 Farzan Solimani
Farzan Solimani Katharina Meier
Katharina Meier Kamran Ghoreschi
Kamran Ghoreschi- Department of Dermatology, Venereology and Allergology, Charité—Universitätsmedizin Berlin, Berlin, Germany
Accumulating data on cellular and molecular pathways help to develop novel therapeutic strategies in skin inflammation and autoimmunity. Examples are psoriasis and atopic dermatitis, two clinically and immunologically well-defined disorders. Here, the elucidation of key pathogenic factors such as IL-17A/IL-23 on the one hand and IL-4/IL-13 on the other hand profoundly changed our therapeutic practice. The knowledge on intracellular pathways and governing factors is shifting our attention to new druggable molecules. Multiple cytokine receptors signal through Janus kinases (JAKs) and associated signal transducer and activators of transcription (STATs). Inhibition of JAKs can simultaneously block the function of multiple cytokines. Therefore, JAK inhibitors (JAKi) are emerging as a new class of drugs, which in dermatology can either be used systemically as oral drugs or locally in topical formulations. Inhibition of JAKs has been shown to be effective in various skin disorders. The first oral JAKi have been recently approved for the treatment of rheumatoid arthritis and psoriatic arthritis. Currently, multiple inhibitors of the JAK/STAT pathway are being investigated for skin diseases like alopecia areata, atopic dermatitis, dermatomyositis, graft-versus-host-disease, hidradenitis suppurativa, lichen planus, lupus erythematosus, psoriasis, and vitiligo. Here, we aim to discuss the immunological basis and current stage of development of JAKi in dermatology.
Introduction
The classification of skin diseases and their treatment options are becoming more and more complex. While for a long period of time the morphology of diseased skin was prominent for disease classification and therapeutic procedures, we now have the methodologies for a deep analysis of molecular processes and immunological pattern analysis responsible for the pathophysiological alterations. These advances enlarged our therapeutic repertoire in dermatology remarkably. Deep analysis of skin biopsies allows us to define pathophysiological factors like cytokines, receptors or signaling molecules that are ultimately present at different levels in distinct skin diseases (1–3). This ultimately leads to the identification of new targets and, if these targets are druggable, to new treatments. One striking example of the aforementioned process is the development of a therapeutic variety with monoclonal antibodies (mabs) and small molecules in the treatment of psoriasis (PSO). Traditionally, PSO was treated with topical corticosteroids or dithranol, with phototherapy or immunosuppressive drugs such as cyclosporine. Nowadays, patients with this chronic inflammatory disease are frequently treated with biologicals, either of the first generation (anti-TNF) or the second generation (anti-IL-17/anti-IL-23) (4–7). In recent years, our better understanding of further inflammatory skin diseases such as lupus erythematosus (LE), lichen planus (LP) or atopic dermatitis (AD) also resulted in the development of novel treatment strategies (8–11). At the same time, we have gained more insight into the role of proteins propagating the intracellular effects of activated cytokine receptors (12). Again, when we pay attention to PSO, small molecules like dimethylfumarate or apremilast are good examples of intracellularly acting compounds that interfere directly or indirectly with signaling pathways (13). Most recently, first inhibitors of signaling proteins directly linked to cytokine receptors have been introduced into the clinics for treating patients with psoriatic arthritis (tofacitinib) and rheumatoid arthritis (tofacitinib and baricitinib). These inhibitors target so-called Janus kinases (JAKs), a family of four proteins: JAK1, JAK2, JAK3, and TYK2 (12). These proteins modulate the inflammatory process by activation of intracytoplasmic transcription factors called signal transducer and activator of transcription (STAT). Once activated, these proteins form dimers, translocate into the nucleus and either positively or negatively modulate the expression of thousands of different genes (12, 14) (Figure 1). Since JAK inhibition is not only restricted to systemic drugs administrated orally but has also been developed as a topical treatment option, it is not surprising that inhibitors of JAKs are receiving growing attention from dermatologists and are tested as systemic and/or topical treatment options in various skin diseases (15).
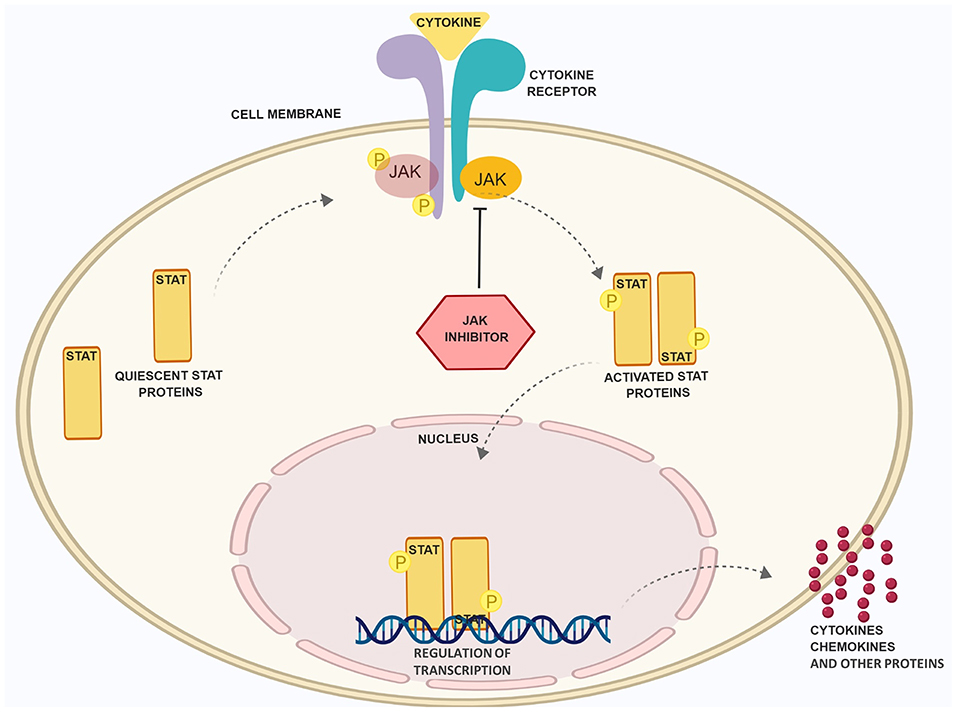
Figure 1. Schematic presentation of the JAK/STAT pathway and the role of JAK inhibitory drugs. Binding of cytokines to receptors, which rely on the JAK/STAT pathway for signal transduction, leads to phosphorylation of JAK and STAT proteins. The latter dimerize, translocate into the nucleus and regulate the expression of inflammatory factors. JAK inhibitors (JAKi) prevent JAK phsophorylation and STAT activation. Figure was created with the help of Biorender.com.
The JAK Family and Their Function
The importance of protein kinases and their crucial enzymatic activity was initially determined in 1966 by the work of Krebs and Fischer who showed the essential role of phosphorylation as a mechanism of cell physiology (16). The primary function of protein kinases is to transfer phosphate groups from adenosine triphosphate (ATP) or guanosine triphosphate (GTP) to the hydroxyl groups of amino acids of their protein targets (12). This mechanism is also important for cytokine receptors, which lack intrinsic enzymatic activity (Figure 2). In principle, the binding of cytokines to their receptors typically initiates an inflammatory signal (Figure 1). A large group of cytokines composed of central interleukins (IL) such as IL-2, IL-6, IL-12, IL-21, IL-22, IL-23, or interferons such as IFN-γ interacts with so-called type I and II cytokine receptors (Figure 2). Both of these receptor types lack intrinsic enzyme activity and strongly rely on JAKs for signal transduction (17). After binding, recruited JAKs initiate a signaling pathway from cellular membrane that ultimately should end in the nucleus: cytokine receptors of type I and II undergo oligomerization, leading to the recruitment of JAKs, which (auto-)phosphorylate tyrosine residues including such within the receptor chains (Figure 1). Successively, STAT proteins will be recruited, which then bind to the phosphorylated residues and become activated due to phosphorylation by JAKs. The now activated STAT proteins undergo dimerization. This last step enables the translocation of STAT proteins into the nucleus and the modulation of gene expression (14). Interestingly, most of cytokine receptors use a combination of JAKs for their activity; this could therefore hamper the idea of targeting single JAKs and instead favor a rationale for the use of pan-JAKi under certain settings. However, one should consider that toxicities may limit a strong and ubiquitous JAK blockade (Table 1).

Figure 2. Selectivity of JAKi toward type I and type II cytokine receptors. JAKi display different capacities to block cytokine receptor signaling. Pan-JAK inhibitors for example have a broad inhibitory effect, while drugs, which target selectively JAK3 or TYK2 have a more limited mode of action (+, inhibition; -, no inhibition). Figure was created with the help of Biorender.com.
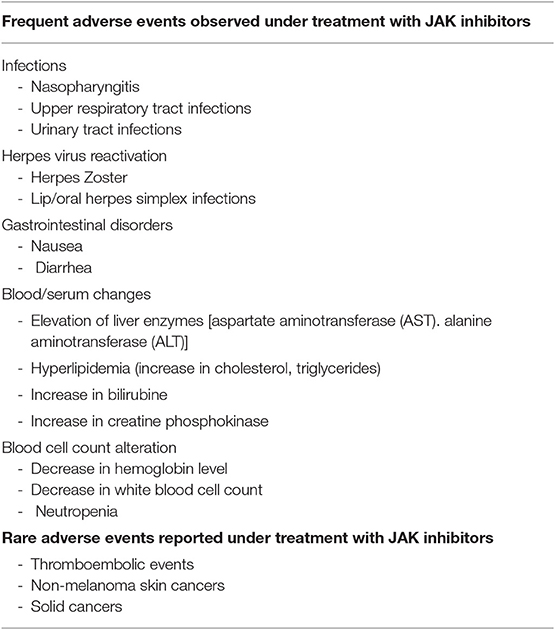
Table 1. Selected adverse effects observed under treatment with JAK inhibitors.
JAK Deficiency in Humans and Mice
The crucial role of JAKs and the relevance of their function can be observed in patients with mutations in genes encoding JAKs or in mice carrying JAK mutations. Here, we will briefly summarize some loss of function (LOF) mutations of JAKs found in humans or by genetic manipulation of mice. Functional deletion of JAK1 or JAK2 in mice results in perinatal or embryonic lethality, respectively. Likewise, reports regarding individuals with deficiency of either JAK1 or JAK2 proteins have not been described, indicating that functional JAK1 and JAK2 are required for embryonic development and survival. In contrast, JAK3 knockout (KO) mice and TYK2 KO mice are both viable. JAK3 is widely expressed in immune cells. Consequently, JAK3 KO mice present a strong reduction of T and B cell numbers and residual immune cells show an impaired activity, exposing mice to multiple infections (18). In agreement with these findings, mutations within the JAK3 gene can present in humans as severe combined immunodeficiency syndrome (SCID) (19, 20). Patients with SCID lack T, NK and diverse B cell populations in the peripheral blood. As a result of this mutation, these patients are constantly exposed to a risk of bacterial, fungal or viral infections (21). The devastating effect on lymphocytes explains the crucial role of JAK3 in lymphocyte biology. JAK3 is the only JAK protein capable of phosphorylating receptors carrying the γc receptor and this receptor chain is exclusively used by receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 (20). LOF mutations within the TYK2 gene are even rarer than JAK3 mutations (22). The few patients reported carrying the mutation show an increased susceptibility for infections, like severe infections of the skin. TYK2 LOF mutations essentially block the signal transduction of the receptors for IL-12, IL-23, and type I IFN (IFN-α/β), resulting in impaired IFN-γ+ Th1 responses and possibly IL-17+ Th17 responses (23). Similarly, TYK2 KO mice present a defective response to aforementioned stimuli and have an impaired macrophage activity with increased risk for viral and bacterial infections (24). Based on the selective expression and function of JAK3 in the hematopoietic system, early research focused on the generation of selective JAK3 inhibitors. However, the first inhibitors brought to clinical practice were less selective than expected. The JAK3 inhibitor tofacitinib showed additional activities against JAK1 and to some extent toward JAK2. In contrast, baricitinib is a JAK1/JAK2 inhibitor. However, even these less selective JAKi seem to have acceptable toxicities. Yet, recent efforts in the field of pharmacological chemistry demonstrated that it is possible to generate JAKi with improved selectivity. For instance, second generation JAK3 inhibitors with high selectivity have been synthesized in the last years (25–28). Accumulating data from published studies and ongoing clinical trials will show which type of JAK blockade—low or highly selective compounds—will improve skin diseases without inducing major side effects.
