table 2). The size of the spleen (SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mice: 6.1 ± 1.5, 5.3 ± 1.1, 7.3 ± 1.6, and 7.0 ± 1.8 × 107 cells; P > 0.05 for all) and the proportion of total T cells, B cells, dendritic cells (DCs), neutrophilic granulocytes and monocytes were not affected in SIRT3/5−/− mice (Table 3). Small, statistically significant, differences were noticed between SIRT3/5+/+ and SIRT3/5−/− mice, i.e., reduced percentages of effector memory CD4+ T cells (23.1 ± 3.3 vs. 17.2 ± 2.3% of CD4+ T cells), CD4−CD8− T cells (4.2 ± 0.9 vs. 3.0 ± 0.2% of CD3+ T cells) and conventional DC1 (cDC1, 35.4 ± 6.3 vs. 27.2 ± 3.0% of CD11c+ DCs) (Table 3). Like SIRT3/5−/− mice, SIRT3−/− mice showed a reduced percentage of cDC1, while SIRT5−/− mice showed an increased percentage of Ly6Clow (alternative) monocytes at the expense of Ly6Chigh (inflammatory) monocytes. Collectively, these results suggested that the dual deletion of SIRT3 and SIRT5 had a subtle impact on the development of immune cells.
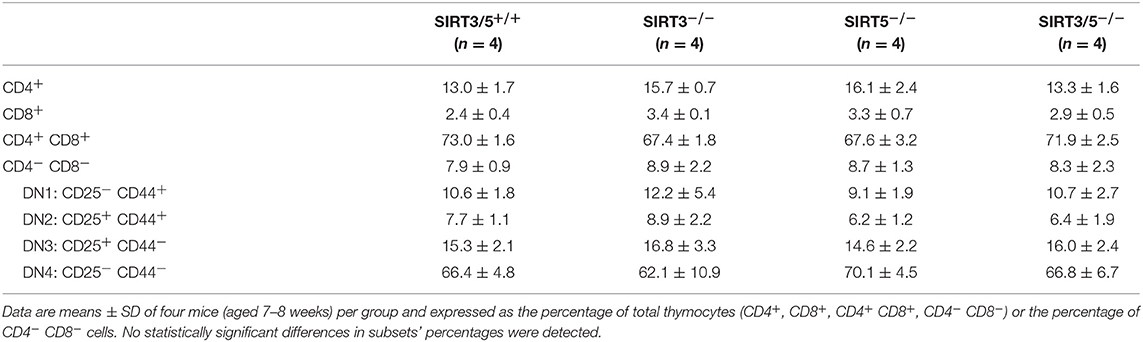
Table 2. Thymic cell subsets.
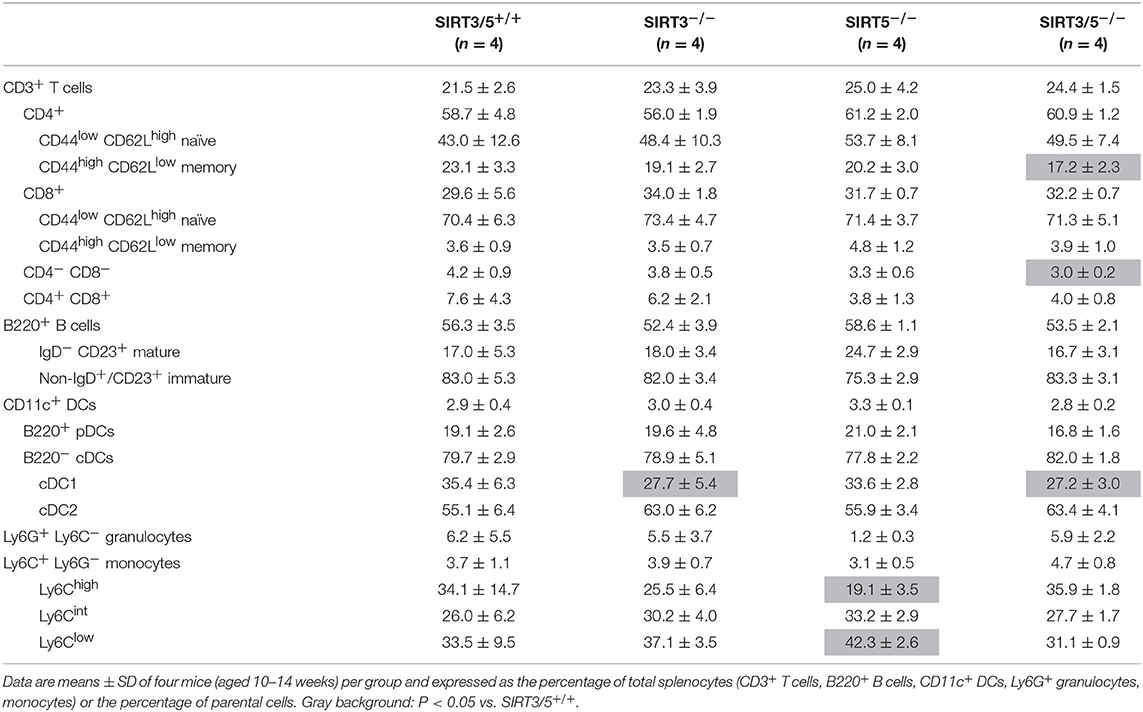
Table 3. Splenic cell subsets.
SIRT3/5 Deficiency Increases the Inflammatory Profile of Macrophages and the Killing Activity of Neutrophils
Macrophages and neutrophils are proficient at sensing microbial products and play key defense roles during infections. SIRT3 and SIRT5 single deficiency did not influence antimicrobial host defenses (37, 38). Therefore, we asked whether dual deficiency of SIRT3 and SIRT5 would reveal a phenotype unseen in single knockouts. SIRT3/5+/+ and SIRT3/5−/− BMDMs were exposed to LPS, CpG, and poly(I:C), which are sensed through TLR4, TLR9, and TLR3, respectively. SIRT3/5−/− BMDMs produced higher levels of TNF, IL-6, and IL-12p40 (as a trend for CpG-induced IL-12p40) and lower levels of IL-10 than SIRT3/5+/+ BMDMs in response to LPS and CpG, while SIRT3/5+/+ and SIRT3/5−/− BMDMs produced similar levels of TNF and IL-6 in response to poly(I:C) (Figure 3A).
