 Wenxiang Sun
Wenxiang Sun Hongsheng Wang
Hongsheng Wang Chen-Feng Qi
Chen-Feng Qi Silvia Bolland
Silvia Bolland- Laboratory of Immunogenetics, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rockville, MD, United States
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by increased production of autoantibodies, which commonly target nuclear antigens, and concomitant deposition of immune complexes that cause inflammation in tissues. SLE is often associated with increased systemic expression of type I interferons, in some cases due to dysregulation in nucleic acid-sensing innate pathways. There is strong genetic evidence for a link between cytoplasmic RNA sensing pathways (RIG-I/MDA5) and SLE, both in human patients and murine models, however questions still remain regarding pathway initiation, cell types involved and downstream effects. Here we show that MAVS, an essential adaptor for RIG-I/MDA5 signaling, is necessary for all symptoms of autoimmune disease that develop spontaneously in the lupus model FcγRIIB−/− mice. This effect was independent of type I interferon signaling, TLR7 expression or STING, all three factors that have been connected to autoimmunity. Mixed bone marrow reconstitution experiments showed reduced occurrence in autoimmune germinal centers and diminished autoantibody production by MAVS-deficient B cells. Thus, MAVS plays a B cell intrinsic role in autoreactive B cell activation that is independent of its anti-viral functions and independent of elevated type I interferon expression.
Introduction
Dysregulation of a number of innate immune pathways has been associated with the onset and pathogenesis of systemic lupus erythematosus (SLE) (1–3). Studies over the last decade have revealed critical roles for intracellular DNA and RNA sensing pathways in the production of type I interferons (IFNs), a double-edge sword in antiviral defense and autoimmunity (1–3). Among the innate immune pathways that recognize viral RNA and have been linked to SLE are members of the RIG-I-like receptor (RLR) group. This group of RNA sensors includes RIG-I, MDA5 or LGP2 (4–6), which activate the downstream adaptor molecule MAVS (also known as IPS-1, VISA, and Cardif). MAVS undergoes polymerization, described as prion-like filament structures on the outer membrane of mitochondria (7), and recruits numerous adaptor molecules and kinases (8–14). MAVS initiates two well-characterized signaling cascades: 1) the interferon regulatory factors (IRFs) 3 and 7 pathway that leads to the expression of type I/III IFNs and 2) the NF-kB pathway that promotes the expression of proinflammatory proteins (15).
Among pathways upstream of MAVS, multiple genetic studies have linked functional alterations of MDA5 with autoimmune disorders in humans and mice. A missense mutation (G821S) of Ifih1 (encoding MDA5) results in a constitutively active form of MDA5 that causes mice to develop lupus-like symptoms including glomerulonephritis and a skin rash (16). A similar gain-of-function mutation of R779H in IFIH1 was reported in a patient with severe early-onset SLE (17). Our previous studies showed that increased copy number of the Ifih1 gene in mice elevated systemic IFN expression, which was insufficient to induce autoimmune symptoms by itself, but accelerated disease when combined with the lupus prone background of the FcγRIIB deficiency (18).
Genetic studies on the MAVS adaptor itself have identified a loss-of-function mutation of MAVS (C79F) in a subset of SLE patients (19). The mutation was associated with reduced expression of type I IFNs and other inflammatory factors but has not yet proved to have any effect on the pathogenesis of SLE in these patients (19). To better understand the mechanism by which MAVS and related pathways increase SLE susceptibility and to determine the cell types that contribute to this phenotype, we have crossed Mavs−/− mice to the autoimmune prone Fcgr2b−/− background. We have compared them to Fcgr2b−/− mice carrying the STINGgt mutation because STING has regulatory roles in development of autoimmune diseases in several models including antigen-induced arthritis (20), autoimmune encephalomyelitis (21), and SLE (22). Deficiency of FcγRIIB in mice is a well-established murine model of SLE and has been shown to induce lupus-like symptoms including increased production of autoantibodies, proteinuria, and glomerulonephritis (23). Mixed bone marrow reconstitution experiments showed that MAVS deficiency abrogated these autoimmune manifestations in a B cell intrinsic manner, which points to a role for MAVS in B cell activation that goes beyond increasing systemic type I IFN expression.
Materials and Methods
Mice
R2−/− mice were obtained from the Taconic National institute of Allergy and infectious Diseases colony. STINGgt/gt and CD45.1 mice were purchased from Jackson Laboratories. Mavs−/− mice were kindly provided by Dr. James Z. Chen (15). Mice were bred and housed in a specific pathogen-free facility in the National Institute of Allergy and Infectious Diseases, National institutes of Health. Littermate mice were used. Notes regarding genetic backgrounds: MAVS deficient mice were generated in 129sv background and backcrossed an unknown number of generations to C57BL/6J. FcγRIIB deficient mice were generated in 129sv background and backcrossed 12 generations to C57BL/6N. When these two strains were bred together, the background was a mix of C57BL/6N and 6J. Heterozygous breeders were used to ensure littermates that were MAVS deficient and sufficient. The IgHa allotype was introduced by breeding congenic mice B6.Cg-Gpi1aThy1aIgha/J (Jackson laboratory line 1317) to FcγRIIB deficient mice. The Ifnar1−/− mice on C57BL/6J background were obtained from the Jackson Laboratory.
Animal studies were approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
TLR7 Stimulation Assay
For in vivo stimulation, B6 and Mavs−/− mice were injected i.v. with 50 μg of imiquimod (Invivogen) in 100 μl of PBS per animal. Six hours later the serum levels of IL-6 and TNF-α were measured by Mouse ELISA kits (ThermoFisher) according to the manufacturer's instructions. For in vitro stimulation, splenic B cells were purified by depletion of CD43+ and CD9+ cells using an EasySep Mouse PE Positive Selection Kit II (Stemcell Technologies). The purity of cells was determined by flow cytometry to be >97% Supplemental Figure 1. The cells were labeled with 5 μM of CFSE and cultured at 1 × 106/ml with complete RPMI 1640 medium in the presence or absence of 2 μg/ml of imiquimod or various concentrations of R848 for 3 days. Then the cells were stained with 7AAD and analyzed by flow cytometry.
Flow Cytometry and Antibodies
Spleen, lymph nodes, and BM single cell suspensions were prepared and stained with antibodies listed in Table 1. Biotinylated antibodies were revealed by streptavidin-conjugated fluorochromes. Cells were analyzed with a FACS LSR II or LSRFortessa X-20 (BD Biosciences) and FlowJo software (Treestar).
