 Silvia Rodriguez-Fernandez1
Silvia Rodriguez-Fernandez1 Marta Murillo2
Marta Murillo2 Adrian Villalba1
Adrian Villalba1 David Perna-Barrull1
David Perna-Barrull1 Mary Cano-Sarabia3
Mary Cano-Sarabia3 Laia Gomez-Muñoz1Eva Aguilera4
Laia Gomez-Muñoz1Eva Aguilera4 Daniel Maspoch3,5Federico Vazquez4Joan Bel2
Daniel Maspoch3,5Federico Vazquez4Joan Bel2 Marta Vives-Pi1,6*
Marta Vives-Pi1,6*- 1Immunology Section, Germans Trias i Pujol Research Institute, Autonomous University of Barcelona, Badalona, Spain
- 2Pediatrics Section, Germans Trias i Pujol University Hospital, Badalona, Spain
- 3Catalan Institute of Nanoscience and Nanotechnology, CSIC and the Barcelona Institute of Science and Technology, Bellaterra, Spain
- 4Endocrinology Section, Germans Trias i Pujol University Hospital, Badalona, Spain
- 5Catalan Institution for Research and Advanced Studies (ICREA), Barcelona, Spain
- 6Biomedical Research Center in Diabetes Network and Associated Metabolic Diseases (CIBERDEM), Carlos III Health Institute (ISCiii), Madrid, Spain
Type 1 diabetes (T1D) is prompted by defective immunological tolerance, an event in which dendritic cells (DCs) are crucial as immune response orchestrators. In fact, they contribute to maintaining tolerance to self-antigens, but they can also prompt an immunogenic response against them, leading to autoimmunity. Countless factors can potentially impact on the proper functionality of the DCs, which range from altered subset distribution, impaired phagocytic function to abnormal gene expression. Moreover, in T1D, metabolic dysregulation could impair DC functions as well. Indeed, since T1D clinical course is likely to be more aggressive in children and adolescents and entails severe dysglycemia, the aim of this study was to analyze circulating DCs subpopulations in pediatric T1D at different stages, as well as to characterize their phagocytosis ability and tolerance induction potential. Thus, pediatric patients newly diagnosed with T1D, with established disease and control subjects were recruited. Firstly, DCs subsets from peripheral blood were found quantitatively altered during the first year of disease, but recovered in the second year of progression. Secondly, to study the tolerogenic functionality of DCs, liposomes with phosphatidylserine (PS) were designed to mimic apoptotic beta cells, which are able to induce tolerance, as previously demonstrated by our group in DCs from adult patients with T1D. In this study, monocyte-derived DCs from pediatric patients with T1D and control subjects were assessed in terms of PS-liposomes capture kinetics, and transcriptional and phenotypic changes. DCs from pediatric patients with T1D were found to phagocyte PS-liposomes more slowly and less efficiently than DCs from control subjects, inversely correlating with disease evolution. Nonetheless, the transcription of PS receptors and immunoregulatory genes, cytokine profile, and membrane expression of immunological markers in DCs was consistent with tolerogenic potential after PS-liposomes phagocytosis. In conclusion, T1D progression in childhood entails altered peripheral blood DCs subsets, as well as impaired DCs phagocytosis, although tolerance induction could still function optimally. Therefore, this study provides useful data for patient follow-up and stratification in immunotherapy clinical trials.
Introduction
Type 1 diabetes (T1D) is prompted by defective immunological tolerance to beta cells, resulting in their destruction by autoreactive lymphocytes. Although the triggering factors causing their activation remain elusive, antigen-presenting cells (APCs) are known to contribute to this activation, and so, to the development of the disease. Among APCs, dendritic cells (DCs), as orchestrators of the immune response, play a crucial role in the process. On the one side, they can induce and preserve self-tolerance but, on the other side, they can aid in its breach. Indeed, many factors could potentially impact on the proper functionality of the DCs, ranging from altered subset distribution, impaired phagocytic function, abnormal gene expression to defective functionality. As a case in point, many studies have linked the deficient removal of apoptotic beta cells performed by phagocytes to the initiation of autoimmunity (1). In fact, beta cells experience massive peaks of apoptosis, not only as a consequence of the autoimmune attack (2, 3), but also in physiological islet remodeling during the perinatal period (4, 5).
Plasma membrane integrity is maintained during apoptosis, thus preventing the release of immunogenic signals (6). This process, accompanied by fast apoptotic cell clearance by phagocytes—termed efferocytosis—, allows apoptosis to be immunomodulatory, which is essential in the maintenance of homeostasis. Apoptotic cell removal is mediated by highly conserved receptors and ligands, intracellular signaling, and engulfment mechanisms. One of the most well-characterized hallmarks of apoptosis is the exposure of phosphatidylserine (PS) on the membrane of apoptotic cells (7). PS interacts with several PS receptors —e.g., MerTK, CD36, and MFG-E8— on APCs, which promote efferocytosis, and this process confers biological advantages, such as peripheral self-tolerance induction (8, 9). Indeed, efferocytosis prompts immunomodulatory effects in phagocytes, which drive a tolerogenic antigen presentation and anti-inflammatory mediators production (10). However, deficiencies in apoptotic cell removal promote the turning of apoptotic cells into secondary necrotic cells, whereby they lose their membrane integrity and release danger signals, thus promoting inflammation and contributing to the initiation of autoimmunity (11, 12). In fact, accumulating evidence points to the relevant role of impaired efferocytosis in the development of multiple sclerosis, rheumatoid arthritis and T1D (13, 14).
Interestingly, the gold-standard experimental model of T1D, the non-obese diabetic (NOD) mouse, harbors a deficiency in the clearance of apoptotic cells by macrophages (15). In humans, polymorphisms in the FCGR2A gene, which encodes for the low-affinity IgG Fc region receptor II-a involved in phagocytosis, contribute to its pathogenesis (16). However, no data regarding the phagocytic function of DCs in human T1D are available. In patients with type 2 diabetes, there is growing evidence that impaired phagocytosis in neutrophils (17, 18) and macrophages (19) is related to glycemic control, albeit it can be alleviated with improved metabolic regulation. These data suggest that changes in glucose homeostasis can alter phagocytic processes, with the particularity that impairment of efferocytosis can contribute to the perpetuation of the autoreactivity in T1D.
By exploiting the inherent ability of apoptotic cell clearance to induce tolerance, a synthetic lipid-vesicle strategy that mimics apoptotic beta cells by being enriched in PS and encapsulating insulin was designed (20). This immunotherapy arrested autoimmunity upon administration to NOD mice after PS-liposomes were phagocyted by DCs, thus eliciting tolerogenic features. This finding was replicated in human DCs from adult patients with T1D (21). Therefore, this strategy achieves successful apoptotic mimicry and constitutes a promising strategy for tackling autoimmune diseases.
Unfortunately, T1D incidence is dramatically increasing in children (22, 23), in whom the disease is more aggressive and entails additional management complications. Since severe dysglycemia could impair DCs functionality, the aim of this study was to analyze circulating DCs subpopulations in pediatric T1D at different stages, as well as to characterize phagocytosis in T1D at the onset—hyperglycemic phase— and at established disease—when glucose levels are better controlled—, and to evaluate the role of phagocytosis in tolerance induction.
Materials and Methods
Patients and Control Individuals
Pediatric patients with T1D (n = 61) were visited as outpatients or hospitalized in the Pediatric Section at Germans Trias i Pujol Hospital. Patients were recruited at the onset and with a longer evolution (more than 6 months). Pediatric control individuals (n = 21) were also recruited. Inclusion criteria were 1–18 years of age and normal body mass index (BMI). Exclusion criteria were being under immunosuppressive or anti-inflammatory treatment, the presence of other autoimmune diseases, type 2 diabetes, pregnancy, and compromised kidney or liver function. All the experiments were carried out in strict accordance with the principles outlined in the Declaration of Helsinki for human research and after the approval of the Committee on the Ethics of Research of the Germans Trias i Pujol Hospital (PI-16-083). Signed informed consent was given by the parents of all subjects, and directly by children older than 12 years old.
Analysis of Peripheral Blood DC Subsets
In order to analyze DCs subsets, 2 mL blood samples were collected in EDTA tubes from control subjects and patients with T1D at diagnosis, and at first and second year of disease progression. Blood samples were lysed (Lysing Buffer, BD Biosciences, San Jose, CA, USA) and stained with antibodies to CD45-AF700, CD3-APCH7, CD19-APCH7, CD14-V450, CD16-APC, CD11c-PECy7, CD123-PerCPCy5.5, CD56-PE, HLA-DR-V500, and Slan-FITC (BD Biosciences). A minimum of 10,000 events per sample was acquired using FACS LSRFortessa (BD Biosciences). Percentage and absolute counts (cells/μL) were analyzed using PerfectCount Microspheres (Cytognos SL, Salamanca, Spain) and FACSDiva software (BD Biosciences) following gating strategies based on international consensus (24).
