 Kelsea M. Drall1
Kelsea M. Drall1 Hein M. Tun1†
Hein M. Tun1† Nadia P. Morales-Lizcano2
Nadia P. Morales-Lizcano2 Theodore B. Konya3
Theodore B. Konya3 David S. Guttman2,4
David S. Guttman2,4 Catherine J. Field5Rupasri Mandal6David S. Wishart6Allan B. Becker7
Catherine J. Field5Rupasri Mandal6David S. Wishart6Allan B. Becker7 Meghan B. Azad7
Meghan B. Azad7 Diana L. Lefebvre8Piush J. Mandhane1
Diana L. Lefebvre8Piush J. Mandhane1 Theo J. Moraes9Malcolm R. Sears8
Theo J. Moraes9Malcolm R. Sears8 Stuart E. Turvey10
Stuart E. Turvey10 Padmaja Subbarao8,9
Padmaja Subbarao8,9 James A. Scott2
James A. Scott2 Anita L. Kozyrskyj1*
Anita L. Kozyrskyj1*- 1Department of Pediatrics, University of Alberta, Edmonton, AB, Canada
- 2Department of Cell & Systems Biology, University of Toronto, Toronto, ON, Canada
- 3Dalla Lana School of Public Health, University of Toronto, Toronto, ON, Canada
- 4Centre for the Analysis of Genome Evolution & Function, University of Toronto, Toronto, ON, Canada
- 5Department of Agricultural, Food and Nutritional Science, University of Alberta, Edmonton, AB, Canada
- 6Department of Biological Sciences, University of Alberta, Edmonton, AB, Canada
- 7Department of Pediatrics and Child Health, Children's Hospital Research Institute of Manitoba, University of Manitoba, Winnipeg, MB, Canada
- 8Department of Medicine, McMaster University, Hamilton, ON, Canada
- 9Department of Pediatrics, Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
- 10Department of Pediatrics, BC Children's Hospital, University of British Columbia, Vancouver, BC, Canada
Colonization with Clostridioides difficile occurs in up to half of infants under the age of 3 months, is strongly influenced by feeding modality and is largely asymptomatic. In spite of this, C. difficile's presence has been associated with susceptibility to chronic disease later in childhood, perhaps by promoting or benefiting from changes in infant gut microbiome development, including colonization with pathogenic bacteria and disrupted production of microbial bioactive metabolites and proteins. In this study, the microbiomes of 1554 infants from the CHILD Cohort Study were described according to C. difficile colonization status and feeding mode at 3–4 months of age. C. difficile colonization was associated with a different gut microbiome profile in exclusively breastfed (EBF) vs. exclusively formula fed (EFF) infants. EBF infants colonized with C. difficile had an increased relative abundance of Firmicutes and Proteobacteria, decreased relative abundance of Bifidobacteriaceae, greater microbiota alpha-diversity, greater detectable fecal short chain fatty acids (SCFA), and lower detectable fecal secretory Immunoglobulin A (sIgA) than those not colonized. Similar but less pronounced differences were seen among partially breastfed infants (PBF) but EFF infants did not possess these differences in the gut microbiome according to colonization status. Thus, breastfed infants colonized with C. difficile appear to possess a gut microbiome that differs from non-colonized infants and resembles that of EFF infants, but the driving force and direction of this association remains unknown. Understanding these compositional differences as drivers of C. difficile colonization may be important to ensure future childhood health.
Introduction
Clostridioides (formerly Clostridium) difficile is a bacterium that is present in the intestine of nearly 40% of infants at 1 month of age, and 30% of infants between the ages of 1 and 6 month (1). C. difficile is the main cause of antibiotic-associated diarrhea in adults (2, 3) and although C. difficile may not be accompanied by diarrheal illness in infants, it has been associated with atopy and microbial dysbiosis (4–6). Furthermore, despite the lack of immediate risks related to carriage of C. difficile in infants, this gram-negative spore-forming bacterium is capable of inducing gut inflammation and disrupting the intestinal epithelial barrier (7, 8). As a result, these less than desirable influences on the intestinal environment may impact the succession and abundance of commensal gut microbiota and overall microbial ecology.
Infancy is a critical period for establishment of the gut microbial ecosystem and immune system priming to confer protection against gut microbial dysbiosis and reduce the risk of negative health outcomes. C. difficile is thought to promote colonization of non-commensals and pathogenic bacteria, although this phenomenon has received little attention in infants. In a small group of infants (n = 53) (6), one study found that Ruminococcus gnavus and Klebsiella pneumoniae species were more prevalent in infants colonized with C. difficile, while non-carriers were more frequently colonized by Bifidobacterium longum. Acquisition of C. difficile during infancy has been attributed to several environmental exposures, notably formula feeding (1, 9, 10). Breastmilk bioactive factors, including human milk oligosaccharides and secretory Immunoglobulin A (sIgA), neutralize toxins and bind pathogens, which may account for asymptomatic colonization of the infant gut with C. difficile and/or lower colonization rates in breastfed infants vs. infants not fed human milk (11–13). Consequently, infants colonized with C. difficile may manifest distinct and persistent changes in their gut ecology, including changes in metabolites, secretory proteins and resident microbiota. Hence, the relationship between C. difficile and the infant gut microbiome merits further examination.
In this study, we report the association between C. difficile (family Peptostreptococcaceae) and other gut microbiome components, including composition, metabolites and sIgA, to provide insights into ecological factors related to C. difficile expansion in infancy. We also explored these differences in exclusively breastfed, partially breastfed, and exclusively formula fed infants to examine the gut microbial community and C. difficile colonization infants with distinct diets.
