 Helong Dai
Helong Dai Angus W. Thomson
Angus W. Thomson Natasha M. Rogers
Natasha M. Rogers- 1Department of Urological Organ Transplantation, The Second Xiangya Hospital of Central South University, Changsha, China
- 2Clinical Research Center for Organ Transplantation of Hunan Province, Changsha, China
- 3Department of Surgery, Thomas E. Starzl Transplantation Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 4Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 5Center for Transplant and Renal Research, Westmead Institute for Medical Research, Westmead, NSW, Australia
- 6Renal Division, Westmead Hospital, Westmead, NSW, Australia
- 7Westmead Clinical School, University of Sydney, Camperdown, NSW, Australia
Dendritic cells (DCs) are highly specialized, bone marrow (BM)-derived antigen-processing and -presenting cells crucial to the induction, integration and regulation of innate, and adaptive immunity. They are stimulated by damage-associated molecular patterns (DAMPS) via pattern recognition receptors to promote inflammation and initiate immune responses. In addition to residing within the parenchyma of all organs as part of the heterogeneous mononuclear phagocyte system, DCs are an abundant component of the inflammatory cell infiltrate that appears in response to ischemia reperfusion injury (IRI). They can play disparate roles in the pathogenesis of IRI since their selective depletion has been found to be protective, deleterious, or of no benefit in mouse models of IRI. In addition, administration of DC generated and manipulated ex vivo can protect organs from IRI by suppressing inflammatory cytokine production, limiting the capacity of DCs to activate NKT cells, or enhancing regulatory T cell function. Few studies however have investigated specific signal transduction mechanisms underlying DC function and how these affect IRI. Here, we address current knowledge of the role of DCs in regulation of IRI, current gaps in understanding and prospects for innovative therapeutic intervention at the biological and pharmacological levels.
An Introduction to Dendritic Cells (DCs)
DCs comprise a heterogeneous population of uniquely well-equipped, bone marrow-derived innate immune cells. They are distributed ubiquitously throughout the body and play an important homeostatic and anti-infectious sentinel role. DCs are highly efficient, antigen (Ag)-acquiring, -processing, and presenting cells, that perform crucial roles in the instigation and regulation of acute and chronic inflammatory responses. While they promote self-tolerance in the healthy steady-state and can be targeted by microbes and tumors to evade immunity, DCs integrate innate and adaptive immunity effectively to combat infection and can also be exploited as anti-cancer vaccines. During autoimmunity and transplant rejection, DCs instigate deleterious immune responses that cause disease; on the other hand, they can be harnessed to silence these conditions using novel targeting and adoptive cell therapy approaches. In the context of ischemic tissue injury that adversely affects short- and long-term transplant outcome, DCs appear to play diverse roles in regulation of the inflammatory response.
DC Subsets—Phenotype and Function
Distinct subsets of DCs, including myeloid/conventional DCs (cDCs) and type-I IFN-producing plasmacytoid DCs (pDCs) have been described extensively elsewhere (1–3) and are summarized in Table 1. Classical DCs, but not monocyte-derived antigen (Ag)-presenting cells (APCs), are critical for central and peripheral regulatory T cell (Treg) induction and the development of tolerance (4) as well as shaping effector T cell responses to Ag. Beyond the classical characterization of DCs, new phenotypic and functional subsets of DCs continue to emerge (5, 6). Moreover, the discovery of new lineage markers and introduction of innovative imaging technologies (including use of reporter mice) have helped to distinguish classical DC subsets from other myeloid cells, particularly macrophages, in tissues such as the kidney (7).

Table 1. DC subsets in mouse and human: phenotype, localization, and function.
According to their maturation and functional status, DCs can be divided into immature, mature, and regulatory populations. Regulatory DCs (DCregs) have been extensively investigated in transplantation (8–11) and autoimmune disease (12, 13), ranging from pre-clinical models to pilot human clinical trials. Infusion of donor-derived DCregs prior to transplantation has been shown to prolong kidney allograft survival and inhibit donor-reactive CD8+ memory T cell responses in non-human primates (14, 15). First-in-human phase I/II clinical trials of adoptive DCreg therapy in living donor renal and liver transplantation have recently been instigated (8, 16).
Ischemia-Reperfusion Injury (IRI)
IRI is a common clinical condition triggered by various physiological derangements (sepsis, cardiogenic shock, vascular surgery, organ retrieval for transplantation). Its pathogenesis has been comprehensively described elsewhere (17–21) but is essentially characterized by endothelial dysfunction, reactive oxygen species (ROS) production, secretion of pro-inflammatory mediators, and recruitment of inflammatory cells which exacerbate/perpetuate tissue injury.
Inflammatory Cells Characterize IRI
Inflammatory cell infiltration after IRI is rapid, peaking 24 h following reperfusion (22). Gr-1+ neutrophils, which release ROS and proteolytic enzymes, and NK1.1+CD161+ NKT cells (CD56+ in humans) which elaborate pro-inflammatory cytokines, are the predominant early inflammatory cells that impair organ function within hours of IRI (23–26). However, neutrophil-depleted animals are not protected following ischemic insult to the kidney (27, 28). Neutrophils, NKT cells and DCs intercommunicate to enhance tissue injury through chemokine/cytokine secretion, as well as cell-cell contact (29, 30). These interactions are depicted in Figure 1 in the context of the early period following renal IRI. While DCs are thought to be crucial to the pathogenesis of IRI, their role in kidney IRI remains unclear, since depletion may be protective (22), deleterious (31), or of no benefit (32). In situ targeting of DCs with the vitamin D analog paricalcitol can induce intrahepatic tolerogenic DCs and alleviate CD4+ T cell responses to attenuate hepatocellular damage (33). In contrast, pDC-released type I IFN promotes tissue injury through induction of hepatocyte IFN regulatory factor-1 (IRF-1) to induce apoptosis (34).