Human DC Generation
DCs were generated as described (21). Briefly, peripheral blood (10 mL) was obtained and monocytes were magnetically isolated using the StraightFrom Whole Blood CD14 MicroBeads kit (Miltenyi Biotech, Bergisch Gladbach, Germany). Monocytes were cultured at 106 cells/mL in X-VIVO 15 media (Lonza, Basel, Switzerland), supplemented with 2% male AB human serum (Biowest, Nuaillé, France), 100 IU/mL penicillin (Normon SA, Madrid, Spain), 100 μg/mL streptomycin (Laboratorio Reig Jofré, Sant Joan Despí, Spain), and 1,000 IU/mL IL-4 and 1,000 IU/mL GM-CSF (Prospec, Rehovot, Israel). After 6 days, DCs differentiation yield was assessed by CD11c-APC staining (Immunotools, Friesoythe, Germany) and viability was determined by annexin V-PE (Immunotools) and 7-AAD (BD Biosciences) using FACSCanto II (BD Biosciences).
Preparation of Liposomes
Liposomes consisted of 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (Lipoid, Steinhausen, Switzerland), 1,2-didodecanoyl-sn-glycero-3-phosphocholine (Lipoid), and cholesterol (Sigma-Aldrich). Liposomes were prepared using the thin-film hydration method from a lipid mixture of 1,2-dioleoyl-sn-glycero-3-phospho-L-serine, 1,2-didodecanoyl-sn-glycero-3-phosphocholine and cholesterol at 1:1:1.33 molar ratio, respectively, as described (20). Particle diameter and surface charge (ζ-potential) were measured by dynamic light scattering using Malvern Zetasizer (Malvern Instruments, Malvern, United Kingdom). Fluorescent liposomes (PSOG488-liposomes) were generated adding lipid-conjugated fluorescent dye Oregon Green 488 (OG488) 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (ThermoFisher Scientific, Waltham, MA, USA) and had a lipid concentration of 30 mmol/L, a diameter size of 836 ± 217 nm (mean±SD), and a ζ-potential of −38.90 ± 2.52 mV. PS-liposomes encapsulating human insulin chains A (PSA-liposomes) and B (PSB-liposomes), were prepared as described (21) with the following physicochemical features: PSA-liposomes were 30 mmol/L of lipid concentration, 783 ± 104 nm of diameter and −38.15 ± 1.20 mV of ζ-potential; PSB-liposomes had 30 mmol/L of lipid concentration, 1040 ± 71 nm of diameter and −38.05 ± 0.07 mV of ζ-potential.
Phagocytosis Assay
To determine whether DCs from patients with T1D had a defect in phagocytosis in the context of apoptotic cell removal, we tested DCs for their ability to capture synthetic PS-liposomes that mimic apoptotic beta cells. Then, DCs from control subjects, recent-onset patients and patients with established disease were cultured with 100 μmol/L PSOG488-liposomes at 37°C from 5 min to 6 h. As a negative control, the same assay was performed at 4°C to confirm that liposomes were captured by phagocytosis. DCs were extensively washed in cold PBS to remove liposomes attached to the cell membrane. Liposome capture was assessed by flow cytometry (FACSCanto II, BD Biosciences).
Gene Expression Analysis
To determine gene expression changes after phagocytosis, DCs were cultured in basal conditions (immature DCs, iDCs) and with PS-liposomes encapsulating human insulin chains A (PSA-liposomes) and B (PSB-liposomes), as described (21). A mixture of PSA-liposomes and PSB-liposomes (PSAB-liposomes, 1 mmol/L) were simultaneously cocultured with DCs from control subjects, recent-onset patients and patients with established disease at 37°C for 4 h to promote tolerogenic features (tolerogenic DCs, tolDCs). RNA was isolated (RNeasy Micro Kit, QIAGEN, Hilden, Germany) and reverse-transcribed (High Capacity cDNA Reverse Transcription Kit, ThermoFisher Scientific). cDNA synthesis reactions were performed using random hexamers (0.5 mg/mL, BioTools, Valle de Tobalina, Madrid, Spain) and reverse transcriptase Moloney-murine-Leukemia-virus (200 U/mL, Promega, Madison, WI, USA). Targeted cDNA was pre-amplified with TaqMan PreAmp Master Mix (ThermoFisher Scientific), and qPCR was performed with TaqMan universal assays (ThermoFisher Scientific) on a LightCycler® 480 (Roche, Mannheim, Germany) using the following ones: CD36 (Hs00354519_m1), CD68 (Hs00154355_m1), CD274 (PD-L1) (Hs00204257_m1), IDO1 (Hs00984148_m1), IL10 (Hs00961622_m1), LAIR1 (Hs00253790_m1), MERTK (Hs01031979_m1), MFGE8 (Hs00983890_m1), PDCD1LG2 (PD-L2) (Hs00228839_m1), PPARG (Hs01115513_m1), TGFB1 (Hs00998133_m1), TNFAIP3 (Hs00234713_m1), TNFSF14 (Hs00542476_g1), and VEGFA (Hs00900055_m1). Relative quantification was performed by normalizing the expression of each gene to that of the reference gene GAPDH (Hs02758991_g1), as described in the 2−ΔCt method (25).
DCs Phenotype After Phagocytosis
DCs from patients at onset and with established disease were cultured for 24 h with 20 μg/mL human insulin (Sigma-Aldrich, St. Louis, MO, USA) and 1 mmol/L of PSAB-liposomes to determine the effect of insulin chains as autoantigens (tolDCs). As controls, DCs were either cultured with 20 μg/mL human insulin (Sigma-Aldrich) to obtain iDCs or adding a cytokine cocktail —TNF-α (1,000 IU/mL, Immunotools), IL-1β (2,000 IU/mL, Immunotools), and Prostaglandin E2 (PGE2, 1 μmol/L, Cayman Chemical, Ann Arbor, MI, USA)— for 24 h to obtain mature DCs (mDCs). Viability and phenotype were analyzed by flow cytometry (FACSCanto II, BD Biosciences). DCs were stained with 7-AAD (BD Biosciences) and antibodies to CD11c-APC, CD86-FITC, HLA-ABC-FITC, HLA-DR-FITC, CD14-PE and CD40-APC (Immunotools), CD36-APCCy7, TIM4-APC, αvβ5 integrin-PE, CD54-PECy7, TLR2-FITC, CXCR4-APCCy7, CCR2-APC (BioLegend, San Diego, CA, USA). Data were analyzed using FlowJo software (Tree Star Inc, Ashland, OR, USA).
Cytokine Production
The Human Th1/Th2/Th17 kit (CBA system; BD Biosciences) was used to measure cytokine production. Culture supernatants from DCs (n≥3 for control subjects and n≥3 for patients with T1D) were collected and frozen at −80°C until use. IL-2, IL-4, IL-6, IFN-γ, TNF-α, IL-17A, and IL-10 were assessed. Data were analyzed using CBA software. VEGFA secretion by DCs was determined in culture supernatant by ELISA (ThermoFisher Scientific).
Statistical Analysis
Prism 7.0 software (GraphPad Software Inc, San Diego, CA, USA) was used for the statistical analysis. For comparisons of unpaired data, a non-parametric Mann-Whitney test was used; for paired comparisons, the non-parametric Wilcoxon test was used. ANOVA was used for comparisons with several factors. For correlation between parameters, Spearman's test was used. A p value < 0.05 was considered significant.
Results
DCs Subsets From Peripheral Blood Are Altered at the Onset and Early Stages of T1D Progression
As a first approach, different subsets of circulating DCs were assessed in control subjects and patients with T1D at diagnosis, and at first year and second year of progression. Their clinical data are summarized in Table 1. All subjects met the inclusion and exclusion criteria and gave informed consent. Aside from differences in the evolution of the disease, patients at onset showed higher glycated hemoglobin (HbA1c) levels than patients at first year and at second year of progression. Patients at second year of progression required higher insulin doses than patients at onset and at first year of evolution.