Rational for the Use of JAKi in Dermatology
The perpetuation of inflammation in diseased skin strongly relies on the interaction between cytokines, immune, and tissue cells propagating distinct inflammatory cascades. Some of these immunological processes in the skin, as in PSO or AD, have been deeply elucidated in recent years. The findings led to a radical change of therapeutic approaches. Based on our better understanding of the immunological mechanisms in PSO and AD, a variety of mabs targeting cytokines and small molecules interfering with intracellular signaling pathways have been developed. In this context, it is not surprising that JAKi are gaining increasing attention for the treatment of inflammatory skin diseases (15, 29, 30). Differently than biologics, which target cytokines by intravenous or subcutaneous injection, JAKi target cytokine signaling by either oral or topical administration. The latter way of application may minimize the risk of side effects as observed by systemic JAK inhibition (Table 1). Moreover, topical JAKi do not bear the risk of skin atrophy or telangiectasia as observed under long-term use of topical corticosteroids. In the following paragraphs, we aim to present the current position of JAKi in dermatology focusing on inflammatory skin diseases for which JAKi are at least in phase II investigation according to announced trials at clinicaltrials.gov.
Alopecia Areata
Alopecia areata (AA) is the most common immunological cause of hair loss (31). AA can affect both, adults and children. Although certain ethnic groups are more frequently affected than others, the disease does not prefer certain hairs types or color (31). Sudden hair loss is a hallmark of AA. In most of the cases, it appears in a circular well-circumscribed region of the scalp or the beard. AA can eventually lead to the loss of all the hairs of the scalp (alopecia totalis) or, in its most severe form, to the loss of hair of the whole body (alopecia universalis) (31). Frequently, the disease is accompanied by atopic dermatitis or by other autoimmune disorders such as autoimmune thyroiditis (32, 33). AA is determined by the loss of the immune privileged status of the hair follicles, which are then attacked by autoreactive CD8+ T cells and by NK T cells. Remarkably, hair follicles have developed different mechanisms to maintain their privileged immune status. For example, they express molecules such as transforming growth factor (TGF-)β1 and TGF-β2, α melanocyte stimulating hormone (α-MSH) and macrophage migration inhibitory factor (MIF), which hinder activation of T cell and NK T cell functions (31). Additionally, genetic background plays a prominent role in AA. Studies revealed that patients with positive family history for AA have a poorer prognosis and show a recalcitrant disease course often not responding to any treatments (34). Genome-wide-association studies (GWAS) showed the presence of diverse susceptibility loci possibly involved in the pathogenesis of AA like genes encoding for HLA, ULB1, IL12RA, and PTPN22 (35). ULB1 encodes for ligands involved in the activation of NKG2D cells, which in C3H/HeJ mice have been shown to be responsible for the destruction of hair follicles (36). Recent findings increased the evidence that JAKs play a crucial role in the pathogenesis of AA. Recipient mice of skin grafted C3H/HeJ mice were treated with mabs targeting IFN-γ, IL-2, and IL-15 each preventing the development of severe AA (36). Furthermore, Xing et al. showed that AA patients and experimental AA mouse models present increased levels of phosphorylated STAT proteins, specifically STAT1, STAT3, and STAT5. These STAT proteins are activated downstream the signals from IFN-γ, IL-2, and IL-15. When using the experimental model of C3H/HeJ mice, systemic treatment with the JAK1/JAK3i tofacitinib or with the JAK1/JAK2i baricitinib protected from hair loss and topical application of tofacitinib stimulated hair regrowth in C3H/HeJ mice (36, 37). In addition, three AA patients were treated orally with the JAK1/JAK2 inhibitor ruxolitinib. This therapeutic approach led to a decrease of CD8+NKG2D+ cells and a rapid amelioration of AA (36). Further, microRNAs that influence the expression of the IL2RA gene seem to be implicated in AA pathogenesis (38). Although the rationale for treating AA with JAKi is given, the clinical introduction of JAKi for the treatment of AA is still at an early stage (Figure 3) (39). Data from phase 2 and 3 studies are needed to clarify the clinical impact of JAKi in patients with AA (Table 2; Figure 3). Some first clinical experiences with JAKi for the treatment of AA have been published and seem to be promising (40–42). Treatment with oral ruxolitinib showed hair regrowth in 9 out of 12 treated patients without causing severe adverse events. JAK1/JAK2 inhibition by ruxolitinib reduced the expression of cytotoxic markers and IFN-γ expression in lesional skin (43). Similarly, Kennedy-Crispin et al. reported hair regrowth in a subset of patients with AA, AA totalis, or AA universalis treated with tofacitinib 5 mg twice daily. During this study, only low-grade infections were documented (Table 1). However, the positive effect on hair regrowth was lost after treatment discontinuation (NCT02197455 and NCT02312882) (44). A recently published case series also reports from the phenomenon of hair loss rebound in AA patients following discontinuation of tofacitinib (45). These first experiences were confirmed by subsequent studies in adults and children using oral or topical JAKi, respectively (46–49). Currently, various double-blind placebo-controlled phase II and III trials testing the efficacy and safety of oral and topical JAKi in AA are ongoing, underlining the growing interest toward these compounds (Table 2). One caveat of this promising approach in AA is the preliminary experience that the effect of oral JAKi seems to be timely restricted and hair loss has been reported to reappear upon cessation of pharmacological JAK inhibition in a substantial number of patients (50).
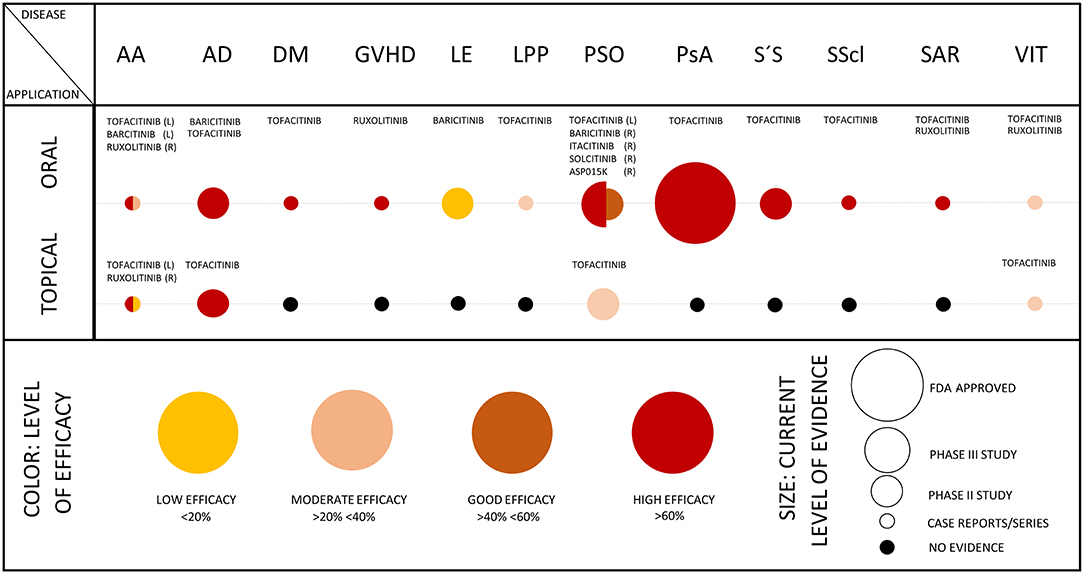
Figure 3. Efficacy of JAKi in dermatology. The scheme summarizes the level of efficacy (as represented by colors) and the level of evidence (as represented by size of circles) in the indicated skin diseases. In diseases, where results from phase II/III studies are available as published, evaluation of JAKi in case series or single case reports was omitted. The scheme was adapted from Eyerich et al. (1). AA, alopecia areata; AD, atopic dermatitis; DM, dermatomyositis; GVHD, graft versus host disease; LE, lupus erythematosus (efficacy on skin lesions); LPP, lichen planopilaris; PSO, psoriasis; PsA, psoriasis arthritis; S'S, Sjögren's syndrome; SScl, systemic sclerosis; SAR, sarcoidosis; VIT, vitiligo; (L), left; and (R), right half of the circle.
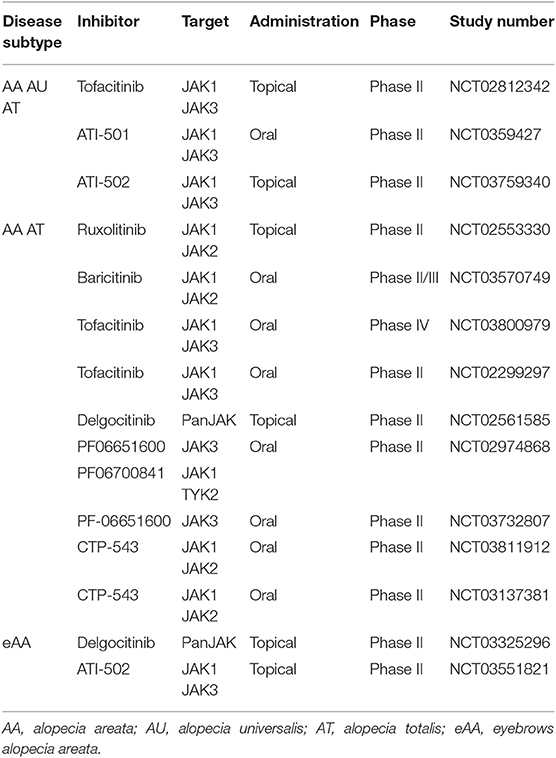
Table 2. Clinical trial program of JAKi in alopecia areata and subtypes according to clinicaltrials.gov.
Atopic Dermatitis
Atopic dermatitis (AD) is a common chronic disease of the skin, which severely impairs patient's quality of life. The prevalence of AD is higher in children and adolescents, but the disease can manifest at any age. Clinically, AD is characterized by the presence of pruriginous eczematous lesions, typically on flexural sites (8). In almost 80% of AD patients, skin integrity is altered due to LOF mutations within the gene encoding filaggrin (FGN). Impaired FGN expression promotes the loss of transepidermal water resulting in xerosis and eczema. The skin barrier dysregulation together with the “atopic” cytokine milieu increases the risk for skin superinfections with bacteria or viruses (51). Historically, AD is thought to be a Th2 dominated disease. Nonetheless, there is growing evidence that the immunological environment of AD is not solely defined by Th2 cells and related cytokines (IL-4, IL-5, IL-10, IL-13, and IL-31) but also by cytokines linked to other Th cell responses such as IFN-γ (Th1), IL17, or IL-22 (Th17) and IL-33 (an alarmin) (8, 52–56). Dupilumab, a mab against IL-4Rα has shown efficacy in a large number of patients and is the first approved biological for AD (57). Nonetheless, the cytokine microenvironment in AD seems to be complex and patients seem to present with different cytokine signatures at different stages. Several mabs are in phase 2/3 development, including mabs neutralizing IL-13 (tralokinumab and lebrikizumab), IL-31 (nemolizumab) (58–60), and IL-33 (etokimab) (61). The neutralization of other cytokines is being tested in patients with AD including secukinumab and ustekinumab, mabs against IL-17A and p40 subunit of IL-23/IL-12, respectively. Given the diversity of cytokines implicated in the inflammatory processes of AD, there is growing interest toward JAKi, which could interfere with the signaling of multiple cytokines simultaneously (62). Tofacitinib and baricitinib are so far the best-studied JAKi in AD (63). The JAK1/JAK3i tofacitinib abrogates IL-4 signaling and the differentiation of Th2 cells (64). Oral tofacitinib has been shown to be effective in patients with moderate to severe AD (65, 66) and topical tofacitinib is effective in mild forms of AD (67). The latter formulation is of special interest in topical AD treatment, which is mostly based on topical corticosteroids or calcineurin inhibitors (67). The JAK1/JAK2i baricitinib is also being tested in AD patients. Guttman-Yassky et al. showed that oral baricitinib at a dose of 2 or 4 mg strongly ameliorates AD, helping patients to spare long-lasting application of topical corticosteroids (68). Other compounds, like the JAK1i oclacitinib or the pan-JAKi JTE-052 showed efficacy when orally administrated in small-sized cohorts (69, 70). Currently, two different phase III studies are investigating the efficacy and safety of oral baricitinib as a monotherapy for AD. Other systemically applied JAKi under clinical investigation for AD include the JAK1 inhibitors upadacitinib (71) and PF-04965842. Topically applied JAKi tested for adult and pediatric AD include the pan-JAKi delgocitinib (LEO124249) (Table 3). Positive results, in absence of severe side effects when using topical JAKi, could enormously help young patients in which the application of topical corticosteroids and systemic immunosuppressive drugs are often precarious.

Table 3. JAKi tested for atopic dermatitis.
Dermatomyositis
Dermatomyositis (DM) is an ab-mediated autoimmune disease, which affects skin and muscles with variating extent (72, 73). A complex auto-ab profile (74) is helpful for diagnosing DM and for estimating the risk for developing distant organ involvement like lung, larynx, gastrointestinal tract, or heart during the disease course. Adult DM is frequently associated with malignancies (75). The pathophysiology is not completely understood, nonetheless multiple studies showed a pivotal role played by IFN-γ producing Th1 cells in DM (72, 76, 77). The standard treatment regimen in DM includes high-dose corticosteroids, non-steroidal immunosuppressants like azathioprine and often requires additional intravenous immunoglobulins (IVIG) (73, 78). In addition, diagnosis and treatment of disease-associated cancer is important. More recently, different case reports and small case studies showed a positive outcome of recalcitrant chronic DM after treatment with either ruxolitinib or tofacitinib (79–82) (Figure 3). Remarkably, JAKi seem to have a positive effect on lung involvement in DM (79, 82–84), which often represents a high-risk-mortality complication. The efficacy of tofacitinib is currently under investigation in a small cohort of 10 patients with recalcitrant DM (NCT03002649) (Table 4). If JAKi can improve non-cancer associated DM with limited toxicities, this treatment option would be of help and spare long-term use of high dose corticosteroids.