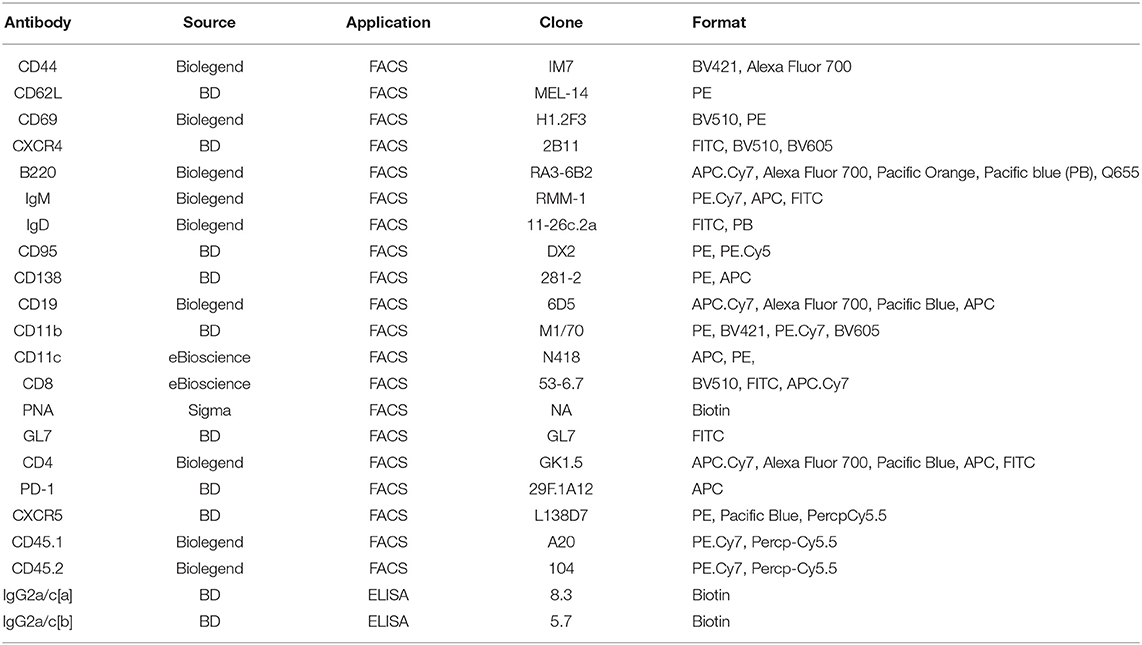
Table 1. List of Antibodies used in FACS and ELISA.
BM Chimera
1 × 107 of BM cells of indicated mice were injected i.v. into lethally irradiated mice that received a dose of 940 rad 1 day earlier. Two months later, the reconstituted mice were analyzed for autoantibody production, tissue pathology, and cellular distribution by flow cytometry.
Proteinuria and ANA Titer Testing
Urinal protein levels were detected with Chemstrip 2GP urine test strips (Roche). Serum ANA titers were determined by the Hep-2 system. Briefly, mice serum samples were diluted at 1:100, 1:300, 1:900, and 1:2,700 with PBS and incubated with Hep-2 substrate slides (MBL, AN-1012) at room temperature for 30 min. After washing twice with PBS for 5 min, the slides were incubated with a secondary anti-mouse IgG-FITC antibody in the dark at room temperature for 30 min. Following washing three times with PBS, the slides were read under a fluorescence microscope and images were taken.
Histology
Mouse kidney tissues were processed for H&E staining by Histoserv, Inc. (Rockville, MD). The glomerulonephritis scoring was done by measuring the diameters of multiple glomeruli as reported previously (18). Splenic sections were stained with PNA and imaged with an Olympus BX41 microscope (10 × and 40 × objectives) equipped with an Olympus DP71 camera.
Quantitative Real-Time PCR (qPCR)
Purified B cells were extracted for total RNA using a RNeasy Mini kit (Qiagen). cDNA was synthesized using a Superscript III First-Strand synthesis system (ThermoFisher), followed by PCR using SYBR Green Supermix and primers of Gapdh and Tlr7 (24) in a CFX Connect Real-Time System (Bio-Rad).
Statistics
Data were analyzed using 2-tailed Student's t test (2-tailed) or One-way ANOVA test. P < 0.05 is regarded as statistically significant.
Results
MAVS Deficiency, but Not the STINGgt Mutation, Ameloriates Kidney Pathology, and Increases Survival in FcγRIIB-Deficient Mice
FcγRIIB-deficient mice (R2−/−) die prematurely due to spontaneous lupus-like autoimmune disease that primarily targets the kidney in the form of lethal glomerulonephritis (23). R2−/− mortality starts at 4 months of age, reaching more than 80% penetrance by 10 months of age [(23) and Figure 1A]. To test the role of cytoplasmic nucleic acid sensors in this autoimmune pathology, R2−/− mice were crossbred either to MAVS-deficient mice (15), or to mice harboring a gt mutation in the sting gene so that it encodes a non-functional STING protein (STINGgt) (25). We observed that MAVS-deficient R2−/− (Mavs−/−R2−/−) mice survived significantly longer than their littermates MAVS-wild type (Mavs+/+R2−/−) or MAVS-heterozygous (Mavs+/−R2−/−) (Figure 1A). This extended survival rate was associated with markedly reduced proteinuria, a sign of kidney failure, and significantly improved histological scores on kidney pathology in Mavs−/−R2−/− mice compared to Mavs+/+R2−/− or Mavs+/−R2−/− (Figures 1B,C). In contrast, the survival or proteinuria score of R2−/− mice was not changed by the presence of the STINGgt mutation (Figures 1D,E).