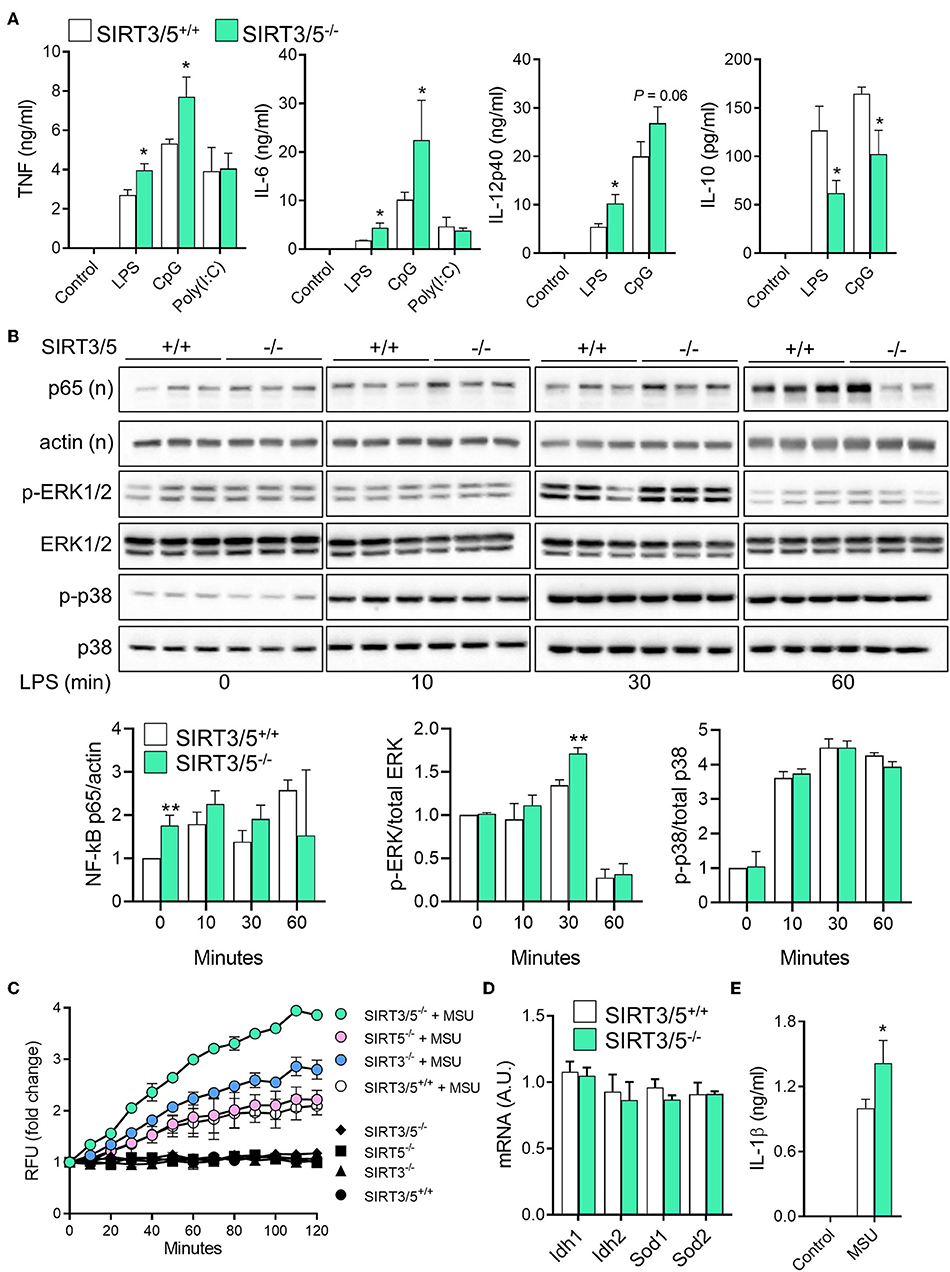
Figure 3. SIRT3/5 deficient macrophages display an enhanced proinflammatory profile. (A) SIRT3/5+/+ and SIRT3/5−/− BMDMs were exposed for 24 h to LPS (10 ng/ml), CpG (1 μg/ml), and poly(I:C) (10 μg/ml). TNF, IL-6, IL-12p40, and IL-10 concentrations in cell culture supernatants were quantified by ELISA. Data are means ± SD from one experiment performed with four mice. (B) SIRT3/5+/+ and SIRT3/5−/− BMDMs were exposed for 0, 10, 30, and 60 min to LPS (10 ng/ml). Nuclear (n) and total protein extracts were used to analyze NF-κB p65, and phosphorylated (p) and total ERK1/2 and p38. Signals were quantified by imaging and results (ratio p65/actin, p-ERK1/2/ERK1/2, and p-p38/p38) expressed relative to the results obtained in resting SIRT3/5+/+ BMDMs set at 1. Data are means ± SD from one experiment performed with three mice. Full blots are presented in Supplementary Figure S1. (C–E) BMDMs were primed with Pam3CSK4 (10 ng/ml) for 18 h and exposed (colored and white symbols) or not (black symbols) to MSU crystals for the indicated time (C) or 6 h (E). mtROS were quantified using MitoSOX (C), Idh1, Idh2, Sod1, and Sod2 mRNA levels by RT-PCR (D) and IL-1β by ELISA (E). Gene expression levels were normalized to actin levels. A.U, arbitrary unit. Data are mean ± SD of four mice aged 10–12 weeks analyzed in triplicate (C–E). *P < 0.05; **P < 0.01.
To address whether the increased inflammation driven by SIRT3/5-deficiency was linked to increased intracellular signaling, we quantified by Western blotting the nuclear translocation of NF-κB p65 and the phosphorylation of ERK1/2 and p38 MAPKs in BMDMs exposed for 0, 10, 30, and 60 min to LPS (Figure 3B). In SIRT3/5−/− BMDMs, there was an increased NF-κB p65 nuclear content at baseline and, albeit not statistically significant, after 30 and 60 min of stimulation. The level of phospho-ERK1/2 was also increased after 30 min of exposure to LPS. Overall, SIRT3/5 deficiency increased inflammatory intracellular signaling pathways and inflammatory cytokine production by BMDMs.
ROS activate the NOD-like receptor pyrin domain-containing-3 (NLRP3) inflammasome that cleaves pro-IL-1β into mature IL-1β that is secreted. Considering that SIRT3 and SIRT5 activate enzymes playing a role in the detoxification process of ROS (i.e., IDH1, IDH2, SOD1, and SOD2) (17–21), we questioned whether SIRT3/5−/− BMDMs produce increased levels of mitochondrial ROS (mtROS) and IL-1β. SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− BMDMs were exposed to monosodium urate (MSU) crystals, a commonly used activator of the NLRP3 inflammasome before measuring mtROS (Figure 3C). MSU crystals induced mtROS equally in SIRT3/5+/+ and SIRT5−/− BMDMs, 1.5-fold more in SIRT3−/− BMDMs and 2-fold more in SIRT3/5−/− BMDMs. mRNA levels of Idh1, Idh2, Sod1, and Sod2 were similar in SIRT3/5+/+ and SIRT3/5−/− BMDMs (Figure 3D), in agreement with the fact that sirtuins target the activity rather than the expression of IDH1, IDH2, SOD1, and SOD2. As expected from the above, SIRT3/5−/− BMDMs secreted higher levels of IL-1β than SIRT3/5+/+ BMDMs (Figure 3E).