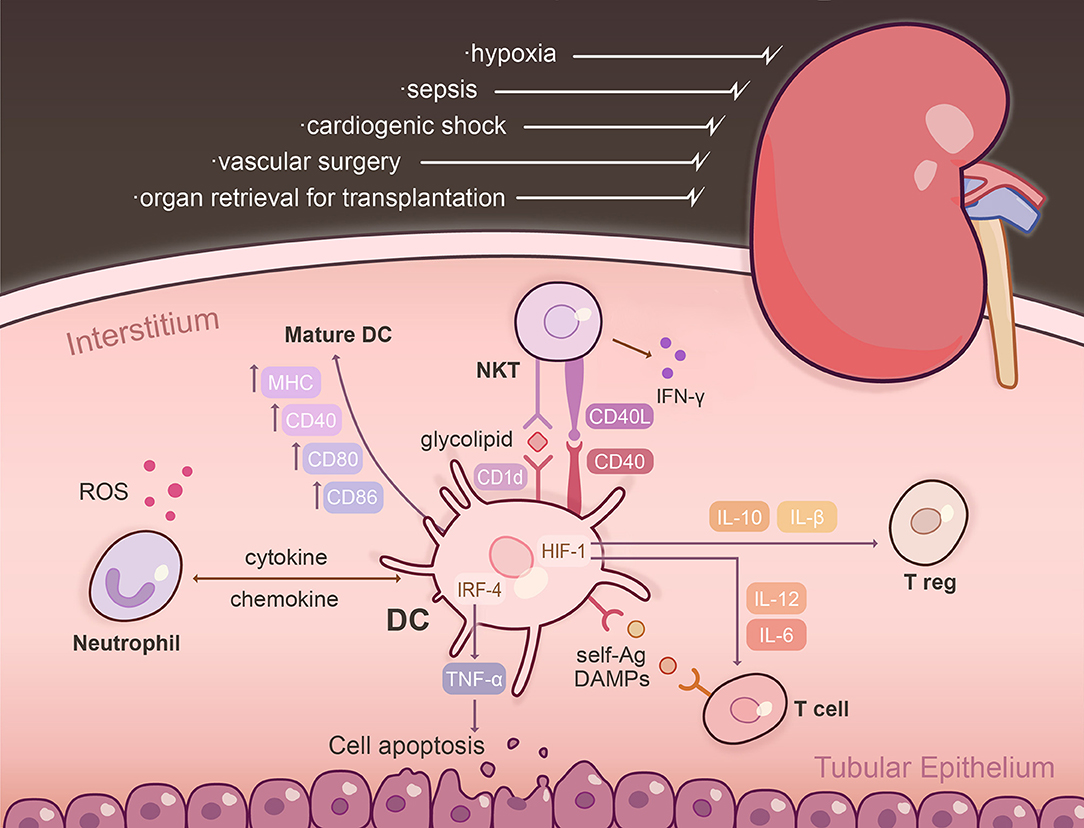
Figure 1. The DC interactome following renal ischemia reperfusion injury (IRI). IRI is a common clinical condition triggered by various physiological derangements including sepsis, cardiogenic shock, vascular surgery, and organ retrieval for transplantation. Following injury, resident, and influxing DCs become activated within the kidney parenchyma and are the dominant TNFα-producing cells. The effect of TNF-α is dependent on the transcription factor IRF-4, promoting renal tubular epithelial cell apoptosis, glomerular endothelial damage, and fibrin deposition. Post-IRI, intra-renal DCs upregulate markers that designate them as mature APCs, including MHC, CD80, CD86, CD40, and CD1d. Activation of NKT cells via CD40 initiates IFN-g to amplify the innate immune response. Renal DCs are capable of presenting self-Ag to a variety of T cells in the context of IRI: CD11c+ DCs cross-present Ag to CD8+ T cells and glycolipids are presented to NKT cells via CD1d. Exposure of DCs to hypoxia/reperfusion augments production of IL-12 and IL-6, and an inflammatory T cell phenotype through HIF-1α transcriptional regulation. Absence of DC-specific HIF-1α limits expression of IL-10 and TGF-β, which are potent inducers of Tregs. DCs intercommunicate with neutrophils and NKT cells to enhance tissue injury through chemokine/cytokine secretion, as well as cell-based contact. Neutrophils release ROS and proteolytic enzymes, and NK1.1+CD161+ NKT cells elaborate pro-inflammatory cytokines and are the predominant early inflammatory cells that impair organ function within hours of IRI. Ag, antigen; DAMPs, damage-associated molecular patterns; HIF-1, hypoxia-inducible factor-1; IRF-4, interferon regulatory factor-4; ROS, reactive oxygen species.
Macrophages, NK cells and adaptive immune cells, including T and B cells, infiltrate injured tissues hours after reperfusion (22). Macrophages have been considered to polarize into M1 (classical) and M2 (alternatively-activated) subsets with pro- or anti-inflammatory function, respectively, although recent reassessment suggests a broader functional repertoire for these cells (35). Heme oxygenase 1 (HO-1) negatively regulates M1 polarization and hepatocellular damage in both mouse liver IRI and human liver transplant biopsies (36). Further evidence shows that HO-1 regulates macrophage activation through the Sirtuin/p53 signaling pathway to drive hepatic death during liver IRI (37). Deletion or inhibition of Dectin-1 can suppress M1 macrophage polarization and alleviate myocardial IRI (38). T cells can continue to localize in injured kidneys for 2 weeks and display an effector-memory and activation phenotype characterized by CD44hiCD62L− and CD69+ expression, respectively (39). Treg function is decreased in aged mice, which contributes to exacerbated liver IRI (40).
Hypoxic Stimuli
In transplantation, immune-mediated injury is a composite of the innate response to IRI and alloimmune reactivity to foreign Ag. Many clinical studies confirm a link between delayed graft function and a higher rate of acute rejection (41–44). We and others have shown that hypoxia activates DCs (45), as evidenced by their phenotypic maturation, pro-inflammatory cytokine production, and enhanced T cell stimulatory and migratory capacity (46–50).
Altered DCs function under hypoxia has been ascribed to changes in hypoxia-inducible factor (HIF)-1α activity. Longer-term hypoxia (>1 h) followed by reperfusion downregulates the G-protein coupled purinergic receptor P2Y11R through HIF-1α transcriptional regulation, resulting in augmented production of IL-12 and IL-6 and an inflammatory T cell phenotype in response to extracellular ATP (51). Hypoxia, and therefore HIF-1α-dependent upregulation of adenosine receptor A2B (52), or triggering receptor expressed on myeloid cells- (TREM)-1 (53, 54) can induce Th2 polarization and pro-inflammatory cytokine release, respectively. BMDCs lacking functional HIF-1α show deficiencies in type I IFN secretion and fail to activate CD8+ T cells (55). Interestingly, the effect of hypoxia on DCs is attenuated by rapamycin (47) and augmented by concurrent exposure to LPS (56).
Together, these data suggest that DCs are reprogrammed by a hypoxic environment to modulate their inflammation-activating repertoire, however discrepant features in many studies may reflect differences in hypoxia duration and severity. In vivo, HIF-1α is crucial for DC-dependent generation of Tregs and Treg homing to inflammatory sites. Absence of DC-specific HIF-1α limits IL-10 and TGF-β expression (both potent inducers of Tregs) and reduces expression of aldehyde dehydrogenase (necessary for catalytic production of retinoic acid) (57). Although HIF-1α is protective against IRI (58), there are no studies assessing its specific effect on DCs in this model.
