 Dieter Mielke1†
Dieter Mielke1† Gama Bandawe1†
Gama Bandawe1† Justin Pollara2
Justin Pollara2 Melissa-Rose Abrahams1Tinashe Nyanhete2
Melissa-Rose Abrahams1Tinashe Nyanhete2 Penny L. Moore3,4,5,6Ruwayhida Thebus1Nicole L. Yates2John C. Kappes7,8
Penny L. Moore3,4,5,6Ruwayhida Thebus1Nicole L. Yates2John C. Kappes7,8 Christina Ochsenbauer7
Christina Ochsenbauer7 Nigel Garrett6,9
Nigel Garrett6,9 Salim Abdool Karim6,10
Salim Abdool Karim6,10 Georgia D. Tomaras2
Georgia D. Tomaras2 David Montefiori2
David Montefiori2 Lynn Morris3,4,5,6
Lynn Morris3,4,5,6 Guido Ferrari2
Guido Ferrari2 Carolyn Williamson1,4,6*
Carolyn Williamson1,4,6*- 1Division of Medical Virology, Institute of Infectious Diseases and Molecular Medicine, University of Cape Town, Cape Town, South Africa
- 2Duke University Medical Center, Durham, NC, United States
- 3HIV Virology Section, Centre for HIV and STIs, National Institute for Communicable Diseases, Johannesburg, South Africa
- 4National Health Laboratory Service, Johannesburg, South Africa
- 5MRC Antibody Immunity Research Unit, University of Witwaterstrand, Johannesburg, South Africa
- 6Centre for the AIDS Programme of Research in South Africa (CAPRISA), University of KwaZulu Natal, Durban, South Africa
- 7Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 8Research Service, Birmingham Veterans Affairs Medical Center, Birmingham, AL, United States
- 9Discipline of Public Health Medicine, School of Nursing and Public Health, University of KwaZulu Natal, Durban, South Africa
- 10Department of Epidemiology, Columbia University, New York, NY, United States
Both neutralization and antibody-dependent cellular cytotoxicity (ADCC) may be required for effective protection against HIV-1 infection. While there is extensive information on the targets of early neutralizing antibody (nAb) responses, much less is known about the targets of ADCC responses, which are more difficult to characterize. In four individuals recruited during acute HIV-infection, ADCC responses were detected 3–7 weeks prior to nAb responses. To determine the relative influence of ADCC and nAb responses on virus evolution, we performed an in-depth investigation of one individual (CAP63) who showed the highest nAb and ADCC responses. Both nAbs and ADCC antibodies targeted the V4 region of the Env, although there were some differences in epitope recognition. We identified accelerated viral evolution in this region concurrent with emergence of nAb activity, but not ADCC activity. Deep sequencing demonstrated that most nAb escape mutations were strongly selected for, however one nAb escape mutation that rendered the virus highly susceptible to autologous ADCC responses, was suppressed despite not affecting viral fitness. This escape mutation also rendered the virus more sensitive to autologous responses, as well as monoclonal antibodies targeting CD4-induced epitopes, compared to the wildtype virus. In conclusion, ADCC responses and nAbs in donor CAP63 recognized overlapping but unique epitopes in the V4 region, and while ADCC activity was present prior to nAbs, it did not drive viral evolution during this time. However, ADCC responses may select against nAb escape pathways that expose other common ADCC epitopes thereby restricting viral replication and expansion.
Introduction
Both neutralization and effector cell functions, mediated through the antibody Fc domain, may be required for effective protection against HIV-1 infection (1). Neutralizing antibody (nAb) responses block viral entry into target cells by binding the HIV-1 envelope glycoprotein (Env) on infectious virions, while there are multiple Fc-mediated effector cell functions, including phagocytosis of opsonized virus particles or infected cells, antibody-dependent complement-mediated lysis, and stimulation of antiviral cytokines/chemokine secretion (2). The effector function antibody-dependent cellular cytotoxicity (ADCC), investigated in this study, mediates NK cell killing of HIV-1 infected cells through the binding of antigen-antibody complexes to the FcγRIIIa receptor (CD16) expressed on NK cells (3). These responses play a role in curbing early SIV viral replication (4–6), are enriched in human HIV-1 infected non-progressors (7), and may contribute to protection from HIV-1 infection, as suggested by the results of the HIV-1 RV144 vaccine trial (8–11). Furthermore, ADCC-mediating, non-neutralizing antibodies in breast milk have been correlated with reduced vertical transmission from viremic mothers (12). It is not known if ADCC responses play a role in the early control of HIV-1 infection, information that would be informative for vaccine design and antibody-based therapeutic studies.
The high mutation rate of HIV makes it possible to use patterns of genetic evolution to evaluate targets and kinetics of immune pressure, providing information on the role of antibodies in controlling viral populations in vivo. However, dissecting the possible role of ADCC antibodies during early stages of HIV-1 infection has proved challenging. While several studies have defined epitopes targeted by ADCC-mediating antibodies [reviewed by (13)], many overlap with neutralizing antibody epitopes, making it difficult to unravel ADCC responses and nAb immune pressure on the virus. Only one study to date, of individuals with chronic infection, has clearly demonstrated HIV-1 escape from ADCC (14). In addition, many methods used to define ADCC epitopes rely on targets coated with peptides or recombinant forms of monomeric Env (13), which do not fully reflect the epitope exposure of the HIV-1 Envelope on a virus infected cell. Therefore, infected cells expressing native Env trimers are preferred to accurately map escape from ADCC responses in natural infection (15).
Here, we investigated the kinetics of early neutralizing and ADCC responses in four HIV-1 subtype C infected individuals. In one individual (CAP63), who developed the earliest and strongest nAb and ADCC responses, we mapped Env targets of neutralizing and ADCC-mediating antibodies, using pseudovirus and infectious molecular clone (IMC)-based assays. Both of these responses targeted the V4 region of the Env, although there were some differences in epitope recognition. In particular, a six amino acid deletion in V4 conferred resistance to nAbs but created sensitivity to ADCC responses; specifically, autologous responses and monoclonal antibodies targeting CD4-induced (CD4i) epitopes. Next-generation sequencing revealed that this deletion was strongly selected against, despite not incurring a replication fitness cost to the virus. These data suggest that nAb escape pathways can be constrained by ADCC activity, illustrating a role of ADCC activity in controlling early replication and an interplay between the neutralizing and effector-mediated functions of early antibody responses.
Results
ADCC Responses Detected Early in Infection Correlate With HIV-Specific Binding Antibodies
We first explored the kinetics of binding antibody and ADCC development in four HIV-1 infected individuals (CAP45, CAP63, CAP210, and CAP239) recruited within 2 to 4 weeks post-infection (WPI), all of whom were infected by a single HIV-1 subtype C variant (16). Binding antibody responses against the subtype C protein 1086cΔ7 gp120 was detected using the BAMA assay and ADCC-mediating antibodies were detected using the ADCC-GTL assay, with 1086cΔ7 rgp120-coated CEM.NKRCCR5 cells as targets. Neutralizing responses were detected using autologous transmitted/Founder (T/F) Env pseudoviruses.
HIV-1 Env specific binding responses and ADCC responses were detected at the enrolment timepoint for all four individuals (Figures 1A,B). The nAb responses against the autologous transmitted/founder (T/F) Env emerged by 7, 9, 13, and 16 wpi in CAP63, CAP45, CAP239, and CAP210, respectively. For all individuals, autologous nAb responses were detected 3–7 weeks after binding responses and ADCC (Figures 1C,E). A strong association was detected between early binding responses and ADCC against the subtype C protein 1086cΔ7 gp120 (rs = 0.884 (95% CI: 0.736–0.951), p < 0.0001; Figure 1D).
