 Catherine F. Hatton
Catherine F. Hatton Christopher J. A. Duncan
Christopher J. A. Duncan- 1Immunity and Inflammation Theme, Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
- 2Department of Infection and Tropical Medicine, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
Viral encephalitis is a rare but clinically serious consequence of viral invasion of the brain and insight into its pathogenesis is urgently needed. Important research questions concern the involvement of the host innate immune response in pathogenesis, key to which is the role played by microglia, resident macrophages of the brain parenchyma. Do microglia have a protective function, by coordinating the innate immune response to viral infection, or do they drive pathogenic neuroinflammation? Here we synthesize recent data from mouse models of acute viral encephalitis, which reveal an unambiguously protective role for microglia. Depletion of microglia, via blockade of colony-stimulating factor 1 receptor (CSF1R) signaling, led to increased viral replication accompanied by more severe neurological disease and heightened mortality. Whilst the underlying mechanism(s) remain to be defined, microglial interactions with T cells and phagocytosis of infected neurones appear to play a role. Paradoxically, the production of inflammatory cytokines was increased in several instances following viral infection in microglia-depleted brains, suggesting that: (i) cells other than microglia mediate inflammatory responses and/or (ii) microglia may exert a regulatory function. Under certain circumstances the microglial antiviral response might contribute negatively to longer-term neurological sequelae, although fewer studies have focused on this aspect in encephalitis models. Understanding regulation of the microglial response, and how it contributes to disease is therefore a priority for future studies. Collectively, these findings demonstrate the central role of microglia in pathogenesis, suggesting the exciting possibility that defects of microglial function might contribute to encephalitis susceptibility and/or outcome in humans.
Introduction
Viral encephalitis is defined as pathological inflammation of the brain parenchyma secondary to viral infection (1). This syndrome is a rare but clinically serious outcome of infection with a range of DNA and RNA viruses. Encephalitis is associated with appreciable mortality and high rates of permanent neurological impairment in survivors, and in most cases there is no available antiviral therapy (1). Annual healthcare expenditure associated with the acute care of patients with encephalitis was estimated in the region of $2 billion (2), although indirect costs are likely much higher. Furthermore, several emerging causes of viral encephalitis are considered by WHO to be a significant threat to global public health (3). Fundamental to addressing this unmet medical need is research to better understand the pathogenesis of viral encephalitis (4).
Host Innate Immunity: Protective or Pathogenic in Viral Encephalitis?
Whilst some viruses are highly neurovirulent (e.g., rabies virus), most people infected with neurotropic viruses do not go on to develop encephalitis, suggesting that rare host and/or viral factors underlie susceptibility. Defining these factors is a major challenge for the field. Despite decades of investigation, there is limited evidence of viral determinants of neurovirulence (1, 5), implicating the host response in pathogenesis. However, it remains unclear whether immunity plays a predominantly beneficial or deleterious role. In other words, do patients develop encephalitis because their response to viral infection is inadequate, impairing antiviral resistance, or excessive, resulting in immunopathology? There is evidence from human studies to support both hypotheses. Although not necessarily mutually exclusive, they imply opposite therapeutic approaches (i.e., cytokine supplementation vs. immunosuppression).
The hypothesis that encephalitis arises through a failure of host resistance is supported by very rare genetic errors of innate antiviral immunity, which confer heightened susceptibility to encephalitis secondary to wild-type (6–14) or attenuated vaccine strain viruses (15, 16). These disorders provide compelling evidence that under normal circumstances innate immunity provides a critical layer of protection against viral encephalitis. The extent to which inborn errors of innate immunity underlie encephalitis more generally, including in adults, remains to be determined.
The alternative hypothesis, that pathogenesis is governed not by a failure to resist viral replication, but rather due to excessive innate immune-mediated damage, is supported by: (i) a lack of correlation between viral loads in cerebrospinal fluid and disease outcome (17–20), suggesting that the extent of viral replication does not alone dictate pathology; and (ii) correlations between biomarkers of innate immune activation and poor outcome (21–23). In addition, phenotypic overlaps are recognized between congenital viral diseases such as cytomegalovirus (CMV) and the genetic disease Aicardi-Goutières syndrome (24), in which the aberrant induction of antiviral innate immunity is considered central to pathogenesis (25); the implication of this shared phenotype is that much of the neurological damage arising from congenital viral infection may be host-derived (26). Nevertheless, there remains no conclusive proof of a causal link between immune activation and neurological outcome, since it is impossible in clinical studies to separate the effects of viral cytopathicity and immunopathology. Insight into this question may come from an ongoing clinical trial of anti-inflammatory corticosteroids in patients with herpes simplex virus encephalitis (https://clinicaltrials.gov/ct2/show/NCT03084783) (27).
Mouse models of viral encephalitis provide an alternative means to resolve these issues. Whilst there are caveats to the use of mice to understand human disease (28, 29), key pathological features of encephalitis are reproduced in these models (30, 31), which have the advantage of being amenable to controlled experimental perturbation. A key theme to emerge from recent studies is the critical role of antiviral immunity within the brain itself.