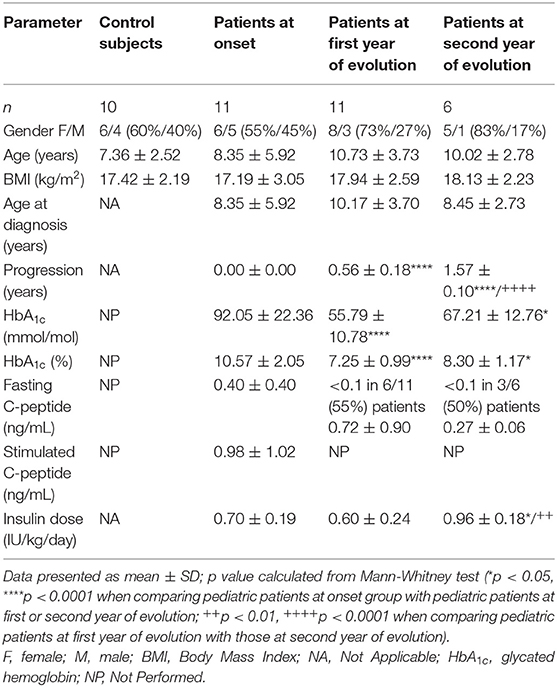
Table 1. Data from the patients with T1D and control subjects included in the peripheral blood DCs subsets analysis.
Thus, four different subsets of DCs in peripheral blood [total DCs, myeloid DCs (myDCs), plasmacytoid DCs (pDCs) and a CD11c−CD123− putative DC subset] were analyzed (Figure 1) following the gating strategy displayed in Figure S1. As for total DCs, percentages and numbers were decreased in patients at first year when compared to control subjects, although patients at second year recovered a normal percentage. The percentage of myDCs was increased in patients at first year when compared to control subjects, patients at onset and at second year of progression, although no differences were observed in their numbers. As for pDCs, patients at first year displayed a higher percentage than patients at onset and patients at second year. The numbers of pDCs were higher in control subjects when compared to patients at onset and at first year. Finally, patients at first year had a decreased percentage of the CD11c−CD123− subset when compared to control subjects, patients at onset and at second year, and their numbers were also lower when compared to control subjects and patients at second year. Therefore, DCs subsets from peripheral blood were found quantitatively altered during the first year of childhood T1D and recovered in the second year of progression.
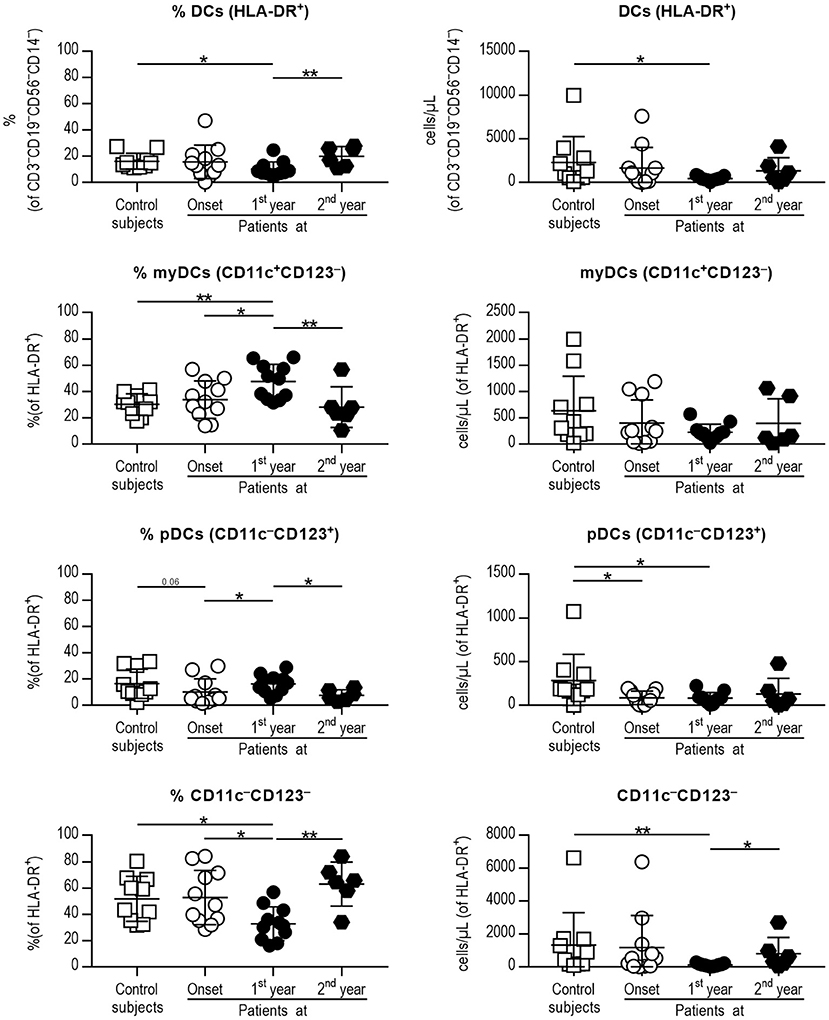
Figure 1. Peripheral blood DC subsets are quantitatively altered at the early stages of T1D. Percentages (left panel) and concentration (cells/μL, right panel) of total DCs (1st row, HLA-DR+ cells of CD3−CD19−CD56−CD14− cells), myeloid DCs (myDCs, 2nd row, CD11c+CD123− cells of HLA-DR+ cells), plasmacytoid DCs (pDCs, 3rd row, CD11c−CD123+ of HLA-DR+ cells) and the CD11c−CD123− subset (4th row, CD11c−CD123− of HLA-DR+ cells). Data are expressed as mean ± SD; white squares are control subjects (n = 10), white circles are patients at onset (n = 11), black dots are patients at first year of progression (n = 11), and black rhombuses are patients at second year of progression (n = 6). Statistically significant differences were found when comparing the groups (*p < 0.05, **p < 0.01, Mann-Whitney test).
DC Differentiation Efficiency Is Similar in Pediatric Patients and Control Subjects
After observing alterations in DCs subsets in pediatric patients with T1D, and in order to assess their functionality, DCs were derived from peripheral blood monocytes from patients at diagnosis and with established disease as well as from control subjects. Their clinical data are summarized in Table 2. The BMI of patients with established disease was significantly higher than that of control subjects and patients at onset. As expected, patients with established disease had a longer evolution and significantly lower HbA1c values than patients at onset. Then, monocytes were magnetically isolated directly from whole blood, and differentiated into DCs in vitro. The number of isolated CD14+ cells (monocytes), viability, and dynamics of the CD11c marker acquisition (differentiation to DCs) at days 1, 4, and 6 of culture showed similar values in the three groups (Table 3).
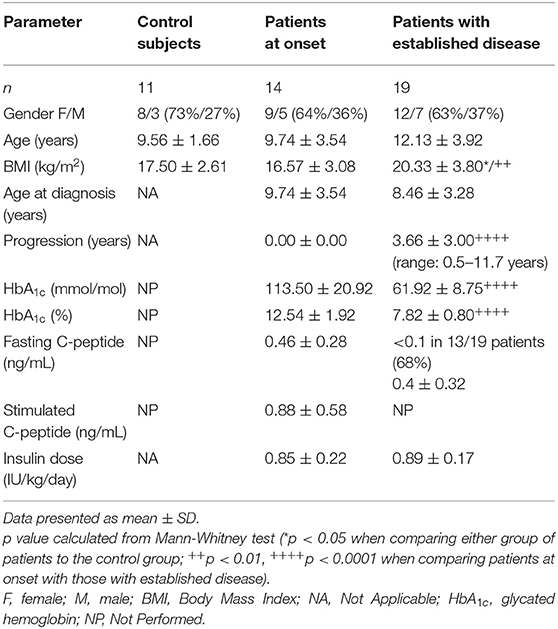
Table 2. Main clinical characteristics and metabolic data of participants included in the study of phagocytosis and tolerance induction.
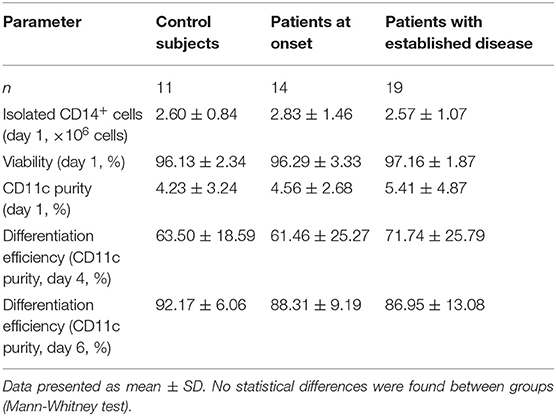
Table 3. Data from monocyte isolation and DC differentiation.