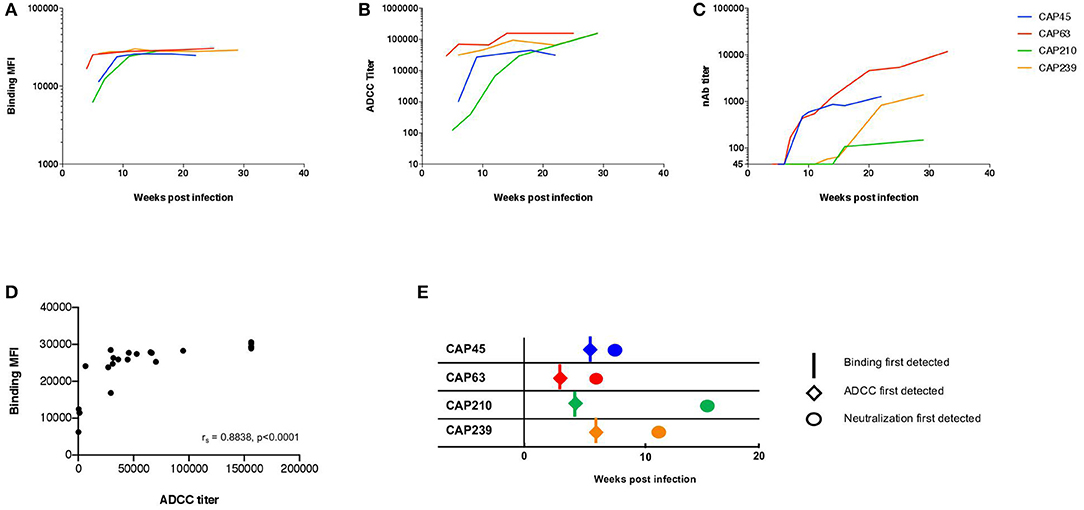
Figure 1. The kinetics of binding, ADCC, and nAb responses to HIV-1 subtype C in four participants of the CAPRISA 002 cohort. Binding (mean fluorescent intensity (MFI) at 1:50 dilution shown) of (A) and ADCC activity (titer, starting at 1:100 dilution) against (B) recombinant 1086cΔ7 gp120-coated CEM.NKRCCR5 cells, and the nAb responses against pseudoviruses with the T/F Envelope incorporated (ID50 titer, starting at a 1:45 dilution) (C) was determined longitudinally for four participants: CAP45 (blue), CAP63 (red), CAP210 (green) and CAP239 (orange). Binding MFI (at 1:50 dilution) and ADCC titer were plotted and a Spearman's r was calculated (rs = 0.8838 (95% CI: 0.7359–0.9512), p < 0.0001) (D). The time of first detection was determined for binding (vertical lines), ADCC (diamonds) and nAb (circles) responses (E).
Previous studies suggest IgG1 and IgG3 are predominantly responsible for ADCC and neutralizing activities in HIV infection, with IgG3 levels declining within the first 6 months (17, 18). We investigated the HIV-specific IgG subclass antibody responses in one participant, CAP63, over the first 20 weeks of infection using an HIV-1 Binding Antibody Multiplex Assay (HIV-1 BAMA) against 17 envelope proteins (gp41/gp120/V1V2/gp140) of various clades and clade consensus sequences.
Moderate gp120/140 IgG1 specific responses were observed at 4 wpi, which increased by 20 wpi (Figure S1A). IgG1 bound most strongly to 1086C gp140 protein, with titers >1,000 at 4 wpi rising to 12,000 by 9 wpi. In comparison, binding of IgG3 to all Env proteins developed after IgG1 and at lower titers (Figure S1B). Both IgG1 and IgG3 HIV-1-specific antibodies were detectable at enrolment. As with other studies, gp41-binding antibody responses were observed at high titers at enrolment and maintained over 20 wpi, with gp41 IgG3 responses declining over the time period (Figures S1C,D) (18). V1/V2-specific binding antibody responses were detected after 13 wpi and only to one V1V2 scaffold (Figures S1E,F). No HIV-specific IgG2 and IgG4 binding was detected at enrolment or over the period studied (data not shown).
Deletions in the V4 Are Prominent in CAP63 Viral Evolution
We then investigated CAP63, who had the earliest and strongest ADCC and nAb responses, in detail. This participant was identified after ~2 weeks of infection (HIV antibody negative and viral load positive) [Fiebig stage 1/2, (19)]. The participant experienced rapid disease progression, characterized by viral loads maintained above 100,000 RNA copies/ml and a rapid CD4+ T cell count decline over 32 weeks of infection (Figure 2) (19). Antiretroviral therapy was initiated at 37 wpi in accordance with prevailing South African Department of Health treatment guidelines.
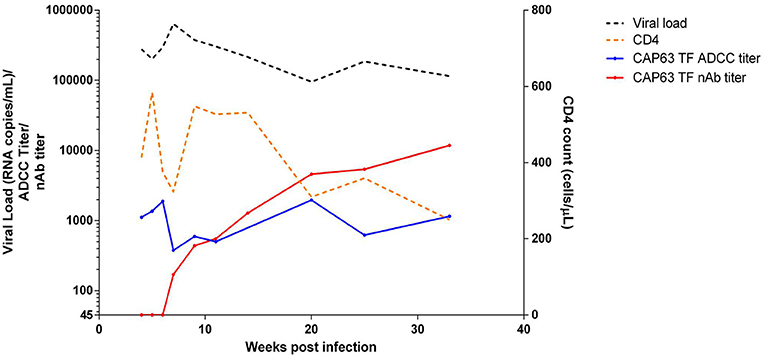
Figure 2. Clinical and humoral response profiles in one CAPRISA 002 participant: CAP63. Viral load (black), CD4 count (orange), ADCC-mediating antibody responses (titer, starting at 1:50 dilution) to CAP63 T/F IMC virus-infected CEM.NKRCCR5 cells (blue) and nAbs (ID50 titer, starting at 1:45 dilution) against CAP63 T/F pseudoviruses (red) were measured over 32 weeks of infection.
In order to investigate regions of the Env protein under antibody selection in CAP63, 90 full length gp160 sequences were generated from samples collected 2 (n = 30), 4 (n = 11), 11 (n = 9), and 29 (n = 40) wpi (Figure 3A). One mutation was observed in the V4 (P397S) in all 4 wpi sequences but not in the 20 sequences at 2 wpi indicating rapid selection of this residue. However, major shifts were observed after detection of nAbs at 7 weeks, and in particular in the V4, the known target of initial nAbs in this individual (20). By 11 wpi, seven of eight sequences contained deletions in the V4 region, while potential N-linked glycosylation site (PNG) shifts were observed throughout infection at three sites: PNG392 (dark green), PNG397 (blue), and PNG406 (red). By 29 wpi, all sequences had combinations of deletions and point mutations (including introductions of PNG sites at 397 and 406) in the V4.
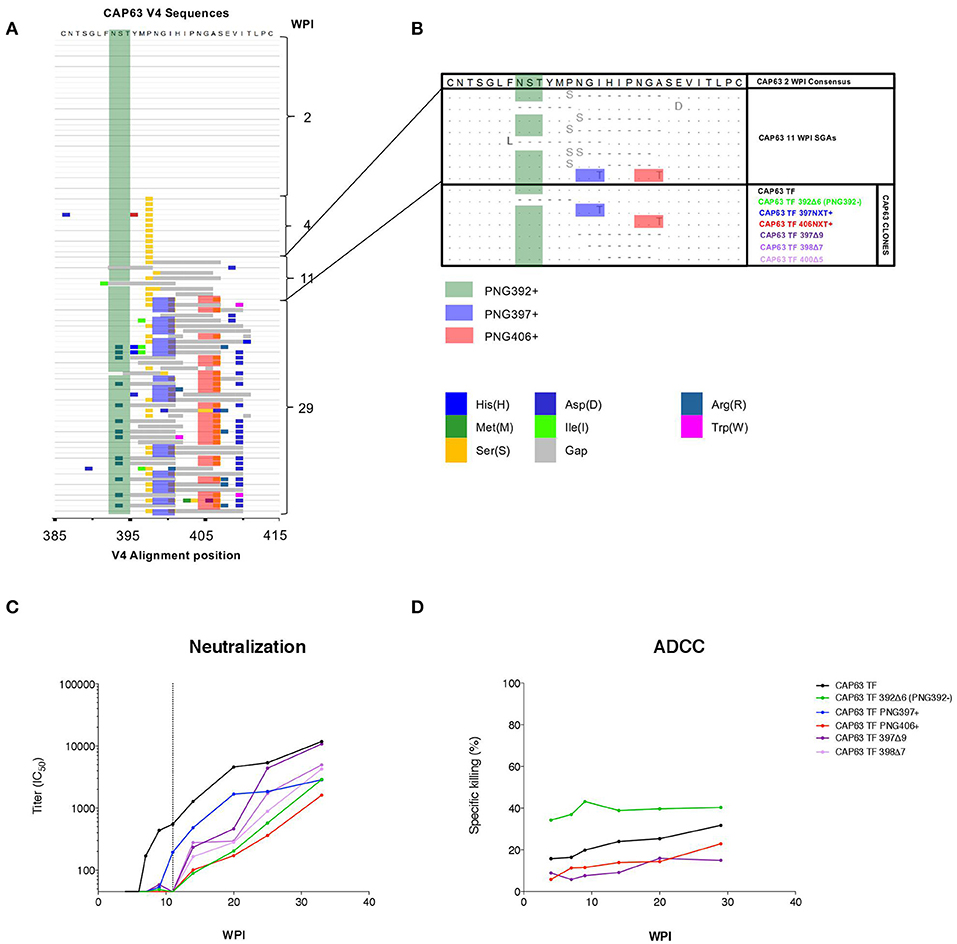
Figure 3. CAP63 V4 viral evolution and antibody escape. A highlighter plot was generated using 91 V4 sequences from 2 (n = 30), 4 (n = 11), 11 (n = 9), and 29 (n = 40) wpi (A). Changes to the PNG profile at three sites are shown: at position 392 (dark green), 397 (blue), and 406 (red). Deletions are shown in gray. Naturally occurring changes in the V4 observed at 11 wpi (B, light blue) were then introduced as mutations in the T/F env and Infectious Molecular clones for use neutralization assays and ADCC assays (B, CAP63 clones). IMC constructs were used to infect CEM.NKRCCR5 target cells for used in an infected-cell elimination assay, while pseudoviruses were used in neutralization assays. The effect of each V4 change on neutralization sensitivity (ID50; starting at 1:45 dilution; dotted line indicates 11 wpi) (C) and ADCC sensitivity (maximum levels of specific killing (%)) (D) was then determined. Figures show the mean of three independent experiments.