The role of sirtuins in the development and functions of granulocytes is scarce and has not been reported for SIRT5 (56, 57). The mRNA expression levels of SIRT3 and SIRT5 decreased gradually from common myeloid progenitors (CMP) to granulocyte-monocyte progenitor (GMP; 1.6-fold) and from GMP to neutrophilic granulocytes (5.3–5.7-fold) (Figure 4A). Compared to SIRT3/5+/+ mice, SIRT3/5−/− mice expressed in the bone marrow statistically significantly more CMP (P = 0.03) but not GMP (P = 0.06) (Figure 4B). Accordingly, SIRT3/5−/− mice expressed normal numbers of neutrophilic granulocytes in the bone marrow and spleen (Figure 2C and Table 3). We then addressed whether SIRT3/5 deficiency affected neutrophil functions. We setup a killing assay in which neutrophils were incubated for 1 h with Listeria monocytogenes before quantifying bacteria. As shown in Figure 4C, 115, 96, 77, and 66% of the starting inoculum were recovered from assays using SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− neutrophils, respectively. Hence, the dual deletion of SIRT3 and SIRT5 promoted the killing of L. monocytogenes by neutrophils. This prompted us to analyze two main mechanisms through which neutrophils kill bacteria, i.e., the production of ROS and the release of neutrophil extracellular traps (NETs). SIRT3/5−/− neutrophils, and to a lesser extent SIRT3−/− and SIRT5−/− neutrophils, produced increased levels of ROS when compared to SIRT3/5+/+ neutrophils (Figure 4D). In contrast, SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− neutrophils produced similar amounts of NETs (Figure 4E). Thus, the proficient killing of L. monocytogenes by SIRT3/5−/− neutrophils was more likely related to an increased generation of ROS than NETs. Lastly, we measured by Luminex the cytokines released by whole blood exposed to heat-killed L. monocytogenes. Fifteen of 17 mediators were produced at measurable levels: CCL2, CCL3, CXCL1, CXCL5, CXCL10, G-CSF, IFNγ, IL-1α, IL-1β, IL-6, IL-10, IL-12p40, IL-17A, IL-18, and TNF. Going well along with an increased inflammatory response of BMDMs (Figure 3), SIRT3/5−/− whole blood produced more G-CSF and showed a trend toward producing more IL-1α, IL-6, and IFNγ than SIRT3/5+/+ whole blood (Figure 4F). The production of other cytokines was not affected.
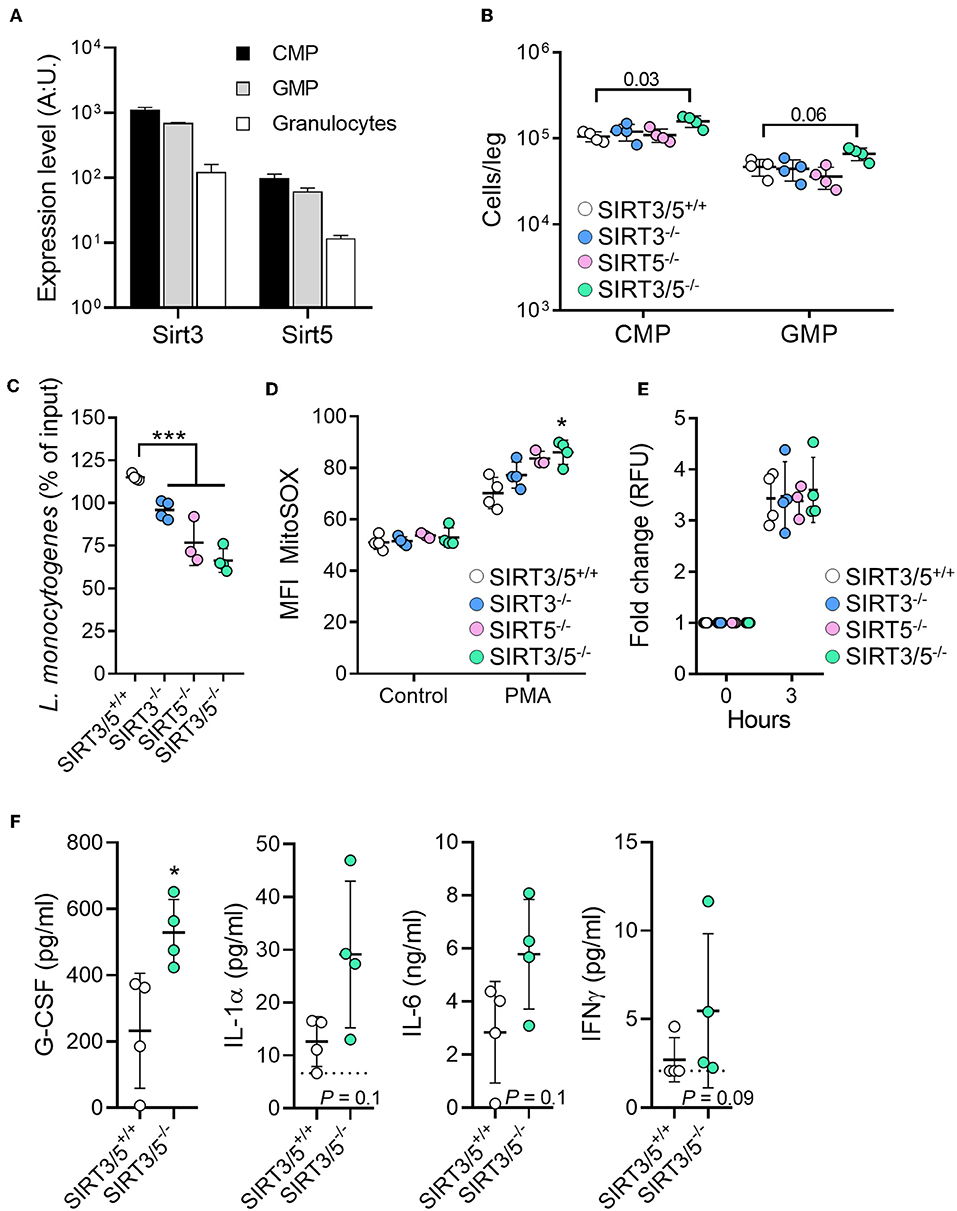
Figure 4. Increased killing of Listeria monocytogenes by SIRT3/5 deficient neutrophils. (A) Sirt3 and Sirt5 mRNA expression levels in common myeloid progenitors (CMP), granulocyte-monocyte progenitor (GMP), and granulocytes. Data were extracted from BioGPS (http://biogps.org). (B) Number of CMP (Lin− c-kit+ CD34+ cells) and GMP (Lin− c-kit+ CD135+ CD34+ cells) per leg of SIRT3/5+/+, SIRT3−/−, SIRT5−/−, and SIRT3/5−/− mice aged 8–9 weeks. The gating strategies are presented in Supplementary Figure S2. (C–E) Killing of bacteria (C) and production of mtROS (D) and NETs (E) by neutrophils. Neutrophils were incubated for 1 h with L. monocytogenes (0.1 cfu/cell) (C), for 50 min with PMA (100 nM) and MitoSOX (D) or for 3 h with PMA (100 nM) and SYTOX (E). Bacteria were enumerated and results expressed relative to the initial inoculum set at 100% (C). *P < 0.05; ***P < 0.005. (F) G-CSF, IL-1α, IL-6, and IFNγ concentrations, determined by Luminex, in blood from SIRT3/5+/+ and SIRT3/5−/− mice incubated with heat-killed L. monocytogenes for 24 h. The concentrations of CCL2, CCL3, CXCL1, CXCL5, CXCL10, IL-1β, IL-10, IL-12p40, IL-17A, IL-18 and TNF were similar for SIRT3/5+/+ and SIRT3/5−/− blood.