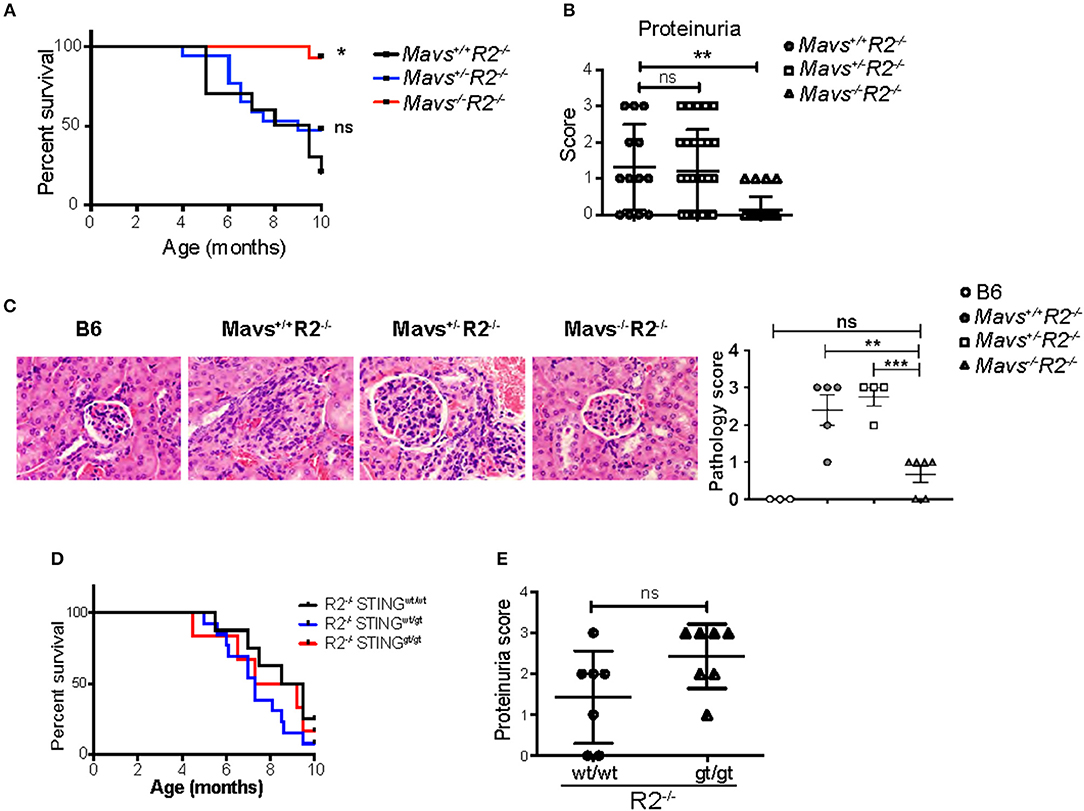
Figure 1. MAVS is required for the autoimmune disease in R2−/− mice. (A) Survival rates of Mavs+/+R2−/− (n = 10), Mavs+/−R2−/− (n = 17), and Mavs−/−R2−/− mice (n = 14). (B) Proteinuria scores of 5-month old mice indicated. Each symbol represents a mouse. (C) H&E staining of kidney sections of mice indicated. Magnification, 20×. The right panel is a summary of pathology scores of multiple mice. Each symbol represents a mouse. (D) Survival rates of mice of different genotypes indicated. n = 6~13. (E) Proteinuria scores of 5-month old mice indicated. Each symbol represents a mouse. *p < 0.05; **p < 0.01; ***p < 0.001 (One-way ANOVA).
MAVS Plays an Essential Role in Promoting Autoreactive Antibodies and Germinal Centers That Goes Beyond the Induction of TLR7 Expression
Serum ANA titers were dramatically reduced in Mavs−/−R2−/− mice compared with MAVS-sufficient R2−/− control mice (Figure 2A). In contrast, the STINGgt mutation (heterozygous or homozygous) (Figure 2B) or deficiency in the receptor for type I IFN (Figure 2C) didn't significantly alter ANA titers in R2−/− mice. Our results with the IFN receptor deficiency did not depend on the age of the mice and are consistent with those previously reported by Richer et al. (26) using the same mouse model.
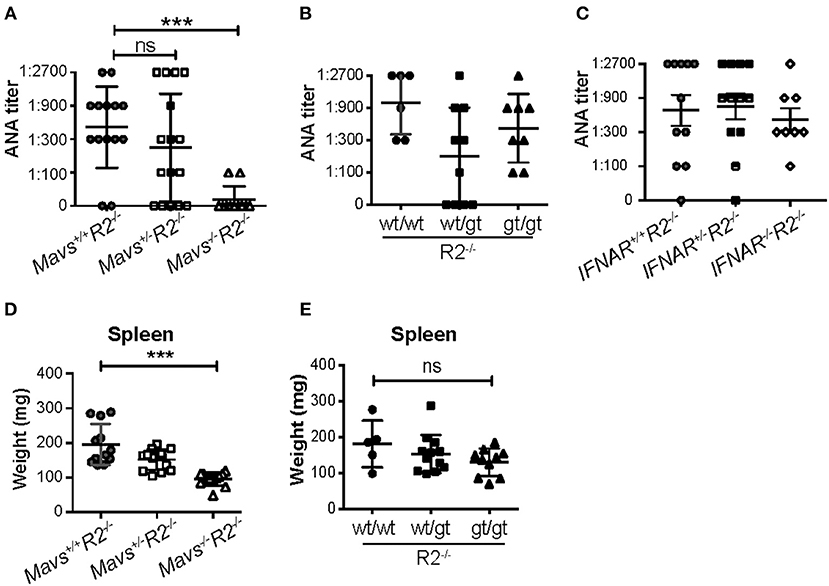
Figure 2. The autoimmune disease in R2−/− mice is dependent on MAVS but independent of type I IFNs and STING. (A–C) Serum ANA levels of the indicated mice (5-month old) were measured by the Hep2 system. (D,E) Spleen weights were measured in 5-month old mice indicated. Each symbol represents a mouse. ***p < 0.001 (One-way ANOVA test); ns, not significant.
MAVS deficiency, and not the STINGgt mutation, ameloriated the splenomegaly typical of R2−/− mice (Figures 2D,E). The range of autoimmune pathology in the various genotypes was also correlated with the extent of immune cell activation. Spleens from Mavs−/−R2−/− mice had reduced numbers of activated B cells (CD69+) and activated T cells (CD45RB−), as well as reduced number of germinal center B cells, plasma cells/plasmablasts and Tfh cells compared to those in MAVS sufficient control R2−/− mice (Figures 3A–D). Immunohistochemical staining of splenic sections dramatically reduced GCs in both the sizes and the numbers (Figure 3E), consistent with the diminished overall pathology score in Mavs−/−R2−/− mice (Figure 1). Thus, we conclude that MAVS deficiency abrogated spontaneous autoimmune GCs and symptoms in autoimmune susceptible R2−/− mice.