Changes in V4 Mediate Escape From Neutralizing and ADCC Antibody Responses
To map epitopes recognized by nAb and ADCC-mediating antibodies, we introduced naturally occurring changes observed at 11 wpi (Figure 3B) into functional CAP63 T/F env and T/F infectious molecular clones (IMCs) (Figure 3B, CAP63 clones). We used IMCs in the ADCC assays as these are more reliable than peptides or proteins for identifying conformational epitopes. Two point mutations in PNGs were introduced individually: I399T (PNG397+) and A408T (406NXT+). Further, four different deletions were introduced: a six-residue deletion from position 392, resulting in the loss of PNGs at 392 [392Δ6(PNG392-)], a nine-residue deletion from position 397 (397Δ9), a seven-residue deletion from position 398 (398Δ7) and a five-residue deletion from position 400 (400Δ5).
The T/F and mutant pseudoviruses were assayed against plasma from multiple time-points between 4 and 37 wpi. All the deletions and both the I399T and A408T mutant viruses (incorporating either PNG397 or PNG406, respectively) observed in the V4 loop resulted in either complete or partial resistance to nAbs at 11 wpi, after which nAbs against these viruses were detected (Figure 3C, shift of curves to the right). In comparison, the T/F IMC was sensitive to plasma ADCC-mediating antibodies from 4 to 29 wpi (Figure 3D). The 400Δ5, 397Δ9, and PNG406+ mutants were not recognized by ADCC-mediating antibodies at all time-points tested suggesting they enabled escape from ADCC responses. These data suggest that nAb escape mutations also mediate escape from ADCC-mediating antibodies.
Interestingly, however, the 392Δ6(PNG392–) mutation resulted in increased sensitivity (in comparison to the T/F virus) to ADCC-mediating antibodies at all the time points tested. This suggests that this deletion, which conferred resistance to nAbs, resulted in increased exposure of epitopes targeted by autologous ADCC-mediating antibodies.
Deletion of the PNG392 (392Δ6) Increases Sensitivity to Antibodies Targeting CD4-Induced Epitopes
We sought to determine if the increased sensitivity of the 392Δ6(PNG392-) mutant to autologous ADCC was due to a change in the Env conformation which exposed otherwise hidden ADCC epitopes, such as the C1C2 epitopes. To do this, we first assessed the ability of autologous plasma to mediate ADCC against, and bind to the surface of, CAP63 virus-infected CD4 positive (CD4+) and negative (CD4-downregulated; CD4–) CEM.NKRCCR5 cells. Autologous ADCC responses against CD4+ infected cells, was higher against the 392Δ6(PNG392–) virus relative to the T/F (Figure 4A). In comparison, the three other mutants, (PNG406+, 397Δ9, and 400Δ5) were less susceptible to ADCC compared to the T/F virus. This observation was supported by surface staining, which showed plasma antibodies bound to the surface of 392Δ6(PNG392–)-infected cells more than the other variants (Figure 4C). In contrast, all the CAP63 mutants were resistant to ADCC and binding responses against infected cells with CD4 downregulated (CD4–), portraying a similar pattern to the neutralization profile (Figures 4B,D).
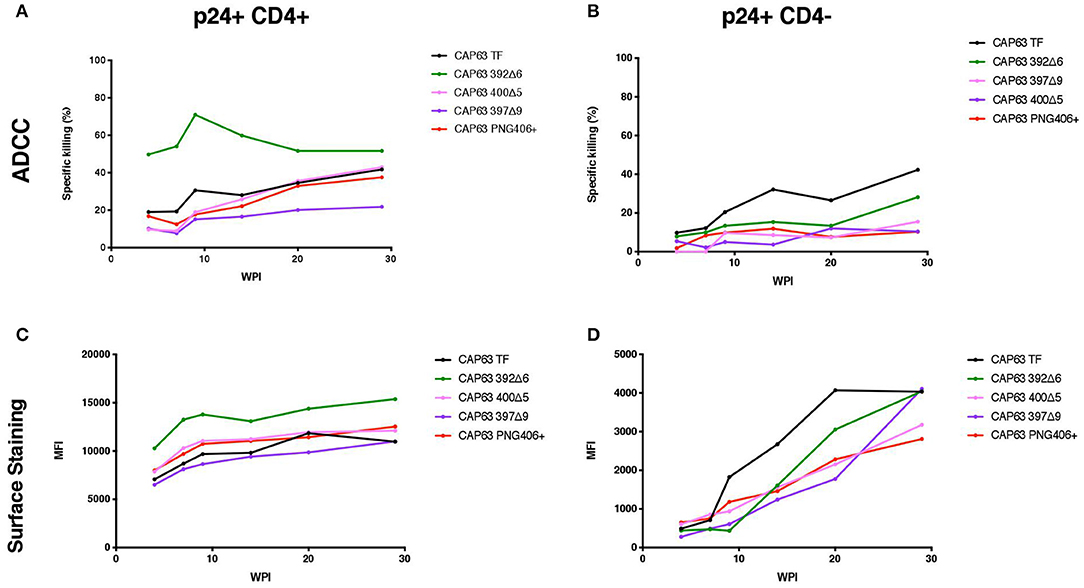
Figure 4. Autologous ADCC responses targeting, and surface staining of CD4+ and CD4– CAP63 IMC-infected CEM.KNRCCR5 cells. Longitudinal plasma was used to test the sensitivity of CD4+ (A) and CD4– (B) CAP63 IMC-infected cells to autologous ADCC responses and the ability of each antibody to bind to the surface of CD4+ (C) and CD4– (D) infected cells. ADCC activity is shown as the specific killing (%) by 1:100 dilution of plasma in an infected-cell elimination assay, and levels of surface binding are depicted by the median fluorescent intensity (MFI) of the secondary antibody. Figures show the mean of three independent experiments.
We hypothesized that the increased sensitivity of the 392Δ6(PNG392–) virus was due to increased susceptibility to CD4i responses, as suggested by the enhanced killing of, and binding to, CD4+ cells. To determine if this mutation changed the structure to expose other common ADCC epitopes, we tested CD4+ infected cells against a panel of anti-HIV-1 mAbs [A32, C11 (CD4i epitopes); PGT151 (gp41-gp120 interface); 3BNC117, VRC01 (CD4 binding site); 10E8 (MPER), and PG9 (V2)]. All Abs had identical Fcs optimized to bind FcγRIIIa to mediate ADCC activity against cells infected with the T/F and mutant IMCs in the infected cell elimination assay and in infected cell surface binding assays (Figure 5). Of the seven mAbs we tested, only 392Δ6(PNG392-) showed increased susceptibility to C11 and A32 ADCC and binding (Figures 5A,C). These data suggest the ADCCs/nAbr mutant (392Δ6[PNG392–]) alters the conformation of the Env in a manner that promoted recognition and binding by CD4i ADCC-mediating antibodies to 392Δ6(PNG392-) Env.
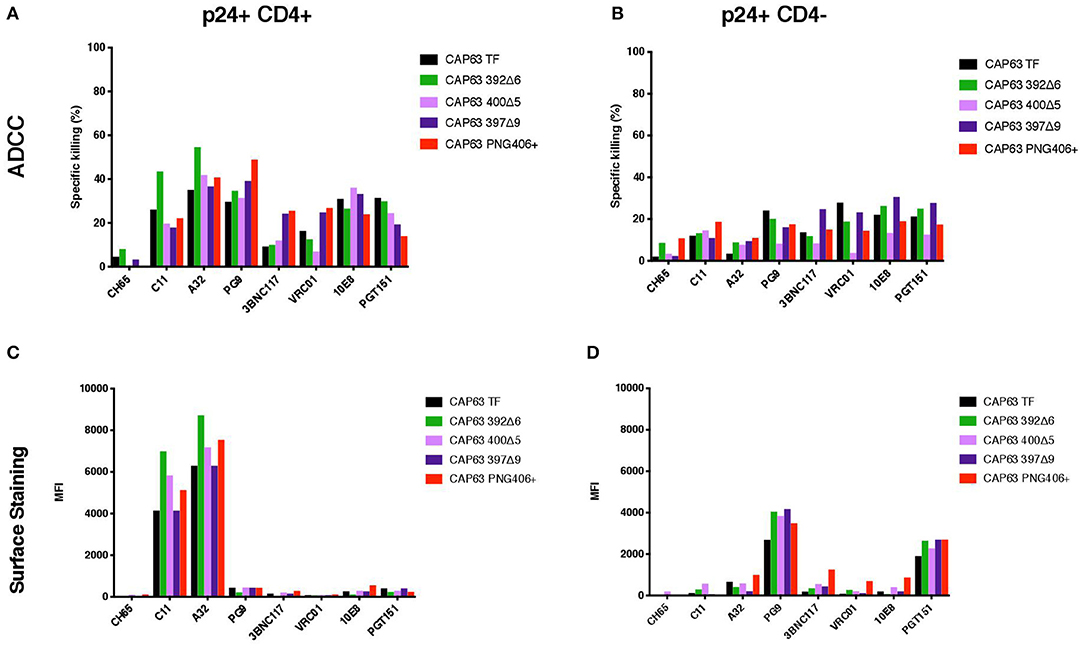
Figure 5. ADCC against, and surface staining of, CAP63 IMC-infected CEM.KNRCCR5 cells by a panel of anti-HIV-1 monoclonal antibodies. A panel of eight anti-HIV-1 monoclonal antibodies were used to test the sensitivity of CD4+ (A) and CD4– (B) CAP63 IMC-infected cells to ADCC and the ability of each antibody to bind to the surface of CD4+ (C) and CD4– (D) infected cells. ADCC activity is shown as the specific killing (%) by 10 μg/mL mAb in an infected-cell elimination assay, and levels of surface binding are depicted by the median fluorescent intensity (MFI) of the secondary antibody. Figures show the mean of three independent experiments.