Ischemic Preconditioning
Ischemic preconditioning (IPC) occurs when brief periods of sublethal ischemia are performed prior to a subsequent prolonged episode and increases organ resistance to IRI (59). However, the underlying mechanism (s) remain elusive. Exposure of renal parenchyma to IPC prior to IRI does not alter the number of infiltrating leukocytes, but reduces CD11c+ DCs (compared to IRI alone), which display lower CCR7 and IL-17 transcript levels, decreased CD80 expression and upregulated IL-10 (60). Elimination of CD11c+ DCs (CD11b+ and CD8+ subsets) using liposomal clodronate is associated with partial loss of preconditioning benefits.
Organ Cross-Talk During IRI
IRI rarely occurs in isolation—systemic release of pro-inflammatory cytokines and cell trafficking to primary and secondary lymphoid tissue ensures widespread modulation of innate immunity. Substantial cross-talk between the injured and remote organs manifests clinically as multi-organ failure. The onset of acute lung injury in the context of acute kidney injury (AKI) occurs more frequently than any other organ combination (61). Experimental models have identified a distinct pulmonary genomic signature during AKI, with differentially expressed pro-inflammatory and pro-apoptotic pathways (62). The intestine can also aggravate the systemic inflammatory response syndrome (63). Increased permeability from gut hypoperfusion, modification of microbiota composition, and blood-borne propagation of toxins contribute to the outcome of AKI. Gut microbiota can also modulate the inflammatory response to injury. Short-chain fatty acids such as acetate, propionate, and butyrate are produced by fermentation of complex carbohydrates and have a broad range of anti-inflammatory effects (64). Concurrent incubation of murine BMDCs with short-chain fatty acids and LPS reduces upregulation of maturation markers CD80, CD86, and CD40 (65). Butyrate specifically inhibits production of IL-12 in human monocyte-derived DCs, limiting development of effector Th1 cells (66, 67). Treatment with short-chain fatty acids also protects against AKI, with lower frequencies of infiltrating macrophages and activated DCs (65) in addition to effects on renal tubular epithelial cell apoptosis and ROS production.
Organ-Specific Roles of DCs in IRI
Tissue-Specific Phenomena
The majority of animal data on the function of DCs in IRI is limited to the liver, heart and kidney, and is best characterized in the latter. Robust models of pulmonary, intestinal and pancreatic IRI are lacking due to technical challenges. Human data is also sparse due to inherent difficulties with DC detection and limited tissue availability. In vivo targeting of organ-specific DCs to limit IRI and innate immune activation is difficult due to the lack of defining cell-surface markers. Tables 1, 2 outline the breadth of available markers, which show significant overlap.

Table 2. Identified DC subsets implicated in IRI.
Kidney
CD11c+ DCs are resident within the kidney parenchyma (86), infiltrate following ischemic insult and are the dominant TNFα-producing cells (77) (Figure 1). TNF-α is crucial to neutrophil influx post-IRI, renal tubular epithelial cell apoptosis, glomerular endothelial damage and fibrin deposition (87–90). The effect of TNF-α depends on the transcription factor IRF-4 (91). Non-specific elimination of DC using liposomal clodronate or administration of etanercept (a decoy receptor for TNF-α) abrogates AKI in IRF-4 deficient mice (91).
Intra-renal DCs post-IRI constitutively express markers that designate them as professional APCs (MHC, CD80, CD86, CD40, CD54, CD1d), but not tissue macrophage markers [CD169, CD204 (77)]. As early as 4 h post-IRI, higher levels of maturation marker expression are observed, favoring the hypothesis that DC maturation occurs in situ rather than by cell replacement (77). Multiple studies have identified a continuum of DC phenotypes that contribute to the innate immune response and IRI. Monocyte subsets migrate to inflamed tissue and differentiate into activated DCs as CCR2+CX3CR1loGR-1+Ly6Chi cells (92). Resident CX3CR1hi monocytes patrolling the parenchymal space also migrate, differentiate, and participate in inflammation. Both CCR2 (93) and CX3CR1 (94) are essential to this process.
The traditional paradigm has been discrete separation of mononuclear phagocytic cell populations into CD11b+ macrophages and CD11c+ DCs that increase following IRI. More recent thinking has focused on phenotypic heterogeneity and functional plasticity of these cells. They are divided broadly into 5 subsets based on intensity of CD11b and CD11c expression and are further characterized by a comprehensive set of cell surface markers and transcription factors (76). All subsets demonstrate phagocytic capacity but differ in their migratory capacity and cytokine profile as well as their ability to stimulate naïve T cells and alter T cell polarization ex vivo. Renal DCs are capable of presenting self-Ag to a variety of T cells in the context of IRI, -CD11c+ DCs cross-present Ag to CD8+ T cells post-IRI (95) and glycolipids are presented to NKT cells via the CD1d cell surface receptor (96). Activation of NKT via CD40 initiates IFN-γ to amplify the innate immune response. Administration of CD1d Ab that blocks NKT-DC interactions or genetic depletion of NKT provides significant protection against renal IRI (96). Renal DCs are also primarily responsible for presenting renal proteins to Ag-specific CD4+ T cells within draining renal lymph nodes. In a model of unilateral IRI where ovalbumin is placed beneath the operated kidney capsule, CD11c+ DCs from the ipsi- or contra-lateral renal lymph nodes induce proliferation of DO11.10 (ovalbumin-restricted) T cells (80). CD11c− fractions failed to induce T cell stimulation.
Naïve rodent kidneys demonstrate additional DC subsets defined by the expression (or absence) of CD103+. The CD103+ subset is primarily involved in Ag cross-presentation after migrating to lymph nodes (97). Transplantation of syngeneic grafts subjected to negligible or prolonged cold storage leads to depletion of CD103+ DCs, regardless of IRI, but only CD103− DCs under the former condition (98). Donor cells are replaced by host DCs, accompanied by increases in CD3+CD4+CD62L− T cells, indicative of effector/effector-memory populations.
Heart
Despite advances in percutaneous coronary intervention and use of statin and antiplatelet agents, the incidence of post-infarct heart failure is rising (99). Adverse ventricular remodeling following myocardial infarction increases mortality by precipitating heart failure. Studies of DC subsets in experimental myocardial IRI are relatively uncommon, mostly due to the technical difficulty, high mortality, and significant variations in infarct size associated with left anterior descending artery ligation (100, 101).