DCs From Patients With T1D Display Impaired Phagocytic Activity in Correlation With Disease Progression
In order to test the ability of DCs to phagocyte lipid vesicles in the context of apoptotic cell removal, fluorescent PS-liposomes were used in a time-course experiment, coculturing PSOG488-liposomes with DCs from each group of subjects. As expected, phagocytosis was not observed at 4°C (Figure 2A). At 37°C (Figures 2A,B), the phagocytosis rate of PSOG488-liposomes by DCs from patients with established disease was decreased when compared to control subjects, starting at 15 min of coculture and throughout the assay. Moreover, DCs from patients with established disease had phagocyted lower levels of PSOG488-liposomes than patients at onset at 2 h of coculture. No significant differences were found when comparing the phagocytosis kinetics of DCs from patients at onset and control subjects. To further dissect this finding, the area under the curve (AUC) of the kinetics of capture was calculated (Figure 2C). The AUC values of patients with established disease were significantly lower than those of patients at onset and control subjects. Also, patients at onset had significantly lower AUC values than control subjects. Therefore, the correlation between AUC values and the time of disease progression was analyzed (Figure 2D), discovering that both parameters were significantly and inversely correlated (Spearman's r = −0.664, p = 0.022). This defect was only observed in children with T1D, and not in adults, as previously reported (21). Thus, and as an additional control, we have included in this study a phagocytosis assay with DCs from adult patients with established disease, which demonstrates that the impairment of phagocytosis is an inherent feature of pediatric T1D (Figure S2 and Table S1).
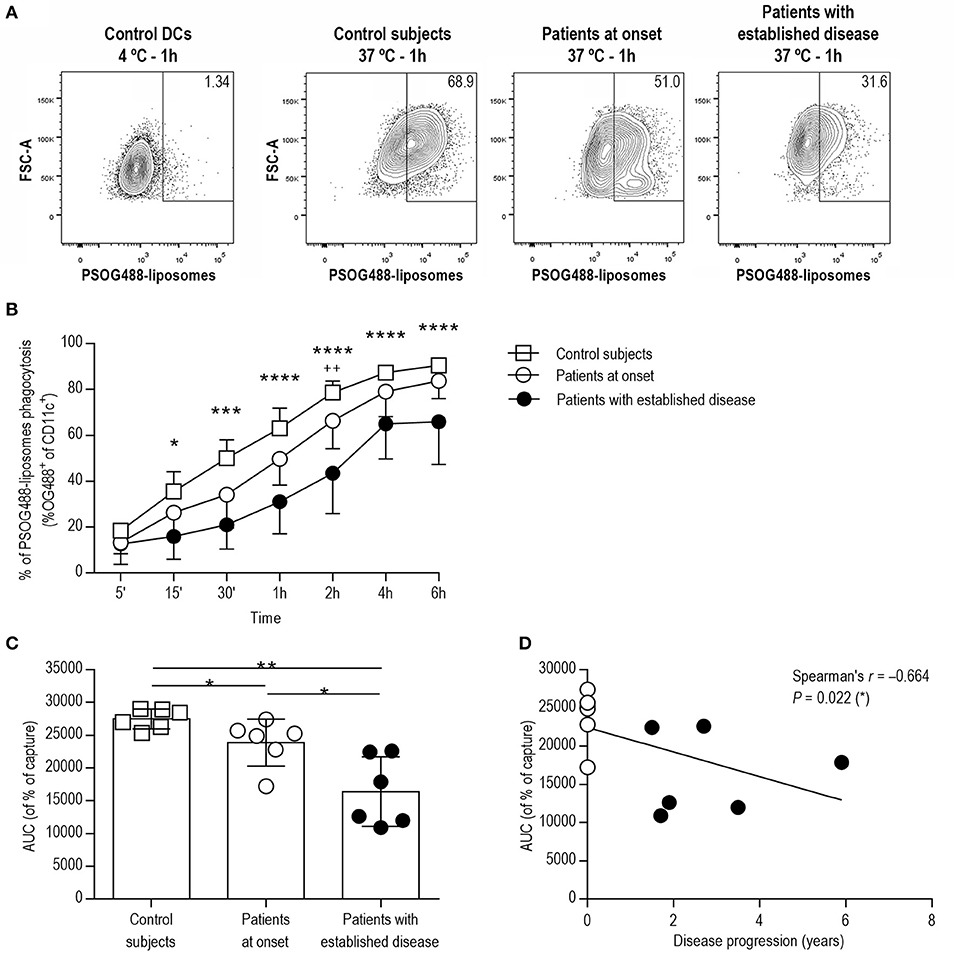
Figure 2. DCs from patients with T1D show impaired phagocytic activity in correlation with the time of disease progression. (A) Flow cytometry contour plots and percentages of the capture of PSOG488-liposomes by DCs (CD11c+). From left to right, control DCs cultured with PSOG488-liposomes at 4°C for 1 h, and DCs from control subjects, patients at onset and patients with established disease cultured with PSOG488-liposomes at 37°C for 1 h. One representative experiment of 6 is shown. (B) Time-course of the capture of PSOG488-liposomes by DCs obtained from control subjects (white squares, n = 6), pediatric patients at onset (white circles, n = 6) and with established disease (black dots, n = 6) at 37°C. Results are mean±SEM. Comparisons between groups showed significant differences (*p < 0.05, ***p < 0.001, ****p < 0.0001 when comparing control subjects with patients with established disease; ++p < 0.01 when comparing patients at onset with patients with established disease, Two-way ANOVA). (C) Area under the curve (AUC) of phagocytosis kinetics curves. Differences were found between groups (*p < 0.05, **p < 0.01, Mann-Whitney test). (D) Correlation between AUC values of phagocytosis kinetics curves and disease progression. A significant correlation was found between both parameters (Spearman's r = −0.664, p = 0.022, Spearman's correlation analysis).
Efferocytosis and Immunoregulation-Related Gene Expression Is Altered in DCs From Patients With T1D
Since the phagocytic capacity of DCs from patients with T1D was altered, the gene expression of specific PS receptors (CD36, CD68, MERTK, MFGE8), and immunoregulatory molecules (PPARG, TNFAIP3, TNFSF14, LAIR1, PDL1, PDL2, TFGB1, and VEGFA) was evaluated in DCs before (iDCs) and 4 h after (tolDCs) coculture with PSAB-liposomes in the three groups: control subjects, patients at onset and patients with established disease (Figure 3). All these genes were expressed in iDCs. Firstly, no significant differences in the expression of CD36, CD68 nor MERTK were found between groups, but a significant reduction in MFGE8 expression was detected in DCs from patients with established disease in comparison to the other groups. Moreover, after liposome capture, the expression levels of genes of PS receptors were preserved or even showed a tendency to increase in comparison to iDCs. In this sense, a tendency for upregulation of MERTK —a feature of tolDCs (26)— was observed in tolDCs from control subjects (p = 0.06).