Methods
Study Design and Population
This study includes a sub-set of 1,562 families enrolled in the CHILD Cohort Study. In this prospective population-based cohort, mothers were recruited and enrolled with informed consent during the second or third trimester of pregnancy between January 2009 and December 2012 from the Vancouver, Edmonton and Manitoba study sites (inclusion and exclusion criteria outlined at www.childstudy.ca) (14). The primary objective of the CHILD Cohort Study was to determine the developmental, environmental, and genetic determinants of later allergy and asthma in childhood (15). All infants included in this subsample provided a fecal sample at 3–4 months of age, which was sequenced by Illumina MiSeq and processed by targeted qPCR to detect C. difficile. Within this study, smaller, yet representative, groups of samples were profiled to describe concentrations of fecal metabolites (n = 467) and secretory IgA (n = 731) (Supplementary Table 1). Gut microbiota compositional findings have previously been described for infants in the CHILD Cohort Study (16), but this paper is the first integration and report of 4 characterizations of the infant gut microbiome and gut immunity from the CHILD Cohort Study. The Human Research Ethics Boards at the University of Manitoba, University of Alberta, and University of British Columbia approved this study.
qPCR for Clostridioides difficile Detection
Fecal samples of 5–10 grams were collected from infant diapers during home-visits conducted at 3–4 months of age by a research assistant or parents according to an approved protocol (Supplementary Figure 1). Samples were aliquoted and stored at −80°C until analyzed. A targeted 16S primer and probe set was used for amplification and quantification of C. difficile and followed the methods set by Penders et al. (17). To minimize differential inhibitory effects due to variable concentrations of genomic template DNA in qPCR, all template DNA samples were first normalized by dilution to 1 ng/μL (18). Then, each multiplex assay was prepared to contain 1X QuantiNova Multiplex PCR Kit (QIAGEN), 0.4 μM of each primer, 0.25 μM of each probe and 1 μL [1 ng/μL] of sample DNA in a final volume of 20 μL. qPCR cycling conditions were as follows: initial denaturation for 2 min at 95.0°C, 40 cycles of denaturation for 5 s at 95°C and annealing/extension/reading for 20 s at 60°C. Oligonucleotides were acquired from IDT (Integrated DNA Technologies Inc, Coralville, IA, USA) and reactions were performed on the MiniOpticonTM Real-Time PCR System (Bio-Rad, Hercules, CA, USA). A standard curve was created and employed to determine the efficiency of the C. difficile primers and probes by performing five 1:10 serial dilutions of C. difficile ATCC 9689D-5 genomic DNA starting at 1 ng/μL. We calculated the lower limit of detection for the multiplex assay to be 1X10-5 ng of DNA or 2 genomes of C. difficile based on the amplification data from the serial dilution and the non-template control. Because each template sample represented a different starting mass of stool, the limit of quantification for the analysis was variable from sample-to-sample, and ranged from 514 to 33,333 genomes/g stool. Infants were classified by C. difficile colonization status (present in fecal sample, yes/no). Amongst colonized infants, median levels of C. difficile (ng/g feces) in infant fecal samples were not different between feeding groups (data not shown).
Fecal Microbiome Analysis
DNA extraction and amplification of bacterial V4 hypervariable region of the bacterial 16S rRNA gene was followed by sequencing and taxonomic classification and was conducted as previously described (19). To summarize, microbial DNA was extracted from the frozen stool samples mentioned above (80 to 200 mg) using the QIAamp DNA Stool Mini kit according to the manufacturer protocol (Qiagen Inc, Valencia CA). Next, the bacterial 16S rRNA genes were amplified at the hypervariable V4 region using PCR with appropriate primers. PCR products were combined for sequencing, performed using the Illumina MiSeq platform (San Diego, CA). Resultant sequences were taxonomically classified and matched at >97% similarity against the Greengenes reference database in QIIME and filtered/excluded if <60% similarity. Finally, microbiota data were rarefied to 13,000 sequences per sample and relative abundances were calculated. At this time, microbiota diversity within samples (alpha diversity) was calculated using standardized estimators of OTU richness and/or evenness: Chao1 and Shannon diversity indices.
Short-Chain Fatty Acid (SCFA) and Other Fecal Metabolites
In a sub-set of fecal samples (N = 467), metabolites were quantified by magnetic resonance spectroscopy (NMR). NMR requires a small quantity of sample for processing and has high reproducibility compared to mass spectrometry (20). Homogenization of 100 mg of sample and subsequent centrifugation were performed as necessary for sample cleaning: Each sample was placed in an Eppendorf tube will 1 mL of ice water, vortexed for 5 min and subjected to sonication for 20 more minutes at 4°C. Samples were then vortexed for another 20 min at 250 rpm. Samples were then centrifuged at 15,000 × g for 1 h at 4°C. The supernatant was removed and placed in a new tube and the process was repeated. The cleaned fecal water was stored at −20°C. After extraction, 280 μL of fecal water was mixed with 70 μL of a standard buffer solution (54% D2O: 46% 750 mM potassium phosphate (mono- and dibasic) pH 7.0 v/v containing 5 mM DSS-d6 (2,2-dimethyl-2-silcepentane-5-sulphonate). The sample (350 μL) was then transferred to 3 mm SampleJet NMR tube for subsequent spectral analysis. All 1H-NMR spectra were collected on a 700 MHz Avance III (Bruker) spectrometer equipped with a 5 mm HCN Z-gradient pulsed-field gradient (PFG) cryoprobe. 1H-NMR spectra were acquired at 25°C using the first transient of the NOESY pre-saturation pulse sequence (noesy1dpr), chosen for its high degree of quantitative accuracy.
