 Cecilie Egholm1†
Cecilie Egholm1† Onur Boyman
Onur Boyman- 1Department of Immunology, University Hospital Zurich, Zurich, Switzerland
- 2Faculty of Medicine, University of Zurich, Zurich, Switzerland
Interleukin-4 (IL-4) receptor (IL-4R) signaling plays a pivotal role in type 2 immune responses. Type 2 immunity ensures several host-protective processes such as defense against helminth parasites and wound repair, however, type 2 immune responses also drive the pathogenesis of allergic diseases. Neutrophil granulocytes (neutrophils) have not traditionally been considered a part of type 2 immunity. While neutrophils might be beneficial in initiating a type 2 immune response, their involvement and activation is rather unwanted at later stages. This is evidenced by examples of type 2 immune responses where increased neutrophil responses are able to enhance immunity, however, at the cost of increased tissue damage. Recent studies have linked the type 2 cytokines IL-4 and IL-13 and their signaling via type I and type II IL-4Rs on neutrophils to inhibition of several neutrophil effector functions. This mechanism directly curtails neutrophil chemotaxis toward potent intermediary chemoattractants, inhibits the formation of neutrophil extracellular traps, and antagonizes the effects of granulocyte colony-stimulating factor on neutrophils. These effects are observed in both mouse and human neutrophils. Thus, we propose for type 2 immune responses that neutrophils are, as in other immune responses, the first non-resident cells to arrive at a site of inflammation or infection, thereby guiding and attracting other innate and adaptive immune cells; however, as soon as the type 2 cytokines IL-4 and IL-13 predominate, neutrophil recruitment, chemotaxis, and effector functions are rapidly shut off by IL-4/IL-13-mediated IL-4R signaling in neutrophils to prevent them from damaging healthy tissues. Insight into this neutrophil checkpoint pathway will help understand regulation of neutrophilic type 2 inflammation and guide the design of targeted therapeutic approaches for modulating neutrophils during inflammation and neutropenia.
Introduction
Neutrophil granulocytes (neutrophils) are the most abundant leukocytes in human blood accounting for ~60–70% of immune cells in circulation at steady state (1). With a very short life span of approximately 5 days, there is a constant need for replenishment of this vast pool (2). Neutrophils are generated in the bone marrow at a rate of 1–2 × 1011 cells per day (3). Their release from the bone marrow is regulated by the signaling of C-X-C chemokine receptors (CXCR) 2 and 4. CXCR2-binding chemokines, CXCL8 in humans and CXCL1 and CXCL2 in mice, promote the mobilization of neutrophils and their egress into the blood stream (4, 5). Conversely, the engagement of CXCR4 with its ligand CXCL12 presented on bone marrow stromal cells keeps the newly-formed granulocytes in their bone marrow niche (6). At steady state, this delicate interplay ensures the maintenance of a stable peripheral blood pool of neutrophils. In case of inflammation or infection, pro-inflammatory mediators, such as granulocyte colony-stimulating factor (G-CSF), are produced by the affected tissue and shift the balance toward increased neutrophil generation and mobilization, for example by increasing CXCR2 and decreasing CXCR4 surface expression on neutrophils (7, 8).
Neutrophils are typically the first non-resident immune cells that arrive at a site of inflammation (9, 10). Upon local activation of the vasculature, endothelial cells present chemokines and cell adhesion molecules at their luminal side (11). These ligands are recognized by their counterparts on the surface of bypassing neutrophils, leading to deceleration and eventually firm adhesion of the leukocytes to the endothelium. The neutrophils then crawl along the vessel wall following fixed gradients of so-called intermediary chemoattractants, particularly CXCR2-binding chemokines, before they transmigrate through the endothelium into the interstitial space. There, they advance to their destination by tracking gradients of other chemotactic stimuli such as N-formylmethionine-leucyl-phenylalanine (fMLP) released by bacteria (12, 13). These end-target chemoattractants override the signals emanating from intermediary chemokines (14). Once they reach the site of infection, neutrophils employ various mechanisms to kill or inactivate pathogens, including phagocytosis, degranulation, production of reactive oxygen species (ROS), and neutrophils extracellular trap (NET) formation (Figure 1; discussed in section Neutrophil Effector Functions).
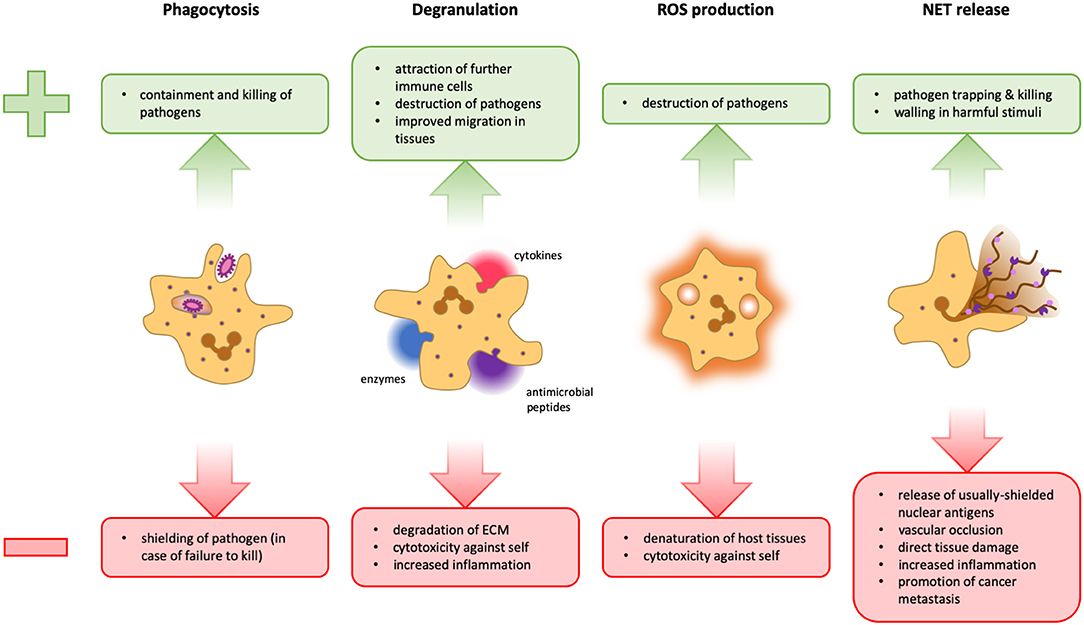
Figure 1. Advantages and disadvantages of neutrophil effector functions. During phagocytosis, microbes are engulfed and degraded in specialized organelles called phagolysosomes. This is a very powerful and clean method to dispose of pathogens because it takes place in a contained space, thus preventing widespread inflammation. However, should a pathogen manage to survive intracellular degradation, it is protected from extracellular factors and other immune cells that could potentially contain it (Left). Neutrophils can release several different cytokines, antimicrobial peptides, and granular enzymes into their surroundings, a response termed degranulation. This mechanism facilitates migration within the tissue, activates and attracts other immune cells and can help fight pathogens that cannot be phagocytosed. However, the release of too many cytokines can lead to an overshooting immune activation. Tissue degradation could aid the spreading of pathogens and destroy the structural basis of organs, and several granular proteins are also toxic for host cells (Middle Left). Neutrophils can release reactive oxygen species (ROS) into phagolysosomes as well as into the extracellular space. These molecules can potentiate pathogen killing, but are not selective and can therefore also damage host cells (Middle Right). The formation of NETs can be an efficient means to trap and kill pathogens and potentially walling off harmful stimuli. However, the release of cytoplasmic and nuclear proteins can cause the formation of autoantibodies, favoring autoimmunity. Furthermore, NETs can obstruct blood vessels and glandular ducts leading to inflammation. Granular proteins attached to the chromatin fibrils damage host tissue and the release of pro-inflammatory mediators may result in overshooting inflammation. Moreover, NETs have also been implicated in the facilitation of tumor metastasis (Right).
Given the abundance and readiness of these heavily-armed immune cells, overshooting neutrophil activity can lead to detrimental tissue damage. Thus, mechanisms need to be in place, which keep neutrophil responses in check but at the same time allow efficient clearance of pathogens. Although neutrophils have traditionally been associated with type 1 and type 3 immunity, they have recently been found to contribute to type 2 immune responses, thus also aiding in the removal of helminth parasites (15, 16). Conversely, neutrophils are often conspicuously absent from tissues where the type 2 cytokines interleukin (IL)-4 and IL-13 dominate (17, 18). In this review, we will propose a mechanism that connects these seemingly contradictory findings. We will first summarize beneficial and harmful effects of neutrophils in type 1, type 2, and type 3 immune responses. We will then focus on a growing body of evidence that suggests involvement of neutrophils in type 2 immunity. Finally, we will discuss recent results from mouse and human neutrophil research that have demonstrated how IL-4 and IL-13 receptor signaling inhibits several neutrophil effector functions (19–21), and we will elaborate on a temporal model unifying the reported findings.