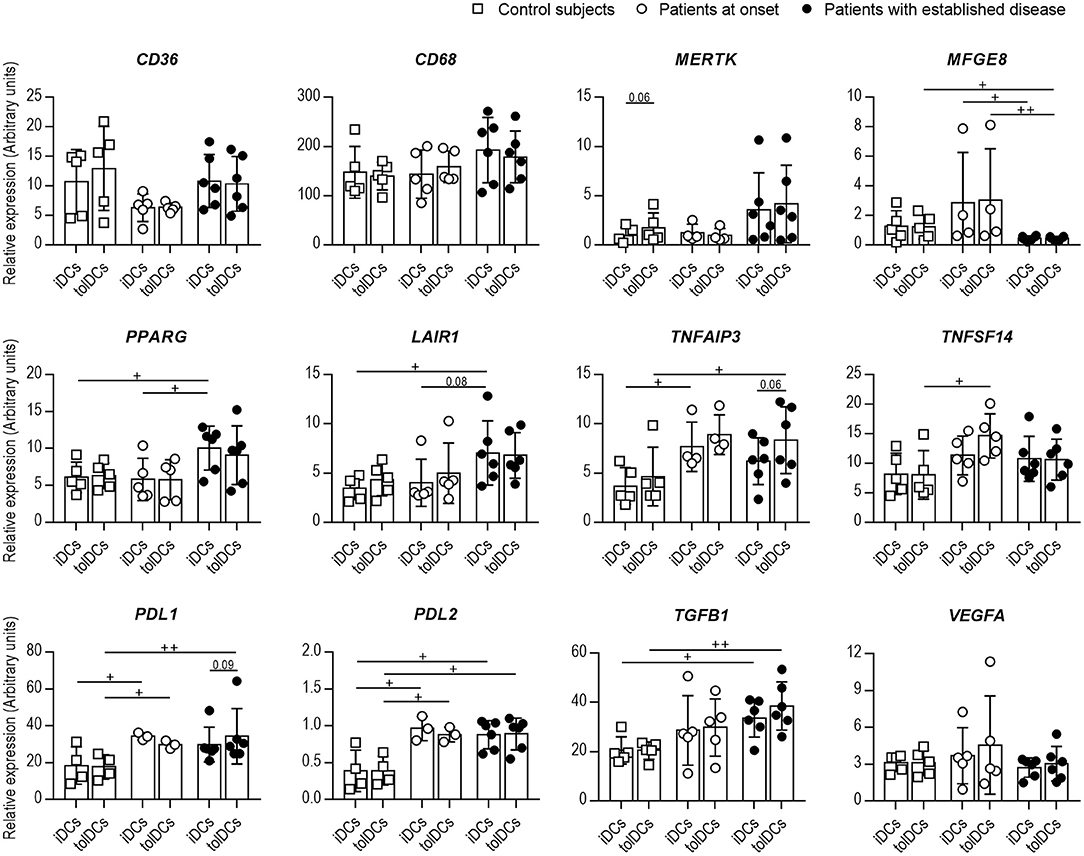
Figure 3. The expression of genes related to efferocytosis and immunoregulation is altered in DCs from patients with T1D. Expression of CD36, CD68, MERTK, MFGE8, PPARG, LAIR1, TNFAIP3, TNFSF14, PDL1, PDL2, TGFB1, and VEGFA genes in immature DCs (iDCs) before and 4 h after (tolDCs) coculture with PSAB-liposomes, analyzed by qPCR. White squares are control subjects (n = 5), white circles are patients at onset (n = 3-5), and black dots are patients with established disease (n = 6). Bars show the mean ± SD. Gene expression signals were normalized to GAPDH expression. No significant differences were found when comparing culture conditions in the same group (Wilcoxon test). Significant differences were found when comparing the same culture conditions in the three groups (+p < 0.05, ++p < 0.01, Mann-Whitney test).
Regarding genes of immunoregulatory molecules, PPARG and LAIR1 expression discerned both stages of T1D, being higher in established disease than at the onset and controls. Patients with T1D also showed higher levels of TNFAIP3, TNFSF14, PDL1, PDL2, and TGFB1 in DCs than controls. Interestingly, tolDCs from patients exhibited a trend for upregulation of the TNFAIP3, TNFSF14, and TGFB1 genes upon PSAB-liposomes capture at different stages of the disease. Finally, VEGFA gene expression did not differ between groups nor after the capture of PSAB-liposomes. Concerning IDO1 and IL10, a low expression was detected in all conditions (Figure S3).
DCs From Pediatric Patients Acquire a Tolerogenic Potential After Phagocytosis of PS-liposomes
In terms of cytokine secretion by DCs, the release of IL-6, TNF-α, IL-10, and VEGFA by DCs from control subjects, recent-onset patients and patients with established disease was consistent with a tolerogenic profile after PSAB-liposomes phagocytosis (Figure 4). Briefly, IL-6 concentration was similar before (iDCs) and after (tolDCs) PSAB-liposomes uptake in all groups. Moreover, IL-6 secretion increased after maturation stimulus in DCs (mDCs) from patients with established T1D, and the same trend was observed in the other groups. Similarly, TNF-α was not increased after liposome uptake, and its secretion tended to increase in pro-inflammatory conditions. IL-10 concentration was detected around the limit of detection in all samples. Remarkably, VEGFA concentration was slightly increased in the supernatant of DCs after liposome uptake when compared to that of iDCs in control subjects and patients with established disease —although patients at onset showed the same tendency (p = 0.06). IL-2, IL-17A, and IFN-γ were not detected in any condition of the assay (data below the detection limit). These results, although not always statistically significant, support the ability of DCs from children with T1D to achieve a tolerogenic phenotype.
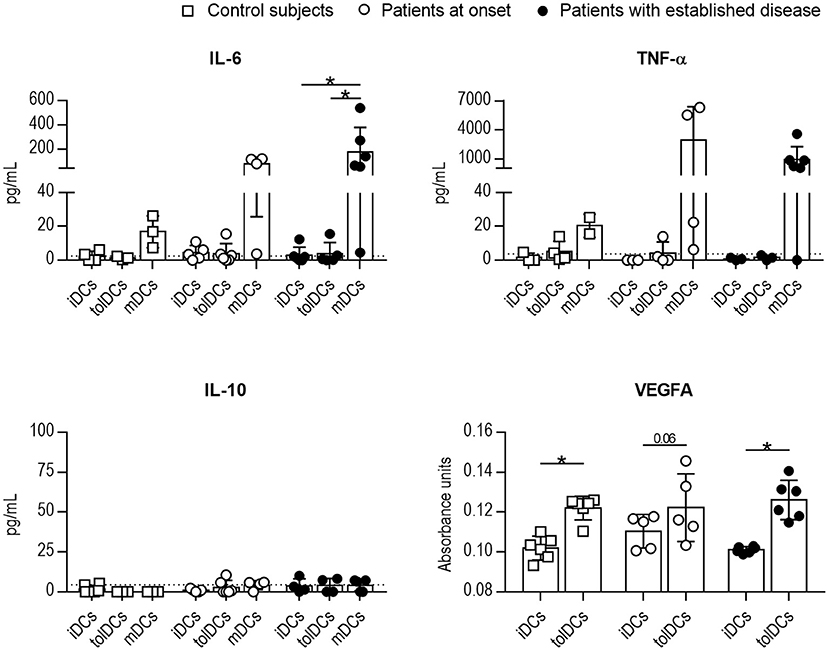
Figure 4. The cytokine profile after the uptake of PSAB-liposomes by DCs points to tolerogenic potential. Concentration of IL-6, TNF-α, IL-10, and VEGFA secreted by DCs obtained from control subjects (white squares, n ≥ 3), patients with T1D at onset (white circles, n ≥ 3) and patients with established disease (black dots, n ≥ 3). Bars represent immature DCs (iDCs), immature DCs 4 h after the capture of PSA-liposomes and PSB-liposomes (tolDCs), or mature DCs (mDCs) induced with a cytokine cocktail. Data presented as mean ± SD. Significant differences were found when comparing the different conditions in the same group of subjects (*p < 0.05, Wilcoxon test). Differences were not found when comparing the same culture condition in the three groups (Mann-Whitney test).
Afterward, we interrogated whether the phenotype of DCs from patients changed toward a tolerogenic profile after 24 h of coculture with PSAB-liposomes (tolDCs), despite the phagocytosis defect. First, the viability of DCs from all groups was higher than 90% in all conditions, thus confirming that PS-liposomes are not toxic to DCs obtained from pediatric subjects (data not shown). No differences were observed in the DCs phenotype between patients at onset or with established disease, under any of the conditions (Figure 5). Membrane expression of PS receptors such as CD36, TIM4 and Integrin αvβ5 was not altered after liposome capture in comparison to iDCs. Then, the expression of molecules related to the immune function of DCs was assessed. Antigen-presenting molecules HLA-ABC displayed a trend for lower expression in tolDCs when compared to mDCs in patients with established disease (p = 0.08); HLA-DR expression did not exhibit any changes. As for adhesion molecules such as CD54, in patients with established disease, tolDCs showed a tendency for higher expression than iDCs (p = 0.06). Regarding costimulatory molecules CD40 and CD86, lower expression was observed in tolDCs when compared to mDCs in patients with established disease, and patients at onset displayed the same pattern. Chemokine receptors CXCR4 and CCR2 were not upregulated after phagocytosis; however, mDCs from patients with established disease tended to express higher levels of these molecules when compared to iDCs and tolDCs (p = 0.06). Finally, no defects in the expression of pattern recognition receptors, TLR2 and CD14, were observed in DCs under any conditions.