A Brain Intrinsic Antiviral Interferon Network Combats Viral Spread in the Brain
Contrary to the long-held view of the brain as an immune-privileged organ, it is now clear that the brain parenchyma poses an intrinsic antiviral network, in which the antiviral cytokines known as type I interferons (IFNs) play a central role (32, 33). Detje et al. were the first to show that the response of neuroectodermal cells (including neurones, astrocytes, and oligodendrocytes) to type I IFNs was essential for the protection of mice against vesicular stomatitis virus (VSV) encephalitis (34). These findings were subsequently reproduced in a model of herpes simplex virus (HSV1) encephalitis (35). All cell types of the central nervous system (CNS) are capable of mounting a type I IFN response, although the relative efficiency of this process appears to vary substantially (33, 36). In VSV models, interferon alpha/beta receptor (IFNAR) signaling within olfactory neurones (37) and astrocytes (38) was necessary to limit viral dissemination throughout the CNS, suggesting that type I IFNs act on viral target cells to control permissiveness and/or onward transmission. However, the cell type(s) responsible for initiating the production of type I IFNs, and the precise intercellular signaling events that underpin protection, remain to be determined. Astrocytes were reported to be the main producers of IFNβ upon infection with model neurotropic RNA viruses (39, 40). Simultaneously, data generated in other encephalitis models have rekindled interest in the role of microglia (32, 41, 42).
Microglia: at the Hub of the Brain's Antiviral Network?
It has long been suspected that microglia—the sole brain-resident immune cells—play an essential role in antiviral defense of the brain (43). However, the tools were not previously available to formally test this hypothesis. Microglia are the “third element” of the CNS, initially described by Ramón y Cajal and further characterized by Del Río-Hortega. These cells are parenchymal resident macrophages of the brain parenchyma, arising from embryonic yolk sac precursors (44, 45) which seed the developing brain around embryonic day 9.5. This period coincides with neuronal birth and is before the formation of the blood-brain barrier and the development of astrocytes or oligodendrocytes (46). Thus, microglia fundamentally shape the developing brain by supporting neurogenesis (47, 48) and synaptic remodeling (49, 50). The essential role of microglia in normal brain development is revealed by humans (51, 52) and mice (44, 53) with genetic deficiencies of microglia due to homozygous deficiency of the colony-stimulating factor 1 receptor (CSF1R).
In the current paradigm, microglia self-renew under homeostatic conditions with minimal contribution from circulating monocytes (44, 54). Like other tissue-resident macrophages, microglia respond rapidly to environmental cues with a broad spectrum of activation states (55). They express relevant endosomal and cytosolic pathogen pattern recognition receptors for detection of viral molecules and are capable of efficiently sensing viral pathogens in vitro (56). Reactive microgliosis, defined an increase in microglial numbers and a change from ramified to amoeboid morphology, is observed in both patients and mice with viral encephalitis (56, 57), and appears to be dependent to some extent on IFNAR signaling (42, 58). Whether microglial reactivity is an appropriate response to viral invasion, or contributes negatively to disease, continues to provoke debate (56, 59). Prior to the availability of efficient methods of microglial depletion, studies reported both protective (41, 60) and pathogenic (61, 62) effects of microglia in encephalitis models. Since then, considerable progress has been made in resolving this controversy through the use of targeted depletion of microglia in mouse models of encephalitis. Methods of microglial depletion have been comprehensively reviewed elsewhere (63, 64) and are briefly summarized below.
Methods of Microglial Depletion
The original approach, developed in the 1980s, was an infusion of clodronate-encapsulated liposomes, which are toxic to macrophages (65). Since liposomes are not capable of penetrating the blood-brain barrier, intracranial or intraventricular administration is necessary to deplete microglia (66, 67). However, even under these circumstances, depletion is incomplete. Various genetic methods were subsequently developed that permitted more efficient targeting of microglia (63), however these have not been used in encephalitis models. By contrast, blockade of CSF1R signaling has been more widely adopted.
As alluded to above, microglia are dependent on signaling through the CSF1R for development and survival (33, 44, 51, 53). Csf1r−/− mice lack microglia as well as all tissue macrophage populations (44, 53). However, these mice also exhibit profound developmental defects and significant perinatal mortality, making them unsuitable for studies of encephalitis. The ligands for CSF1R are CSF1 and IL34. Deletion of either does not lead to complete loss of microglia in the brain, reflecting a degree of redundancy (63). Il34LacZ/LacZ reporter mice (which are effectively IL34 deficient) have been used in a limited number of studies of viral encephalitis (68, 69). Blockade of CSF1R [e.g., with the small molecules such as PLX5622 (70, 71)] efficiently depletes microglia from the brain of intact rodents (72) and has been more frequently employed, presumably due to its technical ease. Collectively, the results of these studies provide a coherent set of data that supports a protective role for microglia in the acute phase of viral encephalitis (Figure 1).