Next-Generation Sequencing Reveals Strong Selection Against ADCCs/nAbr Mutant 392Δ6(PNG392-)-Like Deletions
In order to better understand immune pressure on the V4 region we conducted next-generation sequencing of the C3–C5 region at 2, 4, 7, 9, 11, 14, and 29 wpi. We found a decrease in the V4 loop length after the detection of nAbs at 7 wpi: quasispecies with 1–10 residue deletions were present, with 6, 7, and 9-residue deletions (resulting in loop length reduction from 29 to 23, 22, or 20 residues, respectively) the most predominant (Figure 6). Of these deletions, populations that did not have the PNG392 glycan [392Δ6(PNG392-)-like shown in green] were observed at very low levels by 4 wpi (<1%) and increased in frequency up to 11 wpi (20%), after which they decreased rapidly and were found at very low levels again by 29 wpi (2%). While deletions resulted in the removal of PNG392, point mutations introduced PNGs at positions 397 (blue) and 406 (red), both of which mediate neutralization escape (Figure 3C). PNG in positions 397 and 406 were respectively, detected in 8 and 2% at 7 wpi,11 and 39% at 9 wpi, and then decreased to 2.5 and 6.1% by 14 wpi. However, in contrast with the PNG392- mutations, both PNG sites at 397 and 406 were found at considerably higher frequencies (89 and 33%, respectively) by 29 wpi.
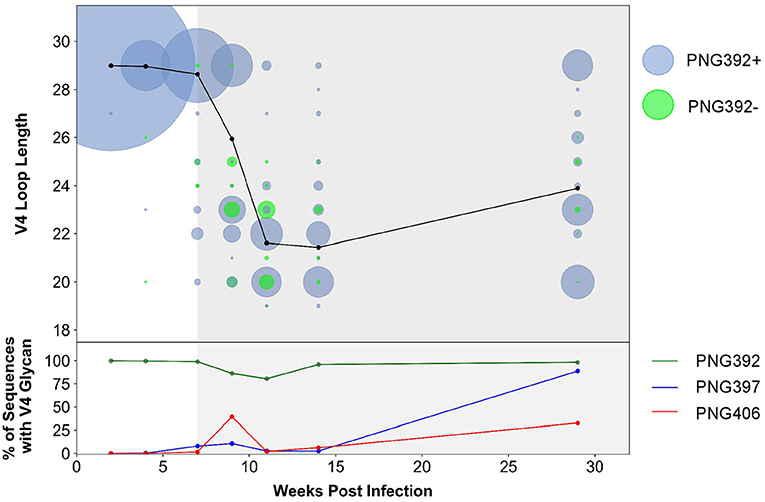
Figure 6. CAP63 V4 loop characteristics based on next-generation sequencing data. Next-generation sequencing of the V4 region at 2, 4, 7, 9, 11, 14, and 29 wpi was performed. The V4 loop length relative to HxB2 and the total number of viral copies at each loop length were calculated using a custom script (top). Viral populations with the PNG392 present are shown in blue and viral populations with the 392NXT PNGS removed are shown in green. The size of each bubble represents the total number of copies in each viral population. The average loop length over time is indicated with a black line. The relative abundance of the three PNG sites was also calculated using a custom script (bottom). Each PNG site is shaded: PNG392 (dark green), PNG397 (blue), and PNG406 (red).
These data suggest the CAP63 viral population initially evaded nAb responses by removing parts of the V4 loop, and incorporating PNG sites at positions 397 (to a lesser extent) and 406 (predominantly). However, escape variants where the V4 deletions also resulted in the loss of the PNG at 392 were selected against. Later in infection, the average loop length increased but frequencies of PNG sites at positions 397 substantially increased, suggesting these PNG sites became the predominant mechanism of nAb escape.
Antibody Escape Mutations Exert Minimal Viral Replicative Fitness Cost in CAP63 Viruses
Deletions in the V4 loop have previously been shown to disrupt Env cell surface display and viral assembly (21), and may therefore impact replicative fitness. Consequently, we sought to determine whether antibody escape mutations in CAP63 were associated with any fitness cost and in particular, whether there was a fitness cost associated with the 392Δ6(PNG392-) mutation, which may account for its lack of persistence.
We evaluated replication of each constructed IMC in PBMCs from two donors over a 14-day period (Figure 7). Overall, replication kinetics of all mutant viruses did not differ appreciably relative to the T/F virus, with the exception of the 397Δ9 virus, which exhibited reduced replicative fitness. Importantly, there was no discernible replicative fitness cost identified in the 392Δ6(PNG392–) mutant virus associated with nAb escape and increased ADCC recognition.
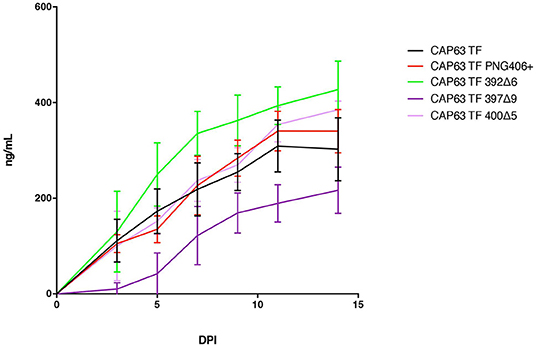
Figure 7. Replication capacity of CAP63 TF and mutant viruses. Average replicative capacities of CAP63 T/F and V4 mutant IMC viruses from 11 weeks post-infection assayed over 14 days in PBMCs. Each data point represents the mean of triplicate wells from two independent experiments.
Discussion
This study showed that viruses with nAb escape mutations that exposed CD4i epitopes resulting in increased ADCC sensitivity, were controlled. ADCC responses in four individuals were detected prior to nAb responses, as shown in other studies (22–24). In one individual (CAP63) who developed potent ADCC and nAb responses to the V4 region, we found that early V4 ADCC responses did not have a noticeable impact on virus sequences, while nAb responses drove rapid escape. However, when nAb escape resulted in a viral variant being more globally susceptible to autologous and heterologous ADCC-mediating antibodies, this escape pathway was blocked. This suggests an indirect role for ADCC responses in controlling viral populations by constraining nAb escape pathways, and potentially a mechanism that keeps HIV in a more ADCC-resistant conformation.
We detected high binding and ADCC responses at enrolment and through early infection, while neutralizing antibodies were detected later in all four participants. This suggests there may be a lower affinity threshold for ADCC-mediating antibodies to bind to the Env and mediate effector functions, while nAbs require further B cell maturation to neutralize virions, which occurs through somatic hypermutation coupled with selection for antigen binding (25). Affinity maturation of nAb lineages has been observed in several longitudinal studies, where binding affinities of the initial antibody to the antigen are substantially lower than subsequent members of a lineage (26, 27).
In addition, in one individual, CAP63, we observed that IgG1 but not IgG3 responses to heterologous gp120 proteins were detected at enrolment, suggesting ADCC responses detected at this time were primarily mediated through IgG1 antibodies, consistent with other studies (28). The magnitude of both of these responses increased substantially around the time nAbs were detected in this individual, although IgG1 responses to a greater extent than IgG3. Both isotypes are known to mediate neutralization and mediate Fc-dependent effector functions including ADCC (17). However, further investigation by isolating monoclonal antibodies from CAP63 will be required to tease apart the role antibody subclasses have had in neutralization and effector functions in vivo.
Although ADCC responses were present before autologous nAbs in CAP63, we only detected V4 escape variants concurrent with, or after, the emergence of the nAbs. Most of these nAb escape mutants also facilitated escape from ADCC-mediating antibody responses: five different mutations/deletions observed in CAP63 viruses resulted in resistance to both ADCC-mediating and neutralizing antibodies, providing the first description of the V4 region as a target for ADCC-mediating antibody responses. It is possible that there is an overlap of antibodies or antibody lineages which mediate both functions, which would not be surprising as many nAbs are able to mediate ADCC activity (13, 29).