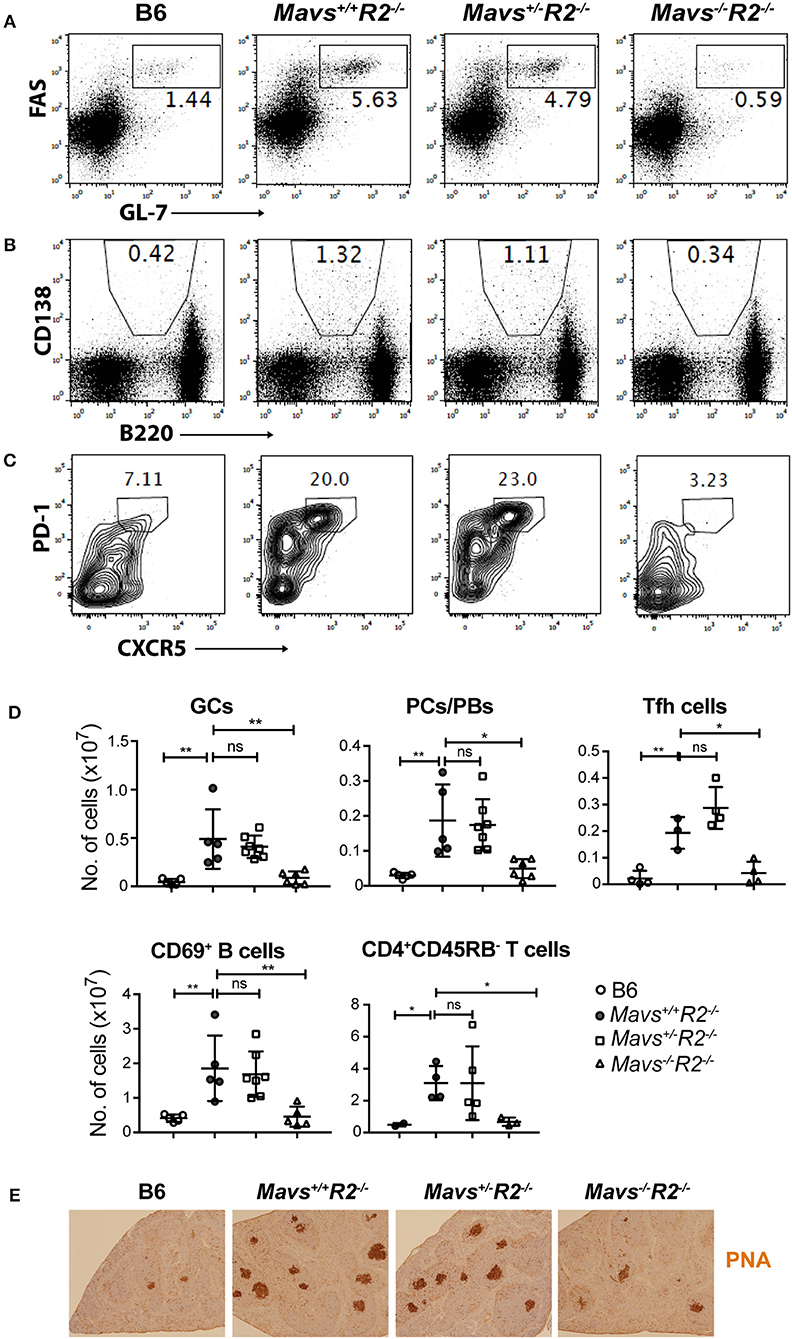
Figure 3. MAVS deficiency prevents spontaneous GCs in R2−/− mice. Splenocytes from the indicated mice were stained with antibodies and analyzed by flow cytometry. (A) GC B cells (FAS+GL-7+) were gated on B220+ lymphocytes. (B) Plasma cells/plasmablasts (PCs/PBs, CD138+B220lo) were gated on lymphocytes. (C) Tfh cells (PD-1+CXCR5+) were gated on CD4+CD62L−CD44+ cells. The numbers are percentages of cells falling in each gate. (D) A summary of multiple mice analyzed as in A–C. Each symbol represents a mouse. *p < 0.05; **p < 0.01 (One-way ANOVA). ns, not significant. (E) Histological analysis of spleen sections for GCs (stained with PNA, dark brown).
Previous studies using an independent MAVS null strain generated by Xu et al. (also referred to as Visa−/−) found that the expression of TLR7 was downregulated in the absence of MAVS (24). Interestingly, the expression of TLR7 in the MAVS knockout line generated by Chen's group (15), as used in this study, was not altered (24). We measured TLR7 expression levels in our cohort and found no significant differences between TLR7 transcripts levels in B cells of Mavs−/−R2−/− and control mice (Figure 4A), consistent with previous analyses (24). Injection of the TLR7 ligand imiquimod into mice resulted in production of IL-6 and TNF-α in serum at equivalent levels among different groups of mice, regardless of MAVS expression (Figure 4B). Furthermore, purified B cells from Mavs−/−R2−/− and control mice proliferated at comparable levels following stimulation with imiquimod (Figure 4C) or R848 in vitro Supplemental Figure 2. We thus conclude that the expression and function of TLR7 was not affected by MAVS deficiency in the particular murine strain deficient in MAVS that we used for our studies.
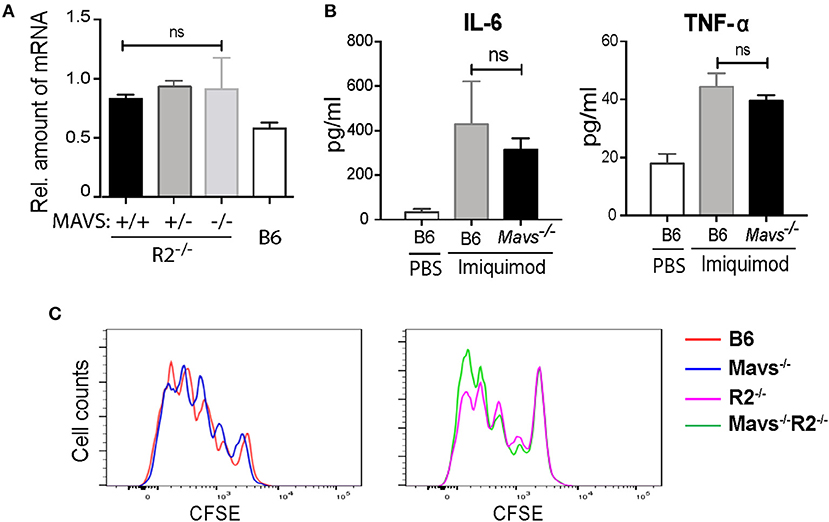
Figure 4. The autoimmune modulatory effect of MAVS in R2−/− mice is independent on the TLR7 signaling. (A) The expression levels of Tlr7 mRNA in purified B cells from the indicated mice were determined by qPCR. (B) Serum levels of IL-6 and TNF-α detected 6 h after a single injection with imiquimod (50 μg per mouse i.v.). The data are means ± SEM of 3 mice. (C) Proliferative response of B cells to imiquimod in vitro. Purified B cells (over 97% purity) were labeled with CFSE and cultured with 2 μg/ml of imiquimod for 3 days. Cells were then analyzed by flow cytometry. Data are representative of at least three independent experiments. ns, not significant.
