 Baila Samreen
Baila Samreen Sha Tao
Sha Tao Karsten Tischer
Karsten Tischer Heiko Adler
Heiko Adler Ingo Drexler
Ingo Drexler- 1Institute for Virology, Düsseldorf University Hospital, Heinrich-Heine-University, Düsseldorf, Germany
- 2Department of Oncology-Pathology, Science for Life Laboratory, Karolinska University Hospital, Stockholm, Sweden
- 3Fachbereich Veterinärmedizin, Institut für Virologie, Freie Universität Berlin, Berlin, Germany
- 4Comprehensive Pneumology Center, Research Unit Lung Repair and Regeneration, Helmholtz Zentrum Muenchen, German Research Center for Environmental Health (GmbH), Member of the German Center of Lung Research (DZL), Munich, Germany
Gammaherpesviruses (γHV) are important pathogens causing persistent infections which lead to several malignancies in immunocompromised patients. Murine γHV 68 (MHV-68), a homolog to human EBV and KSHV, has been employed as a classical pathogen to investigate the molecular pathogenicity of γHV infections. γHV express distinct antigens during lytic or latent infection and antigen-specific T cells have a significant role in controlling the acute and latent viral infection, although the quality of anti-viral T cell responses required for protective immunity is not well-understood. We have generated recombinant modified vaccinia virus Ankara (recMVA) vaccines via MVA-BAC homologous recombination technology expressing MHV-68 ORF6 and ORF61 antigens encoding both MHC class I and II-restricted epitopes. After vaccination, we examined T cell responses before and after MHV-68 infection to determine their involvement in latent virus control. We show recognition of recMVA- and MHV-68-infected APC by ORF6 and ORF61 epitope-specific T cell lines in vitro. The recMVA vaccines efficiently induced MHV-68-specific CD8+ and CD4+ T cell responses after a single immunization and more pronounced after homologous prime/boost vaccination in mice. Moreover, we exhibit protective capacity of prophylactic recMVA vaccination during early latency at day 17 after intranasal challenge with MHV-68, but failed to protect from latency at day 45. Further T cell analysis indicated that T cell exhaustion was not responsible for the lack of protection by recMVA vaccination in long-term latency at day 45. The data support further efforts aiming at improved vaccine development against γHV infections with special focus on targeting protective CD4+ T cell responses.
Introduction
Gammaherpesvirus (γHV) infections are life-long and are associated with several oncological and lymphoproliferative disorders particularly after immune suppression in the host (1–3). Severe diseases in humans like Burkitt's lymphoma, Hodgkin's disease, or nasopharyngeal carcinoma are known to be caused by Epstein-Barr virus (EBV). Kaposi's sarcoma, multicentric Castleman's disease or primary effusion lymphoma are related to Kaposi's sarcoma associated herpesvirus (KSHV) (4–9). Primarily, γHV infection of the mucosal epithelium of the naive host is typically asymptomatic. A subsequent productive lytic replication phase allows the formation of latency reservoirs in B cells, dendritic cells and macrophages (10–12). In a suppressed immune system like in AIDS and transplant patients, the reactivation of latent γHV leads to replication followed by disease. However, human γHV are host-specific, therefore studies on vaccine development and strategies against human γHV infection are limited.
Murine γHV 68 (MHV-68) is a natural rodent pathogen that has extensive pathological similarities with and is genetically homologous to human γHV as EBV or KSHV (13, 14). Currently a surrogate model MHV-68 is engaged to understand the pathogenicity of γHV and to examine the efficacy of vaccination strategies. To date several vaccines including attenuated MHV-68, peptide-pulsed DCs, subunit or recombinant DNA vaccines targeting lytic and/or latent viral antigens have shown reduction of acute infections in lungs but were unsuccessful to affect the establishment of latent infection and host vulnerability to viral tumorigenesis (15–22).
Notably, T cells specific for a wide range of epitopes are fundamental to control MHV-68 infection and display two kinetic pattern for acute or latent MHV-68 infection (23). After mucosal infection, CD8+ T cells diminish the load of infectious virus in lungs, while the primary lytic replication seems to have less impact on the latent viral reservoir (24–26). Since MHC class-II positive B cells represent the major latent virus reservoir, cytotoxic CD4+ T cells play a significant role in controlling γHV infection (27). Interestingly, MHV-68 ORF6 (single-stranded DNA binding protein) and ORF61 antigens (ribonucleotide-reductase large subunit protein) are known to stimulate both CD8+ and CD4+ T cells in C57BL/6 mice. ORF6487 epitope-specific CD8+ T cells predominantly control early infection while ORF61524 epitope-specific CD8+ T cells expand in early latency and circulate at high levels throughout the latent infection period (28–30). Moreover, control of MHV-68 infection is provided by CD4+ T cells specific for ORF6593, ORF61343, and ORF61691 epitopes (31).