Neutrophil Effector Functions
Phagocytosis
The most prominent neutrophil effector function is phagocytosis, the process during which pathogens are recognized, encapsulated and internalized in membranous vesicles, and digested by fusion of the vesicles with lysosomes and granules containing enzymes and antimicrobial peptides. The recognition step usually depends on pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively) as well as opsonization of the pathogen by antibodies or complement factors that bind to Fc or complement receptors on the neutrophil, respectively (22–24). Engagement of these receptors leads to the active engulfment of the pathogen. Subsequently, lysosomal hydrolases and granular enzymes are released into the vesicle by fusion with the respective organelles and start the destruction of the phagocytosed material (25). Additionally, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex is assembled on the phagolysosome membrane, leading to the formation and discharge of superoxide anions () into the lumen. These harm pathogens directly or react further into other ROS, all of which have antimicrobial activity (see below) (26).
Degranulation
Neutrophils possess a large pool of intracellular vesicles (granules) whose contents can be released into the extracellular space or into phagosomes in a process called degranulation. Not all neutrophil granules are the same: There are four very distinct types of secretory organelles that can be distinguished by their content, membrane proteins, function, and time point of mobilization. Primary or azurophilic granules contain the most potent cargo: myeloperoxidase (MPO), defensins, neutrophil elastase (NE), and cathepsins. These proteins are powerful antimicrobial agents and are released only after several checkpoints have been passed and the neutrophil is in closest proximity to the pathogen. Secondary or specific granules also accommodate antimicrobial factors (e.g., lactoferrin, neutrophil-gelatinase associated lipocalin) together with matrix metalloproteases (MMPs) neutrophil collagenase (MMP-8) and leukolysin (MMP-25). The antimicrobial factors provide protection against pathogens while the MMPs can degrade extracellular matrix and thus enable more efficient neutrophil migration through tissues. Tertiary or gelatinase granules predominantly contain their namesake gelatinase (MMP-9) which also facilitates migration. The fourth type are secretory vesicles, which serve as reservoirs of membrane proteins that are needed on the cell surface upon priming, such as the CD11b–CD18 heterodimer (αMβ2-integrin, Mac-1, complement receptor 3), CD35 (complement receptor 1), and the fMLP receptor. They do not contain notable soluble cargo. Besides this multitude of proteins working in situ, neutrophils are also capable of releasing several chemokines, cytokines, and growth factors that recruit other immune cells to a site of inflammation (27–32).
Despite being an essential means of combating infection, degranulation can also cause serious damage if not kept in check. While MMPs may be beneficial for cell migration, they can also destroy the connective tissue, and several constituents of the azurophilic granules not only kill pathogens, but are also cytotoxic to host cells (33) (Figure 1). Moreover, uncontrolled recruitment and activation of immune cells leads to excessive inflammation. It is thus not surprising that over-shooting degranulation of neutrophils has been associated with several inflammatory disorders such as septic shock, severe lung injury, rheumatoid arthritis, and severe asthma (34, 35).
Reactive Oxygen Species
ROS are a group of small, unstable oxygen-based molecules. Owing to their chemical instability, they are highly reactive and can denature or damage proteins, lipids, and DNA (36). In neutrophils, ROS are produced in a process called respiratory or oxidative burst by the multi-unit enzyme complex NADPH oxidase (37). The different subunits of the enzyme are stored in separate compartments of the cell to prevent accidental generation of ROS. Upon activation, the NADPH oxidase assembles on the phagosomal or plasma membrane and produces and releases superoxide anions into the phagosome or extracellular space, respectively (38). Superoxide can react further with protons to hydrogen peroxide (H2O2), which is used by MPO to create hypochlorous acid (HClO) (39). All these types of ROS have microbicidal activity, but also consume protons in their chain of reactions, thereby neutralizing the acidic content from lysosomes and granules (40). This in turn facilitates the liberation of NE and cathepsins, which at low pH are tightly bound to proteoglycans and thus less active (41). In fact, ROS likely participate less in direct pathogen killing, but they rather facilitate proper activation of azurophilic granule enzymes, which cause pathogen killing in the phagosome (42).
Whether ROS are directly or indirectly responsible for pathogen killing, they are a vital part of innate immunity. In fact, bacterial strains that are able to disarm ROS by producing superoxide dismutase (SOD), which catalyzes the reaction of into O2 and H2O2, and catalase, which in turn catalyzes the decomposition of H2O2 into O2 and H2O, are much more virulent than their SOD- or catalase-negative counterparts (43). Another example is chronic granulomatous disease (CGD), a genetic disorder affecting the NADPH oxidase, which renders patients incapable of producing ROS. These patients suffer from frequent and recurrent infections, also with opportunistic pathogens (44). Despite their importance in combatting infection, unchecked production of extracellular ROS leads to tissue damage by virtue of their lack of pathogen specificity (45) (Figure 1).
Neutrophil Extracellular Traps
NETs are meshes of DNA decorated with antimicrobial peptides that can be released by neutrophils in response to various stimuli. They were first described by Brinkmann et al. as a novel mechanism of how neutrophils can combat infection (46). Pathogens, mainly yeast and bacteria, stick to the DNA fibrils, which prevents them from spreading in the tissue, and they are degraded by the granule proteins that are attached to the chromatin network (47). Since 2004, numerous stimuli have been described to induce NET formation, of which large pathogens seem to be the main trigger (48). The exact process of how NETs form remains an active area of research. The most widely accepted model involves chromatin decondensation including histone citrullination, disintegration of nuclear, and granule membranes, intracellular mixing of the components and, finally, release into the extracellular space (49). Some reports provided evidence that the NADPH oxidase was necessary for NET formation. Interestingly, however, despite their lack of a functional NADPH oxidase CGD patients have been shown to form NETs by using mitochondrial ROS (50). It seems that depending on signal type and strength, NET formation can be fast and non-lytic, leaving behind an intact anuclear cytoplast (51), or slow and lytic, spilling the cell contents while the NET breaks free from the cell membrane (52). Some studies also present the possibility of NET release by living cells using mitochondrial in lieu of nuclear DNA (50, 53).
NETs have been shown to have several beneficial properties. Their main use is in immobilizing and degrading bacteria, fungi, and viruses (54–56). Another less prominent function is the shielding of damaged tissues that might otherwise elicit an unwanted inflammation (57). As helpful these mechanisms may be, NETs have also been implicated as players in a multitude of different diseases. Firstly, the release of nuclear material into the extracellular space provides access to otherwise shielded antigens and may result in the formation of autoantibodies (Figure 1), as suggested for rheumatoid arthritis (RA), systemic lupus erythematodes, anti-phospholipid syndrome, and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (58–61). Secondly, the massive release of proteins can be a double threat. On the one hand, cytokines may drive inflammation leading to tissue damage or atherosclerosis (62). On the other hand, the proteases associated with NETs may degrade cytokines and chemokines, resulting in a possibly unwanted anti-inflammatory effect (63). Moreover, the giant and sticky structures of NETs can occlude blood vessels, leading to thrombosis or sepsis (14). Finally, NETs have also been proposed as players in cancer dissemination and metastasis formation (64, 65) (Figure 1).
Role of Neutrophils in Different Types of Immune Responses
The immune system has evolved different types of effector immune responses to counter the various pathogens. These are commonly referred to as type 1, type 2, and type 3 immunity, and each engage different subtypes of innate lymphoid cells (ILCs) and other innate immune cells, CD4+ helper T (TH) and CD8+ cytotoxic T cells, as well as CD4+ follicular helper T (TFH) cells and antibody responses by B cells, as discussed below (Figure 2).