Prior to spectral analysis, all FIDs (free induction decays) were zero-filled to 250 K data points and line broadened 0.5 Hz. The methyl singlet produced by a known quantity of DSS was used as an internal standard for chemical shift referencing (set to 0 ppm) and for quantification. All 1H-NMR spectra were processed and analyzed using the Chenomx NMR Suite Professional software package version 8.1 (Chenomx Inc., Edmonton, AB) (11). The Chenomx NMR Suite software allows for qualitative and quantitative analysis of an NMR spectrum by manually fitting spectral signatures from an internal database to the spectrum. Typically 90% of visible peaks were assigned to a compound and more than 90% of the spectral area could be routinely fit using the Chenomx spectral analysis software. Most of the visible peaks are annotated with a compound name. We sought to identify all metabolites relevant to microbial production or substrate use. Metabolites were quantified as μmol/gram feces. In this study, we report on a subset of metabolites measured, specifically the SCFAs acetate, butyrate, and propionate, in addition to other metabolites in the metabolic pathways of C. difficile including para-cresol, succinate, and glutamate (Supplementary Figure 2).
Fecal Secretory IgA
A sub-sample of fecal samples were assayed for sIgA (N = 731) using the Secretory IgA ELISA (enzyme-linked immunosorbent assay) kit (ELISA, Immundiagnostik AG assay, Bensheim, Germany). Approximately 14 mg of fecal sample was used for the sIgA analyses. Samples were run in duplicate according to the manufacturer's protocol, as previously described (21), and quantified as the average milligram of sIgA per gram wet weight feces (mg/g). To summarize, a fecal sample aliquot for each infant was thawed and an IDK Extract buffer was used to extract fecal sIgA. Samples were then diluted (1:125) with a wash buffer and placed in a microtiter plate along with controls and standards. Wells were aspirated, washed and 100 μL of conjugate was added and allowed to incubate at room temperature. Samples were then shaken on a horizontal mixer, washed with TMB substrate and incubated in the dark (20 min). An ELISA reader was used to measure the absorption at 450 nm (620 nm reference). The reads were multiplied by 12,500 and compared against a standard curve, created using standards provided with the assay kit, for quantification.
Covariate Data
Breastfeeding status was determined through self-report questionnaires administered to mothers at 3–4 months postpartum (N = 1,554). A 3-category variable was created for infant breastfeeding status at the time of stool sample collection and questionnaire administration: (1) exclusively breastfed (EBF), (2) partially (i.e., mixed) breastfed (PBF), and (3) exclusively formula fed (EFF). Complete feeding data were missing in 8 infants, leaving a total of 1554 infants (not the full N = 1,562 with available C. difficile and microbiome data) that were stratified by feeding mode.
Statistical Analysis
All statistical analysis was conducted using Stata (version 13), RStudio (version 1.1.456), and the Galaxy platform (MaAslin) between September 2018 and March 2019. Non-parametric (Mann-Whitney U or Kruskal–Wallis test) and parametric (student's t-test) tests were used where appropriate (Supplementary Figure 3) to compare alpha diversity indices, fecal metabolites, and fecal sIgA according to colonization status. Differences in taxon relative abundance (outcomes) according to C. difficile colonization status (predictor) were determined using the multivariate association with linear models method developed by the Huttenhower lab (MaAslin) (22) (available at: https://huttenhower.sph.harvard.edu/galaxy/). Spearman correlations were computed in Supplementary Table 2, and heatplots were generated using the gplots package and the heatmap.2() command in R. Scatter bar graphs were generated using the ggplots2 package and the geom_boxplot() and geom_beeswarm() commands. Statistical significance was defined as a two-sided p or q-value < 0.05, after FDR correction for multiple comparisons.
Results and Discussion
The prevalence of C. difficile colonization among all study infants was 30.9% (n = 482/1562), which aligns with previously reported estimates (1). These colonization rates differed between feeding groups: 22.6% for EBF, 36.0% for PBF and 49.6% for EFF infants (χ2: 76.71, p < 0.001, Figure 1A, N = 1,554). The mean Shannon and Chao1 indices for EBF and PBF infants were lower for infants who lacked C. difficile compared to infants colonized with C. difficile, suggesting that the richness and abundance of the infant gut microbiota are greater and more equally distributed in the presence of C. difficile (p < 0.05, Figures 1B,C). No differences in alpha diversity were detected with C. difficile colonization in EFF infants. These differences across feeding modality could not be attributed to the normal progression of microbiota development since infant age [median (IQR)] in each of the feeding groups was similar: 3.29 months (1.03) for EBF, 3.33 months (0.94) for PBF, and 3.20 months (1.10) for EFF, p = 0.27.