SIRT3/5 Deficiency Provides a Modest Protection to Listeriosis
Myeloid cells play a crucial role in protecting from L. monocytogenes infection (58, 59). Considering that SIRT3/5 deficiency increased L. monocytogenes killing by neutrophils (Figure 4C) and cytokine production by macrophages and to some extent by whole blood (Figures 3A, 4F), we tested the relevance of these observations in vivo using a model of listeriosis. Mice challenged intravenously with a high inoculum of L. monocytogenes (0.9–1.5 × 105 cfu) exhibited signs of disease 36–40 h post-infection and were severely sick after 65 h as shown by elevated severity scores in most animals. SIRT3/5−/− mice were not as ill as SIRT3/5+/+ mice (P = 0.01; Figure 5A) and had 2.3-fold less L. monocytogenes in blood collected 48 h after infection (SIRT3/5+/+ vs. SIRT3/5−/−: 3.5 ± 0.9 × 103 cfu/ml vs. 1.5 ± 0.3 × 103 cfu/ml; median ± SEM; P = 0.005) (Figure 5B). In line with these observations, SIRT3/5−/− mice had a modest, statistically significant, delayed mortality rate compared to SIRT3/5+/+ mice (median survival of SIRT3/5+/+ vs. SIRT3/5−/−: 3.0 vs. 3.12 days; P = 0.01) (Figure 5C). Going well along with an improved response to infection, SIRT3/5−/− mice had significantly higher blood concentrations of TNF at day 1 (P = 0.001), TNF and IL-1β at day 2 (P = 0.004 and 0.03) and KC/CXCL1 at day 3 (P = 0.05). Albeit not statistically significant, the levels of G-CSF, IL-1α, MCP-1/CCL2, MIP-2/CXCL2 at day 1, G-CSF, KC, MCP-1, and MIP-2 at day 2, and IL-1α, IL-1β, IL-6, IL-10, MCP-1, MIP-2, and TNF at day 3 post-infection were 1.5–4.4-fold higher in SIRT3/5−/− than in SIRT3/5+/+ mice (Figure 5D). As expected, 2 days after the onset of infection, listeriosis induced a dramatic drop of circulating leukocytes including B cells, T cells, neutrophils and Ly6Chigh inflammatory monocytes that rebounded at day 3 (Figure 5E). No differences were observed between SIRT3/5+/+ and SIRT3/5−/− mice, suggesting that the reactivity rather than the number of blood leukocytes conferred some protection to SIRT3/5−/− mice during listeriosis. Interestingly, there was no statistically significant difference of mortality when SIRT3/5−/− and SIRT3/5+/+ mice were challenged with a low inoculum (7.3 × 103 cfu) of L. monocytogenes responsible for <25% death (Figure 5C). Finally, we tested the mouse lines in a model of endotoxemia induced by an intraperitoneal challenge with 10 mg/kg LPS. Surprisingly, there was no statistically significant difference between the SIRT3/5+/+ and SIRT3/5−/− groups (n = 21–22 mice per group; P = 0.1).
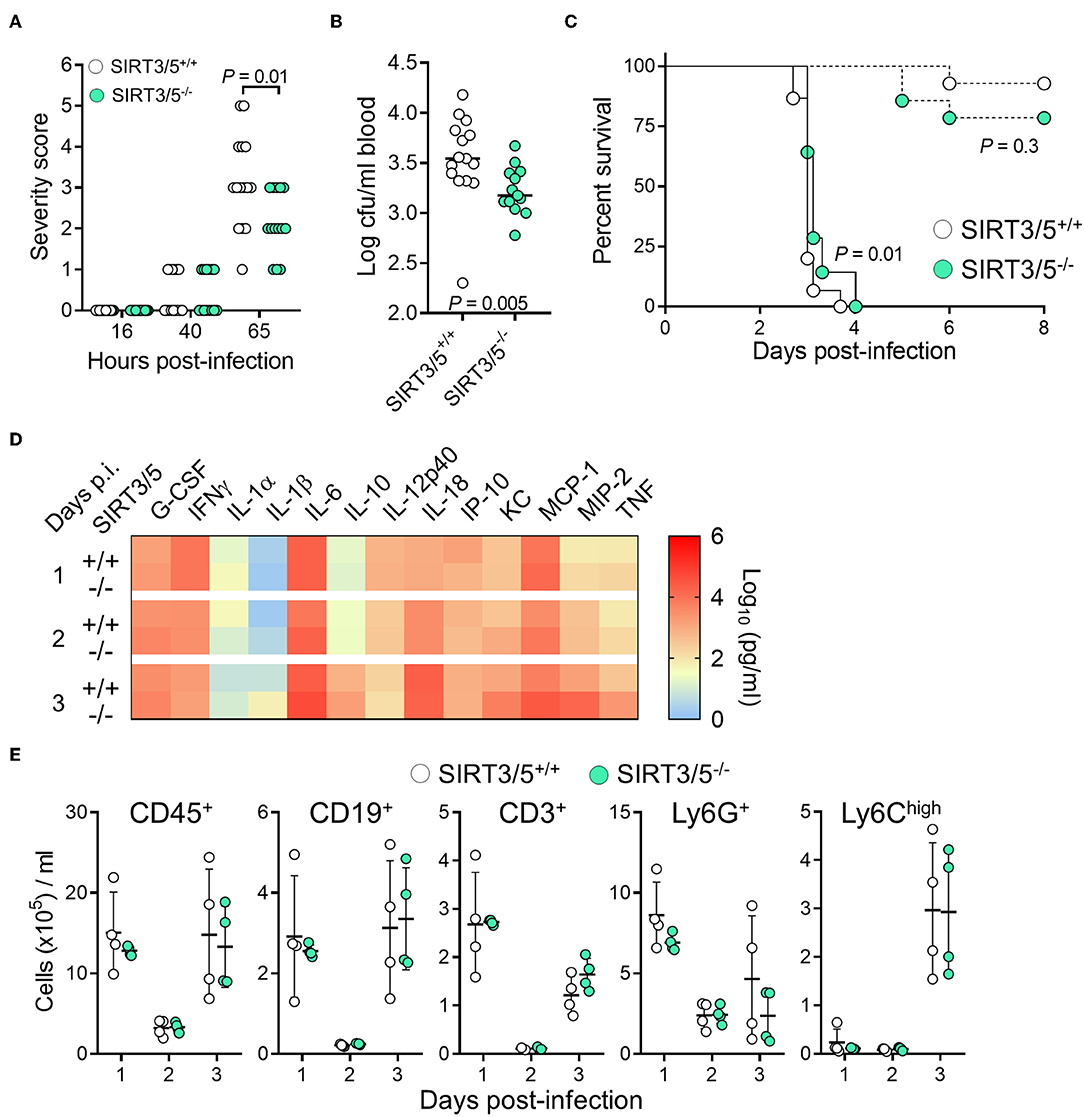
Figure 5. SIRT3/5 deficiency confers some protection to listeriosis. (A–E) SIRT3/5+/+ and SIRT3/5−/− mice (n = 14–15 per group, aged 8–12 weeks) were injected intravenously with 7.3 × 103 cfu (C, dashed lines) or 0.9–1.5 × 105 cfu L. monocytogenes (A–E, plain lines in C). (A) Severity scores were recorded. (B) L. monocytogenes in blood collected 2 days post-infection. (C) Survival of mice. (D) Cytokines, quantified by Luminex, in blood collected 1, 2, and 3 days post-infection (p.i.). The horizontal bar represents the median. Data are mean ± SD of four mice per group. P = 0.001 for TNF at day 1 p.i. 0.004 and 0.03 for TNF and IL-1β at day 2 p.i. and 0.05 for KC at day 3 p.i. ENA-78, IL-3, IL-17A, and MIP-1α were not detected. (E) CD45+, CD19+, CD3+, Ly6G+ and Ly6Chigh leukocytes in blood collected 1, 2, and 3 days post-infection. Each dot represents a mouse, and line and bars mean ± SD.
Discussion
This is the first report about the impact of the dual deficiency of SIRT3 and SIRT5 on immune cell development and antimicrobial host defenses. Double knockout mice developed normally and showed subtle, minor alterations of immune cell subpopulations and host responses to infection. Together with the fact that SIRT3−/− and SIRT5−/− mice are susceptible to bacterial sepsis like wild-type mice (37–40), these observations strengthen the development of pharmacological modulators of the activity of mitochondrial sirtuins for clinical purposes.