DC subsets are found within the CD45+ leukocyte population in healthy myocardium, particularly within the right atrium (68). Conventional CD11c+MHC II+ DCs (cDC1s) have been defined by CD103 or CD11b expression (or neither) (68). These 2 subsets are also classified by XCR1 or CD172 (SIRP-α) expression, and levels of transcription factors IRF8 and IRF4, respectively (102). Under homeostatic conditions, cDC1s present cardiac self-Ag to α myosin heavy chain-specific CD4+ T cells in mediastinal lymph nodes to induce Treg formation. pDCs (CD11bloMHCII−CD11cloPDCA-1+Ly6C+) have also been detected.
DCs infiltrate infarcted myocardium (103), increasing 10-fold, particularly the CD11b+ subset expressing maturation markers such as CD40 (68). Post-myocardial infarction cDCs upregulate CCR7 (the chemokine receptor required for lymph node migration) and CD40 expression; specific activation of the cDC2 subset induces CD86 expression as well as CD4+ T cell proliferation and accompanies IFN-γ and IL-17 production (102). Although mature DC numbers correlate with LV dysfunction (70) and depletion of cDCs reduces infarct size and adverse cardiomyocyte hypertrophy in the border zone (68), a deleterious contribution of DCs is disputed. Prolonged DC ablation leads to impaired LV remodeling, sustained expression of pro-inflammatory cytokines and altered monocyte/macrophage recruitment (69). The administration of liposomal clodronate, which depletes both DCs and macrophages, also impairs wound healing (104). pDC numbers also increase following infarction, but their depletion appears not to affect LV function (68).
DCregs have therapeutic potential in post-infarct healing, modulating Tregs and macrophages. “Tolerogenic DCs” primed with TNF-α and mouse cardiac tissue Ags reduced infarct size, improved LV ejection fraction and increased post-infarct survival (105). This correlated with increased FoxP3+ Tregs in inguinal and mediastinal lymph nodes, as well as in the post-infarct heart. Adoptive transfer of these Tregs into mice post-MI also improved wound healing. Interestingly, troponin- or myosin-primed DCs failed to recapitulate the protective effects seen with tissue-primed DCs.
Liver
Similar roles for DCs have been demonstrated in hepatic IRI. Interestingly, cDCs are depleted from the liver parenchyma, rather than enriched, during maximal injury (12 h post-reperfusion). These DCs display a mature phenotype, with marked CD86 upregulation, but are also necrotic and apoptotic. Depletion of CD11c+ DCs in CD11c-DTR mice worsens tissue damage and the pro-inflammatory cytokine profile following liver IRI. Adoptive transfer of cDCs into CD11c-depleted mice mitigates this injury, dependent on intrinsic TLR9 activation (from hepatocyte DNA release) and subsequent IL-10 production (73). This is also thought to modulate CD11bintLy6Chi inflammatory monocyte cytokine production and ROS generation. Augmenting DC numbers with GM-CSF increases susceptibility to liver IRI (106). Post-reperfusion, these DCs exhibited a mature phenotype and enhanced allostimulatory capacity. This effect required release of high motility box group 1 that upregulates DC-expressed TLR4 and interacts with both TLR9 and the receptor for advanced glyosylation end-products (RAGE) to activate DCs (107).
Liver transplant recipients are more likely to develop a tolerant phenotype compared to recipients of other solid organs. This may be due to a comparatively high density of DCs to parenchymal cell populations (108) and the refractory behavior of liver DCs compared with DCs from other tissues in response to TLR ligation (109, 110). Expression of CD39, which drives ATP hydrolysis, is increased on murine liver mDCs but not pDCs, and the levels exceed those in splenic DCs. Not unexpectedly, CD39KO syngeneic liver transplants exhibit worse tissue injury compared to WT grafts, with accompanying upregulation of CD80, CD86, and MHC II, and downregulation of PD-L1 on hepatic mDCs (109). Notably, CD39 expression on freshly isolated human hepatic CD45+Lin−BDCA-1+ DCs is higher than on peripheral blood mDCs.
The discrepant findings suggesting a dichotomous role for DCs in liver IRI imply that tissue-resident and recruited DCs may play distinct roles in the response to injury. DCs are highly motile, and are recruited into the liver in response to macrophage inflammatory peptide-1 (111) (MCP-1 = CCL2), which is produced following IRI. Administration of Flt3L to WT mice increases the DC-resident population 10-fold (112) and enhances liver parenchymal injury; injury is significantly reduced in Flt3L KO mice (74). Adoptive transfer of CD11c+ mDCs or PDCA-1+ pDCs into Flt3L KO mice at the time of IRI significantly worsens injury. To distinguish the contribution of hepatic-resident vs. infiltrating DCs, a liver transplant model between congenic mice has been used. Donor-derived hepatic DCs upregulated maturation markers to a higher level than on infiltrating (recipient) DCs. Use of Flt3L KO liver donors increased ischemic injury, suggesting a protective effect of liver-resident DCs.
The role of pDCs in liver IRI is poorly defined. IFN-α is produced predominantly by pDCs (113) in response to sensing of self-DNA and TLR9 ligation, which drive the inflammatory response in IRI. IFN-α promotes IRF-1 expression by hepatocytes to induce pro-inflammatory cytokine and death receptor expression (34). Depletion of pDCs using anti-PDCA-1 Ab or use of IFN-α blocking Ab protected the liver against IRI.
The Therapeutic Potential of DC
Despite decades of research and multiple clinical trials, no pharmacological agents are in clinical use for IRI. Cell-based therapy, which capitalizes on the capacity of regulatory cells to modulate adverse immune responses, represents a potential therapeutic option. DCs can exhibit a protective phenotype to modulate pathogenesis of IRI. However, unlike adoptive transfer of Tregs, which can change to a proinflammatory/effector phenotype, depending upon the microenvironment (114, 115), DCregs are maturation-resistant, phenotypically stable and do not need to persist in vivo as functional cells to mediate immune response change (116). Pharmacological or genetic manipulation produces a regulatory phenotype (DCregs), secreting low levels of Th1-driving cytokines (particularly bioactive IL-12), and comparatively high levels of anti-inflammatory cytokines (such as IL-10) (117). They are weak T cell stimulators and possess the capacity to induce or expand Tregs. While the beneficial effect of DCregs has been established in multiple pre-clinical models of allograft rejection [reviewed in Thomson et al. (118)], their testing in IRI has been minimal. Elegant experimental work has demonstrated that following adoptive transfer, allogeneic DCregs are rapidly killed by host immune cells (predominantly NK cells), and their effect is mediated by recipient DCs (119, 120). Even in the absence of alloAg, adoptively transferred DCs can direct T cells to produce a regulatory response and mitigate IRI. Thus, while lack of the sphingosine-1-phosphate receptor 3 (S1pr3) protects against renal IRI (121), infusion of S1pr3−/− DCs also protects against IRI which requires functional Tregs, CD11c+ DCs, and CD169+ macrophages (122). In addition, DCs treated with an adenosine 2A receptor agonist protect WT mice from IRI via suppressing NKT cell activation and IFN-γ production (123).