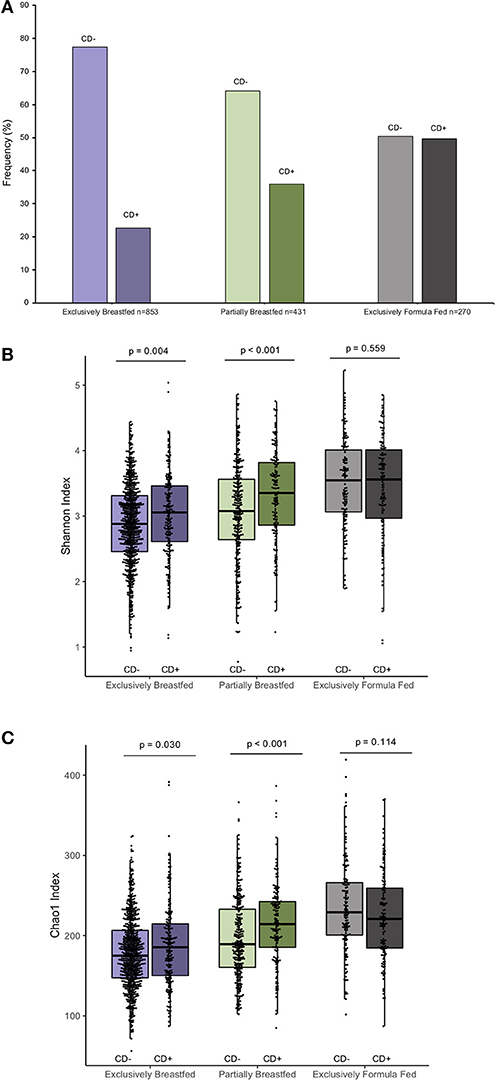
Figure 1. Frequency of C. difficile colonization in our study population and infant microbial alpha-diversity according feeding mode (n = 1,554). Colonization rates differ within feeding groups (A) 22.63% of exclusively breastfed infants (N = 193/853), 35.96% of partially breastfed infants (N = 155/431) and 49.63% of formula fed infants were colonized (N = 134/270) (Fishers' exact p < 0.001). Scatter box-plots of the median (middle line), Q3 and Q1 quartiles (box limits), IQR (whiskers) and outlying values (dots). Data were normally distributed (Supplementary Figure 3) and thus two-sided p-values were calculated with students t-test within infant feeding groups, comparing colonized and non-colonized infants at a significance threshold of α = 0.05. Higher α-diversity was observed for infants colonized with C. difficile (CD+) and breastfed (either exclusively or partially) than non-carriers (CD–) on the same diet. This was the case for both the Shannon diversity index (B) and Chao1 species richness index (C). Purple represents EBF, green for PBF and gray for EFF.
EBF is generally associated with low microbial alpha diversity due to the dominance of Bifidobacterium spp. (19, 23). Bifidobacteria thrive on human milk oligosaccharides but their growth is reported to be suppressed with C. difficile colonization (6, 24). Accordingly, our regression models revealed that Bifidobacterium spp. were less abundant in EBF infants colonized with C. difficile than EBF infants who were not colonized (transformed β = −0.06, q = 0.021, Figure 2). Bifidobacteria are well-known acetate producers (24, 25) and their presence was positively correlated with this metabolite (R = 0.56, p < 0.01, Supplementary Figure 4). Despite an observed lowered relative abundance of Bifidobacterium, we measured higher absolute concentrations of fecal acetate among EBF infants colonized with C. difficile (p = 0.01, Supplementary Figure 2). Many other microbiota produce acetate (26); thus, the greater diversity of microbes we observed in EBF C. difficile positive infants likely contributed to higher fecal acetate levels. In our study, acetate concentrations were also positively correlated with the members of the Campylobacteraceae (R = 0.38, p > 0.10), Peptostreptococcaceae (R = 0.55, p = 0.05) and Clostridiaceae (R = 0.58, p > 0.10) families (Supplementary Figure 4) which were enriched in EBF infants positive for C. difficile (q < 0.05, Figure 2).
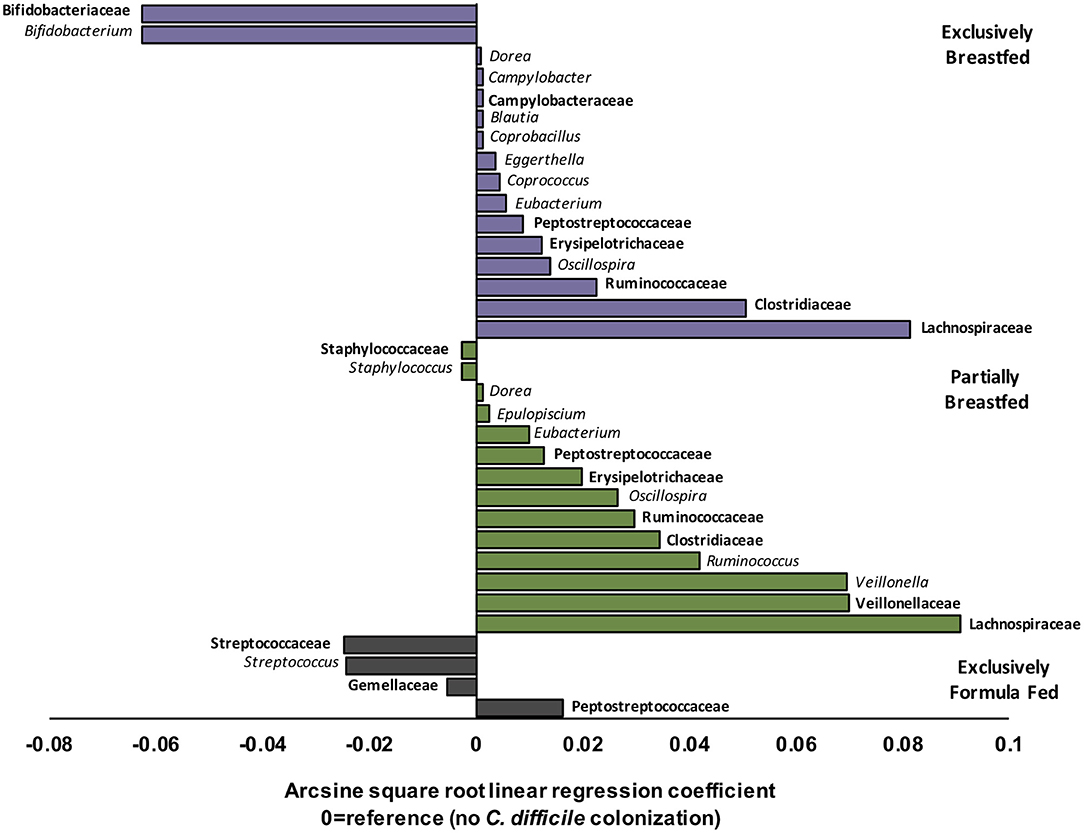
Figure 2. Relative differences in microbiota composition between C. difficile carriers and non-carriers across infant feeding groups (n = 1,554). Multivariate linear regression results (MaAslin) for family (bolded) and genus level taxa that are differentially associated with C. difficile colonization at 3–4 months of age. Values on the x-axis represent arcsine square root transformed regression coefficients of microbiota relative abundances for each linear model, adjusted for multiple comparisons (FDR correction) to determine which taxa are uniquely associated with C. difficile colonization. Each model had a reference of infants without C. difficile colonization at 3–4 months. Data shown only for taxa with FDR corrected two-sided q-value < 0.05. Coefficients > 0 (positive values) represent taxa that enriched in C. difficile carriers, while coefficients < 0 (negative values) represent taxa that were depleted in C. difficile carriers. P-values for each regression can be found in Supplementary Tables 3–5. Purple represents EBF (N = 853, 193 CD+), green for PBF (N = 431, 155 CD+) and gray for EFF (N = 270, 134 CD+).