Notwithstanding that SIRT3 and SIRT5 orchestrate metabolism and oxidative stress responses, SIRT3 and SIRT5 whole body knockout mice have no macroscopic abnormalities (41, 42). The SIRT3/5−/− mouse line we generated here developed normally. No problem of fertility, sex distribution and growth were noticed. Surprisingly, the metabolism of SIRT3/5−/− BMDMs was similar to that of SIRT3/5+/+ BMDMs. Yet, the impact of SIRT3 and SIRT5 on metabolism was mainly demonstrated in cells or tissues such as the liver and the heart that are rich in mitochondrial sirtuins when compared to macrophages (38, 41, 60). Another SIRT3/5−/− mouse line has been recently generated. In line with our observations, no developmental defects were reported. Moreover, these SIRT3/5−/− mice were susceptible to streptozotocin-induced hyperglycemia like controls, while showing only a modest inner retinal dysfunction (61, 62).
Studies on the role of sirtuins in hematopoiesis and immune cell development are scarce. SIRT3/5−/− mice had a normal pool of HSCs and MPPs and a slightly increased number of CMP (and GMP as a trend) in their bone marrow. The primary and secondary immune organs of SIRT3/5−/− mice were largely unaffected, according to absolute numbers and proportions of immune cell subpopulations. There was only a slight reduction of thymus size, which did not impact the proportion of thymocyte subpopulations. This reminds the phenotype of SDHD-ESR mice with deletion of the succinate dehydrogenase, subunit D gene encoding for one of the subunits of the mitochondrial complex II (63). SDHD mice have a thymic atrophy without perturbation of thymocyte development. Overall, deficiencies in SIRT3, SIRT5, and SIRT3/5 do not seem to have a dramatic impact on immune cell development and/or functions [(37, 38) and present study]. Nonetheless, a role for these enzymes might come to light under stress or stimulatory conditions, or in aged mice. For example, SIRT1 shaped the T helper (Th) and T regulatory (Treg) responses of naïve T cells (64–68). Furthermore, in SIRT1−/− mice, the percentages of CD4+, CD8+, and CD4+CD8+ T cell subpopulations were normal, but thymocytes were at increased sensitivity to ionizing radiation induced DNA damaging (69). Finally, SIRT3−/− mice of 18–24 months had a lower frequency of bone marrow hematopoietic progenitors than mice of 12 weeks (70).
SIRT3/5−/− macrophages exposed to TLR agonists produced more inflammatory cytokines and less IL-10 than SIRT3/5+/+ macrophages, contrary to SIRT3−/− and SIRT5−/− macrophages that behaved like wild-type cells (37, 38). Accordingly, NF-κB and MAPK signaling pathways were increased in resting and/or LPS-stimulated SIRT3/5−/− macrophages. These data somehow support the possibility that SIRT3 and SIRT5 compensate each other in single knockout animals. Sirtuins are generally considered to drive anti-inflammatory responses. However, as nicely reviewed recently for SIRT1 (68), sirtuins may promote proinflammatory and anti-inflammatory effects depending on the context and whether myeloid or lymphoid cells are considered. For example, SIRT5 deficiency was associated with both increased and decreased innate inflammatory response in vivo (33, 71). More generally, contrasting observations have been reported for most sirtuins (SIRT1-3, SIRT5-6). Methodological differences may explain these differences when studying monocytes/macrophages (37, 38, 46): the origin/fate of the cells (BMDMs vs. peritoneum macrophages vs. established macrophage cell lines, growth factors used for differentiation and maturation state of macrophages), strategies to delete or overexpress sirtuins or to modulate sirtuin activity (siRNA, shRNA, expression vectors, full or cell-specific knockouts, pharmacological activators, and inhibitors), readouts, and subtle variations in NAD+ concentrations and circadian rhythm known to affect sirtuin activity or expression.