Modified vaccinia virus Ankara (MVA) is an attenuated strain of vaccinia virus. Due to the strong clinical safety record MVA has been utilized as a viral vaccine vector targeting recombinant antigens in order to induce T cell responses in immunotherapeutic approaches against cancer and infectious diseases (32–34). We generated recombinant MVA (recMVA) vaccines stably expressing full length MHV-68 ORF6 and ORF61 genes. We choose en passant recombineering for insertion of the transgene expression cassette into a self-excisable bacterial artificial chromosome (BAC) containing the MVA genome and allowing for the removal of the selection marker in bacteria (35, 36). Following the rescue of infectious MVA from the self-excisable MVA-BAC, the BAC cassette is efficiently removed from the viral genome resulting in markerless infectious virus progeny. To date, vector vaccine strategies based on recombinant target gene expression were able to control lytic but not latent MHV-68 infection proficiently. Our data show that MVA-based vaccines expressing MHV-68 antigens ORF6 and ORF61 were immunogenic and induced strong CD8+ and CD4+ T cell responses. MVA-ORF6 and MVA-ORF61 proved to be effective in a prophylactic MHV-68 challenge model and were able to protect from MHV-68 early latency by significantly reducing the latent virus reservoir. However, the homologous prime/boost approach failed to protect from latency during the later course of infection despite the presence of antigen-specific CD8+ T cells in high frequencies.
Materials and Methods
Cell Lines and Viruses
DF-1 (ATCC CRL 12203), HeLa (ATCC CCL-2), NIH3T3 cells (ATCC CRL 1658), EL4 cells (ATCC TIB-39), and DC2.4 cells (a kind gift of Kenneth L. Rock, University of Massachusetts, USA) were grown in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin/streptomycin. BHK-21 (ATCC CCL-10) cells were grown in RPMI 1640 supplemented with 5% FCS, 5% tryptose phosphate broth, 100 U/mL penicillin/streptomycin. For bone marrow-derived dendritic cells (BMDCs), bone marrow was collected from tibiae and femurs of C57BL/6 mice. Cells were grown in RPMI 1640 containing 10% FCS, 100 U/mL penicillin/streptomycin and 10% granulocyte-macrophage colony-stimulating factor (GM-CSF) described as previously (37). Working stocks of MHV-68 were prepared by infection of BHK-21 cells as described previously (38). MVA (cloned isolate F6) at 582nd passage on chicken embryo fibroblasts (CEF) was routinely propagated and titered following standard methodology (39).
Peptides
MHV-68 specific (ORF6487−495, ORF61524−531, ORF6593−607, ORF61343−357, ORF61691−705) and control peptides (OVA265−280, B546−60, βgal96−103, and B820) were produced by peptides & elephants GmbH (Hennigsdorf, Germany). Peptides were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 1 μg/μl.
Plasmid Construction
In order to generate MVA transfer plasmids encoding ORF6 or ORF61 MHV-68 genes, respective DNA sequences were PCR amplified by using modified primers designed to generate full length cDNAs of ORF6 and ORF61 including a HA tag sequence at the C-terminal end of each transgene. The cDNAs were cloned in MVA transfer plasmid PH5-dVI-MVA by utilizing BamHI+AflII restriction sites. The expression of both, ORF6 and ORF61, was under the control of the modified PH5-vaccinia virus strong early and late promoter. The linearized transgene expression cassette was excised by PacI restriction enzyme and recombined in deletion VI region of MVA by utilizing 50 bp homologous flanking regions following a two step red-recombination protocol in GS1783 E. coli harboring the GFP-expressing MVA-BAC genome resulting in a recMVA-BAC as described previously (40).
Reconstitution of Recombinant MVA
Rescue of recMVA from BAC was done in DF-1 cells (41). After transfection of recMVA-BAC DNA using turbofect according to the manufacture's protocol (Thermo scientific), rabbit fibroma virus (RFV) (MOI 0.1) was added as helper virus to the cell monolayer. After 72 h, viral plaques (CPE) were monitored by GFP fluorescence. Cells were harvested and pelleted at 4,000 rpm for 10 min at 4°C. Supernatant was discarded and cells resuspended in 1 ml DMEM containing 10% FCS followed by three times freeze-thawing and ultra sonification for 30 s. Supernatant was stored at −80°C. BAC cassette free recMVAs were further identified by limiting dilution on DF-1 cells performed in a 96-well plate. Wild-type MVA-F6, MVA-ORF6, and MVA-ORF61 viruses were propagated and titrated by determining the 50% tissue culture infectious dose (TCID50) in CEF- (39). All viruses were purified by two consecutive ultracentrifugation steps through a 36% (wt/vol) sucrose cushion. Recombinant MVAs were characterized for recombinant ORF6 and ORF61 protein synthesis by western blotting by using monoclonal anti-HA antibody (Sigma) and for replication capacity by in vitro low-multiplicity growth kinetics as previously described (42). Briefly, confluent monolayers from one well of six-well tissue culture plates were used per time point. After virus adsorption, the inoculum was removed, cells were washed and further incubated with fresh medium. At multiple time-points post-infection (p.i.), infected cells were harvested and virus was released by freeze±thawing and brief sonication. Serial dilutions of the resulting lysates were plated on confluent CEF monolayers grown in 96-well plates as replicates of eight. At day 7, microscopic analysis monitoring for wells containing viral plaques (CPE) allowed the determination of virus titers by end point dilution as TCID50/ml.