Other microbes that were differentially abundant in the presence of C. difficile were members of the Lachnospiraceae and the Ruminococcaceae families, and both were enriched with C. difficile colonization among EBF and PBF infants (q < 0.05, Figure 2). Among EBF infants, we also observed higher absolute concentrations of non-acetate SCFAs (i.e., butyrate and propionate, p < 0.05, Supplementary Figure 2) when they were colonized with C. difficile. The relative abundance of Ruminococcaceae [e.g., Oscillospira spp. which are butyrate producers (27)] was positively correlated with butyrate (R = 0.35, p < 0.01, Supplementary Figure 4) and with p-cresol (R = 0.27, p = 0.08, Supplementary Figure 4), a known product of C. difficile amino acid metabolism (28). The fecal concentrations of p-cresol were higher in all infants colonized with C. difficile, regardless of infant feeding group (p < 0.01, Supplementary Figure 2). Lachnospiraceae was weakly correlated with propionate concentrations (R = 0.18, p < 0.01). Propionate production by Lachnospiraceae is through the 1,2-propanediol and acrylate pathways, which are possessed by Blautia, Eubacterium, and Coprococcus (29), all genera that were enriched in EBF C. difficile carriers (q < 0.05 for each, Figure 2).
Correlations between microbial relative abundance and butyrate concentrations involved a greater number of gut microbiota in PBF than EBF infants colonized with C. difficile. Specifically, Ruminococcaceae (R: 0.25, p < 0.01), Lachnospiraceae (R: 0.28, p < 0.01) and Clostridiaceae (R: 0.46, p < 0.01) were all positively correlated with butyrate and enriched in PBF infants (q < 0.05 for each, Figure 2). In contrast to EBF infants, Lachnospiraceae taxa in PBF C. difficile positive infants were inversely correlated with propionate levels (R = −0.57, p < 0.01). Since Bacteroidaceae are more abundant with any formula feeding (16), irrespective of C. difficile status in the current study, and they predominantly produce propionate (26), these microbiota likely out-competed Lachnospiraceae in the fermentation of substrates in PBF infants to produce propionate via the succinate pathway. Consistently, we observed a positive correlation between propionate concentrations and relative abundance of Bacteroidaceae among PBF and EFF infants, which was absent in EBF infants and independent of C. difficile status (Supplementary Figure 4).
Unique to PBF infants colonized with C. difficile was a higher relative abundance of Veillonella spp. (family Veillonellaceae, q = 0.002, Figure 2). Also, the relative abundance of Staphylococcus spp. (family Staphylococcaceae, q < 0.001, Figure 2) was lower in PBF infants positive for C. difficile than non-carriers. Fewer compositional differences were detected with C. difficile colonization among EFF infants, relative to breastfed (exclusive and partial) infants and equally no differences were detected in fecal metabolites. The sole family of microbes whose relative abundance was significantly higher in EFF C. difficile carriers was its own family, the Peptostreptococcaceae (q = 4.80E−24, Figure 2). As also expected, the Peptostreptococcaceae family were enriched in EBF and PBF infants colonized with C. difficile (q < 0.001, Figure 2).
Other metabolites measured in our study include glutamate and succinate. Glutamate, a metabolite shown to play a role in the establishment of C. difficile in vivo (30), was not differentially associated with C. difficile colonization in any of the feeding groups (Supplementary Figure 2). This metabolite is essential for C. difficile pathogenesis but may not be required for asymptomatic colonization in infants. Further, unlike glutamate dehydrogenase, a protein marker of C. difficile colonization (30), glutamate is an intermediary metabolite which may be consumed in several microbiota cross-feeding pathways. In fact, fecal levels of glutamate correlated with key microbes that differed by C. difficile status in all feeding groups (Supplementary Figure 4). Similarly, C. difficile utilizes succinate for its expansion and has the ability to ferment succinate to butyrate (31). Consistent with the succinate pathway, succinate concentrations were lower and concentrations of butyrate higher with C. difficile colonization in EBF infants and PBF infants (p = 0.05, Supplementary Figure 2). Since succinate is not easily absorbed by colonic cells (32), as suggested by our findings, levels may be further lowered from cross-feeding by succinate-utilizing members of the “Negativicutes” branch of Firmicutes clade (e.g. Veillonella spp.) (32, 33). Indeed, succinate was negatively correlated with Veillonellaceae in PBF infants (Supplementary Figure 4).