The expression levels of SIRT3 and SIRT5 decreased gradually from CMP to GMP and from GMP to granulocytes, which mirror the decline of mitochondrial mass and mitochondrial DNA during hematopoietic differentiation (72). Granulopoiesis relies on the expression of CCAAT/enhancer binding protein (C/EBP). The expression of SIRT1, which deacetylates C/EBPε and represses neutrophil terminal differentiation (73), declines during granulopoiesis (727 ± 13, 643 ± 20, and 307 ± 61 mRNA arbitrary units in CMP, GMP, and granulocytes, respectively). Thus, the downregulation of sirtuins seems to be a general feature associated with neutrophil development. The fact that neutrophil counts were normal and not increased in SIRT3/5−/− mice suggests either the implementation of compensatory mechanisms, possibly through SIRT1, or that SIRT3 and SIRT5 have a modest influence on granulopoiesis.
Neutrophils produce cytotoxic compounds that target pathogenic bacteria and fungi but are harmful for host tissues (74). Contrary to SIRT3/5+/+ neutrophils, and better than SIRT3−/− and SIRT5−/− neutrophils, SIRT3/5−/− neutrophils killed L. monocytogenes, which was associated with an augmented production of ROS but not of NETs. In comparison, neutrophils deficient in SIRT3 had a mild increase of intracellular ROS but performed either normal or increased NETosis (56, 57). SIRT1 deficiency did not impact on neutrophil functions, and the role of the remaining sirtuins has not been reported. Whereas, SIRT3−/− mice had increased neutrophil infiltration in lungs during sterile injury (36, 75) and mycobacterial infection impairing the survival of mice (76), SIRT5−/− had reduced inflammation and ischemia/reperfusion brain injury (77). Finally, SIRT3−/− and SIRT5−/− mice behaved like wild-type mice in models of sepsis requiring neutrophils to fight the infectious agents (37–40). Interestingly, SIRT3/5−/− mice resisted better than SIRT3/5+/+ mice to acute listeriosis, showing decreased signs of morbidity, reduced blood bacterial loads and significant albeit modest delayed mortality. SIRT3/5−/− mice expressed higher concentrations of cytokines but normal counts of leukocytes in blood, suggesting that the reactivity rather than the number of leukocytes protected SIRT3/5−/− mice from listeriosis. Interestingly, SIRT3/5−/− mice were not more resistant to mild listeriosis than their wild type counterparts, and behaved like wild type mice in a model of endotoxemia. These observations support the assumption that drugs targeting SIRT3 and SIRT5 should not have a deletary impact on host defenses, which would contrast with drugs targeting classical HDACs that strongly impaired innate immune defenses against infections in preclinical models and clinical settings (78–84).
Overall, the double deficiency in SIRT3 and SIRT5 had rather modest and subtle impacts on immune cell development and anti-microbial host defenses. It might be that SIRT4, the remaining mitochondrial sirtuin in SIRT3/5−/− mice, compensated for SIRT3 and SIRT5 absence. Unfortunately, whether SIRT4 affects immune responses has not been reported. Considering the link between sirtuins, metabolism and age-associated pathologies, it is possible that phenotypes will emerge in aged mice or in mice submitted to metabolic stress. SIRT3/5−/− mice should be tested in other preclinical models of sepsis. Nonetheless, putting together the data from the present study together with the fact that single deficiencies in SIRT3 and SIRT5 had no impact in a large panel of experimental sepsis (37–40), one may foresee that therapies directed against mitochondrial sirtuins or concomitant targeting of SIRT3 and SIRT5 activity should have no deep impact on antibacterial host defenses.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The animal study was reviewed and approved by Service des Affaires Vétérinaires, Direction Générale de l'Agriculture, de la Viticulture et des Affaires Vétérinaires (DGAV), état de Vaud (Epalinges, Switzerland; authorizations 876.9 and 877.9).
Author Contributions
TH and EC performed in vitro experiments. TH, EC, and DLR performed in vivo experiments. TR and TH conceived the project, designed the experiments and wrote the paper. All the authors revised the paper.
Funding
TR was supported by the Swiss National Science Foundation (SNSF, grant number 145014, 149511, and 173123) and received an interdisciplinary grant from the Faculty of Biology and Medicine of the University of Lausanne (Switzerland). TH received a scholarship from the Société Académique Vaudoise (Lausanne, Switzerland).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are indebted to Prof. Johan Auwerx (Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) for giving access to SIRT3−/− and SIRT5−/− mouse lines. We thank Yoann Müller, Frédéric Droux, Giovanni Zanchi, Elena Santia, and Vincent Carlen for technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02341/full#supplementary-material
Supplementary Figure S1. Full blots used to extract the panels shown in Figures 1B, 3B.
Supplementary Figure S2. Gating strategies for flow cytometry analysis.CD117 encodes for c-kit.
Supplementary Figure S3. (A) Extracellular acidification rate (ECAR) by SIRT3/5+/+ and SIRT3/5−/− BMDMs exposed to LPS (10 ng/ml) measured using the Seahorse technology. (B) Oxygen consumption rate (OCR) and ECAR by SIRT3/5+/+ and SIRT3−/− BMDMs measured using the Seahorse technology. Data are means ± SD from three mice aged 10–12 weeks analyzed in quadruplicate.