Current Gaps in Knowledge
Variations and limitations in methodology, both for identification and ex vivo generation of DCs, have made it difficult to determine whether phenotype and function are consistent between organs affected by IRI. Which self-Ags DCs respond to in IRI is not well-defined and clearly differs according to damaged organ segment, cell type or cellular component (glomerulus vs. tubule, hepatocyte vs. endothelia, myosin heavy chain vs. tubulin). In addition, there is still no consensus to clearly distinguish tissue DCs and macrophages (124, 125). Cell-surface molecules such as CD11c and CX3CR1 are expressed on various myeloid cells and are imperfect DC markers (126). A further caveat is that ex vivo-generated DCs do not truly represent a physiological subset of tissue-resident DCs. The majority of studies to date have been conducted in animal models of IRI, and the paucity of human data highlights limitations regarding generalizability of results.
DCregs are currently being tested as cell therapy in clinical liver and kidney transplantation, and the results of these trials are eagerly awaited. If DCregs are to be considered as therapy, it will be necessary to determine cell type (autologous vs. allogeneic vs. donor Ag-loaded), dosage, timing, and frequency of infusion (s) and cost. Other aspects, such as potential sensitization of recipients (with the use of banked allogeneic DCs) and comparison of DCregs with other regulatory cell types, have also not been addressed. It is also unclear whether the relative efficacy of DC therapy also depends on the organ undergoing IRI. Published research has also focused on the effect of DCs given prior to the onset of IRI, whereas their influence post-injury is not known.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Science Foundation of China (81800664), Natural Science Foundation of Hunan Province of China (2019JJ50842), and Huxiang Young Talents of Hunan Province (2019RS2013) to HD, National Institutes of Health grants R01 AI118777, U19 AI131453, and U01 AI137799 to AT, and National Health and Medical Research Council Career Development Fellowship (GNT1158977) and Project Grant (GNT1138372) to NR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Teteris SA, Engel DR, Kurts C. Homeostatic and pathogenic role of renal dendritic cells. Kidney Int. (2011) 80:139–45. doi: 10.1038/ki.2011.129
2. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
3. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
4. Esterhazy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T (reg) cells and tolerance. Nat Immunol. (2016) 17:545–55. doi: 10.1038/ni.3408
5. Bryant C, Fromm PD, Kupresanin F, Clark G, Lee K, Clarke C, et al. A CD2 high-expressing stress-resistant human plasmacytoid dendritic-cell subset. Immunol Cell Biol. (2016) 94:447–57. doi: 10.1038/icb.2015.116
6. Rahmatpanah F, Agrawal S, Scarfone VM, Kapadia S, Mercola D, Agrawal A. Transcriptional profiling of age-associated gene expression changes in human circulatory CD1c+ myeloid dendritic cell subset. J Gerontol A Biol Sci Med Sci. (2019) 74:9–15. doi: 10.1093/gerona/gly106
7. Brahler S, Zinselmeyer BH, Raju S, Nitschke M, Suleiman H, Saunders BT, et al. Opposing roles of dendritic cell subsets in experimental GN. J Am Soc Nephrol. (2018) 29:138–54. doi: 10.1681/ASN.2017030270
8. Thomson AW, Humar A, Lakkis FG, Metes DM. Regulatory dendritic cells for promotion of liver transplant operational tolerance: rationale for a clinical trial and accompanying mechanistic studies. Hum Immunol. (2018) 79:314–21. doi: 10.1016/j.humimm.2017.10.017
9. Thomson AW, Ezzelarab MB. Regulatory dendritic cells: profiling, targeting, and therapeutic application. Curr Opin Organ Transplant. (2018) 23:538–45. doi: 10.1097/MOT.0000000000000565
10. Thomson AW, Zahorchak AF, Ezzelarab MB, Butterfield LH, Lakkis FG, Metes DM. Prospective clinical testing of regulatory dendritic cells in organ transplantation. Front Immunol. (2016) 7:15. doi: 10.3389/fimmu.2016.00015
11. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. (2007) 7:610–21. doi: 10.1038/nri2132
12. Obregon C, Kumar R, Pascual MA, Vassalli G, Golshayan D. Update on dendritic cell-induced immunological and clinical tolerance. Front Immunol. (2017) 8:1514. doi: 10.3389/fimmu.2017.01514
13. Hilkens CM, Isaacs JD, Thomson AW. Development of dendritic cell-based immunotherapy for autoimmunity. Int Rev Immunol. (2010) 29:156–83. doi: 10.3109/08830180903281193
14. Ezzelarab MB, Lu L, Guo H, Zahorchak AF, Shufesky WF, Cooper DK, et al. Eomesodermin (lo) CTLA4 (hi) alloreactive CD8+ memory T cells are associated with prolonged renal transplant survival induced by regulatory dendritic cell infusion in CTLA4 immunoglobulin-treated nonhuman primates. Transplantation. (2016) 100:91–102. doi: 10.1097/TP.0000000000000871
15. Ezzelarab MB, Zahorchak AF, Lu L, Morelli AE, Chalasani G, Demetris AJ, et al. Regulatory dendritic cell infusion prolongs kidney allograft survival in nonhuman primates. Am J Transplant. (2013) 13:1989–2005. doi: 10.1111/ajt.12310
16. Moreau A, Alliot-Licht B, Cuturi MC, Blancho G. Tolerogenic dendritic cell therapy in organ transplantation. Transpl Int. (2017) 30:754–64. doi: 10.1111/tri.12889
17. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. (2011) 121:4210–21. doi: 10.1172/JCI45161
18. Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. (2016) 67:293–307. doi: 10.1146/annurev-med-050214-013407
19. Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. (2018) 46:1650–67. doi: 10.1159/000489241
20. Collard CD, Gelman S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology. (2001) 94:1133–8. doi: 10.1097/00000542-200106000-00030
21. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
22. Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. (2010) 30:268–77. doi: 10.1016/j.semnephrol.2010.03.005
23. Mizuno S, Nakamura T. Prevention of neutrophil extravasation by hepatocyte growth factor leads to attenuations of tubular apoptosis and renal dysfunction in mouse ischemic kidneys. Am J Pathol. (2005) 166:1895–905. doi: 10.1016/S0002-9440(10)62498-4
24. Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. (2010) 120:331–42. doi: 10.1172/JCI38702
25. Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev. Nephrol. (2015) 11:88–101. doi: 10.1038/nrneph.2014.180
26. Hashimoto S, Honda M, Takeichi T, Sakisaka M, Narita Y, Yoshii D, et al. Intravital imaging of neutrophil recruitment in intestinal ischemia-reperfusion injury. Biochem Biophys Res Commun. (2018) 495:2296–302. doi: 10.1016/j.bbrc.2017.12.140