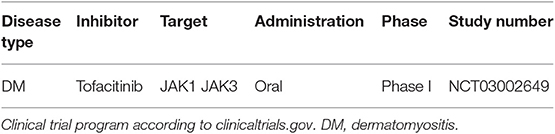
Table 4. JAKi trial in patients with dermatomyositis.
Graft-Versus-Host Disease
Graf-versus-Host Disease (GVHD) is a serious though common systemic reaction following hematopoietic stem cell transplantation or donor lymphocyte infusion (85). It is caused by donor T lymphocytes activated by host antigens. Different organs may be affected during GVHD and the skin is one of the most commonly targeted organs. Cutaneous GVHD presents in an acute and/or a chronic form. Acute (aGVHD) is clinically characterized by maculopapulous rash, disseminated erythematous skin areas, which can eventually converge in a generalized erythroderma (86). The chronic GVHD (cGVHD) shows a heterogeneous clinical presentation and can develop from the acute form. Lesions often present as LP-like or scleroderma-like lesions, nonetheless the disease can present differently and resemble PSO or keratosis pilaris (86). Immunologically, the aGVHD of the skin shows a T cell infiltration made up mostly by Th2 type cells. In contrast, in cGVHD the skin contains an infiltrate rich in type 1 and type 17 cells. In peripheral blood, cGVHD patients present lower numbers of Treg cells and the expression of their transcription factor FOXP3 is strongly reduced (87, 88). Glucocorticosteroids, both as topical and in oral form together with other immunosuppressants and phototherapy or extracorporeal photopheresis are the mainstay treatment for cGVHD (89). Given the important role of T cells and their associated cytokines, the application of JAKi may theoretically be of benefit for patients with GVHD. Experimental studies showed that pro-inflammatory mediators such as IL-6 and IFN-γ play a major role in the pathogenesis of GVHD (90). Furthermore, polymorphisms on genes encoding for IL-6 and IFN-γ have been associated with disease severity (91, 92). Accordingly, Choi et al. showed that IFN-γ receptor (IFN-γR)-deficient allogeneic Tconv reduced the risk for GVHD in mice (93). They tested the efficacy of ruxolitinib in wild type T cells in two murine MHC mismatched models, showing that inhibition of JAK proteins induce an effect similar to the genetic loss of IFN-γR in vitro and in vivo (93). Other experimental studies confirmed that JAKi like ruxolitinib (94) or tofacitinib improve or even prevent severe GVHD (95). The benefit of ruxolitinib in preclinical models was translated to humans and, also here, showed improvement of GVHD in six patients (96). After preliminary encouraging results regarding the effect of ruxolitinib in cGVHD in humans (97) (Figure 3), different studies are now investigating the possibly beneficial role of JAK inhibition in aGVHD and cGVHD. A multicenter, randomized phase 2 trial has been initiated, to test the efficacy of oral ruxolitinib in steroid-refractory aGVHD (98). The GRAVITAS-301 study (NCT03139604) is investigating the efficacy of itacitinib (a JAK1 inhibitor) in acute GVHD in a randomized, double-blind placebo-controlled phase 3 study. This compound as well as ruxolitinib and baricitinib are currently under investigation in cGVHD (Table 5). Most recently, itacitinib even entered two phase I studies for prophylactic use to prevent GVHD after cell stem transplantation (Table 5).
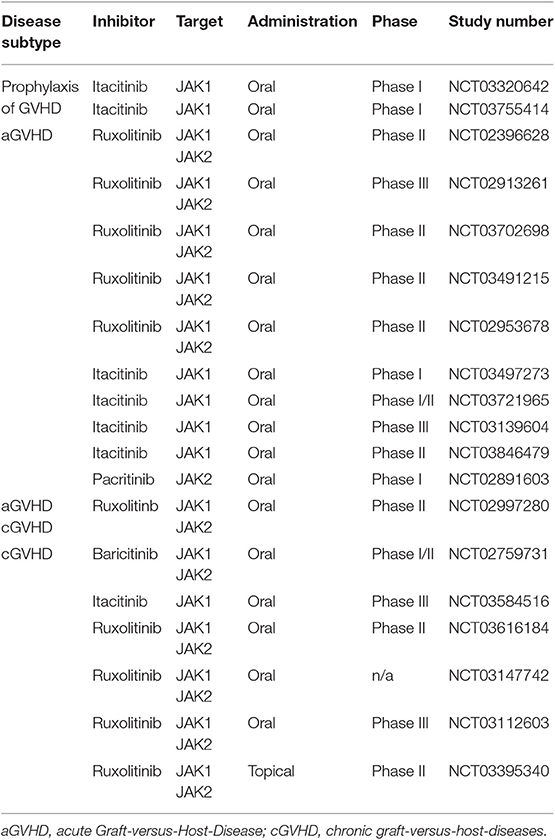
Table 5. Clinical trials using JAKi in Graft-versus-Host-Disease according to clinicaltrials.gov.
Hidradenitis Suppurativa
Hidradenitis suppurativa (HS) (also designated as acne inversa) is a chronic debilitating inflammatory skin disease frequently occurring in skin areas with substantial presence of follicles (99). The disorder is characterized by occlusion of follicular ducts with consequent increase in local bacteria. This process evolves in cyst rupture and generalized local tissue inflammation (99). Of note, it is probable that a sublatent skin inflammatory process precedes bacterial accumulation; this could be triggered e.g., by behavioral factors like smoking and obesity, which typically worsen the course of HS (99). Moreover, HS patients often carry mutations in the γ-secretase encoding gene PSEN (100). Due to the important role played by bacteria in HS, first line treatments are based on antibiotic therapy with anti-inflammatory properties (101, 102). Nonetheless, recent studies elucidated the role of cytokines present in the inflammatory milieu of HS skin showing an overexpression of IL-17A, IL-26, IFN-γ, IL-27, and IL-β, and, a concomitant downregulation of IL-22 (103, 104). This moved the focus of researchers from the role of bacteria to the function of cytokines in HS. Adalimumab, a mab against TNF, is the first biological approved for the treatment of HS (105). Targeting cytokines like IL-1 or IL-17/IL-23 have been reported to have mixed results (37, 106) or are presently under intensive clinical investigation. Given that cytokines are crucial in HS pathogenesis, inhibition of the JAK/STAT pathway could help to regulate the expression of inflammatory factors like IL-6 or IL-23 simultaneously. Clinical studies like NCT03607787 and NCT03569371 are evaluating the safety of INCB054707, a JAK1 inhibitor in HS (Table 6).
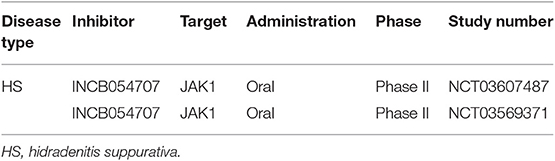
Table 6. Trials on JAKI in hidradenitis suppurativa according to clinicaltrials.gov.
Lichen Planus
LP is a common inflammatory disease of the skin and mucous membranes, clinically characterized by polygonal papules presenting a characteristic white, reticulate on their surface (Wickham striae). Frequently, the oral cavity is affected by LP (OLP) (107). Rarely, LP can affect the scalp, LP planopilaris (LPP), or the nails (nail LP) (107). A classic feature of this inflammatory disorder is the histological presence of a dense T cellular infiltrate disposed in a band like pattern (lichenoid infiltrate) (10). The need of new therapeutic options is given, since LP often presents a challenging chronic recalcitrant course (especially OLP) and the traditional therapeutic options are of limited efficacy. Treatment of LP is mainly based on oral or topical glucocorticosteroids, phototherapy, retinoids or immunosuppressive drugs such as cyclosporine or methotrexate (107). There is growing evidence that LP is a Th1 driven disease (10, 108, 109), although Th17-associated cytokines have also been reported in LP (110–112). A recent study showed that the T cell infiltrate presents high numbers of Th1/Tbet+ cells and the presence of IL-17A+ cells, disposed beneath the basal cells of the epidermis (10). Peripheral Th1/Th17 cell reactive against skin autoantigens further underlines the proposed autoreactive pathophysiology of LP (10). Finally, therapeutic blockade of IL-17 or of IL-23 seems to be a promising approach for the treatment of LP (11). The dominance of IFN-γ in LP skin suggests that JAK inhibition could be a therapeutic option. A small retrospective study consisting of 10 patients with LPP treated with oral tofacitinib as monotherapy or as adjunctive therapy showed mixed clinical efficacy in 8/10 patients (113). According to clinicaltrials.gov, the study NCT03697460 is testing the efficacy of ruxolitinib cream in LP (Table 7). It is likely, that JAKi will be a therapeutic option for patients with steroid-refractory LP variants. Given the beneficial effect of tofacitinib on nail PSO (114), a possibly similar effect on nail LP is conceivable. This should be addressed in the future, since nail LP is very painful and cicatrial.
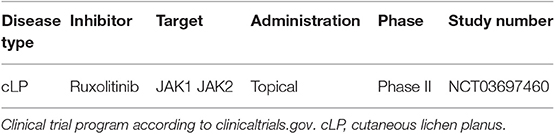
Table 7. Trial on JAKi in lichen planus.
Lupus Erythematosus
Lupus erythematosus (LE) is a severe, chronic autoimmune ab-mediated disorder with variable clinical presentation and difficult-to-treat clinical course. The disease is widely known for the presence of a butterfly-like rash on the face of affected patients with systemic LE (SLE) (115). Based on the diverse subtypes, LE can be restricted to the organ skin or present a systemic course with manifestation at multiple organs (115). Both environmental and behavioral factors (such as smoking or sun exposure) and genetic factors are meant to play a role in the pathogenesis of LE (115–117). Possibly, gut pathobionts and in general, bacterial and viral stimuli could trigger the onset of the disease (118). In LE there is evidence that multiple cytokines such as IL-16, IL-17, IL-18, and TNF are implicated in the inflammatory process. In any event, type I IFNs are thought to play a major role in LE pathogenesis (119). The latter family of cytokines strongly relies on the JAK/STAT pathway for signal transmission (Figure 2). In addition, the role of STAT proteins has been widely investigated (120). The pivotal role of STAT1 in LE has been analyzed in mice and in humans. In the MRL/lpr mouse model, STAT1 is overexpressed in both B and T cells. STAT1 gene silencing (STAT1−/−) in this mouse model is accompanied by a decrease of CD4+ producing IFN-γ T cells, a milder course of nephritis and a remarkable decrease of auto-ab levels (121–123). In humans diagnosed with LE as in mice, STAT1 is overexpressed in T and B cells. STAT1 levels correlate with clinical disease activity and with the level of expression of IFN-γ-induced genes (124–126). In addition, STAT3 and STAT4 seem to be relevant in the pathogenesis of LE. The lack of STAT3 seems to be protective in a LE murine model (127, 128). In MRL/lpr LE prone mice treatment with tofacitinib led to a decrease of auto-ab production and amelioration of nephritis and skin inflammation. Similarly, two different LE mouse models showed a decrease of several cytokines (IL-12, IL-17A, IFN-γ, and TNF), and a decrease of antinuclear auto-ab levels upon treatment with CEP-33779, a JAK2 inhibitor (129). However, in humans, the JAK1 inhibitor GSK2586184 showed to be ineffective in a small size study of patients with SLE (130). Conversely, baricitinib has been tried in patients with SLE in a double blind, randomized, placebo-controlled phase 2 trial (NCT02708095) (Figure 3) (131). Baricitinib was administered at 2 or 4 mg daily. Results showed that the 4 mg dose leads to a consistent clinical improvement in SLE, especially on arthritis. Of note, adverse events were almost comparable to the placebo group (132). At the time of this writing, different compounds targeting different JAKs are under clinical investigation for the treatment of SLE or discoid LE (Table 8).
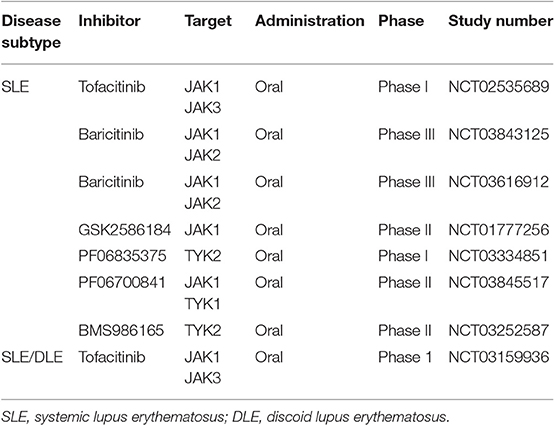
Table 8. Clinical trials of JAKi in systemic and/or discoid lupus erythematosus according to clinicaltrials.gov.