References
1. Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. (2013) 13:551–65. doi: 10.1038/nri3479
2. Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol. (2013) 4:387. doi: 10.3389/fimmu.2013.00387
3. Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. (2014) 289:35237–45. doi: 10.1074/jbc.R114.619304
4. Pope SD, Medzhitov R. Emerging principles of gene expression programs and their regulation. Mol Cell. (2018) 71:389–97. doi: 10.1016/j.molcel.2018.07.017
5. O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
6. Diskin C, Palsson-McDermott EM. Metabolic modulation in macrophage effector function. Front Immunol. (2018) 9:270. doi: 10.3389/fimmu.2018.00270
7. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. (1999) 13:2570–80. doi: 10.1101/gad.13.19.2570
8. Sauve AA, Youn DY. Sirtuins: NAD(+)-dependent deacetylase mechanism and regulation. Curr Opin Chem Biol. (2012) 16:535–43. doi: 10.1016/j.cbpa.2012.10.003
9. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. (2012) 13:225–38. doi: 10.1038/nrm3293
10. van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. (2017) 23:320–31. doi: 10.1016/j.molmed.2017.02.005
11. Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. (2016) 167:985–1000.e21. doi: 10.1016/j.cell.2016.10.016
12. Chen Y. Quantitative analysis of the Sirt5-regulated lysine succinylation proteome in mammalian cells. Methods Mol Biol. (2016) 1410:23–37. doi: 10.1007/978-1-4939-3524-6_2
13. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. (2011) 334:806–9. doi: 10.1126/science.1207861
14. Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. (2014) 19:605–17. doi: 10.1016/j.cmet.2014.03.014
15. Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci USA. (2013) 110:6601–6. doi: 10.1073/pnas.1302961110
16. Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. (2013) 49:186–99. doi: 10.1016/j.molcel.2012.10.024
17. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. (2010) 12:662–7. doi: 10.1016/j.cmet.2010.11.015
18. Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. (2012) 287:14078–86. doi: 10.1074/jbc.M112.355206
19. Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun. (2013) 441:191–5. doi: 10.1016/j.bbrc.2013.10.033
20. Li F, He X, Ye D, Lin Y, Yu H, Yao C, et al. NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell. (2015) 60:661–75. doi: 10.1016/j.molcel.2015.10.017
21. Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. (2016) 17:811–22. doi: 10.15252/embr.201541643
22. Guedouari H, Daigle T, Scorrano L, Hebert-Chatelain E. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim Biophys Acta Mol Cell Res. (2017) 1864:169–76. doi: 10.1016/j.bbamcr.2016.10.015
23. Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. (2009) 137:560–70. doi: 10.1016/j.cell.2009.02.026
24. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18:920–33. doi: 10.1016/j.cmet.2013.11.013
25. Shi L, Yan H, An S, Shen M, Jia W, Zhang R, et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol Oncol. (2019) 13:358–75. doi: 10.1002/1878-0261.12408
26. Bringman-Rodenbarger LR, Guo AH, Lyssiotis CA, Lombard DB. Emerging roles for SIRT5 in metabolism and cancer. Antioxid Redox Signal. (2017) 28:677–90. doi: 10.1089/ars.2017.7264
27. Torrens-Mas M, Oliver J, Roca P, Sastre-Serra J. SIRT3: oncogene and tumor suppressor in cancer. Cancers (Basel). (2017) 9:90. doi: 10.3390/cancers9070090
28. Gertz M, Steegborn C. Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol Life Sci. (2016) 73:2871–96. doi: 10.1007/s00018-016-2180-7
29. Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. (2018) 27:497–512. doi: 10.1016/j.cmet.2018.01.016
30. Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radic Biol Med. (2016) 100:164–74. doi: 10.1016/j.freeradbiomed.2016.04.197
31. Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. (2015) 88:73–81. doi: 10.1016/j.yjmcc.2015.09.005
32. Liu L, Peritore C, Ginsberg J, Shih J, Arun S, Donmez G. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson's disease. Behav Brain Res. (2015) 281:215–21. doi: 10.1016/j.bbr.2014.12.035
33. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
34. Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci Rep. (2016) 6:33201. doi: 10.1038/srep33201
35. Zhang Y, Wang XL, Zhou M, Kang C, Lang HD, Chen MT, et al. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp Mol Med. (2018) 50:21. doi: 10.1038/s12276-017-0002-0
36. Kurundkar D, Kurundkar AR, Bone NB, Becker EJ Jr, Liu W, Chacko B, et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight. (2019) 4:120722. doi: 10.1172/jci.insight.120722
37. Ciarlo E, Heinonen T, Lugrin J, Acha-Orbea H, Le Roy D, Auwerx J, et al. Sirtuin 3 deficiency does not alter host defenses against bacterial and fungal infections. Sci Rep. (2017) 7:3853. doi: 10.1038/s41598-017-04263-x
38. Heinonen T, Ciarlo E, Theroude C, Pelekanou A, Herderschee J, Le Roy D, et al. Sirtuin 5 deficiency does not compromise innate immune responses to bacterial infections. Front Immunol. (2018) 9:2675. doi: 10.3389/fimmu.2018.02675
39. Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem. (2015) 290:396–408. doi: 10.1074/jbc.M114.566349
40. Kim YR, Kim JS, Yun JS, Kim S, Kim SY, Jang K, et al. Toxoplasma gondii GRA8 induces ATP5A1-SIRT3-mediated mitochondrial metabolic resuscitation: a potential therapy for sepsis. Exp Mol Med. (2018) 50:e464. doi: 10.1038/emm.2017.308
41. Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep. (2012) 2:425. doi: 10.1038/srep00425
42. Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, et al. Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep. (2013) 3:2806. doi: 10.1038/srep02806
43. Ciarlo E, Heinonen T, Herderschee J, Fenwick C, Mombelli M, Le Roy D, et al. Impact of the microbial derived short chain fatty acid propionate on host susceptibility to bacterial and fungal infections in vivo. Sci Rep. (2016) 6:37944. doi: 10.1038/srep37944
44. Ciarlo E, Roger T. Screening the impact of sirtuin inhibitors on inflammatory and innate immune responses of macrophages and in a mouse model of endotoxic shock. Methods Mol Biol. (2016) 1436:313–34. doi: 10.1007/978-1-4939-3667-0_21
45. Pazar B, Ea HK, Narayan S, Kolly L, Bagnoud N, Chobaz V, et al. Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro. J Immunol. (2011) 186:2495–502. doi: 10.4049/jimmunol.1001284
46. Ciarlo E, Heinonen T, Theroude C, Herderschee J, Mombelli M, Lugrin J, et al. Sirtuin 2 deficiency increases bacterial phagocytosis by macrophages and protects from chronic staphylococcal infection. Front Immunol. (2017) 8:1037. doi: 10.3389/fimmu.2017.01037
47. Roger T, Ding X, Chanson AL, Renner P, Calandra T. Regulation of constitutive and microbial pathogen-induced human macrophage migration inhibitory factor (MIF) gene expression. Eur J Immunol. (2007) 37:3509–21. doi: 10.1002/eji.200737357
48. Tawadros T, Alonso F, Jichlinski P, Clarke N, Calandra T, Haefliger JA, et al. Release of macrophage migration inhibitory factor by neuroendocrine-differentiated LNCaP cells sustains the proliferation and survival of prostate cancer cells. Endocr Relat Cancer. (2013) 20:137–49. doi: 10.1530/ERC-12-0286