In addition to examining fecal metabolites, we also measured fecal sIgA levels as a marker of intestinal homeostasis and mucosal immunity (34). As we previously reported, C. difficile was associated with lower sIgA concentrations among EBF infants (p = 0.047, Figure 3) (11). Since infant secretion of sIgA has been positively correlated with breastmilk sIgA levels and breastmilk microbiota, maternal factors may contribute to lower concentrations in the infant (35, 36). Notably, animal models have shown that offspring nursed by mothers who are sIgA-deficient have a different gut microbiota composition than those receiving sIgA through breastmilk (37, 38). Similar to what we observed in EBF C. difficile positive infants, reduced fecal sIgA was associated with compositional differences that included an increased relative abundance of Lachnospiraceae and pro-inflammatory microbiota. Previous work from the CHILD Cohort Study has shown that sIgA in breastmilk may be depleted due to factors such as depression (21) or an altered maternal milk microbiota (36), which may predispose the infant to colonization by C. difficile and related dysbiosis. Although sIgA can bind enteric pathogens (34), there is a lack of evidence suggesting that C. difficile contributes to the destruction of sIgA or reduce production of this protein.
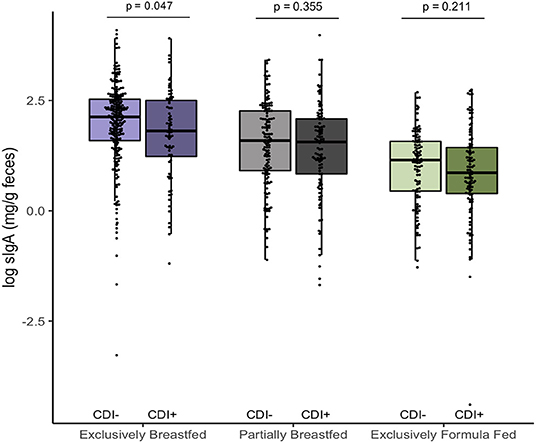
Figure 3. Log transformed measures of fecal secretory IgA, according to infant colonization and feeding mode (n = 731). Scatter box-plots of the median (middle line), Q3 and Q1 quartiles (box limits), IQR (whiskers) and outlying values (dots). Two-sided p-values were calculated with Mann–Whitney U-test of log transformed fecal sIgA (mg/g) comparing colonized and non-colonized infants within the same diet group. Exclusively breastfed infants colonized with C. difficile (CD+) had lower median fecal sIgA than non-carriers (CD–) on the same diet. Purple represents EBF (N = 290, 72 CD+), green for PBF (N = 237, 104 CD+) and gray for EFF (N = 204, 101 CD+).
Finally, some of our findings suggest that the gut microbiota of breastfed (both EBF and PBF) infants colonized with C. difficile resembles the gut microbial composition of adults (e.g., increased relative abundance of Firmicutes such as Eubacterium spp.) (39). Meta-analytic evidence from cohorts worldwide documents similarity between the gut microbiota of EFF infants and that of adults (23). Extending this evidence, our study suggests that the gut microbiome of breastfed infants colonized with C. difficile is compositionally similar to that observed in EFF infants (Supplementary Figure 5).
In our large population cohort study, we were not able to categorize infants according to the proportional intake of breastmilk vs. formula, as others have (40). Since our study did not employ culture-based methodology, another study limitation was inability to detect the strains and toxigenic properties of C. difficile. Should our study findings continue to align with previous findings, we might expect a prevalence of toxigenic strains to be <10% among infants with C. difficile positive samples (12, 41). We are also unable to determine the direction of observed associations: whether C. difficile caused gut microbial dysbiosis, or whether gut dysbiosis increased infant susceptibility to C. difficile colonization. This could be improved by measuring the C. difficile colonization status of infants longitudinally (at more than one time point) to assess if C. difficile colonization is transient or persistent and whether the microbiome changes precede or follow colonization. However, with enhanced characterization of the gut microbiome beyond taxon composition, our study provides evidence for a putative role of C. difficile colonization on the gut microbial ecology of young, full-term infants from a large, general population in North America.
Conclusion
We observed a distinct gut microbiome in young infants colonized with C. difficile and this distinction depended on the breastfeeding status of the infant. The most noticeable microbiome differences with C. difficile colonization, especially depletion of Bifidobacterium spp., were among EBF infants. Similar compositional differences among members of the Firmicutes phylum were seen in EBF and PBF infants. However, unique to PBF infants was enrichment of Veillonellaceae. These findings highlight the differential relationship of C. difficile colonization on EBF vs. PBF vs. EFF infants, which should be considered in future studies of infants feeding modality and disease risk. In summary, we found differences in the infant gut microbiome with C. difficile colonization, but it remains unclear whether C. difficile causes these differences or if external factors in early infancy create a niche that is more permissive to colonization. Newer cohorts with available multi-omics data could validate these findings and explore the hypothesized relations between various microbiota and C. difficile to further understand colonization of this microbe in infancy and its implications in later childhood health.
Data Availability Statement
The data and analysis code that support the findings of this study can be made available from the corresponding author and CHILD Cohort Study coordinators upon reasonable request. These data, including study participant data, are securely stored in the CHILDdb.ca database.