Psoriasis Vulgaris
PSO is a common inflammatory skin disease with well-defined pathogenesis. Based on the research of recent years numerous targeted treatments have been developed for PSO (4). In its classic appearance, this chronic inflammatory disorder presents with erythematous scaly plaques, which are preferentially disposed at extensor sites and in areas of mechanic stress. In addition, scalp, nails and inverse regions are frequently affected. In around 20–30% of cases, patients with PSO also suffer from joint involvement (psoriatic arthritis, PSA) (4). There is a wide range of treatment possibilities for PSO. Topical treatments include glucocorticosteroids, vitamin D derivatives, and dithranol. While phototherapy is becoming less important, oral drugs like methotrexate, acitretin, dimethylfumarate, or apremilast are widely used (4). Cyclosporine should be avoided due to its toxicities compared to the numerous less-toxic alternatives. Besides oral drugs, various biologics have been established for PSO (6). The first generation of antipsoriatic biologics targets TNF. Since PSO is widely accepted as the prototypic Th17 driven disease, multiple second-generation biologics have been approved in the meanwhile. These either neutralize IL-17A, bind its receptor or target IL-23. Although the neutralization of IL-17A is effective in most patients, some patients do not respond. The implication of multiple cytokines like IL-6, IL-22, IL-23, or IFN-γ in psoriasis pathogenesis suggests that the inhibition of JAKs could be a powerful and more profound treatment than a treatment with a single mab. Of note, STAT3 is a key factor in IL-23/Th17 signaling. Active JAKs and active STAT3 are typically found in psoriatic skin (133, 134). The role of IL-23 and Th17 cells has also been studied in the imiquimod (IMQ) PSO mouse model. In this model, silencing of TYK2 by gene targeting abrogated skin inflammation (135). Likewise, the use of JAKi successively ameliorated skin inflammation with a concomitant decrease of cytokine levels in the IMQ PSO mouse model (136). In humans, Krueger et al. showed in a small cohort (n = 12) that treatment with tofacitinib (10 mg twice daily) ameliorated PSO and this was accompanied at a molecular level by the decrease of phosphorylated STAT1 and STAT3. Similarly, tofacitinib decreased epidermal thickness, reduced the number of T cells infiltrating the skin and suppressed the IL-23/Th17 pathway (137). These preliminary findings paved the way for JAKi application in humans with PSO. A phase III randomized double-blind placebo-controlled study (NCT01815424) demonstrated the efficacy of tofacitinib at doses of 5 or 10 mg twice daily in patients with moderate to severe PSO (138). The oral PSO trial (OPT) pivotal I and II studies confirmed these positive results by oral tofacitinib in chronic plaque PSO (Figure 3). Importantly, only 6% of treated patients experienced adverse events (139). Notably, treatment discontinuation was associated with a risk of relapse; however, re-initiation of the treatment rapidly resolved psoriatic inflammation (140). Another phase III randomized multicenter study showed that the efficacy of tofacitinib 10 mg twice daily is similar to the efficacy of etanercept 50 mg twice weekly in PSO (141). Tofacitinib (5 or 10 mg daily) is also beneficial in nail PSO (114). The OPAL study showed that the JAK1/JAK3i is highly effective in the control of PSA in patients not responding to anti TNF-α treatment. In 2018, tofacitinib 5 mg twice daily was approved by the FDA for the treatment of PSA (142). Under this therapeutic regimen tofacitinib seems to have an acceptable safety profile without severe adverse events even during long-term application (143, 144). Based on the experience with tofacitinib, numerous JAKi are tested as oral drugs or as topical formulation for PSO (Table 9) (145–147). So far, the efficacy of topical JAKi for psoriasis is not convincing (148–150). In a double-blind study, topical application of the JAK1/JAK2i ruxolitinib (1 or 1.5%) showed comparable reduction of skin inflammation as calcipotriene 0.005% cream or betamethasone dipropionate 0.05% cream (151). Oral JAKi are more promising. In a randomized, double blind, placebo controlled phase IIb study the JAK1/JAK2i baricitinib at 2, 4, 8, or 10 mg daily dose showed encouraging results in the treatment of moderate to severe PSO (152). Lastly, selective inhibition of TYK2 with the oral compound BMS-986165 seems to be another very promising therapeutic option for the treatment of PSO. In a phase II trial BMS-986165 given at 3, 6, 9, or 12 mg daily over a period of 12 weeks resulted in an impressive clearing of PSO compared to placebo (153). According to clinicaltrials.gov, the number of registered studies on JAKi for PSO is rapidly growing (Table 9). Both oral and topical compounds are currently under clinical investigation for PSO. These include the oral compounds solcitinib, filgotinib, upadacitinib, and delgocitinib (LEO124249), a topical pan-JAKi (154). The selective TYK2 inhibitors PF-06826647 and BMS-986165 are under investigation for PSO or PSO/PSA, respectively (Table 9). The expected results from these clinical trials will be a major step toward extending the therapeutic spectrum of PSO and PSA by oral compounds.
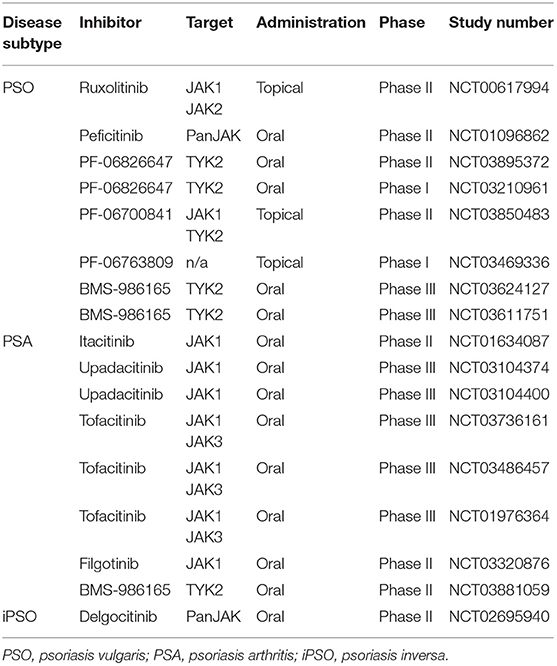
Table 9. JAKi trials in psoriasis vulgaris, psoriatic arthritis and psoriasis inversa according to clinicaltrials.gov.
Vitiligo
Vitiligo is a skin disease with severe psychological impact on patients. The disease is characterized by the presence of white depigmented skin spots due to melanocytic destruction by self-reactive CD8+ T cells (155). There is an enormous need for effective treatment options since presently the limited treatment modalities are only effective in some patients. Currently, patients with vitiligo are either treated with topical glucocorticosteroids, topical calcineurin inhibitors (off-label), or with phototherapy (narrowband UVB). In addition, systemic administration of glucocorticosteroids or other immunosuppressive drugs are used. Mechanistically, type I immune responses seem to be responsible for the development of vitiligo (156–159). In lesional skin, overexpression of IFN-γ, which translates its intracellular signal through STAT1, and associated chemokines like CXCL10 and its receptor CXCR3 are found (158). The biological relevance of these clinical findings was confirmed in mouse models of vitiligo. For instance, transfusion of CXCR3−/− PMEL T cells in vitiligo prone mice did not evolve to phenotypic alterations. Similarly, interfering with the CXCL10-CXCR3 interaction through targeting of CXCL10 with mabs results in vitiligo reversal in mice with established disease (160). These and other findings helped to elucidate the copious immunological players dominating vitiligo and underlined the crucial role of the JAK/STAT pathway and their related factors in the pathogenesis of this disorder. First small-size case series report from the efficacy of JAKi in vitiligo (161). Rothstein et al. used ruxolitinib 1.5% cream over a period of 20 weeks in a small group of 12 patients with vitiligo. They reported successful repigmentation of facial spots, while repigmentation of other anatomical sites showed mixed results (162). Similar results with better responses of facial spots than non-facial spots were reported by a recently published cohort study with 16 patients receiving 2% tofacitinib cream (163). A retrospective case study of 10 patients treated with oral tofacitinib 5 or 10 mg once daily suggests that a combination of phototherapy with JAKi is therapeutically more effective than JAKi monotherapy (164) (Figure 3). These reports indicate that JAKi in combination with UV exposure are required for stimulating repigmentation. Currently, different JAKi are under investigation in phase II trials for topical application in patients with vitiligo (Table 10).
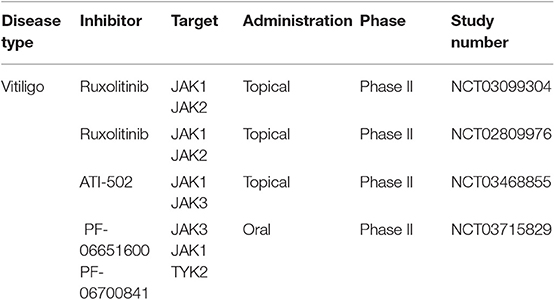
Table 10. Clinical trials on JAKi for treating patients with vitiligo according to clinicaltrials.gov.
Possible Future Applications
The introduction of JAKi is enlarging the therapeutic repertoire of dermatologists and is proving efficacy in numerous inflammatory diseases. Disorders like vitiligo or AA are in high need for efficient treatments. Increasing evidence confirming the beneficial role of systemic or topical JAKi in the treatment of these disorders is accumulating. Since the JAK/STAT signaling pathway plays a crucial role for many cytokines (Figure 2), a variety of inflammatory dermatological disorders may benefit from this new class of immunomodulators. For instance, lichen sclerosus et atrophicans (LSA) is an inflammatory disorder of skin and genital mucosa associated with severe pruritus and scaring. Many patients do not respond adequately to topical steroids (165). Due to its frequent recalcitrant course, there is an urgent need of new therapeutics for LSA. Inflammatory factors like IFN-γ, CXCL10, and CCR5 are highly expressed in lesional skin, suggesting that LSA is a Th1-dominated disorder (166). Oral or topical application of JAKi could stop the inflammatory circuit in LSA. Case reports on positive effects of JAKi exist for LSA and for patients with other sclerosing skin diseases like morphea, eosinophilic fasciitis, or systemic sclerosis (167, 168). These results are in agreement with experimental findings demonstrating a pivotal role of STAT3 in the activation of profibrotic pathways in vivo and in vitro (169). JAK inhibitors also improve inflammatory bowel diseases (IBD) like ulcerative colitis. Based on this data, it can be argued that IBD-associated skin diseases such as cheilitis granulomatosa (CG), pyoderma gangrenosum (PG) or erythema nodosum (EN) could also benefit from JAKi. CG shows a similar immunological profile as Crohn's disease with overexpression of IFN-γ (170–172). Patients with CG benefit from drugs such as thalidomide or lenalidomide (173, 174), which suppress Th1 cell responses (175). In contrast, analysis of PG and EN lesional skin revealed overexpression of TNF and STAT3, the latter one suggesting a rationale for the application of JAKi in these diseases (176). Other inflammatory diseases, where the use of JAKi seems to be desirable include sarcoidosis (177–180), and Sjögren's syndrome (SS), autoimmune diseases with strong molecular association to the JAK/STAT signaling pathway (181–184). Patients with blistering skin diseases like bullous pemphigoid, dermatitis herpetiformis Duhring and pemphigus vulgaris may also benefit from JAKi (185–187). JAKi have been shown to inhibit auto-ab production in preclinical models (188). Moreover, different studies highlighted the crucial role of STAT1 and STAT3 in transporting the signals derived from IL-6 and IL-21, two crucial cytokines for germinal centers formation. Lack of IL-6 and IL-21 leads to a strong reduction of T follicular helper (TFH) cell levels and consequently to impaired germinal center formation and suppression of IgG levels, since TFH cells provide unavoidable help for B cells' ab production (189, 190). Due to this preliminary data, JAKi could dramatically improve patients' quality of life in auto-ab-mediated bullous skin disorders, where high doses of oral glucocorticosteroids are currently the therapeutic gold standard and where the need of new therapeutic approaches is high, even though the B cell depleting mAb rituximab has been approved recently (191, 192). Another group of skin disorders that could benefit from JAKi are eczema. Delgocitinib, a pan-JAKi is currently under investigation in patients with chronic hand eczema (Table 11) and shows some first promising results (193). In nickel-induced allergic contact dermatitis there is evidence of an important involvement of the JAK/STAT pathway (194). Accordingly, mouse models for contact dermatitis show positive response to treatment with topical tofacitinib (JAK1/JAK3i) (195, 196), thus suggesting a possible application of topical JAKi also in humans. In Behcet's disease, an autoimmune disorder with unsolved pathogenesis experimental studies showed overexpression of Th17 related genes, overexpression of type I IFN-inducible genes and activation of the JAK/STAT pathway (197–199). These first experimental results pave the way for a future tentative application of JAKi in this disorder. Lastly, JAKi could improve autoinflammatory disorders. A patient with synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO), has been successfully treated by a combination of methotrexate and oral tofacitinib (200). Similarly, the clinical trials NCT01724580 and NCT02974595 proved efficacy of baricitinib in a small group of patients affected by CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures) and SAVI (stimulator of IFN-genes associated vasculopaty with onset in infancy) (201, 202).