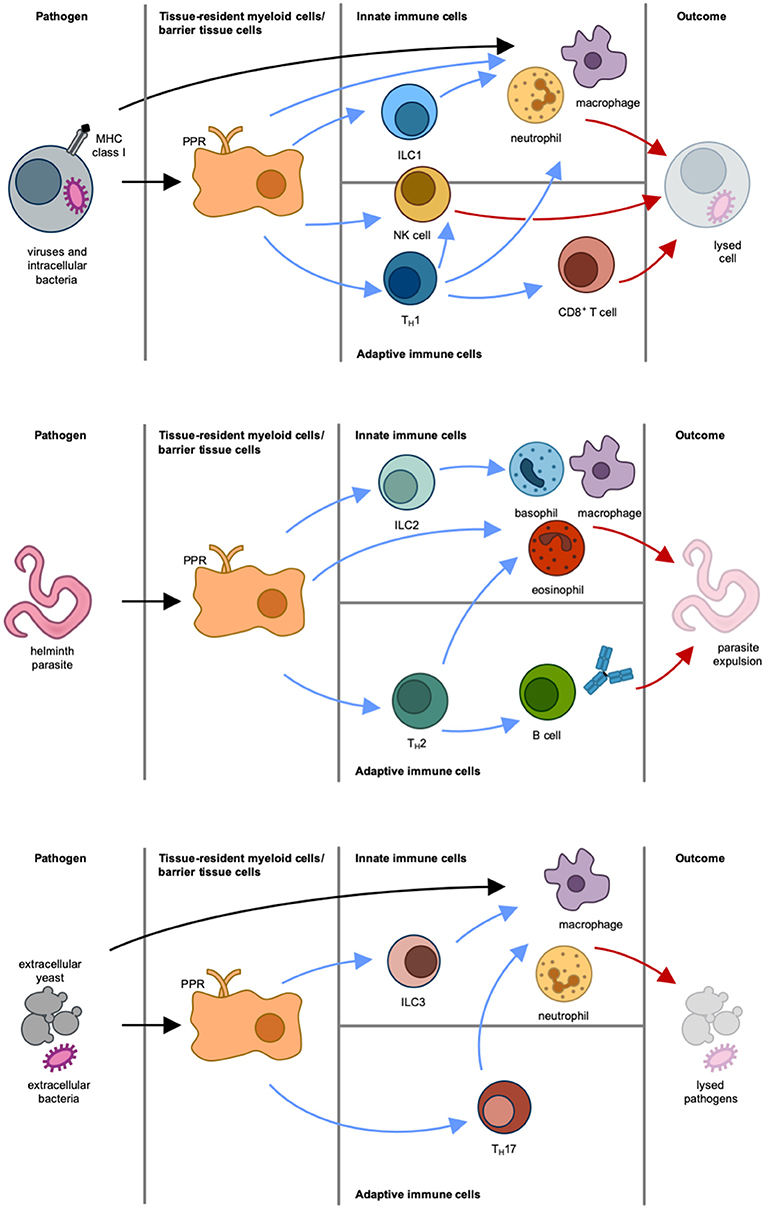
Figure 2. Different types of immune responses. Type 1 immune response (Top): Intracellular pathogens induce differentiation of naïve T cells into TH1 cells via antigen-presenting cells and IL-12. IFNγ is the main effector cytokine that stimulates and actives NK cells, type 1 innate lymphoid cells (ILC1), and TH1 cells, which in turn activate CD8+ cytotoxic T cells, macrophages and neutrophils. NK cells, macrophages and neutrophils can also be directly activated via pattern recognition receptor (PRR)-mediated recognition of pathogen-associated molecular patterns (PAMPs). The result of this immune response is lysis of cell and pathogen. Type 2 immune response (Middle): Parasites induce epithelial damage, which leads to the release of thymic stromal lymphopoietin (TSLP), IL-25, and IL-33 from epithelial cells. This in turn causes both differentiation of naïve T cells into TH2 cells via antigen-presenting cells and stimulation of ILC2, mast cells, basophils and eosinophils. In response to this, the activated immune cells produce IL-4, IL-5, IL-9, and IL-13. These effector cytokines stimulate B cells and induce isotype switching to immunoglobulin E and differentiation of macrophages to alternatively-activated macrophages (also termed M2 macrophages). The result of this immune response is parasite expulsion. Type 3 immune response (Bottom): Extracellular pathogens induce differentiation of naïve T cells into TH17 cells via antigen-presenting cells and IL-23. Production of IL-17 and IL-22 by ILC3 and TH17 cells leads to the activation of macrophages and neutrophils. The result of this immune response is pathogen killing. Initiation of immune response is indicated by black arrows. Blue arrows mark stimulatory signals by cytokines produced by immune cells. Effector responses are indicated by red arrows.
Type 1 Immunity
Type 1 immunity serves to protect against intracellular pathogens by the production of the effector cytokine interferon-γ (IFNγ) and engagement of type 1 ILCs (ILC1), natural killer (NK) cells, type 1 TH (TH1) cells, CD8+ cytotoxic T cells, type 1 TFH cells and immunoglobulin (Ig) G1 and IgG3 antibody responses. Together with these immune cells, also macrophages and neutrophils play a central role in type 1 immunity. These innate cells can directly detect foreign microbes via PAMPs, which they recognize by pattern recognition receptors (PRRs) (66). During infection with the intracellular pathogen Listeria monocytogenes, neutrophils were shown to have an important protective role. This was particularly seen in the liver, which is one of the primary target organs of this bacteria. Here, the early phagocytosis mediated by recruited neutrophils was essential for limiting bacterial spread and controlling infection (67–69). It has also been suggested that IFNγ has a direct effect on neutrophil activation and can induce MHC class II expression, at least ex vivo (70). Although macrophages have a protective role against intracellular pathogens, they are also very susceptible to becoming infected themselves. In the case of Mycobacterium tuberculosis infection of macrophages, it has been reported that neutrophil granules can be transferred to macrophages and facilitate the clearance of chronically infected cells (71).
Type 2 Immunity
Type 2 immunity has evolved to efficiently induce resistance and tolerance to parasitic infections, especially helminthic infestations. Type 2 immunity is typically initiated by the activation of epithelial cells and PRR-expressing myeloid cells that secrete IL-25, IL-33, and thymic stromal lymphopoietin (TSLP). In response to these cytokines, type 2 ILCs (ILC2) begin to produce IL-5 and IL-13, which induce the differentiation of CD4+ T cells to type 2 TH (TH2) cells, which in turn secrete the characteristic type 2 cytokines IL-4, IL-5, IL-9, and IL-13 (72–74). This cytokine milieu fosters the development and proliferation of other cells involved in type 2 immunity, including basophils, eosinophils, mast cells, and NKT cells, and drives the differentiation of type 2 TFH cells and IgE antibody responses. Especially IL-4 and IL-13 are central cytokines in type 2 immunity, and they both signal via the IL-4 receptor (IL-4R) system (75). Two types of IL-4Rs exist (Figure 3): type I IL-4Rs consist of IL-4Rα and the common gamma chain cytokine receptor, whereas type II IL-4Rs are made of IL-4Rα and IL-13Rα1. IL-4 can signal via both type I and type II IL-4Rs, whereas IL-13 only signals via the type II IL-4R. Additionally, IL-13 can bind to IL-13Rα2, which is thought to be a non-signaling decoy receptor of very high affinity to IL-13. Currently, there are several treatment strategies in clinical use that interfere with IL-4R signaling. For targeting both IL-4 and IL-13 signaling Dupilumab, a monoclonal antibody blocking IL-4Rα, and Pitrakinra, an IL-4Rα antagonist, exist. For more selectively blocking of IL-13, Lebrikizumab, a monoclonal antibody targeting IL-13, is of use. Downstream of the IL-4R, the Janus kinases (JAKs) JAK1, JAK2, and JAK3 can the inhibited by the use of the small molecule JAK inhibitors Upadacitinib and Tofacitinib (see also section Biologics and Small Molecules Targeting the IL-4R Signaling Axis).
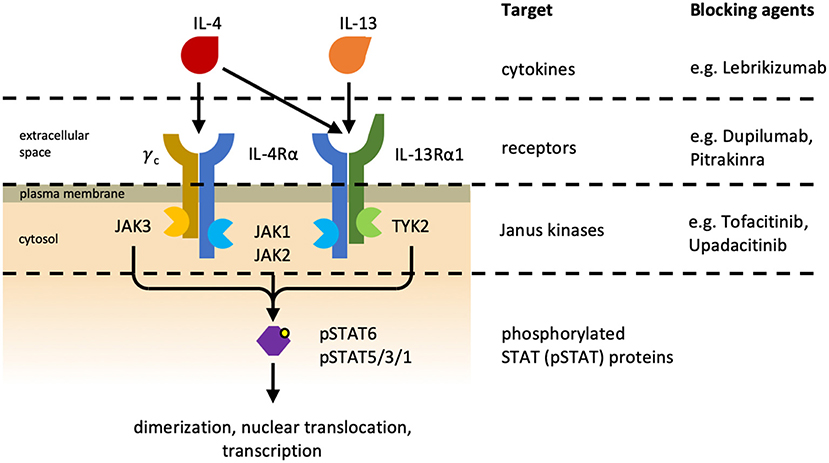
Figure 3. Interleukin-4 receptors. Different heterodimeric IL-4 receptors (IL-4Rs) exist and they share the IL-4Rα subunit. The type I IL-4R (Left) consists of IL-4Rα and the common gamma chain cytokine receptor (γC), and the type II IL-4R (Right) is made of IL-4Rα and IL-13Rα1. IL-4 can associate with and signal via both IL-4Rs, whereas IL-13 can only bind to and signal via the type II IL-4R. Additionally, IL-13 can bind to IL-13Rα2 (not depicted), which is referred to as non-signaling decoy receptor. Cytokine-mediated receptor dimerization leads to the activation of receptor-associated Janus kinases (JAK) and consequently to the phosphorylation and activation of signal transducer and activator of transcription (STAT) proteins. Phosphorylated STAT (pSTAT) proteins subsequently dimerize and translocate to the nucleus where they initiate transcription of their target genes. Different treatment strategies have been developed to inhibit IL-4R signaling. Lebrikizumab is a neutralizing anti-IL-13 antibody. Pitrakinra, an IL-4 antagonist, and Dupilumab, an IL-4Rα-blocking antibody, are both preventing cytokine–receptor interaction. JAKs can also be inhibited pharmaceutically by small molecules. Upadacitinib is a selective JAK1 inhibitor currently being investigated in clinical trials, whereas Tofacitinib inhibits both JAK1 and JAK3 and is approved for the treatment of rheumatoid arthritis and psoriatic arthritis (76).