MAVS Promotes Autoimmunity Through Its Function in Bone Marrow-Derived Cells
To determine the cell types that were responsible for protecting autoimmunity in Mavs−/−R2−/− mice, we generated bone marrow (BM) chimera mice through two different approaches: (i) as a simple BM reconstitution from donor to recipient to test the involvement of bone marrow cells vs. radiation resistant populations, and (ii) a mixed bone marrow reconstitution in which donor cells are comprised of two genotypes in various proportions. For the first approach, we transferred BM cells isolated from Mavs+/+R2−/− or Mavs−/−R2−/− mice into lethally irradiated wild type C57BL/6J (B6) recipient mice. To test the role of MAVS in radiation-resistant populations, we transferred the BM cells of Mavs+/+R2−/− mice into lethally irradiated C57BL/6J. Mavs−/− recipients. BM cells of R2−/− mice efficiently induced splenomegaly (Figure 5A left) and produced a large amount of serum ANA in B6 recipients (Figure 5B left), similar to that seen in non-manipulated R2−/− mice (Figure 2A left). In contrast, BM cells of the Mavs−/−R2−/− origin generated normal sizes of spleens in reconstituted B6 mice (Figure 5A middle) and significantly lower levels of serum ANA (Figure 5B middle). Consistent with this result, the numbers of GC B cells (Figure 5C), plasma cells/plasmablasts, CD69+ activated B cells and CD45RB− activated/memory T cells (Figure 5D) of the R2−/− origin were higher than those originated from the Mavs−/−R2−/− donor BM. Immunohistochemical staining of spleen sections revealed far fewer and smaller GCs in recipients of the Mavs−/−R2−/− donor BM than those of the Mavs+/+R2−/− donor BM (Figure 5E, top panel). The pathology scores of glomerulonephritis were significantly improved in recipients of Mavs−/−R2−/− BM (Figure 5E, lower and right panels). When testing the effect of MAVS in radiation resistant populations, we observed that MAVS deficiency in recipient mice did not alter the splenomegaly (Figure 5A), autoantibodies (Figure 5B), immune activation (Figures 5C,D) or kidney pathology (Figure 5E right panel) when receiving R2−/− BM. Collectively, these data suggested that BM-derived immune cells were responsible for the autoimmune protective effect in MAVS-deficient R2−/− mice.
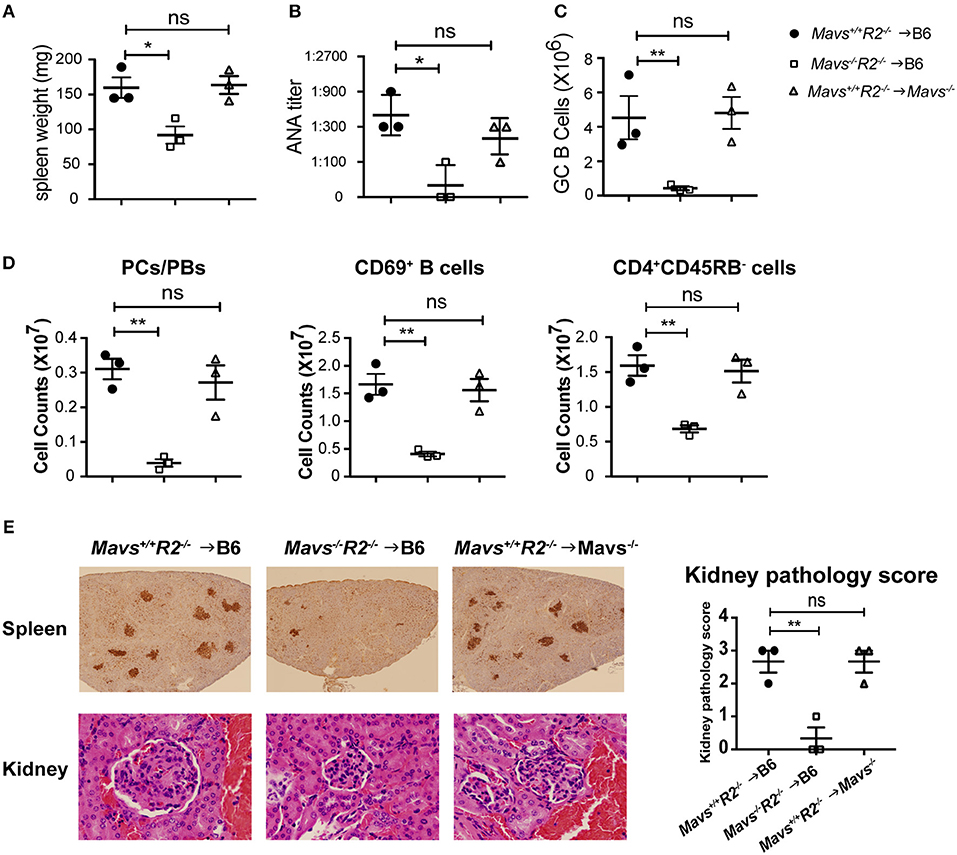
Figure 5. The regulatory effect of MAVS on autoimmunity is bone marrow dependent. (A–E) BM chimera mice were generated using donors of Mavs+/+R2−/− or Mavs−/−R2−/− mice and recipients of B6 or Mavs−/−. Two months after reconstitution, spleen weights (A) and serum ANA levels (B) of recipients were measured. (C,D) Splenic GCs, plasma cells/plasmablasts (PCs/PBs), CD69+ B cells, and CD45RB− CD4 T cells were analyzed by flow cytometry. Each symbol represents a mouse. (E) Histological analysis of spleen and kidney sections for GCs (top panel, stained with PNA, dark brown) and glomeruli (bottom panel, stained with H&E). The kidney pathology scores of multiple mice are shown in right panel. Each symbol represents a mouse. *p < 0.05; **p < 0.01 (One-way ANOVA test).