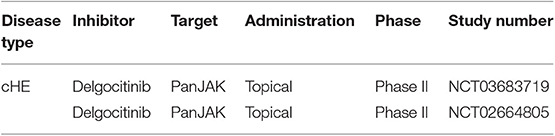
Table 11. Clinical trials of JAKi in chronic hand eczema. cHE, chronic hand eczema according to clinicaltrials.gov.
Concluding Remarks
The introduction of JAKi in dermatology will revolutionize the therapeutic outcome of various inflammatory skin diseases. There is growing evidence that the JAK/STAT signaling pathway is a key pathway in numerous skin disorders. However, we are facing several hurdles. First, the development of selective JAKi was harder than initially thought. First generation JAKi, like tofacitinib and baricitinib, which were thought to be highly selective, target more than one single JAK. This is not necessarily a disadvantage regarding efficacy, but of course may bear a higher risk for toxicities (17). For instance, significant inhibition toward JAK2 leads to anemia. Some unexpected adverse events were also observed when using first generation JAKi (203–205). One example is the increase in cholesterol levels by tofacitinib (144, 206–208), which could be a paradox effect induced by the reduction of inflammation (209). The more recently developed JAKi seem to have improved selectivity (25, 27, 210). The other caveat for treating skin disorders via topical route is to generate formulations that allow small molecules like JAKi to penetrate the skin. Here, improvements like nanocarriers may be helpful (211–213). We still have a high therapeutic need for several skin diseases like vitiligo, AA, AD, LPP, LSA, and others. JAKi as topical formulation or systemic drug could be an outstanding improvement without side effects typically observed when using topical or systemic glucocorticosteroids. Yet, in the majority of the aforementioned skin diseases, we urgently need more evidence and larger double-blind placebo-controlled studies to confirm efficacy and safety of JAKi. The safety profile of JAKi reported so far seems to be acceptable, at least when used as monotherapy. One important question, which could favor the introduction of JAKi in dermatological daily routine, is the comparison of these with the well-established application of mabs. Even though studies directly comparing JAKi with biologicals are widely missing for dermatological indications, the anti-inflammatory properties of JAKi are conceivably profound. Nonetheless, the safety profile of these new compounds compared to the safety profile of mabs seem not to be very different (214–216). Experience from the here discussed studies show that severe adverse events are rare. Common adverse events like infections of the upper respiratory tract or common disturbances such as headache or diarrhea can be frequent, but easy to manage (217–219) (Table 1; Supplementary Tables 1–3). The reactivation of herpes zoster infection seems to be more frequent in patients treated with JAKi compared to those receiving biologicals and therefore vaccination prior to treatment initiation should be discussed (220–223). Blood count alterations seem to be reversible and normalize after withdrawal (224, 225) (Table 1). Published studies with different JAKi indicate an increased risk for bacterial and viral infections (226–230). From an economic point of view the lower production costs of JAKi compared to mabs may favor their application in dermatology (231). However, the simultaneous inhibition of multiple cytokines by systemic JAKi bears the risk for fatal outcomes during severe infections and possibly also the risk for cancer development on the long run (232). Moreover, the dose and half-life of JAKi is an important issue. It will be crucial to lower the threshold of JAK/STAT activation but not to permanently block this pathway. Notably, another major issue which must be addressed in the future and which is presently unknown is the impact of these drugs on long-term treatments setting. First data from non-dermatological studies will be available soon, but data from longer observational periods after approval for dermatological indications are needed (233–236). This will be important for calculating the risk for both, infectious and tumor diseases (237). In conclusion, JAK inhibitory drugs are emerging as a new and effective treatment for various diseases. So far, the results arouse enthusiasm among dermatologist; nonetheless, further studies will decide about the real-world efficacy and safety profiles of this new class of immunomodulators.
Author Contributions
FS and KG designed the manuscript and designed figures and tables. FS, KM, and KG drafted the manuscript and approved the final version of the manuscript. KG revised critically the final version of the manuscript.
Funding
The work was funded by grants of the Deutsche Forschungsgemeinschaft (DFG) FOR 2497/TP02 (GH133/2-1 to KG).
Conflict of Interest
KG has received honoraria or travel expenses for lecture and research activities from Abbvie, Almirall, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Delenex, Eli Lilly, Galderma, Janssen-Cilag, Medac, MSD, Novartis, Pfizer, and UCB Pharma. KM has received honoraria or travel expenses for lecture and research activities from Abbvie, Celgene, Janssen-Cilag, and UCB Pharma.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02847/full#supplementary-material
References
1. Eyerich K, Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. (2018) 32:692–703. doi: 10.1111/jdv.14673
2. Yazdi AS, Rocken M, Ghoreschi K. Cutaneous immunology: basics and new concepts. Semin Immunopathol. (2016) 38:3–10. doi: 10.1007/s00281-015-0545-x
3. Sabat RWK, Loyal L, Döcke WD, Ghoreschi K. T cell pathology in skin inflammation. Semin Immunopathol. (2019) 41:359–77. doi: 10.1007/s00281-019-00742-7
5. Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature. (2015) 521:222–6. doi: 10.1038/nature14175
6. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
7. Eberle FC, Bruck J, Holstein J, Hirahara K, Ghoreschi K. Recent advances in understanding psoriasis. F1000Res. (2016) 5:770. doi: 10.12688/f1000research.7927.1
8. Weidinger S, Novak N. Atopic dermatitis. Lancet. (2016) 387:1109–22. doi: 10.1016/S0140-6736(15)00149-X
9. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
10. Schmidt T, Solimani F, Pollmann R, Stein R, Schmidt A, Stulberg I, et al. TH1/TH17 cell recognition of desmoglein 3 and bullous pemphigoid antigen 180 in patients with lichen planus. J Allergy Clin Immunol. (2018) 142:669–72.e7. doi: 10.1016/j.jaci.2018.02.044
11. Solimani F, Pollmann R, Schmidt T, Schmidt A, Zheng X, Savai R, et al. Therapeutic targeting of Th17/Tc17 cells leads to clinical improvement of lichen planus. Front Immunol. (2019) 10:1808. doi: 10.3389/fimmu.2019.01808
12. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. (2009) 228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x
13. Bruck J, Dringen R, Amasuno A, Pau-Charles I, Ghoreschi K. A review of the mechanisms of action of dimethylfumarate in the treatment of psoriasis. Exp Dermatol. (2018) 27:611–24. doi: 10.1111/exd.13548
14. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
15. Welsch K, Holstein J, Laurence A, Ghoreschi K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol. (2017) 47:1096–107. doi: 10.1002/eji.201646680
16. Fischer EH, Krebs EG. Relationship of structure to function of muscle phosphorylase. Fed Proc. (1966) 25:1511–20.
17. Ghoreschi K, Laurence A, O'Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. (2009) 10:356–60. doi: 10.1038/ni.1701
18. Nosaka T, Vandeursen JMA, Tripp RA, Thierfelder WE, Witthuhn BA, Mcmickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science. (1995) 270:800–2. doi: 10.1126/science.270.5237.800
19. Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O'Shea JJ. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol Rev. (2005) 203:127–42. doi: 10.1111/j.0105-2896.2005.00220.x
20. Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. (1995) 270:797–800. doi: 10.1126/science.270.5237.797
21. Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. (1995) 377:65–8. doi: 10.1038/377065a0
22. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. (2006) 25:745–55. doi: 10.1016/j.immuni.2006.09.009
23. Kreins AY, Ciancanelli MJ, Okada S, Kong XF, Ramirez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. (2015) 212:1641–62. doi: 10.1084/jem.20140280
24. Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. (2000) 13:549–60. doi: 10.1016/S1074-7613(00)00054-6
25. Forster M, Chaikuad A, Dimitrov T, Doring E, Holstein J, Berger BT, et al. Development, optimization, and structure-activity relationships of covalent-reversible JAK3 inhibitors based on a tricyclic imidazo[5,4- d]pyrrolo[2,3- b]pyridine scaffold. J Med Chem. (2018) 61:5350–66. doi: 10.1021/acs.jmedchem.8b00571
26. Forster M, Chaikuad A, Bauer SM, Holstein J, Robers MB, Corona CR, et al. Selective JAK3 inhibitors with a covalent reversible binding mode targeting a new induced fit binding pocket. Cell Chem Biol. (2016) 23:1335–40. doi: 10.1016/j.chembiol.2016.10.008
27. Dai J, Yang LX, Addison G. Current status in the discovery of covalent Janus kinase 3 (JAK3) inhibitors. Mini Rev Med Chem. (2019) 19:1531. doi: 10.2174/1389557519666190617152011
28. Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, et al. PF-06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem Biol. (2019) 14:1235–42. doi: 10.1021/acschembio.9b00188
29. Szilveszter KP, Nemeth T, Mocsai A. Tyrosine kinases in autoimmune and inflammatory skin diseases. Front Immunol. (2019) 10:1862. doi: 10.3389/fimmu.2019.01862
30. Hosking AM, Juhasz M, Mesinkovska NA. Topical Janus kinase inhibitors: a review of applications in dermatology. J Am Acad Dermatol. (2018) 79:535–44. doi: 10.1016/j.jaad.2018.04.018
31. Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. (2012) 366:1515–25. doi: 10.1056/NEJMra1103442
32. Barahmani N, Schabath MB, Duvic M National Alopecia Areata R. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. (2009) 61:581–91. doi: 10.1016/j.jaad.2009.04.031
33. Chu SY, Chen YJ, Tseng WC, Lin MW, Chen TJ, Hwang CY, et al. Comorbidity profiles among patients with alopecia areata: the importance of onset age, a nationwide population-based study. J Am Acad Dermatol. (2011) 65:949–56. doi: 10.1016/j.jaad.2010.08.032
34. Goh C, Finkel M, Christos PJ, Sinha AA. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. (2006) 20:1055–60. doi: 10.1111/j.1468-3083.2006.01676.x
35. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. (2010) 466:113–7. doi: 10.1038/nature09114
36. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
37. Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. (2002) 296:1653–5. doi: 10.1126/science.1071545
38. Tafazzoli A, Forstner AJ, Broadley D, Hofmann A, Redler S, Petukhova L, et al. Genome-wide microRNA analysis implicates miR-30b/d in the etiology of alopecia areata. J Invest Dermatol. (2018) 138:549–56. doi: 10.1016/j.jid.2017.09.046
39. Phan K, Sebaratnam DF. JAK inhibitors for alopecia areata: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. (2019) 33:850–6. doi: 10.1111/jdv.15489
40. Liu LY, Craiglow BG, Dai F, King BA. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. (2017) 76:22–8. doi: 10.1016/j.jaad.2016.09.007
41. Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. (2017) 76:29–32. doi: 10.1016/j.jaad.2016.09.006
42. Almutairi N, Nour TM, Hussain NH. Janus kinase inhibitors for the treatment of severe alopecia areata: an open-label comparative study. Dermatology. (2019) 235:130–6. doi: 10.1159/000494613
43. Mackay-Wiggan J, Jabbari A, Nguyen N, Cerise JE, Clark C, Ulerio G, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. (2016) 1:e89790. doi: 10.1172/jci.insight.89790
44. Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. (2016) 1:e89776. doi: 10.1172/jci.insight.89776
45. Gordon SC, Abudu M, Zancanaro P, Ko JM, Rosmarin D. Rebound effect associated with JAK inhibitor use in the treatment of alopecia areata. J Eur Acad Dermatol Venereol. (2019) 33:e156–e7. doi: 10.1111/jdv.15383
46. Jabbari A, Sansaricq F, Cerise J, Chen JC, Bitterman A, Ulerio G, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. (2018) 138:1539–45. doi: 10.1016/j.jid.2018.01.032
47. Bayart CB, DeNiro KL, Brichta L, Craiglow BG, Sidbury R. Topical Janus kinase inhibitors for the treatment of pediatric alopecia areata. J Am Acad Dermatol. (2017) 77:167–70. doi: 10.1016/j.jaad.2017.03.024
48. Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, Espinosa JM. Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep. (2019) 5:365–7. doi: 10.1016/j.jdcr.2019.02.007
49. Craiglow BG. Topical tofacitinib solution for the treatment of alopecia areata affecting eyelashes. JAAD Case Rep. (2018) 4:988–9. doi: 10.1016/j.jdcr.2018.07.018
50. Gilhar A, Keren A, Paus R. JAK inhibitors and alopecia areata. Lancet. (2019) 393:318–9. doi: 10.1016/S0140-6736(18)32987-8
51. Gao L, Bin L, Rafaels NM, Huang L, Potee J, Ruczinski I, et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol. (2015) 136:1591–600. doi: 10.1016/j.jaci.2015.06.047
52. Czarnowicki T, Krueger JG, Guttman-Yassky E. Novel concepts of prevention and treatment of atopic dermatitis through barrier and immune manipulations with implications for the atopic march. J Allergy Clin Immun. (2017) 139:1723–34. doi: 10.1016/j.jaci.2017.04.004
53. Werfel T, Allam JP, Biedermann T, Eyerich K, Gilles S, Guttman-Yassky E, et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J Allergy Clin Immun. (2016) 138:336–49. doi: 10.1016/j.jaci.2016.06.010
54. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. (2007) 87:971–8. doi: 10.1038/labinvest.3700663
55. Saluja R, Ketelaar ME, Hawro T, Church MK, Maurer M, Nawijn MC. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol Immunol. (2015) 63:80–5. doi: 10.1016/j.molimm.2014.06.018
56. Kamijo H, Miyagaki T, Hayashi Y, Akatsuka T, Watanabe-Otobe S, Oka T, et al. Increased interleukin-26 expression promotes Th17 and Th2-associated cytokine production by keratinocytes in atopic dermatitis. J Invest Dermatol. (2019) doi: 10.1016/j.jid.2019.07.713. [Epub ahead of print].