Generation of T Cell Lines
All T cell lines were established by peptide stimulation of splenocytes obtained from vaccinated mice and maintained by periodical restimulation. For the generation of the CD8+ T cell lines, C57BL/6 mice were vaccinated intraperitoneally (i.p.) once with MVA-ORF6 and MVA-ORF61, respectively, and sacrificed at 8 dpi. For the first round of stimulation, lipopolysacharide (LPS)-activated B cells (LPS-blasts) were generated from splenocytes from naive mice treated with 25 μg/ml LPS and 7 μg/ml dextransulfate for 3 days at 37°C, 5% CO2, and 90% humidity. LPS-blasts were irradiated (30 Gy), pulsed with peptide (250 ng/ml) for 30 min at 37°C and cocultivated with 7 × 106 splenocytes from MVA-ORF6 or MVA-ORF61 vaccinated mice per well in 24-well plates with RPMI 1640 containing 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin for 7 days. For maintenance of T cell lines, the cultures were restimulated every 7 days according to the following scheme. Irradiated (100 Gy), peptide-pulsed (1 μg/ml for 30 min at 37°C) EL4 cells were adjusted to 1 × 106 cells/ml. Irradiated (30 Gy) splenocytes from naive mice were adjusted to 12 × 106 cells/ml. T cells were adjusted to 5 × 105 cells/ml. Finally, 0.5 ml of peptide pulsed EL-4 cells, 0.5 ml of splenocytes, 0.5 ml of 5% TCGF [conditioned medium as supernatant from rat splenocytes stimulated with 5 μg/ml concanavalin A; (37)] and 0.5 ml CD8+ T cells were added in one well of 24 well plate and cocultivated. In order to generate CD4+ T cell lines, C57BL/6 mice were vaccinated twice i.p. with recMVA in a short term prime-boost regimen (prime day 0, boost day 5) as recently shown for CD8 + T cells (43) and sacrificed at 6 d post boost. Spleens were processed as described earlier (37). For maintenance, CD4+ T cell lines were restimulated every 7 days for 20 weeks and thereafter every 14 days as described (37).
In vitro T Cell Assays (T Cell Lines)
BMDCs were infected in 6-well plates with MVA-F6 and recMVA (MOI 5) for 15 h, and MHV-68 (MOI 10) for 48 h. Infected BMDCs 4 × 105/100 μl were incubated with 100 μl of Brefeldin-A (1 μg/ml) and co-cultured with 2 × 105/100 μl of antigen-specific T cells in RPMI 1640 medium supplemented with 10% FCS, 100 U/mL penicillin/streptomycin at 37°C. For peptide-pulsed control cells, 100 μl of peptide were used for CD8+ T cell assays at 1ng/ml and at 1000 ng/ml peptide for CD4+ T cell assays. Briefly, BMDCs were incubated for 30 min at 37°C in the respective peptide solutions, washed and then resuspended in 100 μl of Brefeldin-A (1 μg/ml) and co-cultured with 2 × 105/100 μl of antigen-specific T cells. Intracellular cytokine staining was carried out after 4 h incubation with CD8+ T cells or 15 h with CD4+ T cells as published earlier (37).
Immunization of Mice
Mouse husbandry was conducted under specific-pathogen-free conditions according to the Federation of European Laboratory Animal Science Associations protocols (FELASA) at the “Zentrale Einrichtung für Tierforschung und wissenschaftliche Tierschutzaufgaben (ZETT).” Experiments were performed in accordance with German animal care and ethics legislation and with the approval of the responsible animal welfare authority. C57BL/6N (6–8 weeks old) female mice were purchased from Janvier. Mice were vaccinated intraperitoneally (i.p) with 1 × 108 IU wild type MVA-F6, MVA-ORF6, or MVA-ORF61 in 200 μl PBS or PBS only as control. Vaccination was performed either as prime only or in a short term prime-boost regimen with a boost i.p. at day 5 (43). Spleens were harvested at day 8 after prime or day 6 after prime-boost to measure T cell responses by intracellular cytokine staining (ICS).
Ex vivo T Cell Analysis (Splenocytes)
For ex vivo CD8+ T cell analysis, spleens were removed from vaccinated mice and homogenized with a syringe plunger over metal grid with cell culture medium. Erythrocytes were lysed with 3 ml TAC buffer and washed. Cells were filtered by 70 μm cell strainer and counted. For a short T cell restimulation, 4 × 106 splenocytes were further incubated with respective peptides (1 μg/ml) for 4 h for CD8+ T cells and for 15 h for CD4+ T cells in the presence of BFA. As a control, T cells were stimulated in a non-antigen-specific manner using anti-mouse CD3e antibody (clone 500A2, BD Pharmingen 553238) at 1.25 μg/ml for CD8+ T cells for 4 h and for CD4+ T cells for 15 h in presence of BFA.
MHV-68 Challenge
To analyze the protective capacity of MVA-ORF6 and MVA-ORF61, mice were prime-boost immunized i.p. with 1 × 108 IU of either recMVA in 200 μl PBS or MVA-F6 or PBS only as controls. Four weeks after boosting, mice were anesthetized and infected intranasally with 5 × 104 PFU of MHV-68 in 20 μl RPMI containing 5% FCS. Mice were sacrificed at day 17 or day 45 after challenge. Spleens were harvested to measure weight and then processed for ex vivo T cell analysis (ICS) or used for viral genomic load measurements by qPCR or virus reactivation assays.