However, one mutant with a six-residue deletion resulting in the loss of a glycan at 392 [392Δ6(PNG392–)], was resistant to nAbs but highly sensitive to autologous ADCC-mediating antibody responses, as well as a panel of anti-HIV-1 mAbs. Deep sequencing revealed that this mutation was strongly selected against and was found at very low frequency by 29 wpi. Sunshine et al. showed that dynamic escape processes only resolve with the selection of mutations that confer escape with little/no fitness cost to the virus (30). We showed there was no replicative fitness cost incurred by incorporation of the 392Δ6(392NXT–) deletion, suggesting other pressures such as ADCC-mediating antibodies played a role in controlling viral populations through constraining nAb escape pathways.
We previously showed that non-neutralizing antibodies to CD4i epitopes develop to high titers in early infection in the CAPRISA cohort (31). Loss of the PNG392 glycan in CAP63 viruses unmasked a cluster of these epitopes on infected CD4+ cells, as evidenced by the increased sensitivity of this mutant to autologous ADCC responses and CD4i-targeting monoclonal antibodies. Deletions in V4 region have been shown to disrupt gp160 folding (32–34), and consequently may result in exposure of the CD4i cluster of epitopes or other epitopes. ADCC epitopes such as the A32 and C11 clusters, described by Lewis et al. (35), are a major target of potent effector function-mediating non-neutralizing antibodies (34) and have been correlated with decreased disease progression in a recent transmission study (36). These epitopes are often conformationally masked on virion-associated unliganded Env trimers but become exposed upon engagement of gp120 to CD4 and during virus budding, rendering infected cells susceptible to killing by A32- and C11-like antibodies (35, 37).
Importantly, non-neutralizing antibodies targeting these epitope clusters are easily inducible by vaccination and have provided protection in non-human primate models (38). However, a further structural study is required to better understand if the removal of the PNG392 glycan in CAP63 results in the exposure of an entirely novel epitope, or resulted in the unmasking of known epitopes through receptor-dependent conformational changes.
Understanding the interplay between selective forces driving the emergence of viral variants, particularly in early infection, may help identify targets for vaccine design or approaches aimed to eliminate the acute viral reservoir. The limited impact of ADCC-mediating antibodies on viral evolution prior to the detection of nAbs supports the conclusion that there is stronger selective pressure exerted by nAbs compared to ADCC-mediating antibodies. However, ADCC responses may play a supplementary role by limiting nAb escape mutation pathways to those which do not increase susceptibility to non-neutralizing antibody-mediated responses. Here, in one individual we demonstrated a mechanism through which ADCC-mediating antibody responses apply selective pressure by constraining a viable nAb escape pathway. We will continue to study virus-humoral immune responses in early infection in participants of the CAPRISA 002 cohort to define how generalizable these findings are.
Materials and Methods
Ethics Statement
Written informed consent was obtained from all participants. This study received ethical approval from the University of the Witwatersrand Human Research Ethics Committee, University of KwaZulu-Natal Biomedical Research Ethics Committee, and University of Cape Town Human Research Ethics Committee. All subjects were adult, and provided written informed consent.
Study Participant Samples
Samples were obtained from the CAPRISA 002 Acute HIV Infection cohort (Durban, South Africa) (39). Samples were collected at enrolment, weekly for 3 weeks, fortnightly until 3 months and monthly thereafter up to 6 months post-infection. CD4 T-cell counts were assessed using a FACSCalibur flow cytometer, and HIV viral loads were measured using the COBAS AMPLICOR HIV-1 Monitor test, v1.5 (Roche Diagnostics). EDTA blood plasma samples were stored at −70°C until use. Date of infection was estimated as the midpoint between last negative and first positive HIV antibody test or as 14 days prior to a positive RNA result for individuals negative to HIV antibody tests.
Sequence Analysis
PCR amplification of HIV-1 env genes was done using the SGA approach previously described (16, 40) or a limiting dilution PCR. Sequence alignments, amino acid identity plots and participant consensus sequences were generated using BioEdit version 7.0.8.0 (41). The transmitted full-length envelope virus sequence for each participant was taken as the consensus of single genome amplification (SGA) derived sequences (15–42 per participant) generated from the earliest sampled time-point (16). Maximum likelihood trees were generated using Mega version 4 (42). The Los Alamos Highlighter tool (www.hiv.lanl.gov/content/sequence/HIGHLIGHT/highlighter_top.html) was used to generate plots of synonymous and non-synonymous nucleotide changes.
Viral RNA Extraction and cDNA Synthesis Using the Primer ID (PID) Method
RNA extraction, cDNA synthesis and subsequent amplification were carried out as described previously (43, 44), with the following modifications: cDNA synthesis primers were designed to bind to the C5 (HxB2: 7655-7632) region of the HIV-1 envelope gene. First-round amplification primers were designed to bind to the C3 (HxB2: 7114-7135) region. Raw reads were processed using a custom pipeline housed within the University of Cape Town High Performance Computing core, as previously described (44). The resulting consensus sequences were then used to generate codon aligned nucleotide and amino acid alignments using MACSE (45) and viewed using Bioedit (Version 7.1.11) (41). Amino acid alignments were analyzed for N-glycosylation sites using N-GlycoSite (46), available online at http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html.
Cloning and Generation of Pseudoviruses
The second round PCR reaction was repeated using the high fidelity Phusion Hot Start DNA Polymerase (Finnzymes), together with, 0.2 mM dNTPs (Roche), 4 μM of env 1A-Rx (5′ CAC CGG CTT AGG CAT CTC CTA TAG CAG GAA GAA 3′) and envN (5′ CTG CCA ATC AGG GAA AGT AGC CTT GT 3′) in a final volume 50 μL. The amplicons were gel purified and cloned into the mammalian expression vectors pcDNA3.1D/V5-His-TOPO (Invitrogen) or pTarget (Promega, US) according to the manufacturer's instructions. Functional envelope clones were selected using a 96-well plate format pseudovirion entry efficiency assay based on relative luminescence units (RLU) that is 2.5 times above background.
Cell Lines
TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program Division of AIDS, NIAID, NIH (contributed by Drs. Kappes and Wu). 293T/17 cells were obtained from Dr. George Shaw (University of Pennsylvania, Philadelphia, PA). Both cell lines were cultured in D-MEM (Gibco BRL Life Technologies) containing 10% heat-inactivated fetal bovine serum (FBS) (Biochrom) and 50 μg/mL gentamicin (Sigma). Cell monolayers were disrupted at confluency by treatment with 0.25% trypsin in 1 mM EDTA. CEM.NKRCCR5 cells from Dr. Alexandra Trkola were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, and NIH.
HIV-1 Env Pseudovirus Production and Titration
Stocks of HIV-1 env pseudovirus were produced by co-transfecting 293T/17 cells (1.7 × 107 cells per T75 flask) with 4 μg of an HIV-1 rev/env expression plasmid and 8 μg of an env-deficient HIV-1 backbone plasmid (pSG3ΔEnv) (47) using the PolyFect Transfection Reagent (QIAGEN). Pseudovirus-containing supernatants were harvested 48 h following transfection and clarified by 0.45 μm filtration and adjusted to 20% FBS. Single-use aliquots (1.0 mL) were stored at −80°C. The 50% tissue culture infectious dose (TCID50) for each pseudovirus preparation was determined by infection of TZM-bl cells as previously described (31, 47).
Neutralizing Antibody Assays
Neutralization was measured as described (31) by a reduction in luciferase gene expression after single-round infection of TZM-bl cells with Env-pseudotyped viruses. Titers were calculated as the reciprocal plasma dilution (ID50) causing 50% reduction of relative light units (RLU).
Coating of Cells With Recombinant gp120 HIV-1 Proteins
Recombinant gp120 HIV-1 protein representing the envelope of the HIV-1 subtype C isolate 1086.cΔ7 (GenBank No. DQ435682; Immune Technology, New York, NY, United States), was used to coat CEM.NKRCCR5 target cells by incubating 1 × 106 cells in 1 mL RPMI media with 5 μg/mL gp120 for 75 min at 37°C.
ADCC-GranToxiLux Assay
ADCC activity was detected according to the previously described ADCC-GranToxiLux (GTL) procedure using recombinant gp120 coated CEM. NKRCCR5 as target cells and cryopreserved PBMC from a HIV-seronegative donor as effector cells (48). The results of the GTL assay were considered positive if % Granzyme B activity after background subtraction was ≥8% for the infected target cells. The titer of ADCC-mediating antibodies present in the plasma was calculated by interpolating the reciprocal of the last plasma dilution that yielded positive % Granzyme B activity (≥8%).