57. Simpson EL, Akinlade B, Ardeleanu M. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. (2017) 376:1090–1. doi: 10.1056/NEJMc1700366
58. Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol. (2019) 143:135–41. doi: 10.1016/j.jaci.2018.05.029
59. Kabashima K, Furue M, Hanifin JM, Pulka G, Wollenberg A, Galus R, et al. Nemolizumab in patients with moderate-to-severe atopic dermatitis: Randomized, phase II, long-term extension study. J Allergy Clin Immunol. (2018) 142:1121–30.e7. doi: 10.1016/j.jaci.2018.03.018
60. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taieb A, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. (2018) 78:863–71.e11. doi: 10.1016/j.jaad.2018.01.017
61. Smith D, Helgason H, Sulem P, Bjornsdottir US, Lim AC, Sveinbjornsson G, et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLoS Genet. (2017) 13:1006659. doi: 10.1371/journal.pgen.1006659
62. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. (2015) 136:667–77.e7. doi: 10.1016/j.jaci.2015.03.051
63. He H, Guttman-Yassky E. JAK inhibitors for atopic dermatitis: an update. Am J Clin Dermatol. (2019) 20:181–92. doi: 10.1007/s40257-018-0413-2
64. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. (2011) 186:4234–43. doi: 10.4049/jimmunol.1003668
65. Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. (2015) 73:395–9. doi: 10.1016/j.jaad.2015.06.045
66. Morris GM, Nahmias ZP, Kim BS. Simultaneous improvement of alopecia universalis and atopic dermatitis in a patient treated with a JAK inhibitor. JAAD Case Rep. (2018) 4:515–7. doi: 10.1016/j.jdcr.2017.12.016
67. Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. (2016) 175:902–11. doi: 10.1111/bjd.14871
68. Guttman-Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. (2019) 80:913–21.e9. doi: 10.1016/j.jaad.2018.01.018
69. Haugh IM, Watson IT, Alan Menter M. Successful treatment of atopic dermatitis with the JAK1 inhibitor oclacitinib. Proc (Bayl Univ Med Cent). (2018) 31:524–5. doi: 10.1080/08998280.2018.1480246
70. Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. (2018) 45:701–9. doi: 10.1111/1346-8138.14322
71. Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfo namide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. (2018) 61:1130–52. doi: 10.1021/acs.jmedchem.7b01598
72. Thompson C, Piguet V, Choy E. The pathogenesis of dermatomyositis. Br J Dermatol. (2018) 179:1256–62. doi: 10.1111/bjd.15607
73. Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen AL. Classification and management of adult inflammatory myopathies. Lancet Neurol. (2018) 17:816–28. doi: 10.1016/S1474-4422(18)30254-0
74. Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. (2016) 28:636–44. doi: 10.1097/BOR.0000000000000329
75. Zhang W, Jiang SP, Huang L. Dermatomyositis and malignancy: a retrospective study of 115 cases. Eur Rev Med Pharmacol Sci. (2009) 13:77–80.
76. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. (2005) 57:664–78. doi: 10.1002/ana.20464
77. Greenberg SA, Sanoudou D, Haslett JN, Kohane IS, Kunkel LM, Beggs AH, et al. Molecular profiles of inflammatory myopathies. Neurology. (2002) 59:1170–82. doi: 10.1212/WNL.59.8.1170
78. Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. (1993) 329:1993–2000. doi: 10.1056/NEJM199312303292704
79. Hornig J, Weinhage T, Schmidt LH, Buerke B, Schneider U, Pavenstadt H, et al. Response of dermatomyositis with lung involvement to Janus kinase inhibitor treatment. Z Rheumatol. (2018) 77:952–7. doi: 10.1007/s00393-018-0565-8
80. Hornung T, Janzen V, Heidgen FJ, Wolf D, Bieber T, Wenzel J. Remission of recalcitrant dermatomyositis treated with ruxolitinib. N Engl J Med. (2014) 371:2537–8. doi: 10.1056/NEJMc1412997
81. Kurtzman DJ, Wright NA, Lin J, Femia AN, Merola JF, Patel M, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. (2016) 152:944–5. doi: 10.1001/jamadermatol.2016.0866
82. Wendel S, Venhoff N, Frye BC, May AM, Agarwal P, Rizzi M, et al. Successful treatment of extensive calcifications and acute pulmonary involvement in dermatomyositis with the Janus-Kinase inhibitor tofacitinib - a report of two cases. J Autoimmun. (2019) 100:131–6. doi: 10.1016/j.jaut.2019.03.003
83. Chen Z, Wang X, Ye S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N Engl J Med. (2019) 381:291–3. doi: 10.1056/NEJMc1900045
84. Kurasawa K, Arai S, Namiki Y, Tanaka A, Takamura Y, Owada T, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. (2018) 57:2114–9. doi: 10.1093/rheumatology/key188
85. Aractingi S, Chosidow O. Cutaneous graft-versus-host disease. Arch Dermatol. (1998) 134:602–12. doi: 10.1001/archderm.134.5.602
86. Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. Pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. (2012) 66:515.e1–18. doi: 10.1016/j.jaad.2011.11.960
87. Bruggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, et al. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood. (2014) 123:290–9. doi: 10.1182/blood-2013-07-514372
88. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood. (2005) 106:2903–11. doi: 10.1182/blood-2005-03-1257
89. Ghoreschi K, Thomas P, Penovici M, Ullmann J, Sander CA, Ledderose G, et al. PUVA-bath photochemotherapy and isotretinoin in sclerodermatous graft-versus-host disease. Eur J Dermatol. (2008) 18:667–70. doi: 10.1684/ejd.2008.0517
90. Imamura M, Hashino S, Kobayashi H, Kubayashi S, Hirano S, Minagawa T, et al. Serum cytokine levels in bone marrow transplantation: synergistic interaction of interleukin-6, interferon-gamma, and tumor necrosis factor-alpha in graft-versus-host disease. Bone Marrow Transplant. (1994) 13:745–51.
91. Lin MT, Storer B, Martin PJ, Tseng LH, Gooley T, Chen PJ, et al. Relation of an interleukin-10 promoter polymorphism to graft-versus-host disease and survival after hematopoietic-cell transplantation. N Engl J Med. (2003) 349:2201–10. doi: 10.1056/NEJMoa022060
92. Cavet J, Dickinson AM, Norden J, Taylor PR, Jackson GH, Middleton PG. Interferon-gamma and interleukin-6 gene polymorphisms associate with graft-versus-host disease in HLA-matched sibling bone marrow transplantation. Blood. (2001) 98:1594–600. doi: 10.1182/blood.V98.5.1594
93. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood. (2012) 120:4093–103. doi: 10.1182/blood-2012-01-403196
94. Carniti C, Gimondi S, Vendramin A, Recordati C, Confalonieri D, Bermema A, et al. Pharmacologic inhibition of JAK1/JAK2 signaling reduces experimental murine acute GVHD while preserving GVT effects. Clin Cancer Res. (2015) 21:3740–9. doi: 10.1158/1078-0432.CCR-14-2758
95. Okiyama N, Furumoto Y, Villarroel VA, Linton JT, Tsai WL, Gutermuth J, et al. Reversal of CD8 T-cell-mediated mucocutaneous graft-versus-host-like disease by the JAK inhibitor tofacitinib. J Invest Dermatol. (2014) 134:992–1000. doi: 10.1038/jid.2013.476
96. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. (2014) 123:3832–42. doi: 10.1182/blood-2013-12-543736
97. Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia. (2015) 29:2062–8. doi: 10.1038/leu.2015.212
98. von Bubnoff N, Ihorst G, Grishina O, Rothling N, Bertz H, Duyster J, et al. Ruxolitinib in GvHD (RIG) study: a multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer. (2018) 18:1132. doi: 10.1186/s12885-018-5045-7
99. Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. (2017) 318:2019–32. doi: 10.1001/jama.2017.16691
100. Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma-secretase gene mutations in familial acne inversa. Science. (2010) 330:1065. doi: 10.1126/science.1196284
101. Mendonca CO, Griffiths CE. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. (2006) 154:977–8. doi: 10.1111/j.1365-2133.2006.07155.x
102. Bettoli V, Join-Lambert O, Nassif A. Antibiotic treatment of hidradenitis suppurativa. Dermatol Clin. (2016) 34:81–9. doi: 10.1016/j.det.2015.08.013
103. Hotz C, Boniotto M, Guguin A, Surenaud M, Jean-Louis F, Tisserand P, et al. Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. J Invest Dermatol. (2016) 136:1768–80. doi: 10.1016/j.jid.2016.04.036
104. Wolk K, Warszawska K, Hoeflich C, Witte E, Schneider-Burrus S, Witte K, et al. Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol. (2011) 186:1228–39. doi: 10.4049/jimmunol.0903907
105. Kimball AB, Tzellos T, Calimlim BM, Teixeira HD, Geng Z, Okun MM. Achieving hidradenitis suppurativa response score is associated with significant improvement in clinical and patient-reported outcomes: post hoc analysis of pooled data from PIONEER I and II. Acta Derm Venereol. (2018) 98:932–7. doi: 10.2340/00015555-3012
106. Blok JL, Li K, Brodmerkel C, Horvatovich P, Jonkman MF, Horvath B. Ustekinumab in hidradenitis suppurativa: clinical results and a search for potential biomarkers in serum. Br J Dermatol. (2016) 174:839–46. doi: 10.1111/bjd.14338
107. Le Cleach L, Chosidow O. Clinical practice. Lichen planus. N Engl J Med. (2012) 366:723–32. doi: 10.1056/NEJMcp1103641
108. Wenzel J, Scheler M, Proelss J, Bieber T, Tuting T. Type I interferon-associated cytotoxic inflammation in lichen planus. J Cutan Pathol. (2006) 33:672–8. doi: 10.1111/j.1600-0560.2006.00527.x
109. Shao S, Tsoi LC, Sarkar MK, Xing X, Xue K, Uppala R, et al. IFN-γ enhances cell-mediated cytotoxicity against keratinocytes via JAK2/STAT1 in lichen planus. Sci Transl Med. (2019) 11:eaav7561. doi: 10.1126/scitranslmed.aav7561
110. Chen J, Feng J, Chen X, Xu H, Zhou Z, Shen X, et al. Immunoexpression of interleukin-22 and interleukin-23 in oral and cutaneous lichen planus lesions: a preliminary study. Mediators Inflamm. (2013) 2013:801974. doi: 10.1155/2013/801974
111. Piccinni MP, Lombardelli L, Logiodice F, Tesi D, Kullolli O, Biagiotti R, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. (2014) 20:212–8. doi: 10.1111/odi.12094
112. Shaker O, Hassan AS. Possible role of interleukin-17 in the pathogenesis of lichen planus. Br J Dermatol. (2012) 166:1367–8. doi: 10.1111/j.1365-2133.2011.10793.x
113. Yang CC, Khanna T, Sallee B, Christiano AM, Bordone LA. Tofacitinib for the treatment of lichen planopilaris: a case series. Dermatol Ther. (2018) 31:e12656. doi: 10.1111/dth.12656
114. Merola JF, Elewski B, Tatulych S, Lan S, Tallman A, Kaur M. Efficacy of tofacitinib for the treatment of nail psoriasis: Two 52-week, randomized, controlled phase 3 studies in patients with moderate-to-severe plaque psoriasis. J Am Acad Dermatol. (2017) 77:79–87.e1. doi: 10.1016/j.jaad.2017.01.053
115. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. (2014) 384:1878–88. doi: 10.1016/S0140-6736(14)60128-8
116. Dorgham K, Amoura Z, Parizot C, Arnaud L, Frances C, Pionneau C, et al. Ultraviolet light converts propranolol, a nonselective beta-blocker and potential lupus-inducing drug, into a proinflammatory AhR ligand. Eur J Immunol. (2015) 45:3174–87. doi: 10.1002/eji.201445144
117. Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. (2010) 6:683–92. doi: 10.1038/nrrheum.2010.176
118. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. (2018) 359:1156–61. doi: 10.1126/science.aar7201
119. Kuhn A, Wenzel J, Bijl M. Lupus erythematosus revisited. Semin Immunopathol. (2016) 38:97–112. doi: 10.1007/s00281-015-0550-0