49. Roger T, Schneider A, Weier M, Sweep FC, Le Roy D, Bernhagen J, et al. High expression levels of macrophage migration inhibitory factor sustain the innate immune responses of neonates. Proc Natl Acad Sci USA. (2016) 113:E997–1005. doi: 10.1073/pnas.1514018113
50. Perreau M, Vigano S, Bellanger F, Pellaton C, Buss G, Comte D, et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J Exp Med. (2014) 211:2033–45. doi: 10.1084/jem.20140039
51. Vacher G, Ciarlo E, Savova-Bianchi D, Le Roy D, Hantier G, Niculita-Hirzel H, et al. Innate immune sensing of Fusarium culmorum by mouse dendritic cells. J Toxicol Environ Health A. (2015) 78:871–85. doi: 10.1080/15287394.2015.1051201
52. Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, et al. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci USA. (2009) 106:2348–52. doi: 10.1073/pnas.0808146106
53. Roger T, Delaloye J, Chanson AL, Giddey M, Le Roy D, Calandra T. Macrophage migration inhibitory factor deficiency is associated with impaired killing of gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J Infect Dis. (2013) 207:331–9. doi: 10.1093/infdis/jis673
54. De Kleer I, Willems F, Lambrecht B, Goriely S. Ontogeny of myeloid cells. Front Immunol. (2014) 5:423. doi: 10.3389/fimmu.2014.00423
55. Kurd N, Robey EA. T-cell selection in the thymus: a spatial and temporal perspective. Immunol Rev. (2016) 271:114–26. doi: 10.1111/imr.12398
56. Hayashi H, Cherpokova D, Martinod K, Witsch T, Wong SL, Gallant M, et al. Sirt3 deficiency does not affect venous thrombosis or NETosis despite mild elevation of intracellular ROS in platelets and neutrophils in mice. PLoS ONE. (2017) 12:e0188341. doi: 10.1371/journal.pone.0188341
57. Gaul DS, Weber J, van Tits LJ, Sluka S, Pasterk L, Reiner MF, et al. Loss of Sirt3 accelerates arterial thrombosis by increasing formation of neutrophil extracellular traps and plasma tissue factor activity. Cardiovasc Res. (2018) 114:1178–88. doi: 10.1093/cvr/cvy036
58. Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. (2004) 4:812–23. doi: 10.1038/nri1461
59. Witter AR, Okunnu BM, Berg RE. The essential role of neutrophils during infection with the intracellular bacterial pathogen Listeria monocytogenes. J Immunol. (2016) 197:1557–65. doi: 10.4049/jimmunol.1600599
60. Liang Q, Benavides GA, Vassilopoulos A, Gius D, Darley-Usmar V, Zhang J. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem J. (2013) 454:249–57. doi: 10.1042/BJ20130414
61. Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, et al. NAMPT-mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. (2016) 17:69–85. doi: 10.1016/j.celrep.2016.08.073
62. Lin JB, Lin JB, Chen HC, Chen T, Apte RS. Combined SIRT3 and SIRT5 deletion is associated with inner retinal dysfunction in a mouse model of type 1 diabetes. Sci Rep. (2019) 9:3799. doi: 10.1038/s41598-019-40177-6
63. Bejarano-Garcia JA, Millan-Ucles A, Rosado IV, Sanchez-Abarca LI, Caballero-Velazquez T, Duran-Galvan MJ, et al. Sensitivity of hematopoietic stem cells to mitochondrial dysfunction by SdhD gene deletion. Cell Death Dis. (2016) 7:e2516. doi: 10.1038/cddis.2016.411
64. Legutko A, Marichal T, Fievez L, Bedoret D, Mayer A, de Vries H, et al. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-gamma activity in dendritic cells. J Immunol. (2011) 187:4517–29. doi: 10.4049/jimmunol.1101493
65. Lim HW, Kang SG, Ryu JK, Schilling B, Fei M, Lee IS, et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J Exp Med. (2015) 212:607–17. doi: 10.1084/jem.20132378
66. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. (2010) 115:965–74. doi: 10.1182/blood-2009-02-207118
67. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
68. Chadha S, Wang L, Hancock WW, Beier UH. Sirtuin-1 in immunotherapy: a Janus-headed target. J Leukoc Biol. (2019) 106:337–43. doi: 10.1002/JLB.2RU1118-422R
69. Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. (2003) 100:10794–9. doi: 10.1073/pnas.1934713100
70. Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, et al. SIRT3 reverses aging-associated degeneration. Cell Rep. (2013) 3:319–27. doi: 10.1016/j.celrep.2013.01.005
71. Qin K, Han C, Zhang H, Li T, Li N, Cao X. NAD(+) dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J Autoimmun. (2017) 81:120–9. doi: 10.1016/j.jaut.2017.04.006
72. de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, Snoeck HW. Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell. (2017) 21:725–9.e4. doi: 10.1016/j.stem.2017.11.002
73. Bartels M, Govers AM, Fleskens V, Lourenco AR, Pals CE, Vervoort SJ, et al. Acetylation of C/EBPepsilon is a prerequisite for terminal neutrophil differentiation. Blood. (2015) 125:1782–92. doi: 10.1182/blood-2013-12-543850
74. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
75. Tian YG, Zhang J. Protective effect of SIRT3 on acute lung injury by increasing manganese superoxide dismutase-mediated antioxidation. Mol Med Rep. (2018) 17:5557–65. doi: 10.3892/mmr.2018.8469
76. Kim TS, Jin YB, Kim YS, Kim S, Kim JK, Lee HM, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. (2019) 15:1356–75. doi: 10.1080/15548627.2019.1582743
77. Diaz-Canestro C, Merlini M, Bonetti NR, Liberale L, Wust P, Briand-Schumacher S, et al. Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int J Cardiol. (2018) 260:148–55. doi: 10.1016/j.ijcard.2017.12.060
78. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. (2011) 117:1205–17. doi: 10.1182/blood-2010-05-284711
79. Ciarlo E, Savva A, Roger T. Epigenetics in sepsis: targeting histone deacetylases. Int J Antimicrob Agents. (2013) 42(Suppl.):S8–12. doi: 10.1016/j.ijantimicag.2013.04.004
80. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. (2007) 109:2781–90. doi: 10.1182/blood-2006-05-021873
81. Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. (2009) 32:517–23. doi: 10.1097/SHK.0b013e3181a44c79
82. Mombelli M, Lugrin J, Rubino I, Chanson AL, Giddey M, Calandra T, et al. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J Infect Dis. (2011) 204:1367–74. doi: 10.1093/infdis/jir553
83. Zhang LT, Yao YM, Lu JQ, Yan XJ, Yu Y, Sheng ZY. Sodium butyrate prevents lethality of severe sepsis in rats. Shock. (2007) 27:672–7. doi: 10.1097/SHK.0b013e31802e3f4c
Keywords: sirtuin, innate immunity, cytokine, infection, sepsis, metabolism, macrophage, neutrophil
Citation: Heinonen T, Ciarlo E, Le Roy D and Roger T (2019) Impact of the Dual Deletion of the Mitochondrial Sirtuins SIRT3 and SIRT5 on Anti-microbial Host Defenses. Front. Immunol. 10:2341. doi: 10.3389/fimmu.2019.02341
Received: 17 July 2019; Accepted: 17 September 2019;
Published: 01 October 2019.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Xiaohu Huang, University of Maryland, Baltimore, United StatesChaofeng Han, Second Military Medical University, China
Copyright © 2019 Heinonen, Ciarlo, Le Roy and Roger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thierry Roger, dGhpZXJyeS5yb2dlckBjaHV2LmNo
†ORCID: Thierry Roger orcid.org/0000-0002-9358-0109