27. Paller MS. Effect of neutrophil depletion on ischemic renal injury in the rat. J Lab Clin Med. (1989) 113:379–86.
28. Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J Clin Invest. (2002) 110:1083–91. doi: 10.1172/JCI15623
29. Tittel AP, Heuser C, Ohliger C, Llanto C, Yona S, Hammerling GJ, et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat Methods. (2012) 9:385–90. doi: 10.1038/nmeth.1905
30. Schuster S, Hurrell B, Tacchini-Cottier F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol. (2013) 94:671–5. doi: 10.1189/jlb.1012540
31. Kim MG, Boo CS, Ko YS, Lee HY, Cho WY, Kim HK, et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. (2010) 25:2908–21. doi: 10.1093/ndt/gfq183
32. Lu L, Faubel S, He Z, Andres Hernando A, Jani A, Kedl R, et al. Depletion of macrophages and dendritic cells in ischemic acute kidney injury. Am J Nephrol. (2012) 35:181–90. doi: 10.1159/000335582
33. Funken D, Ishikawa-Ankerhold H, Uhl B, Lerchenberger M, Rentsch M, Mayr D, et al. In situ targeting of dendritic cells sets tolerogenic environment and ameliorates CD4 (+) T-cell response in the postischemic liver. FASEB J. (2017) 31:4796–808. doi: 10.1096/fj.201601358R
34. Castellaneta A, Yoshida O, Kimura S, Yokota S, Geller DA, Murase N, et al. Plasmacytoid dendritic cell-derived IFN-alpha promotes murine liver ischemia/reperfusion injury by induction of hepatocyte IRF-1. Hepatology. (2014) 60:267–77. doi: 10.1002/hep.27037
35. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
36. Zhang M, Nakamura K, Kageyama S, Lawal AO, Gong KW, Bhetraratana M, et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight. (2018) 3:120596. doi: 10.1172/jci.insight.120596
37. Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, et al. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J Hepatol. (2017) 67:1232–42. doi: 10.1016/j.jhep.2017.08.010
38. Fan Q, Tao R, Zhang H, Xie H, Lu L, Wang T, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. (2019) 139:663–78. doi: 10.1161/CIRCULATIONAHA.118.036044
39. Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. (2009) 75:526–35. doi: 10.1038/ki.2008.602
40. Liu R, Zhang S, Ma W, Lu H, Gao J, Gan X, et al. Age-dependent loss of induced regulatory T cell function exacerbates liver ischemia-reperfusion injury. Mol Immunol. (2018) 103:251–6. doi: 10.1016/j.molimm.2018.10.004
41. Quiroga I, McShane P, Koo DD, Gray D, Friend PJ, Fuggle S, et al. Major effects of delayed graft function and cold ischaemia time on renal allograft survival. Nephrol Dial Transplant. (2006) 21:1689–96. doi: 10.1093/ndt/gfl042
42. Seo CH, Ju JI, Kim MH, Jun KW, Ahn SH, Hwang JK, et al. Risk factors and long-term outcomes of delayed graft function in deceased donor renal transplantation. Ann Surg Treat Res. (2015) 89:208–14. doi: 10.4174/astr.2015.89.4.208
43. Wu WK, Famure O, Li Y, Kim SJ. Delayed graft function and the risk of acute rejection in the modern era of kidney transplantation. Kidney Int. (2015) 88:851–8. doi: 10.1038/ki.2015.190
44. Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. (2011) 11:2279–96. doi: 10.1111/j.1600-6143.2011.03754.x
45. Dai H, Watson AR, Fantus D, Peng L, Thomson AW, Rogers NM. Rictor deficiency in dendritic cells exacerbates acute kidney injury. Kidney Int. (2018) 94:951–63. doi: 10.1016/j.kint.2018.06.010
46. Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, et al. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. (2008) 84:1472–82. doi: 10.1189/jlb.0208082
47. Rama I, Bruene B, Torras J, Koehl R, Cruzado JM, Bestard O, et al. Hypoxia stimulus: an adaptive immune response during dendritic cell maturation. Kidney Int. (2008) 73:816–25. doi: 10.1038/sj.ki.5002792
48. Ogino T, Onishi H, Suzuki H, Morisaki T, Tanaka M, Katano M. Inclusive estimation of complex antigen presentation functions of monocyte-derived dendritic cells differentiated under normoxia and hypoxia conditions. Cancer Immunol Immunother. (2012) 61:409–24. doi: 10.1007/s00262-011-1112-5
49. Winning S, Fandrey J. Dendritic cells under hypoxia: how oxygen shortage affects the linkage between innate and adaptive immunity. J Immunol Res. (2016) 2016:5134329. doi: 10.1155/2016/5134329
50. Blengio F, Raggi F, Pierobon D, Cappello P, Eva A, Giovarelli M, et al. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology. (2013) 218:76–89. doi: 10.1016/j.imbio.2012.02.002
51. Chadet S, Ivanes F, Benoist L, Salmon-Gandonniere C, Guibon R, Velge-Roussel F, et al. Hypoxia/reoxygenation inhibits P2Y11 receptor expression and its immunosuppressive activity in human dendritic cells. J Immunol. (2015) 195:651–60. doi: 10.4049/jimmunol.1500197
52. Yang M, Ma C, Liu S, Shao Q, Gao W, Song B, et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol. (2010) 88:165–71. doi: 10.1038/icb.2009.77
53. Pierobon D, Bosco MC, Blengio F, Raggi F, Eva A, Filippi M, et al. Chronic hypoxia reprograms human immature dendritic cells by inducing a proinflammatory phenotype and TREM-1 expression. Eur J Immunol. (2013) 43:949–66. doi: 10.1002/eji.201242709
54. Bosco MC, Pierobon D, Blengio F, Raggi F, Vanni C, Gattorno M, et al. Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood. (2011) 117:2625–39. doi: 10.1182/blood-2010-06-292136
55. Wobben R, Husecken Y, Lodewick C, Gibbert K, Fandrey J, Winning S. Role of hypoxia inducible factor-1alpha for interferon synthesis in mouse dendritic cells. Biol Chem. (2013) 394:495–505. doi: 10.1515/hsz-2012-0320
56. Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, Takala J, et al. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS ONE. (2010) 5:e0010983. doi: 10.1371/journal.pone.0010983
57. Fluck K, Breves G, Fandrey J, Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. (2016) 9:379–90. doi: 10.1038/mi.2015.67
58. Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. (2008) 19:39–46. doi: 10.1681/ASN.2006090998
59. Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury–a review. J Surg Res. (2008) 150:304–30. doi: 10.1016/j.jss.2007.12.747
60. Zhang T, Song N, Fang Y, Teng J, Xu X, Hu J, et al. Delayed ischemic preconditioning attenuated renal ischemia-reperfusion injury by inhibiting dendritic cell maturation. Cell Physiol Biochem. (2018) 46:1807–20. doi: 10.1159/000489366
61. Ricci Z, Ronco C. Pulmonary/renal interaction. Curr Opin Crit Care. (2010) 16:13–8. doi: 10.1097/MCC.0b013e328334b13b
62. Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. (2008) 19:547–58. doi: 10.1681/ASN.2007040469
63. Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. (2007) 28:384–93. doi: 10.1097/shk.0b013e31805569df
64. Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. (2014) 25:657–70. doi: 10.1681/ASN.2013080905
65. Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. (2015) 26:1877–88. doi: 10.1681/ASN.2014030288
66. Wang B, Morinobu A, Horiuchi M, Liu J, Kumagai S. Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cell Immunol. (2008) 253:54–8. doi: 10.1016/j.cellimm.2008.04.016
67. Saemann MD, Parolini O, Bohmig GA, Kelemen P, Krieger PM, Neumuller J, et al. Bacterial metabolite interference with maturation of human monocyte-derived dendritic cells. J Leukoc Biol. (2002) 71:238–46. doi: 10.1189/jlb.71.2.238
68. Lee JS, Jeong SJ, Kim S, Chalifour L, Yun TJ, Miah MA, et al. Conventional dendritic cells impair recovery after myocardial infarction. J Immunol. (2018) 201:1784–98. doi: 10.4049/jimmunol.1800322
69. Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. (2012) 125:1234–45. doi: 10.1161/CIRCULATIONAHA.111.052126
70. Naito K, Anzai T, Sugano Y, Maekawa Y, Kohno T, Yoshikawa T, et al. Differential effects of GM-CSF and G-CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction. J Immunol. (2008) 181:5691–701. doi: 10.4049/jimmunol.181.8.5691
71. Maekawa Y, Mizue N, Chan A, Shi Y, Liu Y, Dawood S, et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: a regulator of bone marrow-derived dendritic cells. Circulation. (2009) 120:1401–14. doi: 10.1161/CIRCULATIONAHA.109.865956
72. Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, et al. CD141 (+) myeloid dendritic cells are enriched in healthy human liver. J Hepatol. (2014) 60:135–42. doi: 10.1016/j.jhep.2013.08.007
73. Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. (2010) 120:559–69. doi: 10.1172/JCI40008
74. Zhang M, Ueki S, Kimura S, Yoshida O, Castellaneta A, Ozaki KS, et al. Roles of dendritic cells in murine hepatic warm and liver transplantation-induced cold ischemia/reperfusion injury. Hepatology. (2013) 57:1585–96. doi: 10.1002/hep.26129
75. Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. (2009) 50:1537–46. doi: 10.1002/hep.23153
76. Kawakami T, Lichtnekert J, Thompson LJ, Karna P, Bouabe H, Hohl TM, et al. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J Immunol. (2013) 191:3358–72. doi: 10.4049/jimmunol.1300342
77. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. (2007) 71:619–28. doi: 10.1038/sj.ki.5002132
78. Kassianos AJ, Wang X, Sampangi S, Muczynski K, Healy H, Wilkinson R. Increased tubulointerstitial recruitment of human CD141hi CLEC9A+ and CD1c+ myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. Am J Physiol Renal Physiol. (2013) 305:F1391–401. doi: 10.1152/ajprenal.00318.2013
79. Leone DA, Kozakowski N, Kornauth C, Waidacher T, Neudert B, Loeffler AG, et al. The phenotypic characterization of the human renal mononuclear phagocytes reveal a co-ordinated response to injury. PLoS ONE. (2016) 11:e0151674. doi: 10.1371/journal.pone.0151674
80. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. (2005) 68:1096–108. doi: 10.1111/j.1523-1755.2005.00502.x
81. Desch AN, Henson PM, Jakubzick CV. Pulmonary dendritic cell development and antigen acquisition. Immunol Res. (2013) 55:178–86. doi: 10.1007/s12026-012-8359-6
82. Masten BJ, Olson GK, Tarleton CA, Rund C, Schuyler M, Mehran R, et al. Characterization of myeloid and plasmacytoid dendritic cells in human lung. J Immunol. (2006) 177:7784–93. doi: 10.4049/jimmunol.177.11.7784
83. von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas JA, Strickland DH, et al. Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J Immunol. (2005) 175:1609–18. doi: 10.4049/jimmunol.175.3.1609
84. Stagg AJ. Intestinal dendritic cells in health and gut inflammation. Front Immunol. (2018) 9:2883. doi: 10.3389/fimmu.2018.02883
85. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev. (2014) 260:86–101. doi: 10.1111/imr.12194
86. Soos TJ, Sims TN, Barisoni L, Lin K, Littman DR, Dustin ML, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. (2006) 70:591–6. doi: 10.1038/sj.ki.5001567
87. Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, et al. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol. (1989) 134:419–30.