Intracellular Cytokine Staining (ICS)
T cell co-cultures with either infected BMDCs [see section In vitro T Cell Assays (T Cell Lines)] or from ex vivo peptide-stimulated splenocytes [see section Ex vivo T Cell Analysis (Splenocytes)] were transferred into 96-well V-bottom plates. Cells were incubated with blocking buffer [PBS having 1% bovine serum albumin (BSA)] containing 1 μg/ml ethidium monoazide bromide (Life technologies GmbH, Germany) for 20 min on ice under light exposure for live/dead discrimination. Afterwards, ICS was performed using BD Cytofix/Cytoperm fixation/permeabilization kit according to the manufacturer's protocol (BD Pharmingen, Germany). Briefly, cells were washed twice with blocking buffer and surface stained with anti-CD8-PB or anti-CD4-PB for 30 min on ice. In addition, cells were stained for CD62L (L-selectin) in order to discriminate between T cells that were specifically reactivated after ex vivo antigen restimulation (CD62L negative) and those that were not reactivated in an antigen-specific manner (CD62L positive). Cells were washed and permeabilized with Cytofix/Cytoperm solution for 15 min on ice. Thereafter, cells were washed, incubated with anti-IFNγ, anti-TNFα or anti-IL-2 for 30 min, washed again, fixed with 2% paraformaldehyde (PFA) and subjected to flow cytometry [BD FACSCanto II (BD Bio Sciences, Germany)].
Ex vivo Reactivation Assay (CPE)
To analyze the frequency of cells carrying viruses reactivating from latency, NIH3T3 cells (104 cell/well) were seeded in 96-well plate. Three-fold dilutions of splenocytes (starting from 1.5 × 105 cells/well) were plated onto NIH3T3 cells with 24 wells per dilution. After 7–14 days, each well was scored for cytopathic effects (CPE) and the frequency of reactivating cells was calculated on the basis of the Poisson distribution by determining the number of cells at which 63.2% of wells scored positive for CPE.
Measurement of Latent Viral Load by Quantitative Real-Time PCR
Measurement of latent viral load in splenocytes of infected mice was determined by quantitative real-time PCR using the ABI 7300 Real Time PCR System (Applied Biosystems, Foster City, CA) as described previously (44). Briefly, a 70-bp region of the MHV-68 glycoprotein B (gB) gene was amplified, and viral DNA copy numbers were quantified. The murine ribosomal protein L8 (rpl8) was amplified in parallel and used to normalize for input DNA between samples. The data are presented as viral genome copy numbers relative to the copy number of L8.
Analysis of Exhaustion Markers
Mice were prime/boost immunized and challenged as described in sections Immunization of Mice and MHV-68 Challenge, respectively. Spleens were removed from vaccinated mice and homogenized with a syringe plunger over metal grid with cell culture medium. Erythrocytes were lysed with 3 ml TAC buffer and washed. Cells were filtered by 70 μm cell strainer, counted and stained with aqua dye (Invitrogen) followed by mouse anti-CD8 PB, anti-CD4 PerCP (eBioscience), anti-PD-1 FITC and anti-CTLA-4 APC (Invitrogen) surface antibody staining as well as anti-Eomes PECy7 and anti-T-bet PE (Invitrogen) intracellular antibody staining as described in section Intracellular Cytokine Staining (ICS).
Statistical Analysis
All data are shown as mean ± SEM of the number of individual mice indicated with n either pooled from or representative for the respective number of independent experiments. The statistical significance was analyzed by an unpaired Student's t-test (two-tailed) using GraphPad Prism 6. p ≤ 0.05 were considered as significant Manuscript Formatting.
Results
Generation and in vitro Characterization of Recombinant MVA Vaccines
Initially, we cloned cDNAs of MHV-68 ORF6 or ORF61 genes into MVA transfer plasmid PH5_dVI_MVA. After the PacI-restricted digestion of transfer plasmids, the linearized transgene expression cassette was transferred into GS1783 E. coli cells containing the MVA-BAC genome including a BAC-GFP cassette residing in the deletion-III region (Figure 1A). The en passant technique based on the red recombination system (40) was applied to recombine the transgene expression cassette into deletion VI of the MVA-BAC (36, 40, 45, 46) (Figure 1A). Moreover, the aphAI (kanamycin) marker gene present in the expression cassette to positively select recMVA-BAC was removed following en passant recombination of homologous sequences of the I-SceI homing endonuclease which flanked the aphAI gene (Figure 1A). Furthermore, the complete BAC expression cassette was removed by homologous recombination after BAC rescue (self-excising BAC) in DF-1 cells (35, 41, 47). Sequencing assured the right insertion as well as correct orientation and sequence of the recombinant expression cassette for both recMVA. Additionally, we analyzed the target protein synthesis by using MVA-ORF6 and MVA-ORF61 infected HeLa cell lysates. Western blot analysis using HA-specific antibodies able to bind the C-terminal HA-tag of the recombinant ORF6 and ORF61 proteins confirmed the expected size of ORF6 (124 kDa) and ORF61 (90 kDa), respectively (Figure 1B, blot image spliced and grouped). Importantly, both recMVA showed similar replication capacity in vitro as compared to wild type MVA-F6 (Figure 1C).