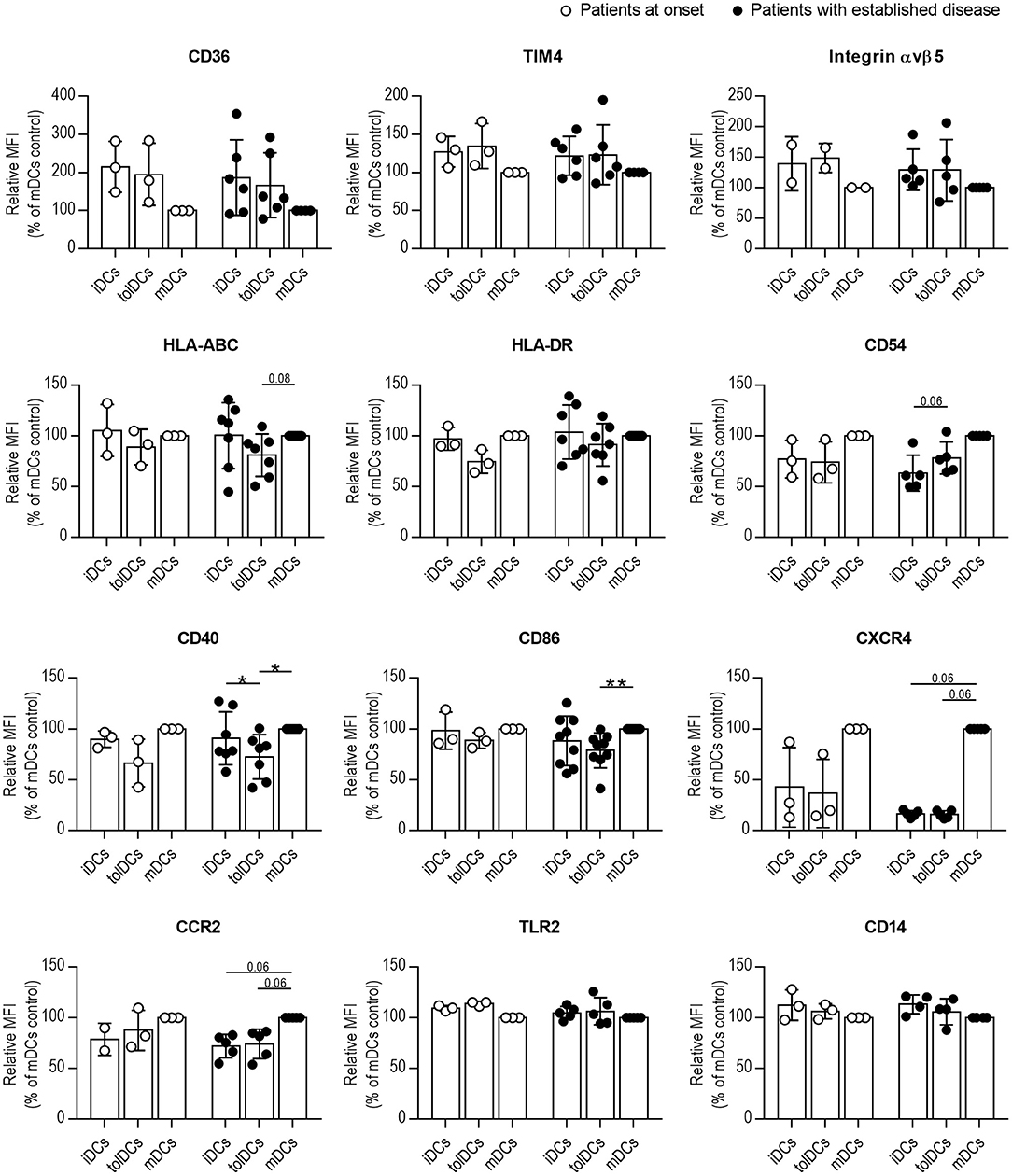
Figure 5. Phagocytosis of PS-liposomes induces a tolerogenic phenotype in DCs from pediatric patients both at onset and established T1D. Membrane expression of CD36, TIM4, Integrin αvβ5, HLA-ABC, HLA-DR, CD54, CD40, CD86, CXCR4, CCR2, TLR2, and CD14 molecules in DCs obtained from patients at onset (white circles, n = 2–3) and with established disease (black dots, n = 4–9). Bars represent immature DCs (iDCs), iDCs after the capture of PSAB-liposomes (tolDCs), and mature DCs (mDCs) induced with a cytokine cocktail, after 24 h. Data are mean ± SD of relative Median of Fluorescence Intensity (MFI), this being MFI of each condition referred to their respective mDCs control. Significant differences were found when comparing culture conditions in the same group (*p < 0.05, **p < 0.01, Wilcoxon test), but not when comparing the same culture conditions in both groups (Mann-Whitney test).
Discussion
It is well-known that DCs, as orchestrators of the immune response with a crucial role in maintaining self-tolerance, are highly involved in the pathogenesis of T1D. Several studies have enumerated the broad spectrum of alterations that DCs from these patients have, ranging from genetic alterations, to phenotypic and functional divergences (16). Fewer studies, however, have focused on the potential of DCs from adult patients to become tolerogenic (27, 28), but no data are available in pediatric patients. To our knowledge, this is the first work that assesses DCs subsets in children with T1D at different stages of the disease and evaluates their tolerogenic potential. Indeed, we believe these are paramount aspects to consider in the near future, when antigen-specific immunotherapies will be available and immunomonitoring of the patients will be required.
Thus, peripheral blood DC subsets were measured to determine quantitative alterations related to disease progression. Interestingly, most of the differences gathered in the first year of evolution were restored in the second year. Briefly, the percentage of myDCs and pDCs was increased at diagnosis when compared to the other time points, whereas the CD11c−CD123− subset was decreased. In this sense, alterations in myDCs and pDCs subsets have been reported in children with T1D at initial stages of disease (29–31), and further efforts are warranted to validate the usefulness of DC subsets as biomarkers in the progression of the disease and to provide further insight into their biological significance.
The alteration in DCs subsets at the early stages of T1D was an early observation that prompted us to study the tolerogenic functionality of DCs. To do so, we employed a nanotherapy imitating apoptotic beta cells consisting of PS-liposomes encapsulating insulin (20). These liposomes were phagocyted by murine DCs, thus arresting autoimmunity in the experimental model of the disease, and also induced tolerogenic features in human DCs from adult patients with T1D (21).
In the present study, three similar groups of pediatric subjects in terms of gender and age were included. Patients with established disease had higher BMI, which was expected according to the recovery of weight after disease onset and to the distribution of BMI related to age (32), but nonetheless healthy, in order to avoid the hypothetical interference of inflammatory processes associated with overweight and obesity. Quite unexpectedly, here we found that the phagocytic activity of monocyte-derived DCs from pediatric patients with T1D was impaired, and this defect deteriorated with the progression of the disease but did not affect the induction of a tolerogenic phenotype after phagocytosis. This immune dysfunction, not previously reported in pediatric patients with T1D, agrees with studies that have reported an impaired phagocytic activity in macrophages from the NOD mouse, both at early ages—before diabetes development—and at adulthood when compared to other strains (15). An attenuated phagocytic activity has also been found in patients with type 2 diabetes, but it was reversed after the improvement of glycemic control (19). Moreover, a reduced capacity for efferocytosis has been reported in other autoimmune diseases unrelated to glucose metabolism (13). In addition, specific PS receptors are crucial in efferocytosis. As expected, mice deficient in PS receptors show impaired efferocytosis and increased inflammation (11). Overall, these data suggest that several factors, e.g., intrinsic defects, dysglycemia and inflammation, may contribute to the impairment of phagocytosis in T1D. It is well-known that efferocytosis is not only a mechanism of apoptotic cell removal but also a crucial process in the maintenance of immunological homeostasis, and so this defect could contribute to the persistent lack of self-tolerance.
Although DCs from pediatric patients with T1D were found to have impaired phagocytosis in correlation with disease progression, DCs from patients at onset and with established disease showed similar expression of PS receptors (CD36, CD68, and MERTK), except for MFGE8. Interestingly, DCs from patients with established disease had a decreased MFGE8 expression, and this defect could contribute to the herein reported phagocytosis impairment. Since several PS receptors work to internalize apoptotic cells (33), the suboptimal expression or function of any of them could explain the altered phagocytosis. In fact, scarce information has been reported about DCs phagocytic activity, since most studies have been conducted with macrophages. Thus, it is plausible that DCs may express the same PS receptors as macrophages but in different proportions, or express entirely different PS receptors (34), which could be accountable for the impaired phagocytosis of DCs.
As for the immunoregulatory profile, DCs from pediatric patients with T1D showed higher expression of PPARG and TGFB1—two genes downstream in the efferocytosis signaling pathway—than control subjects. Moreover, TNFAIP3 displayed higher expression in DCs from patients; interestingly, the expression of Tnfaip3 in murine DCs was reportedly upregulated after efferocytosis (35), thus reducing their rate of apoptotic cell clearance and prompting their acquisition of tolerogenic potential (36). Also, the overexpression of LAIR1—detected in DCs from patients with established disease—has been linked to an inhibition of cholesterol-particle phagocytosis by murine macrophages (37). Thus, the expression of these two genes combined could also provide a plausible explanation for the observed impaired phagocytosis of PS-liposomes. In addition, their higher expression in comparison to control subjects, along with that of PPARG, PDL1, PDL2, and TGFB1 genes, could reflect a continuous attempt of immunoregulation prompted by the ongoing autoimmune reaction (38). Importantly, the expression of tolerogenic markers was also found upregulated in adult DCs after the capture of PSAB-liposomes in our previous RNA-seq analysis (21). However, other factors, such as IDO and IL-10, have been shown to inhibit T cell activation by DCs after efferocytosis, but we did not observe any increase in IDO1 and IL10 expression, in line with similar studies (39).