Upon the establishment of a type 2 cytokine environment a positive feed-back loop is initiated where more naïve T cells differentiate into TH2 cells and stimulation of eosinophils, basophils, and ILC2 takes place. B cells respond to type 2 cytokines by isotype switching to IgE and production of antibodies. Macrophages develop under the influence of type 2 cytokines into alternatively-activated macrophages (also termed M2 macrophages).
At barrier organs, such as the skin, lungs, and intestine, ILC2 are more numerous than in internal organs without barrier function. As ILC2 likely serve to amplify type 2 immune responses initiated by tissue and tissue-resident myeloid cells carrying PRRs, it is conceivable that type 2 immune responses at non-barrier sites differ from those at external and internal barriers. In this context, cobalt chromium microparticles injected intraperitoneally caused a type 2 inflammation dependent on IL-33 release by macrophages, followed by recruitment of neutrophils and production of IL-4, IL-5, IL-13, arginase-1, chitinase-like protein 3 (Chil3 or Ym1), and resistin-like molecule (RELM)-α (77).
In the lung, type 2 immune responses induce goblet cell hyperplasia. These cells produce mucins and anti-nematode protein RELM-β (78, 79). RELM-β together with RELM-α and arginase-1, produced by epithelial cells and fibroblasts, respectively, are also involved in tissue repair and deposition of extracellular matrix, which in the context of helminth infections can serve to encapsulate and trap the parasite. Both IgG and IgE antibodies, produced by B cells, help to limit the motion and fecundity of the worms (80). Antibodies are also crucial for surface labeling of pathogens, which favors opsonization and antibody-dependent cell-mediated cytotoxicity (ADCC). ADCC is mediated via Fc receptors expressed on many innate immune cells. Altogether, these Fc receptor-bearing cells contribute to the reduction of worm fitness, lower transmission, and expulsion, which all serve to protect the body from nematode infections.
Whether and how neutrophils are involved in type 2 immunity and which role the affected barrier vs. non-barrier tissue plays, remains unclear. In mouse models of helminth infection, neutrophils have been reported to have a beneficial role during the early pulmonary stages (15), however this also comes at the price of increased tissue damage (81, 82). Conversely, in human type 2 inflammatory disorders neutrophils have been reported to be absent (17). The role of neutrophils in type 2 immune responses will be further discussed later.
Uncontrolled type 2 immune responses can lead to the development of allergic diseases, including allergic conjunctivitis, allergic rhinitis, allergic asthma, allergic gastrointestinal disorders, and atopic dermatitis (AD). These diseases are characterized by elevated levels of type 2 cytokines and accumulation of the above-mentioned immune cells (83). Dupilumab, a monoclonal antibody blocking IL-4Rα (Figure 3), has been shown to be effective as a treatment of moderate-to-severe AD and moderate-to-severe asthma (84, 85). Thus, interference with this central type 2 cytokine receptor subunit can control some of these allergic diseases.
Type 3 Immunity
Type 3 immunity is typically directed against extracellular bacterial and fungal infections, and it is characterized by the presence of the effector cytokines IL-17 and IL-22, which are prominently synthesized by type 3 ILCs (ILC3) and IL-17-producing TH (TH17) cells (86). Moreover, IL-26, TNF, and granulocyte-macrophage colony-stimulating factor (GM-CSF) are also produced during type 3 immunity. Fibroblasts, epithelial cells, macrophages and, particularly, neutrophils become activated in response to these cytokines. IL-17 and GM-CSF induce extensive recruitment, activation and survival of neutrophils (87). IL-22 promotes epithelial cell homeostasis and has also been found to be important in antimicrobial defense (88).
Type 3 immunity is characterized by extensive neutrophil infiltration. Neutrophils are crucial for protection against and clearance of fungi (89). Also, immunity to certain encapsulated extracellular bacteria (e.g., Staphylococcus aureus) depends on efficient recruitment and activation of neutrophils. The importance of neutrophils in anti-fungal and anti-bacterial immunity is also apparent in subjects suffering from CGD and in patients receiving IL-17-targeting biologic agents (biologics), as these individuals are more susceptible to fungal and staphylococcal infections (90, 91).
A typical pathologic manifestation characterized by a type 3 immune response is the chronic-inflammatory skin disease psoriasis (92). Notably, neutrophil-rich microabscesses (termed Munro's microabscesses) in the uppermost layers of the epidermis are a characteristic hallmark of the skin lesions in plaque-type psoriasis. Inhibition of IL-17 by the use of IL-17-targeting biologics is very effective in psoriasis.
Neutrophils in Type 2 Immune Responses
The Importance of Neutrophils During Helminth Infections
Several groups have shown that neutrophils are important for limiting parasite survival and spreading in mouse models of helminth infections (15, 81, 82). Upon inoculation of mice with infective third-stage larvae of Nippostrongylus brasiliensis, the larvae migrate via the lungs to the intestine. Around the same time as the larvae arrive to the lungs, an increase of lung-infiltrating neutrophils is observed, which most likely represents a rapid mechanism to contain spreading. Thus, both mouse and human neutrophils have been shown to kill helminth larvae in vitro, when collaborating with macrophages (15). In fact, there are a number of studies showing that neutrophils and macrophages collaborate in immobilization and killing of these parasites. In different nematode infectious models, neutrophils and macrophages have been described to cluster together around the pathogen, however, neither of the two cell types was able to kill the larvae efficiently by itself (93–95).
As previously described, neutrophils are professional phagocytes, which is important in situations where the microbe is small. On the contrary, helminths are macropathogens and due to their size impossible for a neutrophil to ingest. Instead, neutrophils take advantage of other effector functions, namely degranulation and NET formation. Guided by virulence factors and chemokines neutrophils and other granulocytes arrive to the site of infection. Interaction between antibodies covering the helminth and Fc receptors on the cells causes ADCC-mediated degranulation (96). Complement factors serve a similar function as antibodies and can lead to complement-dependent cytotoxicity (15). Although eosinophils are acknowledged as the main effector granulocytes in helminth infestations, basophils and neutrophils are also recognized for their importance (97, 98). Both neutrophil granule proteins and NETs are efficient effector mechanisms to trap helminth larvae (99). Exposure of human and mouse neutrophils to Strongyloides stercoralis induces an even faster formation of NETs than the extremely potent NET stimulator phorbol 12-myristate 13-acetate (PMA).
Even though neutrophils have a very short life span, they are capable of initiating immune responses that persist long after they die. One example of this is priming of macrophages. M2 macrophages have been shown to rapidly surround nematodes and facilitate killing and clearance during helminth infection, particularly, during secondary infection (100). This priming of macrophages to become M2 macrophages during an initial infection was, however, only possible in the presence of neutrophils. Without neutrophils, macrophages could not efficiently adapt an M2-like transcriptional phenotype, which also resulted in impaired helminth expulsion.
All these studies support the importance of neutrophils especially in the early stages of a type 2 immune response against helminths. However, with activated neutrophils comes unavoidable tissue damage (82), and it is therefore important that activated neutrophils are kept in check.
Type 2 Cytokines Dampen Neutrophil Functions
Evidence From Mouse Models
In contrast to the apparent importance of neutrophils in helminth infections, there is accumulating evidence that IL-4R signaling by IL-4 and IL-13 inhibits neutrophil effector functions (21). Woytschak et al. found that treatment of mice with IL-4 during cutaneous and systemic bacterial infections increased bacterial and disease burden, while blood neutrophil counts, neutrophil migration, and overall survival of animals decreased (19). The same trend was observed during sterile inflammation induced by using G-CSF, IL-1β, or monosodium urate crystals. Peripheral neutrophils of mice treated with IL-4 were found to have lower CXCR2 expression levels, which fits neatly with their impaired migratory ability. Conversely, bone marrow neutrophils of IL-4-injected animals expressed higher levels of CXCR4, offering an explanation for their decreased egress into the blood stream. All these effects were not observed in IL-4Rα-deficient mice as well as animals treated with a blocking anti-IL-4 monoclonal antibody. Moreover, mice lacking the IL-4Rα were shown to survive a systemic infection with Listeria monocytogenes that was lethal for wild-type control animals (19). Throughout all of the experiments performed in this work, the neutrophils behaved homogenously and there was no observation of duality that could hint at the presence of distinct bona fide neutrophil subpopulations.