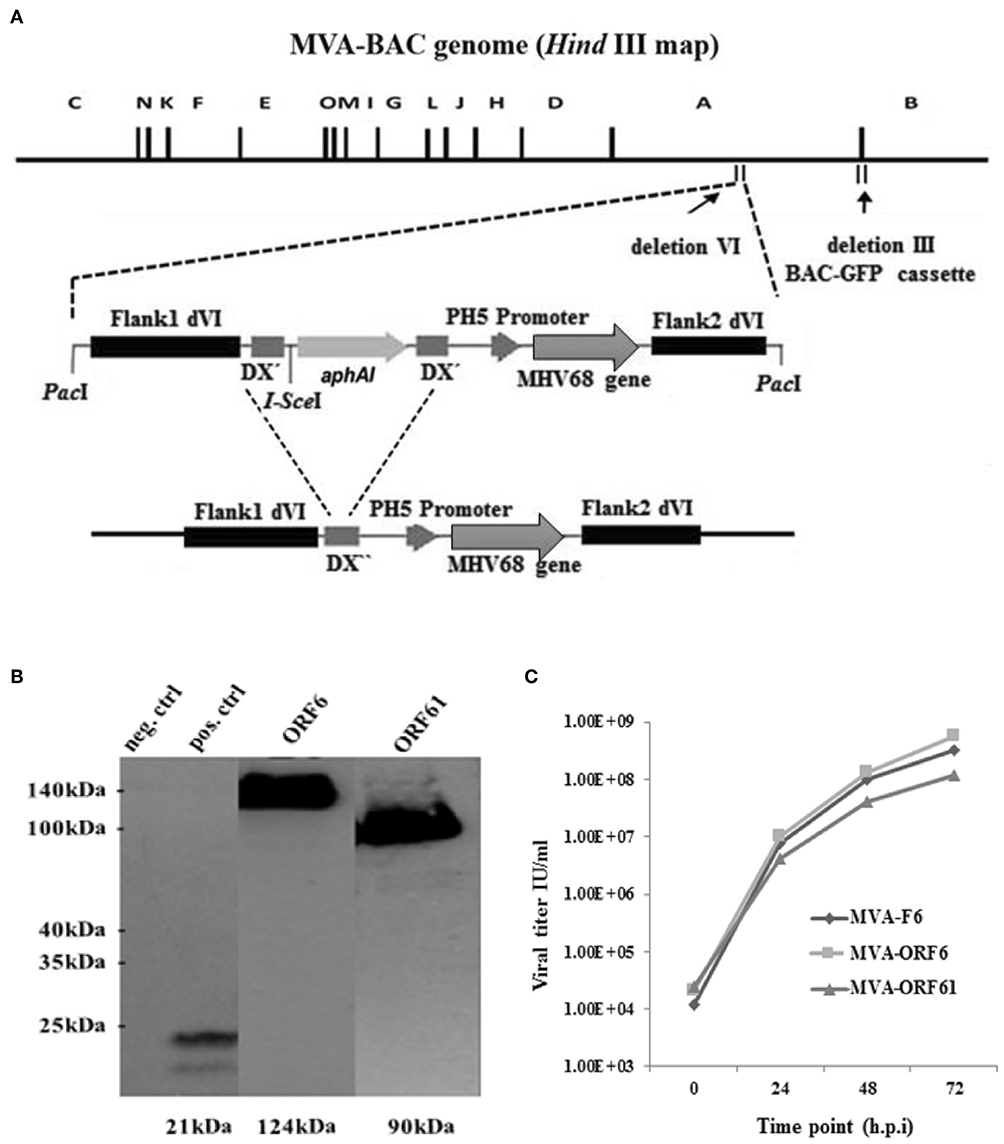
Figure 1. (A) Schematic map of the MVA-BAC genome (HindIII restriction map) including the BAC-GFP cassette as well as the MVA transfer plasmid containing the respective MHV-68 gene (either ORF6 or ORF61). Flank 1 and 2 are homologous sequences necessary to integrate into the site of deletion IV within the MVA genome. I-SceI restriction site is next to aphAI gene, whereas DX″ marks homologous flanks allowing the deletion of aphAI through homologous recombination. The final recombinant MVA with aphAI marker gene deletion is shown at the bottom. (B) Synthesis of recombinant HA-tagged ORF6 (single-stranded DNA binding protein) and ORF61 (ribonucleotide-reductase large subunit protein) (Western blot analysis) after infection of Hela cells with MVA-ORF6, MVA-ORF61, or MVA wild type (as negative control) (MOI-10). For positive control, HeLa cells were transfected with plasmid pIRES-eGFP encoding an HA-tagged HCMV US6 gene and infected with MVA wild type. Cell lysates were harvested after 24 h and run through the SDS 10% PAGE. Immunobloting was done by using HA-specific monoclonal antibodies. The image of the blot was spliced and grouped for editorial purposes. (C) Low-multiplicity growth of MVA-ORF6 and MVA-ORF61. Viruses showed similar growth kinetics compared to MVA wild type (MVA-F6) on CEF cells infected at low MOI of 0.01. Viral titers were determined by end point dilution at indicated hours post infection (h.p.i.) to obtain a 50% tissue culture infectious dose (TCID50).