88. Donnahoo KK, Meng X, Ayala A, Cain MP, Harken AH, Meldrum DR. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am J Physiol. (1999) 277:R922–9. doi: 10.1152/ajpregu.1999.277.3.R922
89. Donnahoo KK, Shames BD, Harken AH, Meldrum DR. Review article: the role of tumor necrosis factor in renal ischemia-reperfusion injury. J Urol. (1999) 162:196–203. doi: 10.1097/00005392-199907000-00068
90. Misseri R, Meldrum DR, Dinarello CA, Dagher P, Hile KL, Rink RC, et al. TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am J Physiol Renal Physiol. (2005) 288:F406–11. doi: 10.1152/ajprenal.00099.2004
91. Lassen S, Lech M, Rommele C, Mittruecker HW, Mak TW, Anders HJ. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol. (2010) 185:1976–83. doi: 10.4049/jimmunol.0904207
92. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
93. Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol. (2003) 14:2503–15. doi: 10.1097/01.ASN.0000089563.63641.A8
94. Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, et al. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. (2008) 294:F264–71. doi: 10.1152/ajprenal.00204.2007
95. Snelgrove SL, Lo C, Hall P, Lo CY, Alikhan MA, Coates PT, et al. Activated renal dendritic cells cross present intrarenal antigens after ischemia-reperfusion injury. Transplantation. (2017) 101:1013–24. doi: 10.1097/TP.0000000000001427
96. Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. (2007) 178:5899–911. doi: 10.4049/jimmunol.178.9.5899
97. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med. (2009) 206:3115–30. doi: 10.1084/jem.20091756
98. Ozaki KS, Kimura S, Nalesnik MA, Sico RM, Zhang M, Ueki S, et al. The loss of renal dendritic cells and activation of host adaptive immunity are long-term effects of ischemia/reperfusion injury following syngeneic kidney transplantation. Kidney Int. (2012) 81:1015–25. doi: 10.1038/ki.2011.458
99. Bahit MC, Kochar A, Granger CB. Post-myocardial infarction heart failure. JACC Heart Fail. (2018) 6:179–186. doi: 10.1016/j.jchf.2017.09.015
100. Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, et al. Myocardial infarct size and ventricular function in rats. Circ Res. (1979) 44:503–12. doi: 10.1161/01.RES.44.4.503
101. Yang XP, Sabbah HN, Liu YH, Sharov VG, Mascha EJ, Alwan I, et al. Ventriculographic evaluation in three rat models of cardiac dysfunction. Am J Physiol. (1993) 265:H1946–52. doi: 10.1152/ajpheart.1993.265.6.H1946
102. Van der Borght K, Scott CL, Nindl V, Bouche A, Martens L, Sichien D, et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. (2017) 18:3005–17. doi: 10.1016/j.celrep.2017.02.079
103. Zhang J, Yu ZX, Fujita S, Yamaguchi ML, Ferrans VJ. Interstitial dendritic cells of the rat heart. Quantitative and ultrastructural changes in experimental myocardial infarction. Circulation. (1993) 87:909–20. doi: 10.1161/01.CIR.87.3.909
104. van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ >Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. (2007) 170:818–29. doi: 10.2353/ajpath.2007.060547
105. Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation. (2017) 135:1444–1457. doi: 10.1161/CIRCULATIONAHA.116.023106
106. Tsung A, Zheng N, Jeyabalan G, Izuishi K, Klune JR, Geller DA, et al. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J Leukoc Biol. (2007) 81:119–28. doi: 10.1189/jlb.0706468
107. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
108. Steptoe RJ, Patel RK, Subbotin VM, Thomson AW. Comparative analysis of dendritic cell density and total number in commonly transplanted organs: morphometric estimation in normal mice. Transpl Immunol. (2000) 8:49–56. doi: 10.1016/S0966-3274(00)00010-1
109. Yoshida O, Kimura S, Jackson EK, Robson SC, Geller DA, Murase N, et al. CD39 expression by hepatic myeloid dendritic cells attenuates inflammation in liver transplant ischemia-reperfusion injury in mice. Hepatology. (2013) 58:2163–75. doi: 10.1002/hep.26593
110. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. (2010) 10:753–66. doi: 10.1038/nri2858
111. Matsuno K, Nomiyama H, Yoneyama H, Uwatoku R. Kupffer cell-mediated recruitment of dendritic cells to the liver crucial for a host defense. Dev Immunol. (2002) 9:143–9. doi: 10.1080/1044667031000137610
112. Steptoe RJ, Fu F, Li W, Drakes ML, Lu L, Demetris AJ, et al. Augmentation of dendritic cells in murine organ donors by Flt3 ligand alters the balance between transplant tolerance and immunity. J Immunol. (1997) 159:5483–91.
113. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
114. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
115. Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, et al. Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS ONE. (2012) 7:e35572. doi: 10.1371/journal.pone.0035572
116. Morelli AE, Thomson AW. Orchestration of transplantation tolerance by regulatory dendritic cell therapy or in-situ targeting of dendritic cells. Curr Opin Organ Transplant. (2014) 19:348–56. doi: 10.1097/MOT.0000000000000097
117. Zahorchak AF, Macedo C, Hamm DE, Butterfield LH, Metes DM, Thomson AW. High PD-L1/CD86 MFI ratio and IL-10 secretion characterize human regulatory dendritic cells generated for clinical testing in organ transplantation. Cell Immunol. (2018) 323:9–18. doi: 10.1016/j.cellimm.2017.08.008
118. Thomson AW, Metes DM, Ezzelarab MB, Raich-Regue D. Regulatory dendritic cells for human organ transplantation. Transplant Rev. (2019) 33:130–6. doi: 10.1016/j.trre.2019.05.001
119. Divito SJ, Wang Z, Shufesky WJ, Liu Q, Tkacheva OA, Montecalvo A, et al. Endogenous dendritic cells mediate the effects of intravenously injected therapeutic immunosuppressive dendritic cells in transplantation. Blood. (2010) 116:2694–705. doi: 10.1182/blood-2009-10-251058
120. Wang Z, Divito SJ, Shufesky WJ, Sumpter T, Wang H, Tkacheva OA, et al. Dendritic cell therapies in transplantation revisited: deletion of recipient DCs deters the effect of therapeutic DCs. Am J Transplant. (2012) 12:1398–408. doi: 10.1111/j.1600-6143.2012.04060.x
121. Bajwa A, Huang L, Ye H, Dondeti K, Song S, Rosin DL, et al. Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury. J Immunol. (2012) 189:2584–96. doi: 10.4049/jimmunol.1200999
122. Bajwa A, Huang L, Kurmaeva E, Gigliotti JC, Ye H, Miller J, et al. Sphingosine 1-phosphate receptor 3-deficient dendritic cells modulate splenic responses to ischemia-reperfusion injury. J Am Soc Nephrol. (2016) 27:1076–90. doi: 10.1681/ASN.2015010095
123. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A (2)AR agonist attenuate acute kidney injury. J Clin Invest. (2012) 122:3931–42. doi: 10.1172/JCI63170
124. Gottschalk C, Kurts C. The debate about dendritic cells and macrophages in the kidney. Front Immunol. (2015) 6:435. doi: 10.3389/fimmu.2015.00435
125. Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol. (2014) 10:625–43. doi: 10.1038/nrneph.2014.170
Keywords: dendritic cells, ischemic injury, kidney, liver, heart
Citation: Dai H, Thomson AW and Rogers NM (2019) Dendritic Cells as Sensors, Mediators, and Regulators of Ischemic Injury. Front. Immunol. 10:2418. doi: 10.3389/fimmu.2019.02418
Received: 13 August 2019; Accepted: 27 September 2019;
Published: 15 October 2019.
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Jerzy Kupiec-Weglinski, David Geffen School of Medicine UCLA, United StatesChristian Kurts, University of Bonn, Germany
Copyright © 2019 Dai, Thomson and Rogers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angus W. Thomson, dGhvbXNvbmF3QHVwbWMuZWR1; Natasha M. Rogers, bmF0YXNoYS5yb2dlcnNAc3lkbmV5LmVkdS5hdQ==