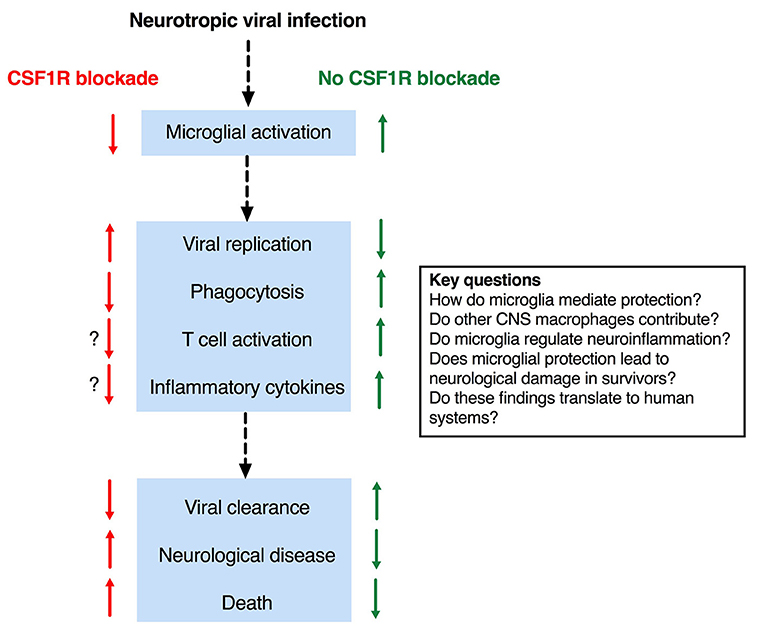
Figure 1. Effects of microglial depletion in mouse models of viral encephalitis and key outstanding questions.
Microglia Are Protective in Encephalitis Disease Models
A range of neurotropic viruses from several families have been studied in microglial depletion models (Table 1). Despite the diversity of viruses investigated, consistent findings have emerged. In virtually all cases, the replication of neurotropic viruses was enhanced by depletion of microglia (58, 73–79), indicating that microglia are essential for viral resistance in the CNS. The only exception was Il34LacZ/LacZ mice, in which viral replication was unaffected (68). In this model, microglia numbers were only modestly reduced in the cerebral cortex (3-fold), and unchanged in the cerebellum (68).
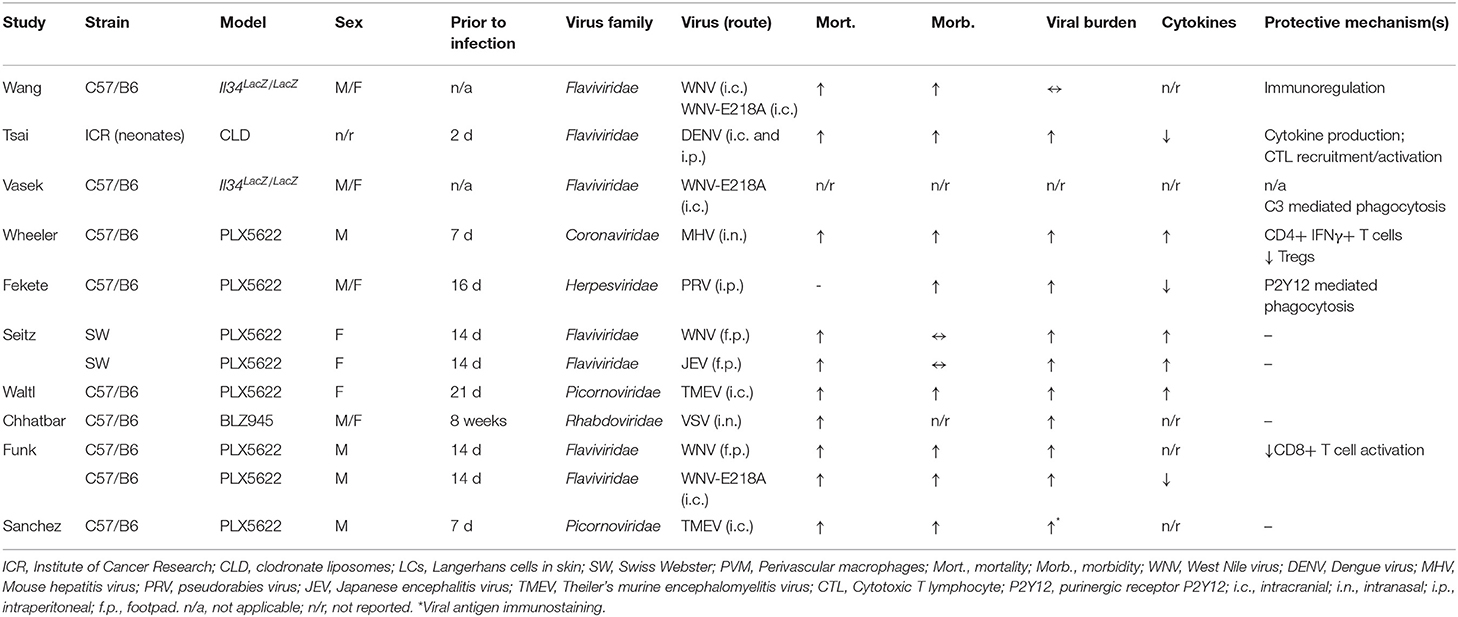
Table 1. Microglial depletion studies in mouse models of viral encephalitis.