Ethics Statement
The studies involving human participants were reviewed and approved by the University of Alberta, University of British Columbia and University of Manitoba Ethics Boards. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
KD and AK conceived the study. KD performed data analysis, prepared figures, and drafted and edited the manuscript. HT generated gut microbiota operational taxonomic unit profiles using QIIME software. TK conducted DNA extraction and sample preparation for sequencing for microbiome analyses. NM-L performed targeted qPCR for C. difficile detection. DW and RM supervised and conducted NMR and fecal metabolite analyses. CF supervised, conducted, and helped interpret the fecal sIgA analyses. MA created the breastfeeding measures. DG, AB, PM, PS, ST, TM, MS, DL, and JS obtained funding and contributed to study design and data collection. AK obtained funding, contributed to data interpretation and critically reviewed the manuscript. All the authors reviewed the manuscript content, provided feedback and approved the final version. AK will serve as guarantor of the manuscript's contents.
Funding
This research was funded by the Canadian Institutes of Health Research (CIHR) Microbiome Initiative (Grant No. 227312). The Canadian Healthy Infant Longitudinal Development study was supported by both the Canadian Institutes of Health Research (CIHR) and the Allergy, Genes and Environment (AllerGen) Network of Centres of Excellence.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge that this work could not have been completed without the cooperation of all members, staff and participants of the CHILD Cohort Study. These include research staff, administrative staff, study families and participants, volunteers, lab technicians, statisticians, and clinical staff.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02866/full#supplementary-material
References
1. Jangi S, Lamont JT. Asymptomatic colonization by Clostridium difficile in infants : implications for disease in later life. J Pediatr Gastroenterol Nutr. (2010) 51:2–7. doi: 10.1097/MPG.0b013e3181d29767
2. Crobach MJT, Vernon JJ, Loo VG, Kong LY, Péchiné S, Wilcox MH, et al. Understanding Clostridium difficile colonization. Clin Microbiol Rev. (2018) 31: e00021–17. doi: 10.1128/CMR.00021-17
3. Tsutaoka B, Hansen J, Johnson D, Holodniy M. Antibiotic-associated pseudomembranous enteritis due to Clostridium difficile. Clin Infect Dis. (1994) 18:982–94. doi: 10.1093/clinids/18.6.982
4. Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA birth cohort study. Gut. (2007) 56:661–7. doi: 10.1136/gut.2006.100164
5. van Nimwegen FA, Penders J, Stobberingh EE, Postma DS, Koppelman GH, Kerkhof M, et al. Mode and place of delivery, gastrointestinal microbiota, and their influence on asthma and atopy. J Allergy Clin Immunol. (2011) 128:948–55.e3. doi: 10.1016/j.jaci.2011.07.027
6. Rousseau C, Levenez F, Fouqueray C, Doré J, Collignon A, Lepage P. Clostridium difficile colonization in early infancy is accompanied by changes in intestinal microbiota composition. J Clin Microbiol. (2011) 49:858–65. doi: 10.1128/JCM.01507-10
8. Fischer M. Clostridium difficileinfection and the role of adaptive immunity in the microbiome. Gastroenterol Hepatol. (2017) 13:301–3.
9. Stoesser N, Eyre DW, Quan TP, Godwin H, Pill G, Mbuvi E, et al. Epidemiology of Clostridium difficile in infants in Oxfordshire, UK: risk factors for colonization and carriage, and genetic overlap with regional C. difficile infection strains. PLoS ONE. (2017) 12:e0182307. doi: 10.1371/journal.pone.0182307
10. Borali E, De Giacomo C. Clostridium difficile infection in children: a review. J Pediatr Gastroenterol Nutr. (2016) 63:130–40. doi: 10.1097/MPG.0000000000001264
11. Bridgman SL, Konya T, Azad MB, Guttman DS, Sears MR, Becker AB, et al. High fecal IgA is associated with reduced Clostridium difficile colonization in infants. Microbes Infect. (2016) 18:543–9. doi: 10.1016/j.micinf.2016.05.001
12. Rousseau C, Lemée L, Le Monnier A, Poilane I, Pons JL, Collignon A. Prevalence and diversity of Clostridium difficile strains in infants. J Med Microbiol. (2011) 60:1112–8. doi: 10.1099/jmm.0.029736-0
13. Nguyen TT, Kim JW, Park JS, Hwang KH, Jang TS, Kim CH, et al. Identification of oligosaccharides in human milk bound onto the toxin a carbohydrate binding site of Clostridium difficile. J Microbiol Biotechnol. (2016) 26:659–65. doi: 10.4014/jmb.1509.09034
14. Subbarao P, Anand SS, Becker AB, Befus AD, Brauer M, Brook JR, et al. The Canadian Healthy Infant Longitudinal Development (CHILD) Study: examining developmental origins of allergy and asthma. Thorax. (2015) 70:998–1000. doi: 10.1136/thoraxjnl-2015-207246
15. Moraes TJ, Lefebvre DL, Chooniedass R, Becker AB, Brook JR, Denburg J, et al. The Canadian healthy infant longitudinal development birth cohort study: biological samples and biobanking. Paediatr Perinat Epidemiol. (2015) 29:84–92. doi: 10.1111/ppe.12161
16. Azad MB, Konya T, Persaud RR, Guttman DS, Chari RS, Field CJ, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG Int J Obstet Gynaecol. (2016) 123:983–93. doi: 10.1111/1471-0528.13601
17. Penders J, Vink C, Driessen C, London N, Thijs C, Stobberingh EE. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol Lett. (2005) 243:141–7. doi: 10.1016/j.femsle.2004.11.052
18. Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. (2002) 148:257–266. doi: 10.1099/00221287-148-1-257
19. Forbes JD, Azad MB, Vehling L, Tun HM, Konya TB, Guttman DS, et al. Association of exposure to formula in the hospital and subsequent infant feeding practices with gut microbiota and risk of overweight in the first year of life. JAMA Pediatr. (2018) 172:e181161. doi: 10.1001/jamapediatrics.2018.1161
20. Emwas AH, Emwas A. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Metabonomics Methods Protocols. (2015) 1277:161–93. doi: 10.1007/978-1-4939-2377-9_13
21. Kang LJ, Koleva PT, Field CJ, Giesbrecht GF, Wine E, Becker AB, et al. Maternal depressive symptoms linked to reduced fecal immunoglobulin A concentrations in infants. Brain Behav. Immun. (2017) 68:1–9. doi: 10.1016/j.bbi.2017.10.007
22. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. (2012) 13:1–18. doi: 10.1186/gb-2012-13-9-r79
23. Ho NT, Li F, Lee-Sarwar KA, Tun HM, Brown BP, Pannaraj PS, et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. (2018) 9:1–13. doi: 10.1038/s41467-018-06473-x
24. Hidalgo-Cantabrana C, Delgado S, Ruiz L, Ruas-Madiedo P, Sánchez B, Margolles A. Bifidobacteria and their health-promoting effects. MicrobiolSpectrum. (2017) 5:1–19. doi: 10.1128/microbiolspec.BAD-0010-2016
25. Bridgman SL, Azad MB, Field CJ, Haqq AM, Becker AB, Mandhane PJ, et al. Fecal short-chain fatty acid variations by breastfeeding status in infants at 4 months: differences in relative versus absolute concentrations. Front. Nutr. (2017) 4:11. doi: 10.3389/fnut.2017.00011
26. Kumari M, Kozyrskyj AL. Gut microbial metabolism defines host metabolism: an emerging perspective in obesity and allergic inflammation. Obes. Rev. (2017) 18:18–31. doi: 10.1111/obr.12484
27. Gophna U, Konikoff T, Nielsen HB. Oscillospira and related bacteria – From metagenomic species to metabolic features. Environ. Microbiol. (2017) 19:835–41. doi: 10.1111/1462-2920.13658
28. Passmore IJ, Letertre MPM, Preston MD, Bianconi I, Harrison MA, Nasher F, et al. Para-cresol production by Clostridium difficile affects microbial diversity and membrane integrity of Gram-negative bacteria. PLoS Pathog. (2018) 14:e1007191. doi: 10.1371/journal.ppat.1007191
29. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. (2017) 19:29–41. doi: 10.1111/1462-2920.13589
30. Girinathan BP, Braun S, Sirigireddy AR, Lopez JE, Govind R. Importance of Glutamate Dehydrogenase (GDH) in Clostridium difficile colonization in vivo. PLoS ONE. (2016) 11:e0160107. doi: 10.1371/journal.pone.0165579
31. Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. Gut Microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe. (2014) 16:770–7. doi: 10.1016/j.chom.2014.11.003
32. Connors J, Dawe N, Van Limbergen J. The role of succinate in the regulation of intestinal inflammation. Nutrients. (2018) 11:1–12. doi: 10.3390/nu11010025
33. Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. (2018) 12:1642–57. doi: 10.1038/s41396-018-0068-2
34. Mantis NJ, Rol N, Corthésy B. Secretory IgA's complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. (2011) 4:603–11. doi: 10.1038/mi.2011.41
35. Pabst O, Cerovic V, Hornef M. Secretory IgA in the coordination of establishment and maintenance of the microbiota. Trends Immunol. (2016) 37:287–96. doi: 10.1016/j.it.2016.03.002
36. Toscano M, De Grandi R, Grossi E, Drago L. Role of the human breast milk-associated microbiota on the newborns' immune system: a mini review. Front Microbiol. (2017) 8:2100. doi: 10.3389/fmicb.2017.02100
37. Elsen LWJ, Van Den Garssen J, Burcelin R, Verhasselt V. Shaping the gut microbiota by breastfeeding: the gateway to allergy prevention? Front Pediatr. (2019) 7:47. doi: 10.3389/fped.2019.00047
38. Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci USA. (2014) 111:3074–9. doi: 10.1073/pnas.1315792111
39. Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. (2016) 8:1–11. doi: 10.1186/s13073-016-0307-y
40. Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. (2017) 171:647–54. doi: 10.1001/jamapediatrics.2017.0378
Keywords: Clostridioides difficile, sIgA, SCFA, infant feeding, microbiome, gut microbiota, metabolites
Citation: Drall KM, Tun HM, Morales-Lizcano NP, Konya TB, Guttman DS, Field CJ, Mandal R, Wishart DS, Becker AB, Azad MB, Lefebvre DL, Mandhane PJ, Moraes TJ, Sears MR, Turvey SE, Subbarao P, Scott JA and Kozyrskyj AL (2019) Clostridioides difficile Colonization Is Differentially Associated With Gut Microbiome Profiles by Infant Feeding Modality at 3–4 Months of Age. Front. Immunol. 10:2866. doi: 10.3389/fimmu.2019.02866
Received: 06 September 2019; Accepted: 22 November 2019;
Published: 11 December 2019.
Edited by:
Maria Carmen Collado, Institute of Agrochemistry and Food Technology (IATA), SpainReviewed by:
Christopher James Stewart, Newcastle University, United KingdomMiguel Gueimonde, Institute of Dairy Products of Asturias (IPLA), Spain
Silvia Turroni, University of Bologna, Italy
Copyright © 2019 Drall, Tun, Morales-Lizcano, Konya, Guttman, Field, Mandal, Wishart, Becker, Azad, Lefebvre, Mandhane, Moraes, Sears, Turvey, Subbarao, Scott and Kozyrskyj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anita L. Kozyrskyj, a296eXJza3lAdWFsYmVydGEuY2E=
†Present Address: Hein M. Tun, HKU-Pasteur Research Pole, Li Ka Shing Faculty of Medicine, School of Public Health, The University of Hong Kong, Hong Kong, China