Moreover, the results of the characterization of the cytokine profile in DCs from pediatric patients were similar to those reported in adults (21), and support the ability of DCs from children with T1D to achieve a tolerogenic phenotype. As in adults, IL-10 was detected at very low levels in all conditions, correlating with the above-mentioned gene expression results, although this could be explained by the timing of the experiment. Nevertheless, it is worth mentioning the profile of VEGFA secretion—a cytokine involved in the inhibition of DCs maturation after apoptotic cell removal (40, 41)—, in which tolDCs from the three groups released higher levels of VEGFA in comparison to iDCs, 4 h after phagocytosis. This further supports that DCs acquire tolerogenic properties after PS-liposomes phagocytosis, even if their phagocytosis ability is impaired. Overall, these results suggest that DCs from patients could mediate immunoregulatory attempts.
Finally, membrane expression of PS receptors and immune-related molecules in DCs from pediatric patients with T1D was also studied to determine whether the activation of tolerogenic signaling pathways was translated to the DC membrane. These PS receptors are expressed in iDCs to allow apoptotic cell phagocytosis. As described previously (21), the expression of PS receptors was maintained after PSAB-liposomes phagocytosis or even showed a tendency to increase in comparison to iDCs, thus preserving the ability to efferocyte. As for the expression of immune-related molecules, tolDCs from pediatric patients, both at onset and with established disease, showed a pattern consistent with tolerogenic potential. Indeed, this phenotype is similar to that displayed by DCs after PS-liposomes uptake in murine models of T1D and multiple sclerosis (20, 42), and also in human DCs from adult patients with T1D (21). However, the main limitation in the studies of tolDCs is the lack of a robust single marker of tolerogenic potential, and so several parameters that altogether point to the tolerogenic potential of DCs are usually determined. To date, tolDCs are accepted as reluctant to maturation and stable in a state of low expression of antigen-presenting and costimulatory molecules, along with a tolerogenic-skewed cytokine profile (43). Thus, the here presented data are consistent with a biological tolerogenic effect in pediatric DCs after liposome uptake.
We are well-aware of the limitations of the study. On the one hand, efferocytosis of apoptotic beta cells was mimicked using previously-described synthetic microvesicles displaying features of apoptotic cells (20). However, the engulfment of these liposomes by DCs induces similar effects to those observed by physiological apoptotic cell phagocytosis and constitutes a good system to test the fitness of DCs. On the other hand, only six PS receptors were explored, and despite their crucial role in efferocytosis, other receptors could be altered in T1D thus impairing DCs phagocytosis. Whether this alteration is instrumental in the progression of the autoimmune attack toward the clinical manifestation of T1D is currently under investigation. In fact, when the clinical diagnosis of T1D takes place, there is already a significant loss of beta cells in the pancreas of the patient. However, in this scenario, autoimmunity to beta cells remains active despite the low numbers of alive or functional beta cells in the pancreatic islets, and it can destroy these remaining beta cells. Thus, the altered phagocytic activity observed in DCs could be translated into impaired phagocytosis of actual apoptotic beta cells, which could facilitate their conversion into secondary-necrotic cells if they are not efficiently removed by phagocytes. This happening would increase the availability of danger signals and feed the autoimmune process against beta cells. Perhaps, children susceptible to the disease may carry a defect in the efferocytosis ability of phagocytes, which could further exacerbate immunogenic responses. Interestingly, the here identified defect in the phagocytic activity of DCs from pediatric patients with T1D was not detected in adult patients (21), so we can speculate that childhood features (growth, puberty, environmental factors) and diabetes (metabolic control, immune response heterogeneity) could influence DCs behavior (44, 45). Nevertheless, the preservation of the tolerogenic potential in DCs from pediatric patients with T1D could help in restraining the autoimmune destruction of beta cells, and in preserving the residual beta cell mass in patients with T1D.
In conclusion, pediatric peripheral DCs subsets are altered in the first stages of T1D and their phagocytic capacity deteriorates with disease progression, although tolerance induction could still function optimally. Despite further studies to draw the intricate picture of this phagocytosis impairment are needed, both the study of peripheral blood DCs and functional DCs studies provide useful data in patient's follow up, and could aid in the stratification of patients in immunotherapy clinical trials, since these results could inform about the immunological status of the patients' DCs and their ability to promote a tolerogenic response.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
All the experiments were carried out in strict accordance with the principles outlined in the Declaration of Helsinki for human research and after the approval of the Committee on the Ethics of Research of the Germans Trias i Pujol Hospital. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
SR-F, FV, and MV-P designed the experiments. MM, EA, FV, and JB selected the patients and control subjects and obtained clinical data. MC-S and DM generated the liposomes. SR-F, AV, DP-B, and LG-M performed the experiments. SR-F and MV-P wrote the manuscript. MM, AV, DP-B, LG-M, and FV reviewed the manuscript and contributed to the discussion. All authors revised the final manuscript and gave final approval of the version to be published.
Funding
This work has been funded by the Spanish Government (FIS PI15/00198 and PI18/00436) co-financed with the European Regional Development funds (FEDER), and by DiabetesCero Foundation. CIBER of Diabetes and Associated Metabolic Diseases (CIBERDEM) is an initiative from Instituto de Salud Carlos III (Spain). SR-F was supported by the Agency for Management of University and Research Grants (AGAUR) of the Generalitat de Catalunya. ICN2 was supported by the Severo Ochoa program from Spanish MINECO (Grant No. SEV-2017-0706).
Conflict of Interest
MC-S, DM, and MV-P are co-founders of Ahead Therapeutics SL, which works towards the clinical translation of the PS-liposomes tolerogenic technology for the treatment of autoimmune diseases.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Ms. N. Real, nurse of the Pediatric Section, and to Dr. E. Martinez-Caceres, Dr. A. Teniente and Ms. Bibiana Quirant, from the Immunology Section of the Hospital Universitari Germans Trias i Pujol, for their help with blood samples. We are grateful to Mr. M. Fernandez (IGTP) for his support in flow cytometry and to Ms. D. Culell-Young for English grammar assistance. We are indebted to the patients and their families.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02811/full#supplementary-material
Abbreviations
DCs, Dendritic Cells; iDCs, Immature Dendritic Cells; mDCs, Mature Dendritic Cells; myDCs, Myeloid Dendritic Cells; PBMCs, Peripheral Blood Mononuclear Cells; PS, Phosphatidylserine; pDCs, Plasmacytoid Dendritic Cells; TolDCs, Tolerogenic Dendritic Cells.