Importantly, microglial depletion was associated with a negative impact on clinically relevant endpoints such as neurological disease and/or death (58, 73–79)—even in the Il34LacZ/LacZ model (68)—implying that microglia are critical to survival in encephalitis. In a study using a nonlethal pseudorabies virus (PRV) model (74), microglial depletion led to the development of overt neurological disease, which correlated with viral burden in the brain (74). Similarly, in other studies where neurological disease was reported, microglia-depleted mice showed increased disease severity prior to death (Table 1). Collectively, these studies establish clear links between viral resistance and negative disease outcomes, consistent with a protective function of microglia.
Depletion of Microglia Impairs the Antiviral Function of Recruited T Cells
The mechanism(s) underlying these protective properties remain uncertain. Preliminary data suggest that the T cell response induced by microglia is involved, however the impact on the quality and magnitude of the antiviral T cell response was inconsistent across different models (76–78), and no impact on T cell immunophenotype was observed in others (74), meaning it is not yet possible to draw definitive conclusions.
Viral clearance and/or disease outcome relies on a functional T cell response in several mouse models of encephalitis (e.g., West Nile virus [WNV], mouse hepatitis virus [MHV], or Theiler's murine encephalomyelitis virus [TMEV]). In the TMEV model, strain-specific differences in disease phenotype are linked to the effectiveness of the CD8+ T cell response, which is subject to Treg suppression, and consequent clinical disease, in susceptible mouse genetic backgrounds (76). Although microglia depletion had no impact on CD8+ T cell recruitment, hippocampal Tregs were increased, and the disease phenotype recalled that seen in susceptible backgrounds (76). In contrast, in a WNV model where CD8+ T cells are also protective, an increased number of CD8+ T cells were recruited to the brain of microglia-depleted mice. However, these CD8+ T cells had a blunted activation phenotype, with reduced proportions of cells expressing the activation markers CD69 or CD160 (78). Whilst it is plausible that this would contribute toward a defect of viral control and/or clearance, due to the increase in overall CD8+ T cell recruitment in PLX5622-treated animals, the net effect was to achieve an equivalent number of activated CD8+ T cells in the brains of both groups (78).
In MHV, both CD8+ and CD4+ T cells are involved in viral clearance (80, 81), whilst CD4+ T cells also contribute to pathogenesis (82). In the MHV model, microglia depletion did not negatively impact CD8+ T cell recruitment, but was associated with significant reductions in the recruitment of CD4+ T cells and Tregs, and importantly in IFNγ expression by CD4+ T cells (77)—in complete contrast to TMEV (76). While the overall impact of these changes to both “effector” CD4+ T cell and Treg populations is difficult to reconcile, the authors concluded that the observed reduction in Treg recruitment might contribute to T cell immunopathology, thereby worsening disease outcome.
The collective implication of these studies is that microglia contribute positively to aspects of T cell recruitment and/or activation, however the precise nature of the protective T cell response induced by microglia requires further examination.
CNS Intrinsic Protection by Microglia: Role of Phagocytosis
A mechanism by which microglia might mediate protection, independently of their effects on the T cell response, was reported in the PRV model. Here microglia were recruited toward and engulfed virus-infected neurones, a process that required microglial P2Y12 signaling (74). As stated above, the depletion of microglia led to the development of overt disease and increased viral replication. Similarly, in a study using an attenuated WNV strain, phagocytosis of presynaptic CA3 neurones in the hippocampus was observed, which depended on deposition of the complement protein C3 (69). Il34LacZ/LacZ mice were included in this study to show that the attrition of presynaptic neurones was microglia-dependent. Since these findings were associated with defects of spatial orientation in recovered wild-type mice, the authors concluded that microglia contribute to pathogenesis. However, the outcome of infection in microglia-depleted Il34LacZ/LacZ mice was not reported (69), whereas in a previous study using the same model there was significantly enhanced mortality in Il34LacZ/LacZ (microglia-depleted) mice (68). Thus, it seems that while microglial phagocytosis may prevent mortality in the acute phase (68, 74), this might come at later stages at a cost of permanent neurological damage (69). Microglia respond to insults with a broad spectrum of activation states, and are implicated in not just the initiation, but also the resolution of inflammatory responses (55). As for phagocytosis, other aspects of the microglial response to infection might contribute to tissue damage in encephalitis models, as they do in models of sterile neuroinflammation (83, 84). Key questions, yet to be addressed in viral encephalitis models, are: (i) what is the effect of microglial depletion in the resolution phase, and (ii) does a “goldilocks zone” of microglial activity exist, in which just enough reaction is sufficient for protection without incurring permanent neurological damage, but where both too little and too much might contribute adversely to encephalitis outcome. In this scenario, therapeutic approaches to either boost or suppress microglial responses, at different stages of disease, might prove beneficial.
Enhanced or Reduced Inflammation in Microglia Depleted Brains?