Primary CD8+ and CD4+ T Cell Responses After recMVA Vaccination
ORF6- and ORF61-specific CD8+ T cells dominate the MHV-68-directed CD8+ T cell response (25). In addition, ORF6 and ORF61 antigens induce a substantial CD4+ T cell response during MHV-68 infection (31). Thus, we examined the capacity of MVA-ORF6 and MVA-ORF61 vaccines to elicit ORF6- and ORF61-specific CD8+ and CD4+ T cells. T cell responses were determined at day 8 after vaccination (Figure 2A) and showed efficient priming of ORF6- and ORF61-specific CD8+ (Figures 2B,C) and CD4+ T cells (Figures 2D,E) using MVA-ORF6 or MVA-ORF61. About 2% of ORF6487-specific and 1% ORF61524-specific CD8+ T cells produced IFNγ and to a lesser extend TNFα (Figures 2B,C). ORF6 and ORF61-specific primary CD4+ T cells produced IFNγ and IL-2 after vaccination with MVA-ORF6 or MVA-ORF61, respectively (Figures 2D,E). T-cell responses against MHV-68 antigens accompanied those to dominant MHC class I or II restricted vaccinia virus (VACV) epitopes such as B820 or B546, respectively. B8 and B5 antigens are expressed by various VACV strains including MVA and served as infection controls and helped to validate the efficacy of recMVA vaccination comparative to MVA-wt for CD8+ and CD4+ T cells, respectively (48). Although MVA-ORF6 and MVA-ORF61 were considered to be immunogenic and elicited ORF6- and ORF61-specific CD4+ and CD8+ T cells, some epitope specificities such as ORF6593-specific CD4+ T cells were of low frequency after a single vaccination. This was particularly obvious in the memory phase at day 35 p.i. in which CD8+ and CD4+ T cell responses strongly contracted, yet to still detectable levels (Supplementary Figure 1).
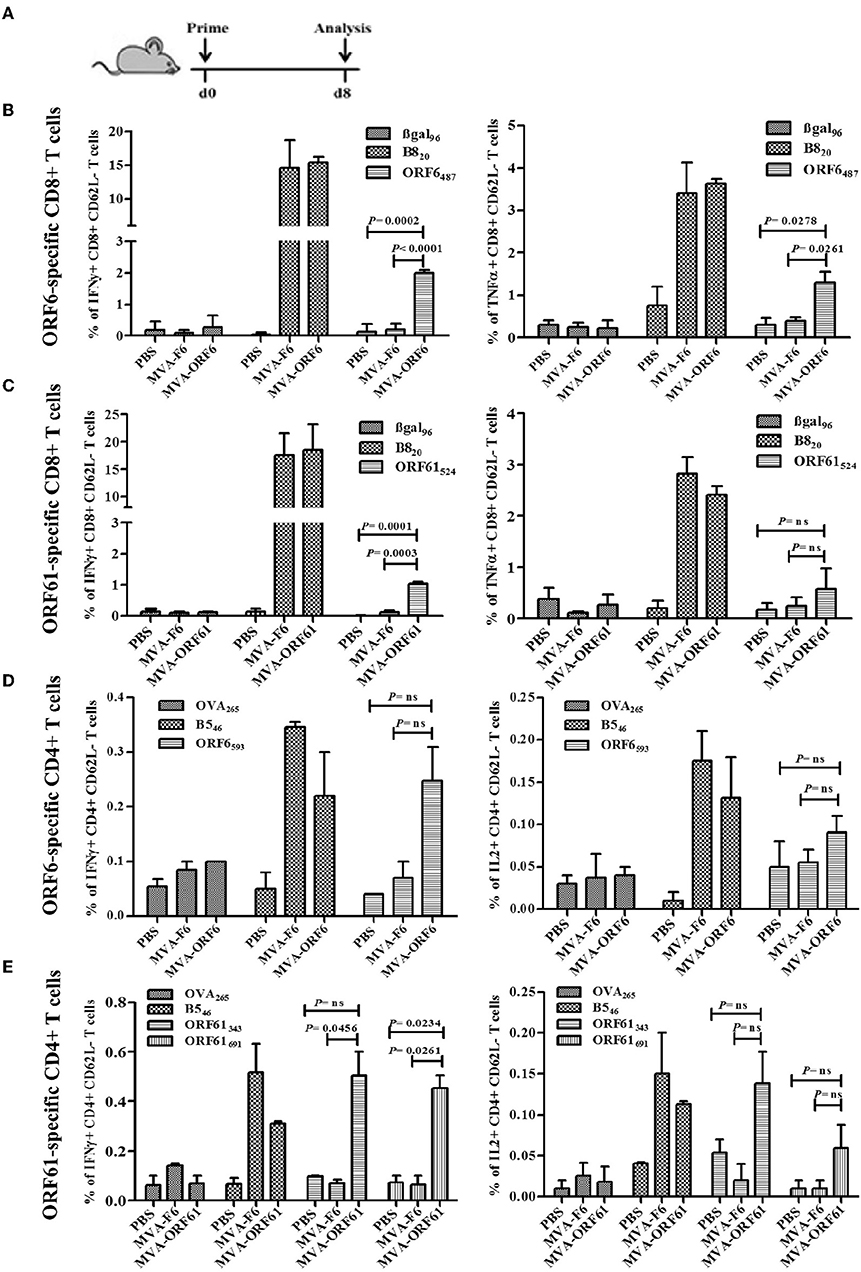
Figure 2. Primary CD8+ and CD4+ T cell response after recMVA vaccination. (A) C57BL/6 mice were vaccinated i.p. using 1 × 108 IU/200 μl of MVA-F6 (wildtype), MVA-ORF6 or MVA-ORF61 or 200 μl of PBS as a control group. For ex vivo T cell analysis (ICS followed by FACS) splenocytes were prepared from immunized mice at 8 dpi. (B,C) CD8+ T cell response. (B) Frequencies of activated (CD62L-) ORF6487- or (C) ORF61524 specific CD8+ T cells producing IFNγ (left panel) or TNFα (right panel). βgal96 or B820 peptides were used as unrelated or MVA-specific controls, respectively (D,E) CD4+ T cell response. (D) Frequencies of activated (CD62L-) ORF6593- or (E) ORF61343- and ORF61691-specific CD4+ T cells producing IFNγ (left panel) or IL2 (right panel). OVA265 or B546 peptides were used as unrelated or MVA-specific controls, respectively. Data shown are mean ± SEM of n = 6 mice per group, pooled from 3 (B,C) or two independent experiments (D,E).