HIV-1 Specific Binding Antibody Assay
Plasma HIV-1 specific antibodies to HIV-1 gp120/gp140 proteins and V1/V2 scaffolds were measured by HIV-1 binding antibody multiplex assay for IgG subclasses (IgG1, IgG3) as previously described (11, 23, 49) with the following modifications. Antibody titers (area under the curve, AUC) were determined by serial dilutions of plasma (1:50, 5-fold). IgG subclasses were detected with the following reagents: IgG1 (4E3, Southern Biotech, Birmingham, AL, United States), IgG2 (HP6002, Southern Biotech, Birmingham, AL, United States), IgG3 (HP6050, Southern Biotech, Birmingham, AL, United States), and IgG4 (HP6025, Southern Biotech, Birmingham, AL, United States). The following antigens (provided by Drs. Liao/Haynes, Duke University unless otherwise indicated; Gp41 (Subtype B) (Immunodiagnostics, Scottsdale, AZ, United States); Group M consensus: ConSgp140CFI, Con6gp120/B; Subtype C Envelopes: 1086Cgp140C_avi, C.con.env03 140CF, CAP45_D11gp120.avi/293F, TV1c8_D11gp120.avi/293F; Subtype B Envelope: B.con.env03 140 CF; Subtype A Envelopes: OOMSA4076gp140, A1.con.env03gp140CF_AVI; V1-V2 Antigens: gp70_B.CaseA2 V1/V2/169K, C.1086C_V1_V2 Tags, MulVgp70_His (empty gp70 scaffold), gp70-CAP210.2.00.E8 V1V2, and gp70-CAP 45.2.00.G3 V1V2 (provided by Dr. Abe Pinter, Rutgers University); V2 Antigens: C.1086 V2 tags 293F; Subtype E Envelopes: A244 gp120 gDneg/293F/mon [as described by Yates et al. (50)].
Construction of Infectious Molecular Clones (IMCs)
An infectious molecular clone representing the transmitted/founder virus sequence of CAP63 was constructed from proviral DNA using the method described in detail by Ochsenbauer et al. (51).
Mutagenesis of Envelope and Infectious Molecular Clones
Site-directed mutagenesis was used to introduce mutations and deletions into the CAP63_T/F IMC using a QuikChange II site-directed mutagenesis kit (Stratagene) and mutations were confirmed by full length sequencing of the pseudovirus (PSV) or IMC. Point mutations and deletions were named based on HXB2 numbering and number of deleted residues (Δn). Mutations I400T, A410T, 393Δ6, 398Δ7, 397Δ9, and 401Δ5 were introduced into the T/F pseudovirus constructs while A410T, 393Δ6, 397Δ9, and 401Δ5 were introduced into the T/F IMC.
Generation of Virus Stocks From IMCs
HIV-1 IMC derived virus stocks were produced by transfecting 293T/17 cells (3 × 106 cells per T75 flask) with 12 μg of an HIV-1 IMC plasmid DNA using the FUGENE Transfection Reagent (Roche). Virus-containing supernatant was harvested 48 h following transfection and clarified by centrifugation and 0.45 μm filtration and adjusted to 20% FBS. Single-use aliquots (1.0 mL) were stored at −80°C. The TCID50 was determined both by infection of TZM.bl cells and CEM.NKRLUC cells as previously described (31, 47). Twenty five microliters of virus stock was placed in a total volume of 100 μL of GM and 11 serial 5-fold dilutions were made in a 96-well plate. Virus dilutions were co-incubated with 60,000 cells in 100 μl RPMI-12%-GM containing 16 μg DEAE-Dextran/mL for 4 days. On day 4, 100 μl of the cell suspension was placed in a 96-well solid white plate. One hundred microliters of BriteLite Plus (Perkin Elmer) was added to the white plate and incubated for 2 min. Luminescence was read in a luminometer using the 1.0 s/well protocol implemented by the computer program Wallac (Perkin Elmer).
HIV-1 IMC-Infection of CEM.NKRCCR5 Cells
HIV-1 IMC virus stocks were titrated to determine the input required for optimal viral gene expression within 72 h post-infection of CEM.NKRCCR5 cells as measured by intra-cellular p24 expression. Stocks were used to infect 2 × 106 cells with each IMC by incubation with the appropriate dose for 30 min at 37°C and 5% CO2 in the presence of DEAE-Dextran (7.5 μg/mL). The cells were subsequently resuspended at 0.5 × 106/mL and cultured for 2 days in complete medium containing 7.5 μg/mL DEAE-Dextran. On assay day, the infection was monitored by measuring the frequency of cells expressing intracellular p24. The assays performed using the IMC-infected target cells were considered reliable if the percentage of viable p24+ target cells on assay day was ≥20%. Assay data generated using infected cells was normalized to the % of target cells positive for intracellular p24.
Infected Cell Elimination Assay
HIV-1-infected or mock-infected CEM.NKRCCR5 cells were used as targets in the ICE assays and cryo-preserved PBMCs rested overnight in R10 supplemented with 10 ng/ml of IL-15 (Miltenyi Biotec) were used as a source of effector cells. Infected and uninfected target cells were labeled with a fluorescent target-cell marker (TFL4; OncoImmunin) and a viability marker (NFL1; OncoImmunin) for 15 min at 37°C, as specified by manufacturer. Cells were washed in R10 and adjusted to a concentration of 0.2 × 106 cells/mL. PBMCs were then added to target cells at an effector/target ratio of 30:1 (6 × 106 cells/mL). The target/effector cell suspension was plated in V-bottom 96-well plates and co-cultured with 10 μg/mL mAb or 1:100 dilution of plasma. Co-cultures were incubated for 6 h at 37°C in 5% CO2.
After the incubation period, cells were washed and stained with anti-CD4-PerCP-Cy5.5 (eBioscience, clone OKT4) at a final dilution of 1:40 in the dark for 20 min at room temperature (RT). Cells were then washed, resuspended in 100 μL/well Cytofix/Cytoperm (BD Biosciences), incubated in the dark for 20 min at 4°C, washed in 1× Cytoperm wash solution (BD Biosciences) and co-incubated with anti-p24 antibody (clone KC57-RD1; Beckman Coulter) to a final dilution of 1:100, and incubated in the dark for 25 min at 4°C. Cells were washed three times with Cytoperm wash solution and resuspended in 125 μL PBS-1% paraformaldehyde. The samples were acquired within 24 h using a BD Fortessa cytometer. The appropriate compensation beads were used to compensate the spill over signal for the four fluorophores. Data analysis was performed using FlowJo 9.6.6 software (TreeStar).
Specific killing was determined by the reduction in % of p24+ cells in the presence of mAbs or plasma after taking into consideration non-specific killing, and was calculated as:
CH65 (an anti-influenza monoclonal antibody) or plasma from a seronegative donor were used as negative controls and A300 (plasma from an HIV-1 chronically-infected individual) was used as a positive control.
Infected Cell Antibody Binding Assay
Infected CEM.NKRCCR5 cells were obtained as described above. Cells incubated in the absence of virus (mock infected) were used as a negative infection control. Following infection, infected and mock infected cells were washed in PBS, dispensed into 96-well V-bottom plates at 2 × 105 cells/well and incubated with 10 μg/mL mAb or 1:100 dilution of plasma for 2 h at 37°C. Subsequently, cells were washed twice with 250 μL/well of wash buffer (1% FBS-PBS; WB), stained with vital dye (Live/Dead Fixable Aqua Dead Cell Stain, Invitrogen) to exclude non-viable cells from subsequent analysis, washed with WB and stained with anti-CD4-PerCP-Cy5.5 (clone OKT4; eBiosciences) to a final dilution of 1:40 in the dark for 20 min at RT. Cells were then resuspended in 100 μL/well Cytofix/Cytoperm (BD Biosciences), incubated in the dark for 20 min at 4°C, washed in 1× Cytoperm wash solution (BD Biosciences) and co-incubated with anti-p24 antibody (clone KC57-RD1; Beckman Coulter) to a final dilution of 1:100 and a secondary FITC-conjugated antibody (goat anti-human IgG(H+L)-FITC, KPL) to a final dilution of 1:100, and incubated in the dark for 25 min at 4°C. Cells were washed three times with Cytoperm wash solution and resuspended in 125 μL PBS-1% paraformaldehyde. The samples were acquired within 24 h using a BD Fortessa cytometer. A minimum of 50,000 total events was acquired for each analysis. Gates were set to include singlet and live events. The appropriate compensation beads were used to compensate the spill over signal for the four fluorophores. Data analysis was performed using FlowJo 9.6.6 software (TreeStar). MFI from wells which included the secondary antibody alone (no mAb or plasma) were subtracted from samples to calculate the MFI specifically due to mAb/plasma binding. CH65 (an anti-influenza monoclonal antibody) or plasma from a seronegative donor were used as negative controls and A300 (plasma from an HIV-1 chronically-infected individual) was used as a positive control.