Chen and colleagues found that neutrophil infiltration into the lungs upon infection of mice with the helminth Nippostrongylus brasiliensis was associated with IL-4R signaling (82). In this model, IL-4Rα-deficient mice presented with increased pulmonary neutrophil infiltration and worse disease scores, while neutrophil depletion led to a decrease of acute lung injury. In a mouse model of RA, a disease where neutrophils are believed to play a major role in the pathogenesis (101), Schmid et al. demonstrated that treatment of mice with IL-4 protected from joint inflammation (102). Other groups made similar observations treating RA with IL-4 and identified neutrophils and macrophages as the main targets of the therapy (103, 104). In a study combining helminth infection and RA, elicitation of a type 2 immune response by the parasite improved joint inflammation (105). In a recent publication, Harris and colleagues showed that IL-4R signaling under hypoxic conditions directly inhibited the survival of human neutrophils (106). These results highlight that IL-4R signaling during hypoxic acute respiratory distress syndrome, protects against neutrophil-induced lung injury. Under normoxic conditions, IL-4 or IL-13 did not appear to increase neutrophil apoptosis (20). Thus, IL-4R signaling seems to have yet another way of limiting neutrophil actions and protect against the harmful consequences of uncontrolled neutrophils.
As discussed earlier, micro- and nanoparticles are potent inducers of type 2 immunity. Since the body cannot degrade them, microparticle-elicited reactions often do not resolve, but lead to repeated waves of inflammation and immune cell influx. Here, IL-4R-mediated dampening of neutrophil responses again may serve as a preservation mechanism because neutrophils will not be able to clear the microparticles, but will only potentiate tissue damage while trying to do so.
Using a unique intravital imaging method, Wang et al. studied the migration behavior of neutrophils after a focal thermal injury in the liver. They found that after a short extensive influx, neutrophils proceeded to reenter the vasculature 12 h after injury and were finally recruited to the bone marrow in a CXCR4-dependent manner (107). Here, neutrophils were found to be responsible for the creation of paths through the tissue which facilitated vascular regrowth and access for other immune cells. Woytschak et al. found that treatment of neutrophils with IL-4 reduced their ability to migrate toward CXCL2 and resulted in downregulation of its receptor CXCR2, but not CXCR4 (19). Therefore, IL-4R engagement on neutrophils may not only dampen their migration toward CXCL2, but also promote reverse migration by shifting the balance toward signaling via CXCR4.
Ma and colleagues showed that the local phenotype of neutrophils change over time after myocardial infarction in mice (108). They found that proinflammatory N1 neutrophils dominated at day 1, whereas anti-inflammatory N2 neutrophils prevailed at days 5 and 7 after injury while they were not detected in peripheral blood. Tissue repair and fibrosis is part of the remodeling taking place after myocardial infarction. It has been shown that IL-4 and IL-13 are key drivers of these processes (109). It is therefore possible that the emerging tissue repair environment with type 2 cytokine milieu shifts the neutrophils from a pro-inflammatory N1 to an anti-inflammatory N2 phenotype. Although neutrophils seem to lose their pro-inflammatory phenotype in a type 2 immune milieu and may even contribute to the resolution of inflammation, they are distinct from conventional myeloid-derived suppressor cells (MDSCs) since the former are mature cells that change their phenotype in response to type 2 cytokines while the latter form newly from myeloid precursors in response to G-CSF/GM-CSF, IL-6, and a variety of other cytokines (110). There have, however, been reports of IL-4R signaling being important for the immunosuppressive nature of MDSCs (111, 112).
Translation to the Human Setting
Evidence of IL-4R signaling inhibiting neutrophils not only covers preclinical studies in mice, but there is also clinical data suggesting that this mechanism is evolutionarily conserved. Impellizzieri et al. recently demonstrated that incubation of human neutrophils with IL-4 or IL-13 significantly reduced their ability to migrate toward CXCL8 and produce NETs (20). These findings were mirrored when using freshly-isolated neutrophils from allergic patients with acute symptoms. Moreover, IL-4- or IL-13-treated neutrophils from healthy donors and neutrophils from allergic individuals showed lower surface expression of CXCR2 and its functional twin CXCR1, compared to untreated neutrophils from healthy donors, whereas CXCR4 expression was unaltered (20). The decrease in migratory ability in vitro upon IL-4 stimulation was confirmed and extended in a humanized mouse model, where an air pouch was induced in the back of NOD-Prkdcscid-Il2rgnull (NSG) mice, followed by triggering of local inflammation by injection of CXCL8 and lipopolysaccharide in the air pouch and intravenous adoptive transfer of human neutrophils. Flow cytometric analysis of the air pouch content revealed significantly lower counts of human neutrophils when the cells were pretreated with IL-4 as opposed to controls, indicating that IL-4R signaling in human neutrophils leads to impaired migration also in vivo. In accordance with the findings of Woytschak et al. with mouse neutrophils, Impellizzieri et al. did not find a duality in neutrophil functionality in their experiments that would hint at different bona fide subpopulations.
Biologics and Small Molecules Targeting the IL-4R Signaling Axis
In a small clinical trial with patients suffering from plaque-type psoriasis, an autoimmune disease characterized by type 3 inflammation and cutaneous neutrophil infiltration, treatment with IL-4 resulted in marked disease improvement (113); this therapeutic effect was likely due to a shift in the cytokine milieu from a type 3 to a type 2 immune response as well as a direct inhibition of neutrophils by IL-4. Conversely, in AD, an inflammatory skin disorder known for its type 2 cytokine signature, afflicted individuals often suffer from recurrent skin infections (18). Here, neutrophils are conspicuously absent in both healthy and lesional skin (17, 114). However, neutrophil chemoattractants were found to be elevated similarly in psoriatic and atopic skin (115), thus the stark difference in skin-infiltrating neutrophils in psoriasis and AD cannot be explained by differences in chemoattractants. Moreover, treatment of moderate-to-severe AD patients with the IL-4Rα-blocking antibody Dupilumab has been shown to not only significantly decrease disease burden, but also lower the incidence of skin infections (84, 116, 117). This protective effect against infections has been observed also for other type 2 immune diseases when treated with biologics targeting the IL-4R complex, such as the anti-IL-13 antibody Lebrikizumab in asthma and AD (118, 119) and the IL-4Rα-blocking agent Pitrakinra in asthma (120). There are also small molecule inhibitors of the IL-4R signaling axis targeting receptor-associated JAKs. Since JAKs are shared between different cytokine receptors, they are less selective for IL-4 or IL-13 signaling, but can also be used to suppress other pathways. Tofacitinib inhibits JAK1 and JAK3 and is approved for the treatment of RA and psoriatic arthritis (76). It has also been investigated for use in AD, both as systemic and topical treatment (121, 122). The selective JAK1 inhibitor Upadacitinib is currently being investigated as an alternative to tofacitinib for treating RA and psoriatic arthritis (76), but it may also be interesting for the treatment of type 2 immune diseases since IL-4Rα also uses JAK1 for signaling (Figure 3).
None of the studies involving IL-4 as a therapeutic agent or inhibitors of the IL-4/IL-13-signaling axis mentioned here, however, directly examined neutrophil activity before, after or during treatment. Therefore, we can only speculate that the amelioration of plaque-type psoriasis upon IL-4 treatment and the decrease in infections in type 2 diseases upon IL-4R signaling blockade is, at least in part, the direct result of increased or decreased IL-4R signaling on neutrophils, respectively. Further investigations focusing on neutrophils in these disease and treatment conditions will reveal to what extent direct IL-4R-mediated neutrophil inhibition contributes to the clinical pictures.
Taken together, the growing body of evidence indicates that IL-4 and IL-13 exert an inhibitory effect on neutrophils. Such inhibition affects neutrophil migration and tissue infiltration as well as neutrophil effector functions. These effects limit neutrophil-mediated tissue damage. Although some of these effects could be the result of the classical type 1 and type 3 vs. type 2 immune regulation, the aforementioned studies clearly prove that cell-autonomous IL-4R signaling directly curtails mouse and human neutrophil effector functions.
Hypothesis of Kinetic Involvement and Exclusion of Neutrophils in Type 2 Immune Responses
Having discussed the evidence suggesting an inhibitory role of IL-4R signaling on neutrophils, it seems awkward that neutrophils have been reported to play a significant role in type 2 immunity against helminth infections. Can these two seemingly contradictory paradigms fit together? We propose a hypothesis that involves a temporal separation of the two events and considers biological, physiological, and clinical aspects of type 2 immune diseases (Figure 4).