MAVS Acts Both in a B Cell Autonomous Manner and in Trans to Regulate Autoreactive GC B Cells
For our second approach, we generated mixed BM chimeric mice to examine whether the protective effect of MAVS deficiency was intrinsic to B cells. First we used a mixture containing 50% of R2−/− [Ighb] BM cells (served as founder cells) and 50% of BM cells from R2−/− [Igha] mice that bore one of 3 different MAVS alleles: wild-type (Mavs+/+, Group 1), heterozygous (Mavs+/−, Group 2), or null (Mavs−/−, Group 3) (Figure 6A). As a genetic background control, we reversed the IgH allotype of the donors in Group 4 mice, which received 50% R2−/− (Igha) BM and 50% Mavs−/−R2−/− (Ighb) BM (Figure 6A). The inclusion of 50% of R2−/− BM cells of either Igha (Group 4) or Ighb (Groups 1–3) background allowed these cells to generate spontaneous autoreactive GCs thereby providing niches for MAVS sufficient or deficient R2−/− B cells to function. The production of autoreactive IgG2a/c was analyzed by ELISA using antibodies distinguishing Igha and Ighb allotypes, allowing us to pinpoint the origin of donor-derived autoantibodies. As shown in Figure 6B, the numbers of splenic B cells with a MAVS-wild type or MAVS-deficient origin were comparable in Group 1 (43% Igha and 57% Ighb). In comparison, the MAVS-null B cells were less represented in both Group 3 and Group 4, independent of allotype: they were 33% of the total in group 3 (Igha cells) and 26% of the total (Igha cells) in group 4 (Figure 6B, left panel), compared with 43 and 39% of the total in Group 1 and Group 2, respectively. The numbers of splenic GCs had a slight bias toward the IgHa allotype but were markedly reduced in cells with the Mavs−/−R2−/− origin (Group 3 and 4) compared to those derived from Mavs+/+R2−/− (Group 1) or Mavs+/−R2−/− (Group 2) donors (Figure 6B, right panel). The overall lower numbers of total GCs in Group 3 and Group 4 indicated that the formation of GCs by the founder cells was suppressed by the presence of MAVS deficient cells. This is reminiscent to what Ravetch et al. published when comparing germinal centers generated in bone marrow reconstitution experiments using a mix of R2−/− and wild type cells (27). Nevertheless, the significantly lower proportional numbers of GC B cells of Mavs−/− (Igha) origin resulted in the total absence of autoreactive IgG2a/c antibodies of the a allotype assessed by Hep-2 cell staining (Figure 6C) and serum ANA scores (Figure 6D, group 3 shows ANA score of 0 compared to score of 2 for MAVS-proficient control). The inverted results obtained when the Mavs−/− cells express the Igha allotype (12% of MAVS-deficient cells in Figure 6B; ANA score of 0 in Figure 6D, group 4) rule out any possible effect of genetic background beyond the MAVS deficiency.
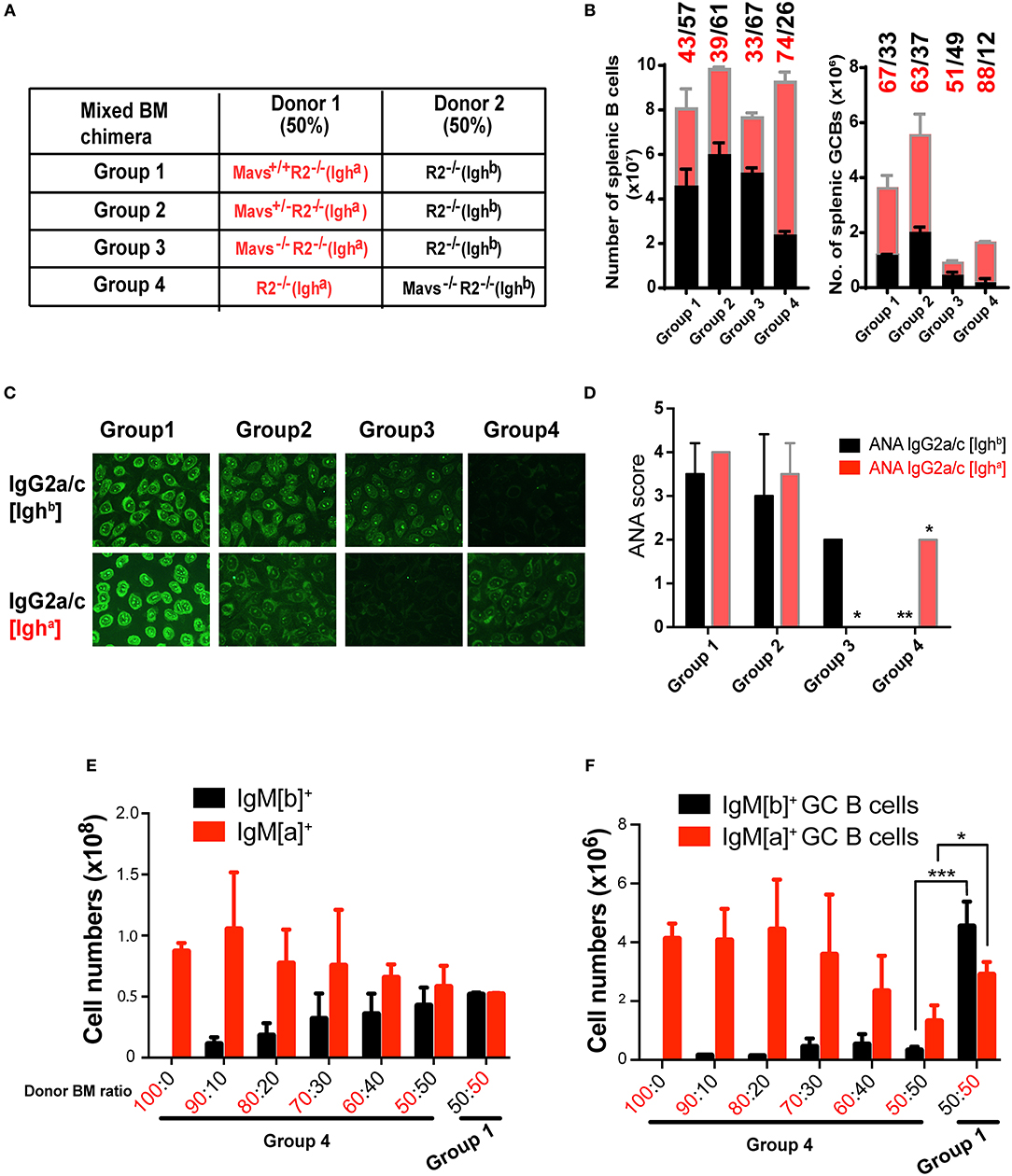
Figure 6. BM chimera mice confirm the B cell intrinsic effect of MAVS on activation and antibody secretion. (A–D) Mixed BM chimera mice were generated with donor BM at a 1:1 ratio. (B) Two months following reconstitution, the numbers of total B (left panel) and GC B cells (right panel) in recipients were analyzed by flow cytometry. (C) The ANA reactivity with an origin of either IgG2a/c [Ighb] (top panel) or IgG2a/c[Igha] (bottom panel) was determined by using the Hep2 system. (D) A summary of ANA scores of multiple mice. (E,F) Donor BM was mixed at the indicated ratio and transferred into recipient mice. The total B cells (E) and GC B cells (F)of the donor origins were analyzed by flow cytometry and enumerated. Data are means ± SEM of 5–8 mice per group (A–F). *p < 0.05; **p < 0.01; ***p < 0.001 (One-way ANOVA test).