As discussed above, clinical studies have reported associations between inflammatory cytokine production and adverse outcomes. Interestingly, despite the suspicion that microglia are central to pathogenic neuroinflammation, microglial depletion was unexpectedly associated in some studies with an increase in the synthesis of cytokines and/or chemokines in the brain of infected animals (75–77), whereas in other studies there was a reduction (73, 74, 78). There is no definitive explanation for these contradictory observations. One possibility is the increased recruitment of circulating monocytes to microglia-depleted brains in circumstances where cytokine/chemokine synthesis was enhanced (76, 77), which was not observed in other studies (58, 74, 78)—including ones in which a reduction in cytokine/chemokine induction was reported (78). However, these factors were not consistently reported, and furthermore, whether this apparent association is causal remains uncertain. Other possible explanations include:
(i) a correlation between increased viral replication and inflammation–noted in one of these studies (77) and previously reported elsewhere (85, 86);
(ii) the activation of CNS-resident cell types, such as astrocytes, which was similarly noted in one study (76), as in other disease models (87, 88).
(iii) the loss of an immunoregulatory function of microglia, as mentioned above.
Overall, further work is needed to clarify the potential relationship between microglial depletion and immune dysregulation in the CNS.
Non-specific Effects of CSF1R Inhibition: a Confounding Factor?
There are also outstanding questions regarding the specificity of CSF1R blockade for microglia. Whilst PLX5622 (71) and BLZ945 (89) are more specific for CSF1R than earlier inhibitors (63), PLX5622 has been shown to deplete macrophages from various tissues, including kidney (90) and peripheral nervous system (91), leaving open the possibility of non-specific depletion of macrophages either in the CNS (e.g., meningeal or perivascular macrophages) or elsewhere. In encephalitis models, depletion of CNS macrophages (CD45hi CD11b+) was reported in some instances (58, 78), although not in several others (74, 76, 77, 79). Again, this inconsistency is unexplained, but one possibility is the use of CD45/CD11b expression to distinguish CNS macrophages from microglia (84, 92). Increased microglial CD45 expression has been noted during VSV encephalitis (58), which might confound assessment of the effect of PLX5622 on CNS macrophages. Importantly, any depletion observed was minor compared to the effects on microglia (58, 78). The use of more specific markers of microglia (e.g., TMEM119) or perivascular macrophages (e.g., CD206) might help to resolve this issue (93, 94).
In the studies which quantified effects on systemic myeloid populations (58, 76–78) there were no reductions in splenic or bone marrow macrophages (74, 76–78), however variable effects on circulating monocytes were reported, with reductions in some studies (76, 78), but no effect in others (58, 74, 77). Any impact on systemic myeloid populations is especially problematic for flavivirus models, where systemic viral replication occurs as a precursor to CNS neuroinvasion (75, 78); systemic depletion of myeloid cells might (i) enhance systemic viral replication, in turn enhancing the load of virus reaching the CNS, and/or (ii) cause CNS-extrinsic disease that might contribute independently to mortality. Funk et al. reported an enhancement of systemic WNV replication and clinical disease in association with PLX5622-induced monocyte depletion (78). To overcome this, they also challenged mice via the intracranial route, confirming a negative impact of microglial depletion (78), consistent with previous findings (68). By contrast, Seitz et al., in the same WNV model, did not detect systemic viral replication (75). The extent to which possible “off-target” effects of CSF1R inhibition might confound data generated in encephalitis models remains uncertain.
Conclusion
Through the recent use of microglial depletion systems, it is evident that microglia play an essential protective function in mouse models of viral encephalitis. Nevertheless, important questions remain about the mechanism(s) by which microglia (and possibly other CNS resident macrophages) mediate these protective effects (Figure 1). An intriguing observation–that microglial depletion leads in some circumstances to enhanced neuroinflammation, hints at a possible regulatory function of microglia in encephalitis and is an important area for future investigation. Another priority area is the possibility that dysregulated microglial responses, whilst protective against mortality in the early stages of encephalitis, might contribute at later stages to permanent neurological sequelae. A key question is whether these various findings in mouse models translate to human systems; the recent development of methods of microglial differentiation from human pluripotent stem cells offers a potential solution to this question [reviewed in (46)]. Collectively, these observations suggest the exciting possibility that (i) defects in the microglial response might underlie encephalitis susceptibility in patients, and (ii) that targeting this response may provide new therapeutic opportunities.
Author Contributions
CD: conceptualization and funding acquisition. CH and CD: literature searching, manuscript writing, and figures and tables.
Funding
This work was supported by the Wellcome Trust (211153/Z/18/Z to CD) and the MRC (MR/N013840/1 to CH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Aidan Hanrath for constructive comments on the manuscript. We apologize to any authors whose work was omitted due to space constraints.