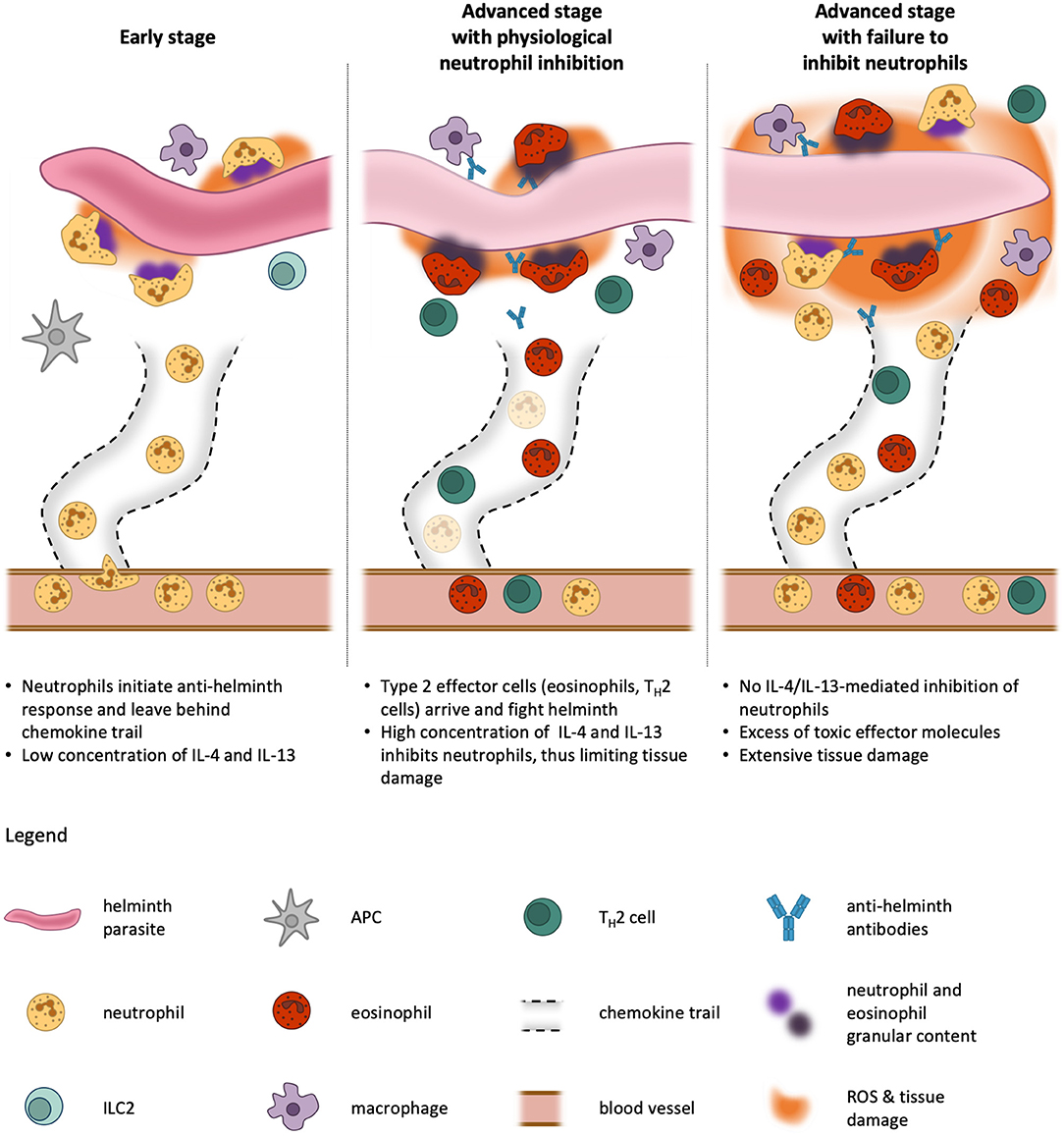
Figure 4. Kinetics of neutrophil activation and inhibition during type 2 immune responses. In an early phase of helminth infestation, neutrophils are quick to be recruited to the site of infection because they are abundant in the blood stream and are primed rapidly. On their way through the tissue, they leave behind channels and trails for other cells to migrate more efficiently toward the parasite. Once they encounter the helminth, the neutrophils release pro-inflammatory cytokines and exert effector functions against the invader. This facilitates the activation of neighboring antigen-presenting cells (APCs). At this stage, only little IL-4 and or IL-13 is produced by tissue-resident cells, such as type 2 innate lymphoid cells (ILC2), and thus neutrophil functions are unblunted (Left). At later stages, APCs migrate to the lymph nodes where they initiate differentiation of TH2 cells, which in turn home to the infested tissue and produce their signature cytokines IL-4, IL-5, and IL-13. These cytokines cause recruitment and activation of professional type 2 effector cells, eosinophils and alternatively-activated macrophages, which use the preformed neutrophil channels to efficiently reach the pathogen. Our model proposes that, in a physiological condition, the presence of IL-4 and IL-13 dampens neutrophil activity and thus prevents excessive tissue damage, as these first responders are not needed anymore at this stage and would do more harm than good (Middle). If this inhibitory mechanism fails, neutrophils continue to fight the helminth simultaneously with eosinophils and macrophages, resulting in profound tissue damage far beyond the necessary immune response to expulse the parasite (Right). The size of the helminth parasite is not to scale, which would be larger in reality.
Neutrophils are abundant in the blood stream and very easily primed for attack of pathogens. Hence, as soon as a helminth parasite invades the body and is recognized as foreign, there are countless neutrophils already at the right place to extravasate and serve as a first line of host defense. By creating an inflammatory environment, neutrophils promote the activation of antigen-presenting cells (APCs). Moreover, pathogens killed by neutrophils in these very early stages may be easier for APCs to take up, digest, and present. Furthermore, starting from the blood vessel, neutrophils create a channel decorated with chemokine trails in the tissue, thus facilitating access for other immune cells (123). At this stage, neutrophils constitute the overwhelming majority of leukocytes in the tissue and there are only low concentrations of IL-4 or IL-13 produced by resident immune cells. Thus, the neutrophils are not inhibited. APCs then migrate to the lymph nodes where they initiate differentiation of antigen-specific naïve T cells to TH2 cells. These in turn home to the infected and inflamed tissue where they produce their signature type 2 cytokines IL-4 and IL-13. Due to clonal expansion, there soon are many activated TH2 cells present and the resulting cytokine milieu leads to inhibition of neutrophils, but also the recruitment and activation of eosinophils which are now taking over the neutrophils' job. Eosinophils are better equipped for fighting parasites and helminths, but take longer to be activated and accumulate in sufficient numbers. Simultaneous action of neutrophils and eosinophils would be detrimental for the surrounding host tissues. Thus, as soon as the type 2 immune response is established, the neutrophils step back and become quiescent due to IL-4R signaling.
In summary, the neutrophils bridge the time between pathogen invasion and the arrival of sufficient numbers of type 2 effector cells by initiating defense and help recruiting other immune cells. In doing so, they also facilitate the establishment of an inflammatory microenvironment. Since overshooting neutrophil activity would be destructive for host tissues, neutrophils are inhibited once they are not needed anymore. We thus postulate that the connection between IL-4R signaling and neutrophil inhibition has evolved as a safety mechanism to protect the body from neutrophil-inflicted harm.
Author Contributions
CE, LH, and OB set the outline of the manuscript. CE and LH prepared the figures and wrote the first draft of manuscript with input from DI. OB revised and edited the manuscript and figures.
Funding
This work was supported by the Swiss National Science Foundation (310030-172978), the Hochspezialisierte Medizin Schwerpunkt Immunologie (HSM-2-Immunologie), and the Clinical Research Priority Program CYTIMM-Z of the University of Zurich (all to OB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the Boyman laboratory for helpful discussions.