References
1. Vives-Pi M, Rodriguez-Fernandez S, Pujol-Autonell I. How apoptotic β-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. (2015) 20:263–72. doi: 10.1007/s10495-015-1090-8
2. Eizirik DL, Grieco FA. On the immense variety and complexity of circumstances conditioning pancreatic β-cell apoptosis in type 1 diabetes. Diabetes. (2012) 61:1661–3. doi: 10.2337/db12-0397
3. Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab. (2017) 6:943–57. doi: 10.1016/j.molmet.2017.06.019
4. Meier JJ, Kohler CU, Alkhatib B, Sergi C, Junker T, Klein HH, et al. Beta-cell development and turnover during prenatal life in humans. Eur J Endocrinol. (2010) 162:559–68. doi: 10.1530/EJE-09-1053
5. Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. (2000) 49:1325–33. doi: 10.2337/diabetes.49.8.1325
6. Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. (2010) 189:1059–70. doi: 10.1083/jcb.201004096
7. Fadok VA, Voelker DR, Campbell PA, Bratton DL, Cohen JJ, Noble PW, et al. The ability to recognize phosphatidylserine on apoptotic cells is an inducible function in murine bone marrow-derived macrophages. CHEST. (1993) 103:102S. doi: 10.1378/chest.103.2_Supplement.102S
8. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. (2018) 9:127. doi: 10.3389/fimmu.2018.00127
9. Henson PM. Cell removal: efferocytosis. Annu Rev Cell Dev Biol. (2017) 33:127–44. doi: 10.1146/annurev-cellbio-111315-125315
10. Szondy Z, Sarang Z, Kiss B, Garabuczi E, Koroskenyi K. Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front Immunol. (2017) 8:909. doi: 10.3389/fimmu.2017.00909
11. Kimani SG, Geng K, Kasikara C, Kumar S, Sriram G, Wu Y, et al. Contribution of defective PS recognition and efferocytosis to chronic inflammation and autoimmunity. Front Immunol. (2014) 5:566. doi: 10.3389/fimmu.2014.00566
12. Colonna L, Lood C, Elkon KB. Beyond apoptosis in lupus. Curr Opin Rheumatol. (2014) 26:459–66. doi: 10.1097/BOR.0000000000000083
13. Abdolmaleki F, Farahani N, Gheibi Hayat SM, Pirro M, Bianconi V, Barreto GE, et al. The role of efferocytosis in autoimmune diseases. Front Immunol. (2018) 9:1645. doi: 10.3389/fimmu.2018.01645
14. Martinez J. Prix Fixe: efferocytosis as a four-course meal. Curr Top Microbiol Immunol. (2017) 403:1–36. doi: 10.1007/82_2015_467
15. O'Brien BA, Geng X, Orteu CH, Huang Y, Ghoreishi M, Zhang Y, et al. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun. (2006) 26:104–15. doi: 10.1016/j.jaut.2005.11.006
16. Creusot RJ, Postigo-Fernandez J, Teteloshvili N. Altered function of antigen-presenting cells in type 1 diabetes: a challenge for antigen-specific immunotherapy? Diabetes. (2018) 67:1481–94. doi: 10.2337/db17-1564
17. Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus. (DM). FEMS Immunol Med Microbiol. (1999) 26:259–65. doi: 10.1111/j.1574-695X.1999.tb01397.x
18. Davidson NJ, Sowden JM, Fletcher J. Defective phagocytosis in insulin controlled diabetics: evidence for a reaction between glucose and opsonising proteins. J Clin Pathol. (1984) 37:783–6. doi: 10.1136/jcp.37.7.783
19. Lecube A, Pachon G, Petriz J, Hernandez C, Simo R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS ONE. (2011) 6:e23366. doi: 10.1371/journal.pone.0023366
20. Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, et al. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS ONE. (2015) 10:e0127057. doi: 10.1371/journal.pone.0127057
21. Rodriguez-Fernandez S, Pujol-Autonell I, Brianso F, Perna-Barrull D, Cano-Sarabia M, Garcia-Jimeno S, et al. Phosphatidylserine-liposomes promote tolerogenic features on dendritic cells in human type 1 diabetes by apoptotic mimicry. Front Immunol. (2018) 9:253. doi: 10.3389/fimmu.2018.00253
22. Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T, et al. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989–2013: a multicentre prospective registration study. Diabetologia. (2019) 62:408–17. doi: 10.1007/s00125-018-4763-3
23. Mayer-Davis EJ, Lawrence JM, Dabelea D, Divers J, Isom S, Dolan L, et al. Incidence trends of type 1 and type 2 diabetes among youths, 2002–2012. N Engl J Med. (2017) 376:1419–29. doi: 10.1056/NEJMoa1610187
24. Robb MA, McInnes PM, Califf RM. Biomarkers and surrogate endpoints: developing common terminology and definitions. JAMA. (2016) 315:1107–8. doi: 10.1001/jama.2016.2240
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C(T)] Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
26. Cabezon R, Carrera-Silva EA, Florez-Grau G, Errasti AE, Calderon-Gomez E, Lozano JJ, et al. MERTK as negative regulator of human T cell activation. J Leukoc Biol. (2015) 97:751–60. doi: 10.1189/jlb.3A0714-334R
27. Phillips BE, Garciafigueroa Y, Engman C, Trucco M, Giannoukakis N. Tolerogenic dendritic cells and T-regulatory cells at the clinical trials crossroad for the treatment of autoimmune disease; emphasis on type 1 diabetes therapy. Front Immunol. (2019) 10:148. doi: 10.3389/fimmu.2019.00148
28. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways forward for tolerance-inducing cellular therapies- an AFACTT perspective. Front Immunol. (2019) 10:181. doi: 10.3389/fimmu.2019.00181
29. Nieminen JK, Vakkila J, Salo HM, Ekstrom N, Harkonen T, Ilonen J, et al. Altered phenotype of peripheral blood dendritic cells in pediatric type 1 diabetes. Diabetes Care. (2012) 35:2303–10. doi: 10.2337/dc11-2460
30. Vuckovic S, Withers G, Harris M, Khalil D, Gardiner D, Flesch I, et al. Decreased blood dendritic cell counts in type 1 diabetic children. Clin Immunol. (2007) 123:281–8. doi: 10.1016/j.clim.2007.03.002
31. Klocperk A, Petruzelkova L, Pavlikova M, Rataj M, Kayserova J, Pruhova S, et al. Changes in innate and adaptive immunity over the first year after the onset of type 1 diabetes. Acta Diabetol. (2019). doi: 10.1007/s00592-019-01427-1. [Epub ahead of print].
32. Swanton S, Choh AC, Lee M, Laubach LL, Linderman JK, Czerwinski SA, et al. Body mass index associations between mother and offspring from birth to age 18: the Fels Longitudinal Study. Obes Sci Pract. (2017) 3:127–33. doi: 10.1002/osp4.90
33. Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. (2014) 468–470:565–80. doi: 10.1016/j.virol.2014.09.009
34. Trahtemberg U, Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol. (2017) 8:1356. doi: 10.3389/fimmu.2017.01356
35. Cummings RJ, Barbet G, Bongers G, Hartmann BM, Gettler K, Muniz L, et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature. (2016) 539:565–9. doi: 10.1038/nature20138
36. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. (2011) 35:82–96. doi: 10.1016/j.immuni.2011.05.013
37. Yi X, Zhang J, Zhuang R, Wang S, Cheng S, Zhang D, et al. Silencing LAIR-1 in human THP-1 macrophage increases foam cell formation by modulating PPARγ and M2 polarization. Cytokine. (2018) 111:194–205. doi: 10.1016/j.cyto.2018.08.028
38. Seyfert-Margolis V, Gisler TD, Asare AL, Wang RS, Dosch HM, Brooks-Worrell B, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes. (2006) 55:2588–94. doi: 10.2337/db05-1378
39. Dixon KO, O'Flynn J, van der Kooij SW, van Kooten C. Phagocytosis of apoptotic or necrotic cells differentially regulates the transcriptional expression of IL-12 family members in dendritic cells. J Leukoc Biol. (2014) 96:313–24. doi: 10.1189/jlb.3A1013-538RR
40. Golpon HA, Fadok VA, Taraseviciene-Stewart L, Scerbavicius R, Sauer C, Welte T, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. (2004) 18:1716–8. doi: 10.1096/fj.04-1853fje
41. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. (2008) 8:579–91. doi: 10.1038/nrc2403
42. Pujol-Autonell I, Mansilla MJ, Rodriguez-Fernandez S, Cano-Sarabia M, Navarro-Barriuso J, Ampudia RM, et al. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine. (2017) 12:1231–42. doi: 10.2217/nnm-2016-0410
43. Ten Brinke A, Hilkens CM, Cools N, Geissler EK, Hutchinson JA, Lombardi G, et al. Clinical use of tolerogenic dendritic cells-harmonization approach in european collaborative effort. Mediators Inflamm. (2015) 2015:471719. doi: 10.1155/2015/471719
44. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes. (2014) 63:3835–45. doi: 10.2337/db14-0365
Keywords: dendritic cells, immune tolerance, type 1 diabetes, phagocytosis, autoimmunity
Citation: Rodriguez-Fernandez S, Murillo M, Villalba A, Perna-Barrull D, Cano-Sarabia M, Gomez-Muñoz L, Aguilera E, Maspoch D, Vazquez F, Bel J and Vives-Pi M (2019) Impaired Phagocytosis in Dendritic Cells From Pediatric Patients With Type 1 Diabetes Does Not Hamper Their Tolerogenic Potential. Front. Immunol. 10:2811. doi: 10.3389/fimmu.2019.02811
Received: 29 August 2019; Accepted: 15 November 2019;
Published: 28 November 2019.
Edited by:
Jordi Ochando, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Daniel Benitez-Ribas, Hospital Clínic de Barcelona, SpainGiada Mondanelli, University of Perugia, Italy
Copyright © 2019 Rodriguez-Fernandez, Murillo, Villalba, Perna-Barrull, Cano-Sarabia, Gomez-Muñoz, Aguilera, Maspoch, Vazquez, Bel and Vives-Pi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Vives-Pi, bXZpdmVzQGlndHAuY2F0