References
1. Venkatesan A, Michael BD, Probasco JC, Geocadin RG, Solomon T. Acute encephalitis in immunocompetent adults. Lancet. (2019) 393:702–16. doi: 10.1016/S0140-6736(18)32526-1
2. Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis-associated hospitalizations in the United States, 1998-2010. Neurology. (2014) 82:443–51. doi: 10.1212/WNL.0000000000000086
3. WHO. List of Blueprint Priority Diseases. (2018). Available online at: https://www.who.int/blueprint/priority-diseases/en/
4. John CC, Carabin H, Montano SM, Bangirana P, Zunt JR, Peterson PK. Global research priorities for infections that affect the nervous system. Nature. (2015) 527:S178–86. doi: 10.1038/nature16033
5. Solomon T, Winter PM. Neurovirulence and host factors in flavivirus encephalitis–evidence from clinical epidemiology. Arch Virol Suppl. (2004) 18:161–70. doi: 10.1007/978-3-7091-0572-6_14
6. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. (2003) 33:388–91. doi: 10.1038/ng1097
7. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. (2006) 314:308–12. doi: 10.1126/science.1128346
8. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. (2007) 317:1522–7. doi: 10.1126/science.1139522
9. Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
10. Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. (2011) 121:4889–902. doi: 10.1172/JCI59259
11. Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. (2012) 209:1567–82. doi: 10.1084/jem.20111316
12. Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol. (2017) 139:1995–7 e9. doi: 10.1016/j.jaci.2016.10.033
13. Ogunjimi B, Zhang SY, Sorensen KB, Skipper KA, Carter-Timofte M, Kerner G, et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest. (2017) 127:3543–56. doi: 10.1172/JCI92280
14. Carter-Timofte ME, Hansen AF, Mardahl M, Fribourg S, Rapaport F, Zhang SY, et al. Varicella-zoster virus CNS vasculitis and RNA polymerase III gene mutation in identical twins. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e500. doi: 10.1212/NXI.0000000000000500
15. Duncan CJ, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med. (2015) 7:307ra154. doi: 10.1126/scitranslmed.aac4227
16. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med. (2019) 6:2057–70. doi: 10.1084/jem.20182295
17. Lakeman FD, Whitley RJ. Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis. (1995) 171:857–63. doi: 10.1093/infdis/171.4.857
18. Ruzek D, Piskunova N, Zampachova E. High variability in viral load in cerebrospinal fluid from patients with herpes simplex and varicella-zoster infections of the central nervous system. Clin Microbiol Infect. (2007) 13:1217–9. doi: 10.1111/j.1469-0691.2007.01831.x
19. Pasquet A, Ajana F, Melliez H, Giurca C, Poissy J, Yazdanpanah Y. Central nervous system HIV replication and HIV-related pachymeningitis in a patient on protease inhibitor monotherapy despite an undetectable plasma viral load. AIDS. (2012) 26:1726–8. doi: 10.1097/QAD.0b013e32835646fb
20. Schloss L, Falk KI, Skoog E, Brytting M, Linde A, Aurelius E. Monitoring of herpes simplex virus DNA types 1 and 2 viral load in cerebrospinal fluid by real-time PCR in patients with herpes simplex encephalitis. J Med Virol. (2009) 81:1432–7. doi: 10.1002/jmv.21563
21. Winter PM, Dung NM, Loan HT, Kneen R, Wills B, Thu le T, et al. Proinflammatory cytokines and chemokines in humans with Japanese encephalitis. J Infect Dis. (2004) 190:1618–26. doi: 10.1086/423328
22. Kamei S, Taira N, Ishihara M, Sekizawa T, Morita A, Miki K, et al. Prognostic value of cerebrospinal fluid cytokine changes in herpes simplex virus encephalitis. Cytokine. (2009) 46:187–93. doi: 10.1016/j.cyto.2009.01.004
23. Griffiths MJ, Ooi MH, Wong SC, Mohan A, Podin Y, Perera D, et al. In enterovirus 71 encephalitis with cardio-respiratory compromise, elevated interleukin 1beta, interleukin 1 receptor antagonist, and granulocyte colony-stimulating factor levels are markers of poor prognosis. J Infect Dis. (2012) 206:881–92. doi: 10.1093/infdis/jis446
24. Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. (1984) 15:49–54. doi: 10.1002/ana.410150109
25. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38. doi: 10.1084/jem.20161596
26. Gresser I, Morel-Maroger L, Riviere Y, Guillon JC, Tovey MG, Woodrow D, et al. Interferon-induced disease in mice and rats. Ann N Y Acad Sci. (1980) 350:12–20. doi: 10.1111/j.1749-6632.1980.tb20602.x
27. Sergerie Y, Boivin G, Gosselin D, Rivest S. Delayed but not early glucocorticoid treatment protects the host during experimental herpes simplex virus encephalitis in mice. J Infect Dis. (2007) 195:817–25. doi: 10.1086/511987
28. Davis MM. A prescription for human immunology. Immunity. (2008) 29:835–8. doi: 10.1016/j.immuni.2008.12.003
29. Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. (2014) 34:433–54. doi: 10.1615/CritRevImmunol.2014011600
30. Holbrook MR, Gowen BB. Animal models of highly pathogenic RNA viral infections: encephalitis viruses. Antiviral Res. (2008) 78:69–78. doi: 10.1016/j.antiviral.2007.10.004
31. Mancini M, Vidal SM. Insights into the pathogenesis of herpes simplex encephalitis from mouse models. Mamm Genome. (2018) 29:425–45. doi: 10.1007/s00335-018-9772-5