References
1. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Ann Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
2. Schouten HC. Neutropenia management. Ann Oncol. (2006) 17(Suppl. 10):x85–9. doi: 10.1093/annonc/mdl243
3. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
4. Ronnefarth VM, Erbacher AI, Lamkemeyer T, Madlung J, Nordheim A, Rammensee HG, et al. TLR2/TLR4-independent neutrophil activation and recruitment upon endocytosis of nucleosomes reveals a new pathway of innate immunity in systemic lupus erythematosus. J Immunol. (2006) 177:7740–9. doi: 10.4049/jimmunol.177.11.7740
5. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. (2010) 120:2423–31. doi: 10.1172/JCI41649
6. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. (2003) 34:70–4. doi: 10.1038/ng1149
7. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
8. Bajrami B, Zhu H, Kwak HJ, Mondal S, Hou Q, Geng G, et al. G-CSF maintains controlled neutrophil mobilization during acute inflammation by negatively regulating CXCR2 signaling. J Exp Med. (2016) 213:1999–2018. doi: 10.1084/jem.20160393
9. Deniset JF, Kubes P. Recent advances in understanding neutrophils. F1000Res. (2016) 5:2912. doi: 10.12688/f1000research.9691.1
10. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. (2011) 12:1035–44. doi: 10.1038/ni.2109
11. Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, et al. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. (1997) 91:385–95. doi: 10.1016/S0092-8674(00)80422-5
12. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
13. Li Jeon N, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat Biotechnol. (2002) 20:826–30. doi: 10.1038/nbt712
14. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17:1381–90. doi: 10.1038/nm.2514
15. Bonne-Annee S, Kerepesi LA, Hess JA, O'Connell AE, Lok JB, Nolan TJ, et al. Human and mouse macrophages collaborate with neutrophils to kill larval Strongyloides stercoralis. Infect Immun. (2013) 81:3346–55. doi: 10.1128/IAI.00625-13
16. Allen JE, Sutherland TE, Ruckerl D. IL-17 and neutrophils: unexpected players in the type 2 immune response. Curr Opin Immunol. (2015) 34:99–106. doi: 10.1016/j.coi.2015.03.001
17. De Benedetto A, Agnihothri R, McGirt LY, Bankova LG, Beck LA. Atopic dermatitis: a disease caused by innate immune defects? J Invest Dermatol. (2009) 129:14–30. doi: 10.1038/jid.2008.259
18. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. (2011) 242:233–46. doi: 10.1111/j.1600-065X.2011.01027.x
19. Woytschak J, Keller N, Krieg C, Impellizzieri D, Thompson RW, Wynn TA, et al. Type 2 interleukin-4 receptor signaling in neutrophils antagonizes their expansion and migration during infection and inflammation. Immunity. (2016) 45:172–84. doi: 10.1016/j.immuni.2016.06.025
20. Impellizzieri D, Ridder F, Raeber ME, Egholm C, Woytschak J, Kolios AGA, et al. IL-4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation. J Allergy Clin Immunol. (2019) 144:267–79.e4. doi: 10.1016/j.jaci.2019.01.042
21. Heeb LEM, Egholm C, Impellizzieri D, Ridder F, Boyman O. Regulation of neutrophils in type 2 immune responses. Curr Opin Immunol. (2018) 54:115–22. doi: 10.1016/j.coi.2018.06.009
22. Kim JS, Kwon HY, Choi WH, Jeon CY, Kim JI, Kim J, et al. Phagocytosis of serum- and IgG-opsonized zymosan particles induces apoptosis through superoxide but not nitric oxide in macrophage J774A.1. Exp Mol Med. (2003) 35:211–21. doi: 10.1038/emm.2003.29
23. Kim MK, Huang ZY, Hwang PH, Jones BA, Sato N, Hunter S, et al. Fcγ receptor transmembrane domains: role in cell surface expression, γ chain interaction, and phagocytosis. Blood. (2003) 101:4479–84. doi: 10.1182/blood.V101.11.4479
24. Van Kessel KPM, Bestebroer J, Van Strijp JAG. Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front Immunol. (2014) 5:467. doi: 10.3389/fimmu.2014.00467
25. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Ann Rev Pathol. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
26. Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. (2008) 112:935–45. doi: 10.1182/blood-2007-12-077917
27. Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. (1997) 89:3503–21.
28. Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol. (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
29. Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J Leukoc Biol. (2010) 87:905–14. doi: 10.1189/jlb.1009676
30. Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. (2003) 5:1317–27. doi: 10.1016/j.micinf.2003.09.008
31. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. (2014) 5:508. doi: 10.3389/fimmu.2014.00508
32. Naegelen I, Beaume N, Plançon S, Schenten V, Tschirhart EJ, Bréchard S. Regulation of neutrophil degranulation and cytokine secretion: a novel model approach based on linear fitting. J Immunol Res. (2015) 2015:1–15. doi: 10.1155/2015/817038
33. Kruger P, Saffarzadeh M, Weber ANR, Rieber N, Radsak M, Von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLOS Pathog. (2015) 11:e1004651. doi: 10.1371/journal.ppat.1004651
34. Wark PAB, Johnston SL, Moric I, Simpson JL, Hensley MJ, Gibson PG. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J. (2002) 19:68–75. doi: 10.1183/09031936.02.00226302
35. Soehnlein O, Oehmcke S, Rothfuchs AG, Frithiof R, Van Rooijen N, Morgelin M, et al. Neutrophil degranulation mediates severe lung damage triggered by streptococcal M1 protein. Eur Respir J. (2008) 32:405–12. doi: 10.1183/09031936.00173207
36. Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
37. Graham DB, Robertson CM, Bautista J, Mascarenhas F, Diacovo MJ, Montgrain V, et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCγ2 signaling axis in mice. J Clin Investig. (2007) 117:3445–52. doi: 10.1172/JCI32729
38. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. (2015) 12:5–23. doi: 10.1038/cmi.2014.89
39. Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. (2008) 4:278–86. doi: 10.1038/nchembio.85
40. Murphy R, Decoursey TE. Charge compensation during the phagocyte respiratory burst. Biochim Biophys Acta Bioenerg. (2006) 1757:996–1011. doi: 10.1016/j.bbabio.2006.01.005
41. Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. (2002) 416:291–7. doi: 10.1038/416291a
42. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
43. Uribe-Querol E, Rosales C. Control of phagocytosis by microbial pathogens. Front Immunol. (2017) 8:1368. doi: 10.3389/fimmu.2017.01368
44. Dinauer MC. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood. (2019) 133:2130–9. doi: 10.1182/blood-2018-11-844563
45. Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des. (2004) 10:1611–26. doi: 10.2174/1381612043384664
46. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
47. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. (2012) 198:773–83. doi: 10.1083/jcb.201203170
48. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
49. Van Avondt K, Hartl D. Mechanisms and disease relevance of neutrophil extracellular trap formation. Eur J Clin Invest. (2018) 48(Suppl. 2):e12919. doi: 10.1111/eci.12919
50. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
51. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
52. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
53. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
54. Schönrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. (2016) 7:366. doi: 10.3389/fimmu.2016.00366
55. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
56. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
57. Boeltz S, Amini P, Anders H-J, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. (2019) 26:395–408. doi: 10.1038/s41418-018-0261-x
58. Van Der Linden M, Van Den Hoogen LL, Westerlaken GHA, Fritsch-Stork RDE, Van Roon JAG, Radstake TRDJ, et al. Neutrophil extracellular trap release is associated with antinuclear antibodies in systemic lupus erythematosus and anti-phospholipid syndrome. Rheumatology. (2018) 57:1228–34. doi: 10.1093/rheumatology/key067
59. Bouts YM, Wolthuis DF, Dirkx MF, Pieterse E, Simons EM, van Boekel AM, et al. Apoptosis and NET formation in the pathogenesis of SLE. Autoimmunity. (2012) 45:597–601. doi: 10.3109/08916934.2012.719953
60. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
61. Nakazawa D, Tomaru U, Ishizu A. Possible implication of disordered neutrophil extracellular traps in the pathogenesis of MPO-ANCA-associated vasculitis. Clin Exp Nephrol. (2013) 17:631–3. doi: 10.1007/s10157-012-0738-8
62. Giaglis S, Stoikou M, Grimolizzi F, Subramanian BY, van Breda SV, Hoesli I, et al. Neutrophil migration into the placenta: good, bad or deadly? Cell Adh Migr. (2016) 10:208–25. doi: 10.1080/19336918.2016.1148866
63. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. (2014) 20:511–7. doi: 10.1038/nm.3547
64. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
65. Sorensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. (2016) 126:1612–20. doi: 10.1172/JCI84538.
66. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
67. Gregory SH, Sagnimeni AJ, Wing EJ. Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J Immunol. (1996) 157:2514–20.