Titration of Viral Stocks by β-Gal Staining
The β-Gal reporter found in TZM-bl cells allows direct enumeration of infectious viral units by counting β-Gal expression-positive infected cell colonies under a microscope, as previously described (52). Briefly, 1 × 105 TZM-bl cells in 500 μL of growth media were seeded onto each well of a 24-well plate overnight. On the second day, IMC virus stock was diluted at 1:10, 1:50, 1:250 1:1,250, and 1:6,250 in DMEM containing 1% FBS and antibiotics. Media was removed from the cells and 200 μL of fresh media was added. Fifty microliters of virus dilution and a no-virus control was added to the wells and incubated for 4 h at 37°C. Five hundred microliters of fresh media was added to each well and samples were incubated for 48 h at 37°C. On day 4, supernatant was removed from the infected cells and 250 μL of fixing solution (PBS with 0.8% Gluteraldehyde and 2% formaldehyde) and incubated at room temperature for 8 min. Fixing solution was removed and the cells washed three times with 500 μL PBS per well. Two hundred and fifty microliters of staining solution (4 mM Potassium Ferricyanide, 4 mM Potassium Ferrocyanide, 400 μg/mL MgCl2, and 400 μg/mL X-gal in PBS) was added to each well and incubated at 37°C for 2 h. The staining solution was removed and 250 μL PBS added to each well. Blue cells were counted under a light microscope and the number of infectious units calculated. Infectivity was calculated by normalizing the virus stock to 7,500 IU per mL and determining the number of IU per unit p24 determined by ELISA.
Replication Assay
The kinetics of viral replication was determined in human PBMC. Cells were stimulated for 72 h in RPMI with 15% FCS (Invitrogen) with 2 mM L-glutamine and gentamycin supplemented with 200 U/mL IL2 and phytoheamaglutanin (PHA) at 37°C in 5% CO2, as previously described (52). The cells were then washed twice in RPMI with 15% FCS with 2 mM L-glutamine and gentamycin and rested overnight in the same media supplemented with 200 U/mL IL-2 (growth media). A 96-well plate was then seeded with 750,000 PHA-IL2 treated cells per well in 100 μL of growth media. Virus was added to triplicate PBMC wells at a multiplicity of infection (MOI) of 0.01 (MOI, IU/cell) in 100 μL of media. Cells were incubated at 37°C in 5% CO2 and 72 μL of culture supernatant was sampled with replacement of media at days 3, 5, 7, 9, 11, and 14 post-infection for determination of p24. Each virus inoculum tested was subjected to the same treatment in the absence of cells to determine background levels of p24. The slope of each growth assay was calculated using the LOGEST function in excel on the logarithmic growth phase of the curve.
p24 ELISA
COSTAR white opaque 96-well ELISA plates (Corning) were coated overnight with affinity purified sheep anti-HIV-1 p24 gag antibody (Aalto Bio Reagents), washed three times with tris-buffered saline (TBS) and allowed to dry, as previously described (52). Plates were stored at −20°C for medium term use. A p24 standard dilution series was prepared in binding buffer made up of TBS with 1% Empigen (Sigma) after reconstituting bacterially expressed p24 (Aalto Bio Reagents) in TBS with 1% FCS. Virus containing sample supernatants were also serially diluted in binding buffer and 100 μL of samples and p24 standards were placed in pre-coated and blocked plates. Plates were covered and incubated for 2 h at room temperature. The plates were then washed three times with TBS. One hundred microliters of a 1 in 12,500 dilution of an alkaline phosphatase conjugated sheep anti-HIV-1 monoclonal antibody BC 1071-AP (Aalto Biosciences) in TBS with 0.1% TWEEN-20 was added and incubated for 1 h at room temperature. Wells were washed four times with a TBS buffer with TROPIX and 0.1% TWEEN. Bound antibodies were measured by placing 100 μL of a solution of CDP Star with Saphire-II (Life Tech) in TROPIX buffer in each well for half an hour and reading in a luminometer. All reactions were run in triplicate and the equation generated from the linear range of the p24 standard curve was used to determine the amount of p24 in each sample based on the results of the luminometer readings.
Data Availability Statement
The deep sequencing datasets generated during this study are available in the NCBI short-read archive repository (Accession number: PRJNA586767), Env sequences used have been deposited in the Genbank repository (Accession numbers: MN635324-MN635400).
Ethics Statement
The studies involving human participants were reviewed and approved by the Human Research Ethics Committee, University of the Witwatersrand; the Biomedical Research Ethics Committee, University of KwaZulu-Natal; and the Human Research Ethics Committee, University of Cape Town. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
DMi, GB, JP, GF, and CW: conceptualization and methodology. DMi, GB, JP, M-RA, RT, TN, and NY: investigation. NG, CO, JK, GT, DMo, GF, CW, and SK: resources. DMi, GB, and CW: writing – original draft and visualization. PM, LM, GF, and CW: supervision and project administration. DMo, SK, GF, and CW: funding acquisition.
Funding
The research was supported by the Poliomyelitis Research Foundation (PRF grant # 12/28) and the Bill and Melinda Gates Foundation's Collaboration for AIDS Vaccine Discovery/Comprehensive Vaccine Immune Monitoring Consortium (Grant ID: 1032144; Grant ID: 38619). The CAPRISA 002 acute infection cohort study has been supported by the National Institute of Allergy and Infectious Disease (NIAID), National Institutes of Health (NIH) (grant# AI51794). DMi received bursaries from the University of Cape Town, the PRF and the Beit Trust. PM is supported by the South African Research Chairs Initiative of the Department of Science and Technology and the NRF (Grant No: 98341). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to CAP63 and other participants in the CAPRISA Acute Infection cohort and to the clinical/laboratory staff at CAPRISA for their continued commitment to the study. We thank Jessica Peel for the expert technical assistance. We would further like to thank the Columbia University-Southern African Fogarty AIDS International Training and Research Programme for providing the opportunity to learn antibody-dependent cellular cytotoxicity assays at Duke University (GB). This manuscript consists, partly, of data published within the PhD thesis of GB.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02875/full#supplementary-material
Figure S1. Kinetics of HIV gp120-specific IgG responses in CAP63. Seventeen envelope proteins (gp41/gp140/gp120/V1V2) of various clades and clade consensus sequences and plasma from CAP63 were used in an HIV-1 binding antibody mulitplex assay (HIV-1 BAMA). Binding assay results for IgG1 (left) and IgG3 (right) are calculated as area under the curve (AUC) for each antigen. A panel of 11 gp120/140 proteins of various clades were used to detect gp120-specific IgG1 (A) and IgG3 (B) binding antibody responses. Gp41-specific IgG1 (C) and IgG3 (D) binding antibody responses were detected from the first time-point tested (4 wpi). In addition, a panel of five V1, V2, or V1V2 scaffolds of different clades were used to test for the presence of V1V2 IgG1 (E) or IgG3 (F) binding antibodies.
References
1. Hessell AJ, Hangartner L, Hunter M, Havenith CEG, Beurskens FJ, Bakker JM, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. (2007) 449:101–4. doi: 10.1038/nature06106
2. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
3. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
4. Hidajat R, Xiao P, Zhou Q, Venzon D, Summers LE, Kalyanaraman VS, et al. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J Virol. (2009) 83:791–801. doi: 10.1128/JVI.01672-08
5. Xiao P, Zhao J, Patterson LJ, Brocca-Cofano E, Venzon D, Kozlowski PA, et al. Multiple vaccine-elicited nonneutralizing antienvelope antibody activities contribute to protective efficacy by reducing both acute and chronic viremia following simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J Virol. (2010) 84:7161–73. doi: 10.1128/JVI.00410-10
6. Xiao P, Patterson LJ, Kuate S, Brocca-Cofano E, Thomas MA, Venzon D, et al. Replicating adenovirus-simian immunodeficiency virus (SIV) recombinant priming and envelope protein boosting elicits localized, mucosal IgA immunity in rhesus macaques correlated with delayed acquisition following a repeated low-dose rectal SIV(mac251) ch. J Virol. (2012) 86:4644–57. doi: 10.1128/JVI.06812-11
7. Lambotte O, Pollara J, Boufassa F, Moog C, Venet A, Haynes BF, et al. High antibody-dependent cellular cytotoxicity responses are correlated with strong CD8 T cell viral suppressive activity but not with B57 status in HIV-1 elite controllers. PLoS ONE. (2013) 8:e74855. doi: 10.1371/journal.pone.0074855
8. Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. (2012) 366:1275–86. doi: 10.1056/NEJMoa1113425
9. Rolland M, Edlefsen PT, Larsen BB, Tovanabutra S, Sanders-Buell E, Hertz T, et al. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature. (2012) 490:417–20. doi: 10.1038/nature11519
10. Tomaras GD, Ferrari G, Shen X, Alam SM, Liao H-X, Pollara J, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci USA. (2013) 110:9019–24. doi: 10.1073/pnas.1301456110
11. Yates NL, Liao HX, Fong Y, DeCamp A, Vandergrift NA, Williams WT, et al. Vaccine-induced Env V1-V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci Transl Med. (2014) 6:228ra39. doi: 10.1126/scitranslmed.3007730
12. Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. (2012) 8:1–13. doi: 10.1371/journal.ppat.1002739
13. Pollara J, Bonsignori M, Moody MA, Pazgier M, Haynes BF, Ferrari G. Epitope specificity of human immunodeficiency virus-1 antibody dependent cellular cytotoxicity [ADCC] responses. Curr HIV Res. (2013) 11:378–87. doi: 10.2174/1570162X113116660059
14. Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, et al. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci USA. (2011) 108:7505–10. doi: 10.1073/pnas.1016048108
15. Isitman G, Stratov I, Kent SJ. Antibody-dependent cellular cytotoxicity and nk cell-driven immune escape in HIV infection: Implications for HIV vaccine development. Adv Virol. (2012) 2012:637208. doi: 10.1155/2012/637208
16. Abrahams M-R, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping L-H, et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol. (2009) 83:3556–67. doi: 10.1128/JVI.02132-08
17. Tomaras GD, Haynes BF. HIV-1-specific antibody responses during acute and chronic HIV-1 infection. Curr Opin HIV AIDS. (2009) 4:373–9. doi: 10.1097/COH.0b013e32832f00c0
18. Yates NL, Lucas JT, Nolen TL, Vandergrift NA, Soderberg KA, Seaton KE, et al. Multiple HIV-1-specific IgG3 responses decline during acute HIV-1. AIDS. (2011) 25:2089–97. doi: 10.1097/QAD.0b013e32834b348e
19. Abrahams M-R, Treurnicht FK, Ngandu NK, Goodier SA, Marais JC, Bredell H, et al. Rapid, complex adaptation of transmitted HIV-1 full-length genomes in subtype C-infected individuals with differing disease progression. AIDS. (2013) 27:507–18. doi: 10.1097/QAD.0b013e32835cab64
20. Moore PL, Gray ES, Choge IA, Ranchobe N, Mlisana K, Abdool Karim SS, et al. The c3-v4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J Virol. (2008) 82:1860–9. doi: 10.1128/JVI.02187-07
21. Yuan T, Li J, Zhang M-Y. HIV-1 envelope glycoprotein variable loops are indispensable for envelope structural integrity and virus entry. PLoS ONE. (2013) 8:e69789. doi: 10.1371/journal.pone.0069789
22. Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J Virol. (2001) 75:6953–61. doi: 10.1128/JVI.75.15.6953-6961.2001
23. Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. (2008) 82:12449–63. doi: 10.1128/JVI.01708-08
24. Asmal M, Sun Y, Lane S, Yeh W, Schmidt SD, Mascola JR, et al. Antibody-dependent cell-mediated viral inhibition emerges after simian immunodeficiency virus SIVmac251 infection of rhesus monkeys coincident with gp140-binding antibodies and is effective against neutralization-resistant viruses. J Virol. (2011) 85:5465–75. doi: 10.1128/JVI.00313-11
25. Doria-Rose NA, and Joyce MG. (2015). Strategies to guide the antibody affinity maturation process. Curr. Opin. Virol. 11, 137–147. doi: 10.1016/j.coviro.2015.04.002.