120. Alunno A, Padjen I, Fanouriakis A, Boumpas DT. Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells. (2019) 8:E898. doi: 10.3390/cells8080898
121. Dong J, Wang QX, Zhou CY, Ma XF, Zhang YC. Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus. (2007) 16:101–9. doi: 10.1177/0961203306075383
122. Hadj-Slimane R, Chelbi-Alix MK, Tovey MG, Bobe P. An essential role for IFN-alpha in the overexpression of Fas ligand on MRL/lpr lymphocytes and on their spontaneous Fas-mediated cytotoxic potential. J Interferon Cytokine Res. (2004) 24:717–28. doi: 10.1089/jir.2004.24.717
123. Yiu G, Rasmussen TK, Ajami B, Haddon DJ, Chu AD, Tangsombatvisit S, et al. Development of Th17-associated interstitial kidney inflammation in lupus-prone mice lacking the gene encoding STAT-1. Arthritis Rheumatol. (2016) 68:1233–44. doi: 10.1002/art.39535
124. Karonitsch T, Feierl E, Steiner CW, Dalwigk K, Korb A, Binder N, et al. Activation of the interferon-gamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum. (2009) 60:1463–71. doi: 10.1002/art.24449
125. Lu MC, Lai NS, Chen HC, Yu HC, Huang KY, Tung CH, et al. Decreased microRNA(miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol. (2013) 171:91–9. doi: 10.1111/j.1365-2249.2012.04676.x
126. Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity. (2016) 44:683–97. doi: 10.1016/j.immuni.2016.02.012
127. Ding C, Chen X, Dascani P, Hu X, Bolli R, Zhang HG, et al. STAT3 signaling in B cells is critical for germinal center maintenance and contributes to the pathogenesis of murine models of lupus. J Immunol. (2016) 196:4477–86. doi: 10.4049/jimmunol.1502043
128. Xu Z, Duan B, Croker BP, Morel L. STAT4 deficiency reduces autoantibody production and glomerulonephritis in a mouse model of lupus. Clin Immunol. (2006) 120:189–98. doi: 10.1016/j.clim.2006.03.009
129. Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol. (2011) 187:3840–53. doi: 10.4049/jimmunol.1101228
130. Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus. (2016) 25:1420–30. doi: 10.1177/0961203316640910
131. Kubo S, Nakayamada S, Tanaka Y. Baricitinib for the treatment of rheumatoid arthritis and systemic lupus erythematosus: a 2019 update. Expert Rev Clin Immunol. (2019) 15:693–700. doi: 10.1080/1744666X.2019.1608821
132. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
133. Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. (2005) 11:43–9. doi: 10.1038/nm1162
134. Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an emerging target for topical treatment of inflammatory skin diseases. PLoS ONE. (2016) 11:e0164080. doi: 10.1371/journal.pone.0164080
135. Ishizaki M, Akimoto T, Muromoto R, Yokoyama M, Ohshiro Y, Sekine Y, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J Immunol. (2011) 187:181–9. doi: 10.4049/jimmunol.1003244
136. Works MG, Yin F, Yin CC, Yiu Y, Shew K, Tran TT, et al. Inhibition of TYK2 and JAK1 ameliorates imiquimod-induced psoriasis-like dermatitis by inhibiting IL-22 and the IL-23/IL-17 axis. J Immunol. (2014) 193:3278–87. doi: 10.4049/jimmunol.1400205
137. Krueger J, Clark JD, Suarez-Farinas M, Fuentes-Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: a randomized phase 2 study. J Allergy Clin Immunol. (2016) 137:1079–90. doi: 10.1016/j.jaci.2015.12.1318
138. Zhang J, Tsai TF, Lee MG, Zheng M, Wang G, Jin H, et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: a phase 3, randomized, double-blind, placebo-controlled study. J Dermatol Sci. (2017) 88:36–45. doi: 10.1016/j.jdermsci.2017.05.004
139. Papp KA, Menter MA, Abe M, Elewski B, Feldman SR, Gottlieb AB, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. (2015) 173:949–61. doi: 10.1111/bjd.14018
140. Bissonnette R, Iversen L, Sofen H, Griffiths CE, Foley P, Romiti R, et al. Tofacitinib withdrawal and retreatment in moderate-to-severe chronic plaque psoriasis: a randomized controlled trial. Br J Dermatol. (2015) 172:1395–406. doi: 10.1111/bjd.13551
141. Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. (2015) 386:552–61. doi: 10.1016/S0140-6736(14)62113-9
142. Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. (2017) 377:1525–36. doi: 10.1056/NEJMoa1615977
143. Valenzuela F, Korman NJ, Bissonnette R, Bakos N, Tsai TF, Harper MK, et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: long-term safety and efficacy in an open-label extension study. Br J Dermatol. (2018) 179:853–62. doi: 10.1111/bjd.16798
144. Wolk R, Armstrong EJ, Hansen PR, Thiers B, Lan S, Tallman AM, et al. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J Clin Lipidol. (2017) 11:1243–56. doi: 10.1016/j.jacl.2017.06.012
145. Bissonnette R, Luchi M, Fidelus-Gort R, Jackson S, Zhang H, Flores R, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J Dermatolog Treat. (2016) 27:332–8. doi: 10.3109/09546634.2015.1115819
146. Papp K, Pariser D, Catlin M, Wierz G, Ball G, Akinlade B, et al. A phase 2a randomized, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus kinase inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2015) 173:767–76. doi: 10.1111/bjd.13745
147. Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br J Dermatol. (2016) 174:985–95. doi: 10.1111/bjd.14399
148. Papp KA, Bissonnette R, Gooderham M, Feldman SR, Iversen L, Soung J, et al. Treatment of plaque psoriasis with an ointment formulation of the Janus kinase inhibitor, tofacitinib: a Phase 2b randomized clinical trial. BMC Dermatol. (2016) 16:15. doi: 10.1186/s12895-016-0051-4
149. Ports WC, Khan S, Lan S, Lamba M, Bolduc C, Bissonnette R, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. (2013) 169:137–45. doi: 10.1111/bjd.12266
150. Bagherani N, Smoller BR. Efficacy of topical tofacitinib, a Janus kinase inhibitor, in the treatment of plaque psoriasis. Dermatol Ther. (2017) 30:e12467. doi: 10.1111/dth.12467
151. Punwani N, Scherle P, Flores R, Shi J, Liang J, Yeleswaram S, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. (2012) 67:658–64. doi: 10.1016/j.jaad.2011.12.018
152. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2016) 174:1266–76. doi: 10.1111/bjd.14403
153. Papp K, Gordon K, Thaci D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. (2018) 379:1313–21. doi: 10.1056/NEJMoa1806382
154. Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, et al. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. (2015) 64:41–51. doi: 10.1007/s00011-014-0782-9
155. Taieb A, Picardo M. Clinical practice. Vitiligo. N Engl J Med. (2009) 360:160–9. doi: 10.1056/NEJMcp0804388
156. Grimes PE, Morris R, Avaniss-Aghajani E, Soriano T, Meraz M, Metzger A. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. (2004) 51:52–61. doi: 10.1016/j.jaad.2003.12.031
157. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. (2012) 132:1869–76. doi: 10.1038/jid.2011.463
158. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra23. doi: 10.1126/scitranslmed.3007811
159. Wankowicz-Kalinska A, van den Wijngaard RM, Tigges BJ, Westerhof W, Ogg GS, Cerundolo V, et al. Immunopolarization of CD4+ and CD8+ T cells to Type-1-like is associated with melanocyte loss in human vitiligo. Lab Invest. (2003) 83:683–95. doi: 10.1097/01.LAB.0000069521.42488.1B
160. Agarwal P, Rashighi M, Essien KI, Richmond JM, Randall L, Pazoki-Toroudi H, et al. Simvastatin prevents and reverses depigmentation in a mouse model of vitiligo. J Invest Dermatol. (2015) 135:1080–8. doi: 10.1038/jid.2014.529
161. Relke N, Gooderham M. The use of Janus kinase inhibitors in vitiligo: a review of the literature. J Cutan Med Surg. (2019) 23:298–306. doi: 10.1177/1203475419833609
162. Rothstein B, Joshipura D, Saraiya A, Abdat R, Ashkar H, Turkowski Y, et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J Am Acad Dermatol. (2017) 76:1054–60.e1. doi: 10.1016/j.jaad.2017.02.049
163. Mobasher P, Guerra R, Jiayang Li S, Frangos J, Ganesan AK, Huang V. Open label pilot study of 2% tofacitinib for the treatment of refractory vitiligo. Br J Dermatol. (2019). doi: 10.1111/bjd.18606
164. Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J Am Acad Dermatol. (2017) 77:675–682.e1. doi: 10.1016/j.jaad.2017.05.043
165. Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus an update. Am J Clin Dermatol. (2013) 14:27–47. doi: 10.1007/s40257-012-0006-4
166. Terlou A, Santegoets LA, van der Meijden WI, Heijmans-Antonissen C, Swagemakers SM, van der Spek PJ, et al. An autoimmune phenotype in vulvar lichen sclerosus and lichen planus: a Th1 response and high levels of microRNA-155. J Invest Dermatol. (2012) 132(3 Pt 1):658–66. doi: 10.1038/jid.2011.369
167. Deverapalli SC, Rosmarin D. The use of JAK inhibitors in the treatment of progressive systemic sclerosis. J Eur Acad Dermatol Venereol. (2018) 32:e328. doi: 10.1111/jdv.14876
168. Kim SR, Charos A, Damsky W, Heald P, Girardi M, King BA. Treatment of generalized deep morphea and eosinophilic fasciitis with the Janus kinase inhibitor tofacitinib. JAAD Case Rep. (2018) 4:443–5. doi: 10.1016/j.jdcr.2017.12.003
169. Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. (2017) 8:1130. doi: 10.1038/s41467-017-01236-6
170. DeFilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. (2015) 172:1487–97. doi: 10.1111/bjd.13493
171. Nasifoglu S, Heinrich B, Welzel J. Successful therapy for pyoderma gangrenosum with a Janus kinase 2 inhibitor. Br J Dermatol. (2018) 179:504–5. doi: 10.1111/bjd.16468
172. Freysdottir J, Zhang S, Tilakaratne WM, Fortune F. Oral biopsies from patients with orofacial granulomatosis with histology resembling Crohn's disease have a prominent Th1 environment. Inflamm Bowel Dis. (2007) 13:439–45. doi: 10.1002/ibd.20023
173. Thomas P, Walchner M, Ghoreschi K, Rocken M. Successful treatment of granulomatous cheilitis with thalidomide. Arch Dermatol. (2003) 139:136–8. doi: 10.1001/archderm.139.2.136
174. Solimani F, Eming R, Juratli HA, Scarsella L, Gschnell M, Pfutzner W. Successful treatment of cheilitis granulomatosa with lenalidomide. J Eur Acad Dermatol Venereol. (2019) 33:e357–e9. doi: 10.1111/jdv.15654
175. McHugh SM, Rifkin IR, Deighton J, Wilson AB, Lachmann PJ, Lockwood CM, et al. The immunosuppressive drug thalidomide induces T helper cell type 2 (Th2) and concomitantly inhibits Th1 cytokine production in mitogen- and antigen-stimulated human peripheral blood mononuclear cell cultures. Clin Exp Immunol. (1995) 99:160–7. doi: 10.1111/j.1365-2249.1995.tb05527.x
176. Vavricka SR, Galvan JA, Dawson H, Soltermann A, Biedermann L, Scharl M, et al. Expression patterns of TNFα, MAdCAM1, and STAT3 in intestinal and skin manifestations of inflammatory bowel disease. J Crohns Colitis. (2018) 12:347–54. doi: 10.1093/ecco-jcc/jjx158
177. Zhou T, Casanova N, Pouladi N, Wang T, Lussier Y, Knox KS, et al. Identification of Jak-STAT signaling involvement in sarcoidosis severity via a novel microRNA-regulated peripheral blood mononuclear cell gene signature. Sci Rep. (2017) 7:4237. doi: 10.1038/s41598-017-04109-6
178. Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med. (2018) 379:2540–6. doi: 10.1056/NEJMoa1805958
179. Damsky W, Thakral D, McGeary MK, Leventhal J, Galan A, King B. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. (2019) doi: 10.1016/j.jaad.2019.05.098. [Epub ahead of print].