We noticed that MAVS deficient B cells had a slight disadvantage in development when tested full B cell numbers in bone marrow reconstitution. As shown in Supplemental Figure 3, Mavs−/− mice have smilar numbers of B cell pupulations in bone marrow, but once in the periphery they show a significant disadvantage in follicular cell numbers and an advantage in marginal zone B cell numbers. To get a clear estimate of the effect of MAVS deficiency in autoreactive germinal center B cells independent of this effect on all B cells, we generated mixed bone marrow chimeras with various proportions of donor cells and then measured their numbers after reconstitution in the whole spleen and in germinal centers. In this experiment, B cells expressing the surface IgMb allotype, which in group 4 are MAVS-deficient, have only a slight disantage in development (Figure 6E) but a much more pronounced disadvantage within GC populations (Figure 6F). This effect is also shown in the inverted experiment in group 1, ruling out other genetic effects beyond the MAVS mutation. Collectively, these data suggested that Mavs−/−R2−/− B cells had an intrinsic defect in differentiation into autoreactive GCs and producing IgG2a/c autoantibodies. However we also detected an additional extrinsic effect of MAVS deficiency, either from neighboring B cells acting in trans, or from other immune populations that might affect GC formation.
Discussion
Our findings reveal a decisive role of MAVS in autoimmune GC responses: the development of autoreactive GCs and autoantibody production in lupus susceptible R2−/− mice was nearly completely abrogated by the MAVS deficiency. As a result, the lupus symptoms of R2−/− mice including glomerulonephritis and proteinuria were significantly improved and the lifespan of the mice was prolonged. Moreover, elevated expression of TLR7 and the STING signaling were not required in this model. At the cellular level, it is the B cells that were mainly responsible for the protective effect in Mavs−/−R2−/− mice, arguing for a role for MAVS in B cell tolerance that goes beyond systemic expression of type I interferon.
We currently don't have a full mechanistic view on how the MAVS pathway might enable autoreactive B cells to differentiate into GC B cells and autoantibody-secreting cells. Recent findings suggest that MAVS may function through a viral independent mechanism, although none of the published findings point to B cell intrinsic effects. It has been shown that MAVS can undergo oligomerization by reactive oxygen species (ROS) generated by oxidative stress (28). Interestingly, peripheral blood lymphocytes of SLE patients contain spontaneous MAVS oligomerization, which correlates with increased production of type I IFNs. Furthermore, cells expressing the loss-of-function mutation of MAVS (C79F) contain lower levels of ROS-dependent MAVS oligomerization (28). It remains unclear, however, how ROS induces MAVS oligomerization. One possibility is that ROS damages endogenous RNA, especially the ribosomal RNA (29), which leads to recognition by MDA5 and then triggers MAVS oligomerization. It has been shown that accumulation of endogenous RNA caused by dysregulated RNA editing (e.g., adenosine deaminase ADAR1 deficiency) indeed induces virus-independent MAVS activation (30). These findings collectively support an anti-viral independent mechanism for MAVS function in B cell biology that is compatible with our findings of a B cell autonomous effect of MAVS in autoimmunity.
MAVS deficiency diminishes even further the spontaneously induced low background interferon expression levels. This can affect expression of genes that are easily induced by type I interferon, one of them being TLR7, which is well-established as susceptibility factor in lupus development, particulary in the context of the R2−/− model and the Yaa autoimmune modifier (31, 32). However, our data points to a TLR7-independent role of MAVS in the development of autoimmunity. Both expression levels and functional testing of TLR7 were not altered in Mavs−/− B cells [(24) and Figure 4]. Furthermore, expression levels of TLR7 are tightly linked to type I IFNs, and our data from mice deficient in IFNAR demonstrated that IFN signaling is dispensable for the phenotype. It is worth to note that another strain of MAVS knockout (VISA−/−) generated by Xu et al. showed a role of MAVS in regulation of TLR7 expression (24). This could be due to differences in genetic backgound, which has increasingly drawn attention from the scientific community (33, 34).
Contrasting the essential role for MAVS in B cell autoimmunity, we found no such role for the cytoplasmic DNA sensor STING, also known to induce type I IFN. The STINGgt mutation did not change the survival rate of FcγRIIB deficient mice or altered autoantibody levels (Figure 2). Previous reports addressing the link of the STING pathway with autoimmunity have provided varied views: STING is essential for the phenotype of Trex1−/− mice in which accumulation of endogenous DNA substrates triggers production of type I IFNs (35). In contrast, STING has a suppressive effect in the phenotype of lpr mice (22). While these two reports connect STING with autoimmunity, albeit in opposite directions, our data points to a non-involvement for this pathway in our mouse model. One explanation for these differences could be that our model is independent of type I IFN expression (Figure 2C) while the Trex1−/− mouse model is completely based on augmented IFN levels (35).