68. Carr KD, Sieve AN, Indramohan M, Break TJ, Lee S, Berg RE. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur J Immunol. (2011) 41:2666–76. doi: 10.1002/eji.201041363
69. Witter AR, Okunnu BM, Berg RE. The essential role of neutrophils during infection with the intracellular bacterial pathogen Listeria monocytogenes. J Immunol. (2016) 197:1557–65. doi: 10.4049/jimmunol.1600599
70. Ellis TN, Beaman BL. Interferon-γ activation of polymorphonuclear neutrophil function. Immunology. (2004) 112:2–12. doi: 10.1111/j.1365-2567.2004.01849.x
71. Tan BH, Meinken C, Bastian M, Bruns H, Legaspi A, Ochoa MT, et al. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J Immunol. (2006) 177:1864–71. doi: 10.4049/jimmunol.177.3.1864
72. Lloyd CM, Snelgrove RJ. Type 2 immunity: expanding our view. Sci Immunol. (2018) 3:eaat1604. doi: 10.1126/sciimmunol.aat1604
73. Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. (2011) 11:375–88. doi: 10.1038/nri2992
74. Boyman O, Kaegi C, Akdis M, Bavbek S, Bossios A, Chatzipetrou A, et al. EAACI IG biologicals task force paper on the use of biologic agents in allergic disorders. Allergy. (2015) 70:727–54. doi: 10.1111/all.12616
75. LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. (2008) 132:259–72. doi: 10.1016/j.cell.2007.12.030
76. Bechman K, Yates M, Galloway JB. The new entries in the therapeutic armamentarium: the small molecule JAK inhibitors. Pharmacol Res. (2019) 147:104392. doi: 10.1016/j.phrs.2019.104392
77. Mishra PK, Palma M, Buechel B, Moore J, Davra V, Chu N, et al. Sterile particle-induced inflammation is mediated by macrophages releasing IL-33 through a Bruton's tyrosine kinase-dependent pathway. Nat Mater. (2019) 18:289–97. doi: 10.1038/s41563-018-0271-6
78. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med. (2011) 208:893–900. doi: 10.1084/jem.20102057
79. Herbert DR, Yang JQ, Hogan SP, Groschwitz K, Khodoun M, Munitz A, et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J Exp Med. (2009) 206:2947–57. doi: 10.1084/jem.20091268
80. McCoy KD, Stoel M, Stettler R, Merky P, Fink K, Senn BM, et al. Polyclonal and specific antibodies mediate protective immunity against enteric helminth infection. Cell Host Microbe. (2008) 4:362–73. doi: 10.1016/j.chom.2008.08.014
81. Sutherland TE, Logan N, Ruckerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol. (2014) 15:1116–25. doi: 10.1038/ni.3023
82. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. (2012) 18:260–6. doi: 10.1038/nm.2628
83. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. (2015) 15:271–82. doi: 10.1038/nri3831
84. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. (2016) 375:2335–48. doi: 10.1056/NEJMoa1610020
85. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. (2018) 378:2486–96. doi: 10.1056/NEJMoa1804092
86. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
87. Nembrini C, Marsland BJ, Kopf M. IL-17-producing T cells in lung immunity and inflammation. J Allergy Clin Immunol. (2009) 123:986–94. doi: 10.1016/j.jaci.2009.03.033
88. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. (2013) 252:116–32. doi: 10.1111/imr.12027
89. Gazendam RP, van de Geer A, Roos D, van den Berg TK, Kuijpers TW. How neutrophils kill fungi. Immunol Rev. (2016) 273:299–311. doi: 10.1111/imr.12454
90. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. (2015) 60:1176–83. doi: 10.1093/cid/ciu1154
91. Boyman O, Comte D, Spertini F. Adverse reactions to biologic agents and their medical management. Nat Rev Rheumatol. (2014) 10:612–27. doi: 10.1038/nrrheum.2014.123
92. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
93. Al-Qaoud KM, Pearlman E, Hartung T, Klukowski J, Fleischer B, Hoerauf A. A new mechanism for IL-5-dependent helminth control: neutrophil accumulation and neutrophil-mediated worm encapsulation in murine filariasis are abolished in the absence of IL-5. Int Immunol. (2000) 12:899–908. doi: 10.1093/intimm/12.6.899
94. Anthony RM, Urban JF Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. (2006) 12:955–60. doi: 10.1038/nm1451
95. Morimoto M, Morimoto M, Whitmire J, Xiao S, Anthony RM, Mirakami H, et al. Peripheral CD4 T cells rapidly accumulate at the host: parasite interface during an inflammatory Th2 memory response. J Immunol. (2004) 172:2424–30. doi: 10.4049/jimmunol.172.4.2424
96. Motran CC, Silvane L, Chiapello LS, Theumer MG, Ambrosio LF, Volpini X, et al. Helminth infections: recognition and modulation of the immune response by innate immune cells. Front Immunol. (2018) 9:664. doi: 10.3389/fimmu.2018.00664
97. Bass DA, Szejda P. Eosinophils versus neutrophils in host defense. Killing of newborn larvae of Trichinella spiralis by human granulocytes in vitro. J Clin Invest. (1979) 64:1415–22. doi: 10.1172/JCI109599
98. Voehringer D. The role of basophils in helminth infection. Trends Parasitol. (2009) 25:551–6. doi: 10.1016/j.pt.2009.09.004
99. Bonne-Annee S, Kerepesi LA, Hess JA, Wesolowski J, Paumet F, Lok JB, et al. Extracellular traps are associated with human and mouse neutrophil and macrophage mediated killing of larval Strongyloides stercoralis. Microbes Infect. (2014) 16:502–11. doi: 10.1016/j.micinf.2014.02.012
100. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. (2014) 15:938–46. doi: 10.1038/ni.2984
101. Spohn G, Arenas-Ramirez N, Bouchaud G, Boyman O. Endogenous polyclonal anti-IL-1 antibody responses potentiate IL-1 activity during pathogenic inflammation. J Allergy Clin Immunol. (2017) 139:1957–65 e3. doi: 10.1016/j.jaci.2016.09.033
102. Schmid AS, Hemmerle T, Pretto F, Kipar A, Neri D. Antibody-based targeted delivery of interleukin-4 synergizes with dexamethasone for the reduction of inflammation in arthritis. Rheumatology. (2018) 57:748–55. doi: 10.1093/rheumatology/kex447
103. Wermeling F, Anthony RM, Brombacher F, Ravetch JV. Acute inflammation primes myeloid effector cells for anti-inflammatory STAT6 signaling. Proc Natl Acad Sci USA. (2013) 110:13487–91. doi: 10.1073/pnas.1312525110
104. Cao Y, Brombacher F, Tunyogi-Csapo M, Glant TT, Finnegan A. Interleukin-4 regulates proteoglycan-induced arthritis by specifically suppressing the innate immune response. Arthritis Rheum. (2007) 56:861–70. doi: 10.1002/art.22422
105. Chen Z, Andreev D, Oeser K, Krljanac B, Hueber A, Kleyer A, et al. Th2 and eosinophil responses suppress inflammatory arthritis. Nat Commun. (2016) 7:11596. doi: 10.1038/ncomms11596
106. Harris AJ, Mirchandani AS, Lynch RW, Murphy F, Delaney L, Small D, et al. IL4Rα signaling abrogates hypoxic neutrophil survival and limits acute lung injury responses in vivo. Am J Respir Crit Care Med. (2019) 200:235–46. doi: 10.1164/rccm.201808-1599OC
107. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–6. doi: 10.1126/science.aam9690
108. Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. (2016) 110:51–61. doi: 10.1093/cvr/cvw024
109. Gieseck RL III, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. (2018) 18:62–76. doi: 10.1038/nri.2017.90
110. Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. (2010) 40:3317–20. doi: 10.1002/eji.201041170
111. Lee CR, Lee W, Cho SK, Park SG. Characterization of multiple cytokine combinations and TGF-β on differentiation and functions of myeloid-derived suppressor cells. Int J Mol Sci. (2018) 19:E869. doi: 10.3390/ijms19030869
112. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
113. Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. (2003) 9:40–6. doi: 10.1038/nm804
114. Dhingra N, Suarez-Farinas M, Fuentes-Duculan J, Gittler JK, Shemer A, Raz A, et al. Attenuated neutrophil axis in atopic dermatitis compared to psoriasis reflects TH17 pathway differences between these diseases. J Allergy Clin Immunol. (2013) 132:498–501 e3. doi: 10.1016/j.jaci.2013.04.043
115. Choy DF, Hsu DK, Seshasayee D, Fung MA, Modrusan Z, Martin F, et al. Comparative transcriptomic analyses of atopic dermatitis and psoriasis reveal shared neutrophilic inflammation. J Allergy Clin Immunol. (2012) 130:1335–43 e5. doi: 10.1016/j.jaci.2012.06.044
116. Fleming P, Drucker AM. Risk of infection in patients with atopic dermatitis treated with dupilumab: a meta-analysis of randomized controlled trials. J Am Acad Dermatol. (2018) 78:62–9 e1. doi: 10.1016/j.jaad.2017.09.052
117. Ou Z, Chen C, Chen A, Yang Y, Zhou W. Adverse events of Dupilumab in adults with moderate-to-severe atopic dermatitis: a meta-analysis. Int Immunopharmacol. (2018) 54:303–10. doi: 10.1016/j.intimp.2017.11.031
118. Korenblat P, Kerwin E, Leshchenko I, Yen K, Holweg CTJ, Anzures-Cabrera J, et al. Efficacy and safety of lebrikizumab in adult patients with mild-to-moderate asthma not receiving inhaled corticosteroids. Respir Med. (2018) 134:143–9. doi: 10.1016/j.rmed.2017.12.006
119. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taieb A, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. (2018) 78:863–71.e11. doi: 10.1016/j.jaad.2018.01.017
120. Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. (2007) 370:1422–31. doi: 10.1016/S0140-6736(07)61600-6
121. Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. (2015) 73:395–9. doi: 10.1016/j.jaad.2015.06.045
122. Wernham AGH, Veitch D, Grindlay DJC, Rogers NK, Harman KE. What's new in atopic eczema? An analysis of systematic reviews published in 2017. Part 1: treatment and prevention. Clin Exp Dermatol. (2019) 44:363–69. doi: 10.1111/ced.14044
Keywords: neutrophil, type 2 immunity, interleukin-4, interleukin-13, interleukin-4 receptor, inflammation, helminth, neutropenia
Citation: Egholm C, Heeb LEM, Impellizzieri D and Boyman O (2019) The Regulatory Effects of Interleukin-4 Receptor Signaling on Neutrophils in Type 2 Immune Responses. Front. Immunol. 10:2507. doi: 10.3389/fimmu.2019.02507
Received: 01 July 2019; Accepted: 07 October 2019;
Published: 24 October 2019.
Edited by:
Miriam Wittmann, University of Leeds, United KingdomReviewed by:
Krzysztof Guzik, Jagiellonian University, PolandLuz Pamela Blanco, National Institutes of Health (NIH), United States
Copyright © 2019 Egholm, Heeb, Impellizzieri and Boyman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Onur Boyman, b251ci5ib3ltYW5AdXpoLmNo
†These authors have contributed equally to this work as first authors