Ex vivo Analysis of CD8+ and CD4+ T Cell Responses After Prime-Boost Vaccination
Next, we determined the antigen-specific T cell responses after prime-boost immunizations with MVA-ORF6 or -ORF61. Mice received a homologous boost vaccination at day 5 post primary immunization (Figure 3A) consistent with a short period prime-boost vaccination protocol (43). This protocol has been demonstrated to result in equally efficient antigen-specific T cell expansion as compared to standard boosting at day 30 post priming. Saving time may be beneficial under certain circumstances in order to allow for earlier protective immunity or to shorten costs. At day 6 post boost, MVA-ORF6 vaccination resulted in higher levels of CD8+ IFNγ+ T cells (6%) specific for ORF6487 (Figure 3B) as compared to a single immunization (Figure 2B, Supplementary Figure 1A). The expansion of CD8+ IFNγ+ T cells in the secondary response to MVA-ORF61 (1.5%) was not significant (Figure 3B) compared to a single immunization (Figure 2C, Supplementary Figure 1C). Likewise, high levels of IFNγ producing ORF6593-, ORF61343-, or ORF61691-specific activated CD4+ T cells (0.25–0.3%) were observed (Figure 3C). Interestingly, ORF6593-specific responses were similar to the primary response in the acute phase (day 8), whereas ORF61343 and ORF61691 epitope-specific responses were comparatively reduced (Figures 2D,E). In comparison to the memory response after single immunization, however, we could still observe a boost effect for all epitopes tested (Supplementary Figures 1B,D). The data indicates good capabilities of MVA-ORF6 and -ORF61 to induce antigen-specific memory T cell responses that could reactivate and expand after boost immunization.
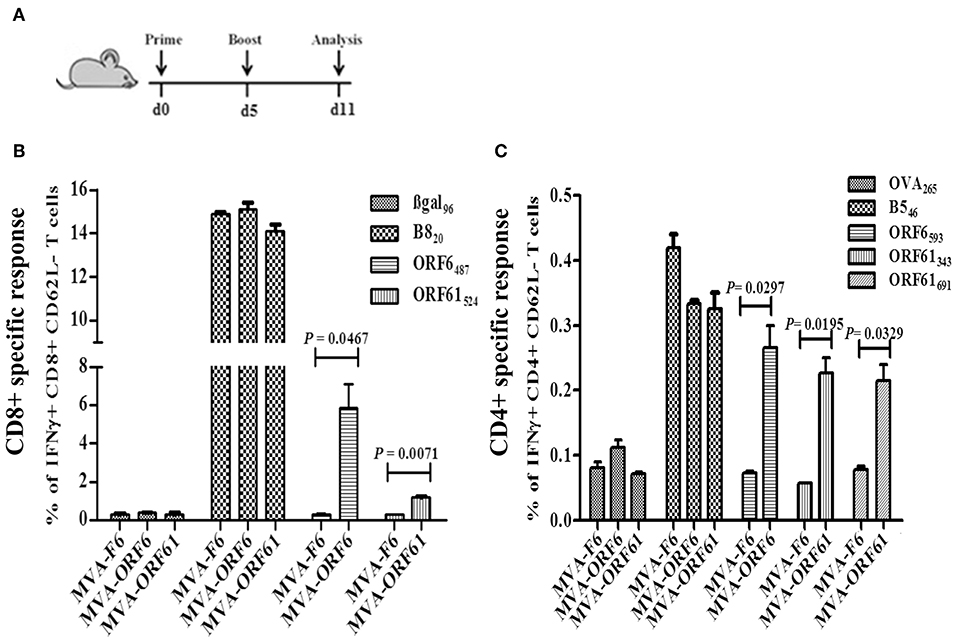
Figure 3. RecMVA prime-boost vaccination (A) Vaccination strategy. C57BL/6 mice were vaccinated i.p. with 1 × 108 IU/200 μl MVA-F6, MVA-ORF6 or MVA-ORF61. At day 6 of post boost, splenocytes were prepapared for ex vivo T cell analysis (ICS/FACS). (B) CD8+ T cells. Frequencies of activated (CD62L-) IFNγ producing ORF6487- and ORF61524 -specific CD8+ T cells. (C) CD4+ T cells. Frequencies of activated (CD62L−) IFNγ producing ORF6593−, ORF61343−, and ORF61691− specific CD4+ T cells. Data shown are mean ± SEM of n = 10 mice per group, pooled from two independent experiments.