26. Bonsignori M, Zhou T, Sheng Z, Chen L, Gao F, Joyce MG, et al. (2016). Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 165, 449–63. doi: 10.1016/j.cell.2016.02.022.
27. Liao H-X, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, et al. (2013). Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–76. doi: 10.1038/nature12053.
28. Ljunggren K, Broliden PA, Morfeldt-Månson L, Jondal M, and Wahren B. (1988). IgG subclass response to HIV in relation to antibody-dependent cellular cytotoxicity at different clinical stages. Clin. Exp. Immunol. 73, 343–7.
29. Bruel T, Guivel-Benhassine F, Amraoui S, Malbec M, Richard L, Bourdic K, et al. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat Commun. (2016) 7:10844. doi: 10.1038/ncomms10844
30. Sunshine JE, Larsen BB, Maust B, Casey E, Deng W, Chen L, et al. Fitness-balanced escape determines resolution of dynamic founder virus escape processes in HIV-1 infection. J Virol. (2015) 89:10303–18. doi: 10.1128/JVI.01876-15
31. Gray ES, Moore PL, Choge IA, Decker JM, Bibollet-Ruche F, Li H, et al. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J Virol. (2007) 81:6187–96. doi: 10.1128/JVI.00239-07
32. Pollard SR, Rosa MD, Rosa JJ, Wiley DC. Truncated variants of gp120 bind CD4 with high affinity and suggest a minimum CD4 binding region. EMBO J. (1992) 11:585–91.
33. Wyatt R, Sullivan N, Thali M, Repke H, Ho D, Robinson J, et al. Functional anf immunological characterization of human immunodeficiency virus type 1 envelope glycoproteins containing deletions of the major variable regions. J Virol. (1993) 67:4557–65.
34. Veillette M, Coutu M, Richard J, Batraville L-A, Dagher O, Bernard N, et al. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol. (2015) 89:545–51. doi: 10.1128/JVI.02868-14
35. Lewis G, Finzi A, DeVico A, Pazgier M. Conformational masking and receptor-dependent unmasking of highly conserved env epitopes recognized by non-neutralizing antibodies that mediate potent ADCC against HIV-1. Viruses. (2015) 7:5115–32. doi: 10.3390/v7092856
36. Tuen M, Bimela JS, Banin AN, Ding S, Harkins GW, Weiss S, et al. Immune correlates of disease progression in linked HIV-1 infection. Front Immunol. (2019) 10:1062. doi: 10.3389/fimmu.2019.01062
37. Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, et al. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci USA. (2013) 110:E69–78. doi: 10.1073/pnas.1217609110
38. Fouts TR, Bagley K, Prado IJ, Bobb KL, Schwartz JA, Xu R, et al. Balance of cellular and humoral immunity determines the level of protection by HIV vaccines in rhesus macaque models of HIV infection. Proc Natl Acad Sci USA. (2015) 112:E992–9. doi: 10.1073/pnas.1423669112
39. van Loggerenberg F, Mlisana K, Williamson C, Auld SC, Morris L, Gray CM, et al. Establishing a cohort at high risk of HIV infection in South Africa: challenges and experiences of the CAPRISA 002 acute infection study. PLoS ONE. (2008) 3:e1954. doi: 10.1371/journal.pone.0001954
40. Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. (2008) 105:7552–7. doi: 10.1073/pnas.0802203105
41. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. in Nucleic acids Symposium Series No. 41, (Oxford, UK).
42. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. (2007) 24:1596–9. doi: 10.1093/molbev/msm092
43. Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci USA. (2011) 108:20166–71. doi: 10.1073/pnas.1110064108
44. Wibmer CK, Gorman J, Anthony CS, Mkhize NN, Druz A, YorkT, et al. Structure of an N276-dependent HIV-1 neutralizing antibody targeting a rare V5 glycan hole adjacent to the CD4 binding site. J Virol. (2016) 90:10220–35. doi: 10.1128/JVI.01357-16
45. Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: multiple alignment of coding SEquences accounting for frameshifts and stop codons. PLoS ONE. (2011) 6:e22594. doi: 10.1371/journal.pone.0022594
46. Zhang M, Gaschen B, Blay W, Foley B, Haigwood N, Kuiken C, et al. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology. (2004) 14:1229–46. doi: 10.1093/glycob/cwh106
47. Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. (2005) 79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005
48. Pollara J, Hart L, Brewer F, Pickeral J, Packard BZ, Hoxie JA, et al. High-throughput quantitative analysis of HIV-1 and SIV-specific ADCC-mediating antibody responses. Cytom. Part A. (2011) 79A:603–12. doi: 10.1002/cyto.a.21084
49. Zolla-Pazner S, DeCamp A, Gilbert PB, Williams C, Yates NL, Williams WT, et al. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS ONE. (2014) 9:e87572. doi: 10.1371/journal.pone.0087572
50. Yates NL, DeCamp AC, Korber BT, Liao H-X, Irene C, Pinter A, et al. HIV-1 envelope glycoproteins from diverse clades differentiate antibody responses and durability among vaccinees. J Virol. (2018) 92. doi: 10.1128/JVI.01843-17
51. Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. (2012) 86:2715–28. doi: 10.1128/JVI.06157-11
Keywords: ADCC, CD4-induced, neutralizing antibody, selection, escape
Citation: Mielke D, Bandawe G, Pollara J, Abrahams M-R, Nyanhete T, Moore PL, Thebus R, Yates NL, Kappes JC, Ochsenbauer C, Garrett N, Abdool Karim S, Tomaras GD, Montefiori D, Morris L, Ferrari G and Williamson C (2019) Antibody-Dependent Cellular Cytotoxicity (ADCC)-Mediating Antibodies Constrain Neutralizing Antibody Escape Pathway. Front. Immunol. 10:2875. doi: 10.3389/fimmu.2019.02875
Received: 02 October 2019; Accepted: 22 November 2019;
Published: 11 December 2019.
Edited by:
Lucia Lopalco, San Raffaele Hospital (IRCCS), ItalyReviewed by:
P. J. Klasse, Weill Cornell Medicine, United StatesMartyn Andrew French, University of Western Australia, Australia
Copyright © 2019 Mielke, Bandawe, Pollara, Abrahams, Nyanhete, Moore, Thebus, Yates, Kappes, Ochsenbauer, Garrett, Abdool Karim, Tomaras, Montefiori, Morris, Ferrari and Williamson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn Williamson, Y2Fyb2x5bi53aWxsaWFtc29uQHVjdC5hYy56YQ==
†These authors have contributed equally to this work