In summary, we have uncovered anti-viral independent roles for MAVS in regulation of autoimmunity. MAVS deficiency correlates with diminished spontaneous autoimmune GCs and autoantibodies in autoimmune susceptible R2−/− mice. Our results parallel the findings in SLE patients who bear a loss-of-function mutation of MAVS with reduced levels of type I IFNs and autoreactive anti-RNA binding protein antibodies. These findings broaden our understandings of MAVS functions in anti-viral immunity and autoimmunity with important implications for future therapeutic strategies to target the MAVS pathway in the control of SLE.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The animal study was reviewed and approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
Author Contributions
WS, HW, C-FQ, JW, and BS performed the experiments and analyzed the data. SB conceived and designed the study and analyzed the data. WS, HW, and SB wrote the paper.
Funding
This work was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. James Z. Chen for providing MAVS deficient mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02452/full#supplementary-material
References
1. Kontaki E, Boumpas DT. Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun. (2010) 35:206–11. doi: 10.1016/j.jaut.2010.06.009
2. Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. (2008) 17:394–9. doi: 10.1177/0961203308090020
3. Weidenbusch M, Kulkarni OP, Anders HJ. The innate immune system in human systemic lupus erythematosus. Clin Sci. (2017) 131:625–34. doi: 10.1042/CS20160415
4. Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science. (2006) 314:994–7. doi: 10.1126/science.1132505
5. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. (2006) 441:101–5. doi: 10.1038/nature04734
6. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. (2005) 175:2851–8. doi: 10.4049/jimmunol.175.5.2851
7. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. (2011) 146:448–61. doi: 10.1016/j.cell.2011.06.041
8. Huang Y, Liu H, Ge R, Zhou Y, Lou X, Wang C. UXT-V1 facilitates the formation of MAVS antiviral signalosome on mitochondria. J Immunol. (2012) 188:358–66. doi: 10.4049/jimmunol.1102079
9. Huang Y, Liu H, Li S, Tang Y, Wei B, Yu H, et al. MAVS-MKK7-JNK2 defines a novel apoptotic signaling pathway during viral infection. PLoS Pathog. (2014) 10:e1004020. doi: 10.1371/journal.ppat.1004020
10. Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA. (2007) 104:14050–5. doi: 10.1073/pnas.0704014104
11. Liu XY, Wei B, Shi HX, Shan YF, Wang C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. (2010) 20:994–1011. doi: 10.1038/cr.2010.103
12. Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. (2008) 28:651–61. doi: 10.1016/j.immuni.2008.03.013
13. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. (2006) 439:208–11. doi: 10.1038/nature04374
14. Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal. (2009) 2:ra47. doi: 10.1126/scisignal.2000287
15. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
16. Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. (2014) 40:199–212. doi: 10.1016/j.immuni.2013.12.014
17. Van Eyck L, De Somer L, Pombal D, Bornschein S, Frans G, Humblet-Baron S, et al. Brief report: IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol. (2015) 67:1592–7. doi: 10.1002/art.39110
18. Crampton SP, Deane JA, Feigenbaum L, Bolland S. Ifih1 gene dose effect reveals MDA5-mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. J Immunol. (2012) 188:1451–9. doi: 10.4049/jimmunol.1102705
19. Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol Med. (2011) 3:142–52. doi: 10.1002/emmm.201000120
20. Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B, et al. Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. J Immunol. (2012) 188:4913–20. doi: 10.4049/jimmunol.1103668
21. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol. (2014) 192:5571–8. doi: 10.4049/jimmunol.1303258
22. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci USA. (2015) 112:E710–717. doi: 10.1073/pnas.1420217112
23. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. (2000) 13:277–85. doi: 10.1016/S1074-7613(00)00027-3
24. Xu LG, Jin L, Zhang BC, Akerlund LJ, Shu HB, Cambier JC. VISA is required for B cell expression of TLR7. J Immunol. (2012) 188:248–58. doi: 10.4049/jimmunol.1100918
25. Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. (2011) 79:688–94. doi: 10.1128/IAI.00999-10
26. Richez C, Yasuda K, Bonegio RG, Watkins AA, Aprahamian T, Busto P, et al. IFN regulatory factor 5is required for disease development in the FcgammaRIIB-/-Yaa and FcgammaRIIB-/- mouse models of systemic lupus erythematosus. J Immunol. (2010) 184:796–806. doi: 10.4049/jimmunol.0901748
27. McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. (2005) 307:590–3. doi: 10.1126/science.1105160
28. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal. (2016) 9:ra115. doi: 10.1126/scisignal.aaf1933
29. Willi J, Kupfer P, Evequoz D, Fernandez G, Katz A, Leumann C, et al. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. (2018) 46:1945–57. doi: 10.1093/nar/gkx1308
30. Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol. (2017) 18:744–52. doi: 10.1038/ni.3766
31. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. (2007) 27:801–10. doi: 10.1016/j.immuni.2007.09.009
32. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. (2006) 312:1669–72. doi: 10.1126/science.1124978
33. Dikiy S, Rudensky AY. Mouse watch: a cautionary tale. Immunity. (2019) 51:10–2. doi: 10.1016/j.immuni.2019.06.019
34. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
Keywords: MAVS, FcγRIIB, lupus, antinuclear autoantibody, germinal center, STING, TLR7, type I interferon
Citation: Sun W, Wang H, Qi C-F, Wu J, Scott B and Bolland S (2019) Antiviral Adaptor MAVS Promotes Murine Lupus With a B Cell Autonomous Role. Front. Immunol. 10:2452. doi: 10.3389/fimmu.2019.02452
Received: 08 August 2019; Accepted: 01 October 2019;
Published: 16 October 2019.
Edited by:
Laurence Morel, University of Florida, United StatesReviewed by:
Keith Elkon, University of Washington, United StatesOnkar Prakash Kulkarni, Birla Institute of Technology and Science, India
Dipyaman Ganguly, Indian Institute of Chemical Biology (CSIR), India
Copyright © 2019 Sun, Wang, Qi, Wu, Scott and Bolland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Bolland, c2JvbGxhbmRAbmlhaWQubmloLmdvdg==
†These authors have contributed equally to this work as share first authorship