32. Drokhlyansky E, Goz Ayturk D, Soh TK, Chrenek R, O'Loughlin E, Madore C, et al. The brain parenchyma has a type I interferon response that can limit virus spread. Proc Natl Acad Sci USA. (2017) 114:E95–104. doi: 10.1073/pnas.1618157114
33. Hwang M, Bergmann CC. Intercellular communication is key for protective ifnalpha/beta signaling during viral central nervous system infection. Viral Immunol. (2019) 32:1–6. doi: 10.1089/vim.2018.0101
34. Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, et al. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol. (2009) 182:2297–304. doi: 10.4049/jimmunol.0800596
35. Rosato PC, Leib DA. Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog. (2015) 11:e1005028. doi: 10.1371/journal.ppat.1005028
36. Sorgeloos F, Kreit M, Hermant P, Lardinois C, Michiels T. Antiviral type I and type III interferon responses in the central nervous system. Viruses. (2013) 5:834–57. doi: 10.3390/v5030834
37. van den Pol AN, Ding S, Robek MD. Long-distance interferon signaling within the brain blocks virus spread. J Virol. (2014) 88:3695–704. doi: 10.1128/JVI.03509-13
38. Detje CN, Lienenklaus S, Chhatbar C, Spanier J, Prajeeth CK, Soldner C, et al. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon Beta producers that protect from lethal encephalitis. J Virol. (2015) 89:2731–8. doi: 10.1128/JVI.02044-14
39. Pfefferkorn C, Kallfass C, Lienenklaus S, Spanier J, Kalinke U, Rieder M, et al. Abortively infected astrocytes appear to represent the main source of interferon beta in the virus-infected brain. J Virol. (2016) 90:2031–8. doi: 10.1128/JVI.02979-15
40. Soung A, Klein RS. Viral encephalitis and neurologic diseases: focus on astrocytes. Trends Mol Med. (2018) 24:950–62. doi: 10.1016/j.molmed.2018.09.001
41. Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. (2016) 7:13348. doi: 10.1038/ncomms13348
42. Nayak D, Johnson KR, Heydari S, Roth TL, Zinselmeyer BH, McGavern DB. Type I interferon programs innate myeloid dynamics and gene expression in the virally infected nervous system. PLoS Pathog. (2013) 9:e1003395. doi: 10.1371/journal.ppat.1003395
43. Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. (2010) 468:253–62. doi: 10.1038/nature09615
44. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
45. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
46. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
47. Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. (2013) 33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013
48. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. (2013) 16:543–51. doi: 10.1038/nn.3358
49. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
50. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
51. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet. (2019) 104:936–47. doi: 10.1016/j.ajhg.2019.03.010
52. Guo L, Bertola DR, Takanohashi A, Saito A, Segawa Y, Yokota T, et al. Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-pyle disease spectrum and degenerative encephalopathy with brain malformation. Am J Hum Genet. (2019) 104:925–35. doi: 10.1016/j.ajhg.2019.03.004
53. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. (2002) 99:111–20. doi: 10.1182/blood.V99.1.111
54. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
55. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
56. Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev. (2004) 17:942–64, table of contents. doi: 10.1128/CMR.17.4.942-964.2004
57. Cagnin A, Myers R, Gunn RN, Lawrence AD, Stevens T, Kreutzberg GW, et al. In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain. (2001) 124(Pt 10):2014–27. doi: 10.1093/brain/124.10.2014
58. Chhatbar C, Detje CN, Grabski E, Borst K, Spanier J, Ghita L, et al. Type I interferon receptor signaling of neurons and astrocytes regulates microglia activation during viral encephalitis. Cell Rep. (2018) 25:118–29 e4. doi: 10.1016/j.celrep.2018.09.003
59. Chen Z, Zhong D, Li G. The role of microglia in viral encephalitis: a review. J Neuroinflammation. (2019) 16:76. doi: 10.1186/s12974-019-1443-2
60. Conrady CD, Zheng M, van Rooijen N, Drevets DA, Royer D, Alleman A, et al. Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol. (2013) 190:2807–17. doi: 10.4049/jimmunol.1203265
61. Quick ED, Leser JS, Clarke P, Tyler KL. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J Virol. (2014) 88:13005–14. doi: 10.1128/JVI.01994-14
62. Quick ED, Seitz S, Clarke P, Tyler KL. Minocycline has anti-inflammatory effects and reduces cytotoxicity in an ex vivo spinal cord slice culture model of west Nile virus infection. J Virol. (2017) 91:e00569-17. doi: 10.1128/JVI.00569-17
63. Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol. (2015) 36:625–36. doi: 10.1016/j.it.2015.08.005
64. Jakel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front Cell Neurosci. (2017) 11:24. doi: 10.3389/fncel.2017.00024
65. Van Rooijen N. The liposome-mediated macrophage 'suicide' technique. J Immunol Methods. (1989) 124:1–6. doi: 10.1016/0022-1759(89)90178-6
66. Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. (2011) 31:12992–3001. doi: 10.1523/JNEUROSCI.2102-11.2011