180. Wei JJ, Kallenbach LR, Kreider M, Leung TH, Rosenbach M. Resolution of cutaneous sarcoidosis after Janus kinase inhibitor therapy for concomitant polycythemia vera. JAAD Case Rep. (2019) 5:360–1. doi: 10.1016/j.jdcr.2019.02.006
181. Davies R, Sarkar I, Hammenfors D, Bergum B, Vogelsang P, Solberg SM, et al. Single cell based phosphorylation profiling identifies alterations in toll-like receptor 7 and 9 signaling in patients with primary sjogren's syndrome. Front Immunol. (2019) 10:281. doi: 10.3389/fimmu.2019.00281
183. Davies R, Hammenfors D, Bergum B, Vogelsang P, Gavasso S, Brun JG, et al. Aberrant cell signalling in PBMCs upon IFN-α stimulation in primary Sjogren's syndrome patients associates with type I interferon signature. Eur J Immunol. (2018) 48:1217–27. doi: 10.1002/eji.201747213
184. Liew SH, Nichols KK, Klamerus KJ, Li JZ, Zhang M, Foulks GN. Tofacitinib (CP-690,550), a Janus kinase inhibitor for dry eye disease: results from a phase 1/2 trial. Ophthalmology. (2012) 119:1328–35. doi: 10.1016/j.ophtha.2012.01.028
185. Juczynska K, Wozniacka A, Waszczykowska E, Danilewicz M, Wagrowska-Danilewicz M, Wieczfinska J, et al. Expression of the JAK/STAT signaling pathway in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflamm. (2017) 2017:6716419. doi: 10.1155/2017/6716419
186. Tavakolpour S. Tofacitinib as the potent treatment for refractory pemphigus: a possible alternative treatment for pemphigus. Dermatol Ther. (2018) 31:e12696. doi: 10.1111/dth.12696
187. Sarny S, Hucke M, El-Shabrawi Y. Treatment of mucous membrane pemphigoid with janus kinase inhibitor baricitinib. JAMA Ophthalmol. (2018) 136:1420–2. doi: 10.1001/jamaophthalmol.2018.3789
188. Onda M, Ghoreschi K, Steward-Tharp S, Thomas C, O'Shea JJ, Pastan IH, et al. Tofacitinib suppresses antibody responses to protein therapeutics in murine hosts. J Immunol. (2014) 193:48–55. doi: 10.4049/jimmunol.1400063
189. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. (2013) 190:3049–53. doi: 10.4049/jimmunol.1203032
190. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. (2012) 119:3997–4008. doi: 10.1182/blood-2011-11-392985
191. Amber KT, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
192. Didona D, Maglie R, Eming R, Hertl M. Pemphigus: current and future therapeutic strategies. Front Immunol. (2019) 10:1418. doi: 10.3389/fimmu.2019.01418
193. Worm M, Bauer A, Elsner P, Mahler V, Molin S, Nielsen TSS. Efficacy and safety of topical delgocitinib in patients with chronic hand eczema: data from a randomised, double-blind, vehicle-controlled Phase 2a study. Br J Dermatol. (2019). doi: 10.1111/bjd.18469
194. Bechara R, Antonios D, Azouri H, Pallardy M. Nickel sulfate promotes IL-17A producing CD4+ T cells by an IL-23-dependent mechanism regulated by TLR4 and Jak-STAT pathways. J Invest Dermatol. (2017) 137:2140–8. doi: 10.1016/j.jid.2017.05.025
195. Fujii Y, Sengoku T. Effects of the Janus kinase inhibitor CP-690550 (tofacitinib) in a rat model of oxazolone-induced chronic dermatitis. Pharmacology. (2013) 91:207–13. doi: 10.1159/000347184
196. Fukuyama T, Ehling S, Cook E, Baumer W. Topically administered Janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. (2015) 354:394–405. doi: 10.1124/jpet.115.223784
197. Puccetti A, Fiore PF, Pelosi A, Tinazzi E, Patuzzo G, Argentino G, et al. Gene expression profiling in behcet's disease indicates an autoimmune component in the pathogenesis of the disease and opens new avenues for targeted therapy. J Immunol Res. (2018) 2018:4246965. doi: 10.1155/2018/4246965
198. Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behcet's disease: a comprehensive review. J Autoimmun. (2015) 64:137–48. doi: 10.1016/j.jaut.2015.08.013
199. Tulunay A, Dozmorov MG, Ture-Ozdemir F, Yilmaz V, Eksioglu-Demiralp E, Alibaz-Oner F, et al. Activation of the JAK/STAT pathway in Behcet's disease. Genes Immun. (2015) 16:170–5. doi: 10.1038/gene.2014.64
200. Yang Q, Zhao Y, Li C, Luo Y, Hao W, Zhang W. Case report: successful treatment of refractory SAPHO syndrome with the JAK inhibitor tofacitinib. Medicine. (2018) 97:e11149. doi: 10.1097/MD.0000000000011149
201. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
202. Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, et al. Pharmacokinetics, pharmacodynamics, and proposed dosing of the Oral JAK1 and JAK2 inhibitor baricitinib in pediatric and young adult CANDLE and SAVI patients. Clin Pharmacol Ther. (2018) 104:364–73. doi: 10.1002/cpt.936
203. Verden A, Dimbil M, Kyle R, Overstreet B, Hoffman KB. Analysis of spontaneous postmarket case reports submitted to the FDA regarding thromboembolic adverse events and JAK inhibitors. Drug Saf . (2018) 41:357–61. doi: 10.1007/s40264-017-0622-2
204. Tarp S, Eric Furst D, Boers M, Luta G, Bliddal H, Tarp U, et al. Risk of serious adverse effects of biological and targeted drugs in patients with rheumatoid arthritis: a systematic review meta-analysis. Rheumatology. (2017) 56:417–25. doi: 10.1093/rheumatology/kew442
205. Scott IC, Hider SL, Scott DL. Thromboembolism with Janus kinase (JAK) Inhibitors for rheumatoid arthritis: how real is the risk? Drug Saf . (2018) 41:645–53. doi: 10.1007/s40264-018-0651-5
206. Sands BE, Taub PR, Armuzzi A, Friedman GS, Moscariello M, Lawendy N, et al. Tofacitinib treatment is associated with modest and reversible increases in serum lipids in patients with ulcerative colitis. Clin Gastroenterol Hepatol. (2019) doi: 10.1016/j.cgh.2019.04.059. [Epub ahead of print].
207. Novikova DS, Udachkina HV, Markelova EI, Kirillova IG, Misiyuk AS, Demidova NV, et al. Dynamics of body mass index and visceral adiposity index in patients with rheumatoid arthritis treated with tofacitinib. Rheumatol Int. (2019) 39:1181–9. doi: 10.1007/s00296-019-04303-x
208. Saeed I, McLornan D, Harrison CN. Managing side effects of JAK inhibitors for myelofibrosis in clinical practice. Expert Rev Hematol. (2017) 10:617–25. doi: 10.1080/17474086.2017.1337507
209. Perez-Baos S, Barrasa JI, Gratal P, Larranaga-Vera A, Prieto-Potin I, Herrero-Beaumont G, et al. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. Br J Pharmacol. (2017) 174:3018–31. doi: 10.1111/bph.13932
210. Hammaren HM, Virtanen AT, Abraham BG, Peussa H, Hubbard SR, Silvennoinen O. Janus kinase 2 activation mechanisms revealed by analysis of suppressing mutations. J Allergy Clin Immunol. (2019) 143:1549–59.e6. doi: 10.1016/j.jaci.2018.07.022
211. Lademann J, Richter H, Meinke MC, Lange-Asschenfeldt B, Antoniou C, Mak WC, et al. Drug delivery with topically applied nanoparticles: science fiction or reality. Skin Pharmacol Physiol. (2013) 26:227–33. doi: 10.1159/000351940
212. Lademann J, Richter H, Schanzer S, Meinke MC, Darvin ME, Schleusener J, et al. Follicular penetration of nanocarriers is an important penetration pathway for topically applied drugs. Hautarzt. (2019) 70:185–92. doi: 10.1007/s00105-018-4343-y
213. Lademann J, Schaefer H, Otberg N, Teichmann A, Blume-Peytavi U, Sterry W. Penetration of microparticles into human skin. Hautarzt. (2004) 55:1117–9. doi: 10.1007/s00105-004-0841-1
214. Fleischmann R, Mysler E, Hall S, Kivitz AJ, Moots RJ, Luo Z, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. (2017) 390:457–68. doi: 10.1016/S0140-6736(17)31618-5
215. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. (2017) 376:652–62. doi: 10.1056/NEJMoa1608345
216. van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, Garcia Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. (2012) 367:508–19. doi: 10.1056/NEJMoa1112072
217. Mueller RB, Hasler C, Popp F, Mattow F, Durmisi M, Souza A, et al. Effectiveness, tolerability, and safety of tofacitinib in rheumatoid arthritis: a retrospective analysis of real-world data from the St. Gallen and Aarau cohorts. J Clin Med. (2019) 8:E1548. doi: 10.3390/jcm8101548
218. Ma C, Lee JK, Mitra AR, Teriaky A, Choudhary D, Nguyen TM, et al. Systematic review with meta-analysis: efficacy and safety of oral Janus kinase inhibitors for inflammatory bowel disease. Aliment Pharmacol Ther. (2019) 50:5–23. doi: 10.1111/apt.15297
219. Winthrop KL, Park SH, Gul A, Cardiel MH, Gomez-Reino JJ, Tanaka Y, et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1133–8. doi: 10.1136/annrheumdis-2015-207319
220. Curtis JR, Xie F, Yun H, Bernatsky S, Winthrop KL. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1843–7. doi: 10.1136/annrheumdis-2016-209131
221. Winthrop KL, Wouters AG, Choy EH, Soma K, Hodge JA, Nduaka CI, et al. The safety and immunogenicity of live zoster vaccination in patients with rheumatoid arthritis before starting tofacitinib: a randomized phase II trial. Arthritis Rheumatol. (2017) 69:1969–77. doi: 10.1002/art.40187
222. Almanzar G, Kienle F, Schmalzing M, Maas A, Tony HP, Prelog M. Tofacitinib modulates the VZV-specific CD4+ T cell immune response in vitro in lymphocytes of patients with rheumatoid arthritis. Rheumatology. (2019) 58:2051–60. doi: 10.1093/rheumatology/kez175
223. Winthrop KL, Yamanaka H, Valdez H, Mortensen E, Chew R, Krishnaswami S, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:2675–84. doi: 10.1002/art.38745
224. Weinhold KJ, Bukowski JF, Brennan TV, Noveck RJ, Staats JS, Lin L, et al. Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Immunol. (2018) 191:10–20. doi: 10.1016/j.clim.2018.03.002
225. Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. (2014) 54:1354–61. doi: 10.1002/jcph.354
226. Chen Y, Gong FY, Li ZJ, Gong Z, Zhou Z, Ma SY, et al. A study on the risk of fungal infection with tofacitinib (CP-690550), a novel oral agent for rheumatoid arthritis. Sci Rep. (2017) 7:6779. doi: 10.1038/s41598-017-07261-1
227. Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. (2018) 93:339–47. doi: 10.1002/ajh.24976
228. Strand V, Ahadieh S, French J, Geier J, Krishnaswami S, Menon S, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res Ther. (2015) 17:362. doi: 10.1186/s13075-015-0880-2
229. Curtis JR, Xie F, Yang S, Bernatsky S, Chen L, Yun H, et al. Risk for herpes zoster in tofacitinib-treated rheumatoid arthritis patients with and without concomitant methotrexate and glucocorticoids. Arthritis Care Res. (2019) 71:1249–54. doi: 10.1002/acr.23769
230. Colombel JF. Herpes zoster in patients receiving JAK inhibitors for ulcerative colitis: mechanism, epidemiology, management, and prevention. Inflamm Bowel Dis. (2018) 24:2173–82. doi: 10.1093/ibd/izy150
231. Kotyla PJ. Are Janus kinase inhibitors superior over classic biologic agents in RA patients? Biomed Res Int. (2018) 2018:7492904. doi: 10.1155/2018/7492904
232. Curtis JR, Lee EB, Kaplan IV, Kwok K, Geier J, Benda B, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. (2016) 75:831–41. doi: 10.1136/annrheumdis-2014-205847
233. Harigai M, Takeuchi T, Smolen JS, Winthrop KL, Nishikawa A, Rooney TP, et al. Safety profile of baricitinib in Japanese patients with active rheumatoid arthritis with over 1.6 years median time in treatment: an integrated analysis of Phases 2 and 3 trials. Mod Rheumatol. (2019) 20:1–8. doi: 10.1080/14397595.2019.1583711
234. Sandborn WJ, Panes J, D'Haens GR, Sands BE, Su C, Moscariello M, et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin Gastroenterol Hepatol. (2019) 17:1541–50. doi: 10.1016/j.cgh.2018.11.035
235. Wollenhaupt J, Lee EB, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther. (2019) 21:89. doi: 10.1186/s13075-019-1866-2
236. Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis. (2017) 76:1253–62. doi: 10.1136/annrheumdis-2016-210457
Keywords: inflammatory skin diseases, JAK (Janus kinase), JAK inhibition, pathophysiology, immunopathogenesis, autoimmune skin diseases, JAK/STAT pathway
Citation: Solimani F, Meier K and Ghoreschi K (2019) Emerging Topical and Systemic JAK Inhibitors in Dermatology. Front. Immunol. 10:2847. doi: 10.3389/fimmu.2019.02847
Received: 07 May 2019; Accepted: 19 November 2019;
Published: 03 December 2019.
Edited by:
Michael P. Schön, University Medical Center Göttingen, GermanyReviewed by:
Raju P. Khubchandani, Jaslok Hospital, IndiaLuise Erpenbeck, University of Göttingen, Germany
Copyright © 2019 Solimani, Meier and Ghoreschi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamran Ghoreschi, a2FtcmFuLmdob3Jlc2NoaUBjaGFyaXRlLmRl