67. Lee JC, Seong J, Kim SH, Lee SJ, Cho YJ, An J, et al. Replacement of microglial cells using Clodronate liposome and bone marrow transplantation in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Biochem Biophys Res Commun. (2012) 418:359–65. doi: 10.1016/j.bbrc.2012.01.026
68. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. (2012) 13:753–60. doi: 10.1038/ni.2360
69. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. (2016) 534:538–43. doi: 10.1038/nature18283
70. Acharya MM, Green KN, Allen BD, Najafi AR, Syage A, Minasyan H, et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci Rep. (2016) 6:31545. doi: 10.1038/srep31545
71. Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. (2015) 12:139. doi: 10.1186/s12974-015-0366-9
72. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
73. Tsai TT, Chen CL, Lin YS, Chang CP, Tsai CC, Cheng YL, et al. Microglia retard dengue virus-induced acute viral encephalitis. Sci Rep. (2016) 6:27670. doi: 10.1038/srep27670
74. Fekete R, Cserep C, Lenart N, Toth K, Orsolits B, Martinecz B, et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. (2018) 136:461–82. doi: 10.1007/s00401-018-1885-0
75. Seitz S, Clarke P, Tyler KL. Pharmacologic depletion of microglia increases viral load in the brain and enhances mortality in murine models of flavivirus-induced encephalitis. J Virol. (2018) 92:e00525-18. doi: 10.1128/JVI.00525-18
76. Waltl I, Kaufer C, Gerhauser I, Chhatbar C, Ghita L, Kalinke U, et al. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav Immun. (2018) 74:186–204. doi: 10.1016/j.bbi.2018.09.006
77. Wheeler DL, Sariol A, Meyerholz DK, Perlman S. Microglia are required for protection against lethal coronavirus encephalitis in mice. J Clin Invest. (2018) 128:931–43. doi: 10.1172/JCI97229
78. Funk KE, Klein RS. CSF1R antagonism limits local restimulation of antiviral CD8(+) T cells during viral encephalitis. J Neuroinflammation. (2019) 16:22. doi: 10.1186/s12974-019-1397-4
79. Sanchez JMS, DePaula-Silva AB, Doty DJ, Truong A, Libbey JE, Fujinami RS. Microglial cell depletion is fatal with low level picornavirus infection of the central nervous system. J Neurovirol. (2019) 25:415–21. doi: 10.1007/s13365-019-00740-3
80. Marten NW, Stohlman SA, Atkinson RD, Hinton DR, Fleming JO, Bergmann CC. Contributions of CD8+ T cells and viral spread to demyelinating disease. J Immunol. (2000) 164:4080–8. doi: 10.4049/jimmunol.164.8.4080
81. Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J Immunol. (2003) 170:3204–13. doi: 10.4049/jimmunol.170.6.3204
82. Anghelina D, Pewe L, Perlman S. Pathogenic role for virus-specific CD4 T cells in mice with coronavirus-induced acute encephalitis. Am J Pathol. (2006) 169:209–22. doi: 10.2353/ajpath.2006.051308
83. Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. (2015) 34:1612–29. doi: 10.15252/embj.201490791
84. Greter M, Lelios I, Croxford AL. Microglia Versus Myeloid Cell Nomenclature during brain inflammation. Front Immunol. (2015) 6:249. doi: 10.3389/fimmu.2015.00249
85. Menasria R, Canivet C, Piret J, Gosselin J, Boivin G. Protective role of CX3CR1 signalling in resident cells of the central nervous system during experimental herpes simplex virus encephalitis. J Gen Virol. (2017) 98:447–60. doi: 10.1099/jgv.0.000667
86. Luo H, Winkelmann ER, Zhu S, Ru W, Mays E, Silvas JA, et al. Peli1 facilitates virus replication and promotes neuroinflammation during West Nile virus infection. J Clin Invest. (2018) 128:4980–91. doi: 10.1172/JCI99902
87. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
88. Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. (2017) 37:2224–36. doi: 10.1177/0271678X17694185
89. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
90. Zimmerman KA, Song CJ, Li Z, Lever JM, Crossman DK, Rains A, et al. Tissue-resident macrophages promote renal cystic disease. J Am Soc Nephrol. (2019) 30:1841–56. doi: 10.1681/ASN.2018080810
91. Lee S, Shi XQ, Fan A, West B, Zhang J. Targeting macrophage and microglia activation with colony stimulating factor 1 receptor inhibitor is an effective strategy to treat injury-triggered neuropathic pain. Mol Pain. (2018) 14:1744806918764979. doi: 10.1177/1744806918764979
92. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. (2014) 211:1533–49. doi: 10.1084/jem.20132477
93. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
Keywords: viral immunity, microglial depletion, CSF1R, PLX5622, IL34, interferon, neuroimmunology, central nervous system
Citation: Hatton CF and Duncan CJA (2019) Microglia Are Essential to Protective Antiviral Immunity: Lessons From Mouse Models of Viral Encephalitis. Front. Immunol. 10:2656. doi: 10.3389/fimmu.2019.02656
Received: 04 September 2019; Accepted: 28 October 2019;
Published: 13 November 2019.
Edited by:
Stipan Jonjic, University of Rijeka, CroatiaReviewed by:
James R. Lokensgard, University of Minnesota, United StatesE. Ashley Moseman, School of Medicine, Duke University, United States
Copyright © 2019 Hatton and Duncan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher J. A. Duncan, Y2hyaXN0b3BoZXIuZHVuY2FuQG5jbC5hYy51aw==