ORF6 and ORF61-Specific T Cell Lines Recognize Infected BMDC in vitro
Since ORF6 and ORF61 encode both, MHC class-I- and II-restricted epitopes, we tested the endogenous presentation of ORF6- and ORF61-specific epitopes in recMVA or MHV-68 infected target cells by determining the recognition by polyclonal epitope-specific CD8+ or CD4+ T cell lines generated from recMVA-vaccinated mice. Interestingly, BMDCs infected with MVA-ORF6, MVA-ORF61 or MHV-68 and co-cultivated with CD8+ T cells specific for ORF6487 and ORF61524 epitopes, activated both CD8+ T cell lines as good as peptide-pulsed BMDCs used as a positive control (Figures 4A,B). Likewise, we investigated MHC class-II-restricted endogenous recognition of infected BMDCs by MHV-68-specific CD4+ T cells (Figures 4C–E) (37). ORF6593-specific CD4+ T cells were activated indicating efficient MHC class II-restricted presentation by MVA-ORF6 or MHV-68-infected BMDCs (Figure 4C). While MVA-ORF61-infected BMDCs activated ORF61343- and ORF61691-specific CD4+ T cells, MHV-68-infected BMDCs failed to stimulate both ORF61-specific CD4+ T cell lines (Figures 4D,E). Of note, we determined a comparable affinity for ORF6487 and ORF61524-specific CD8+ T cells (Supplementary Figure 2A). In contrast to ORF61-specific CD4+ T cells, ORF6593-specific CD4+ T cells displayed higher affinity to their peptide/MHC class II complexes (Supplementary Figure 2B).
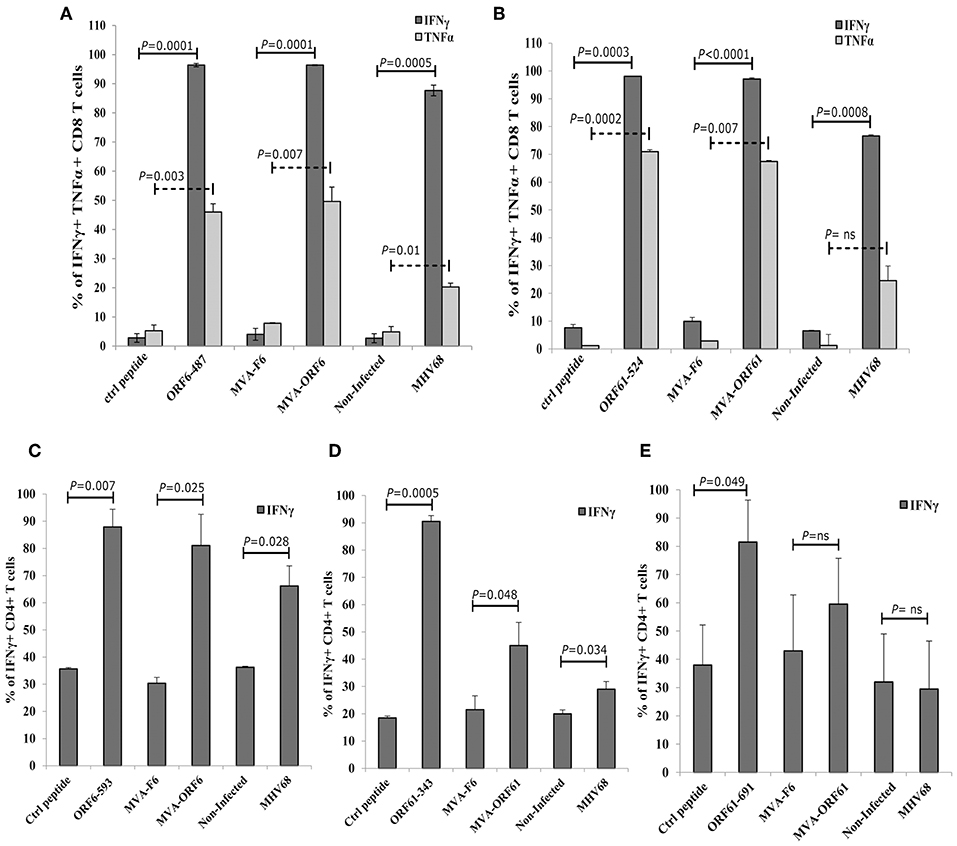
Figure 4. Antigen processing and presentation of ORF6 and ORF61 antigens. After infection with MVA-F6, MVA-ORF6 or MVA-ORF61 (MOI 5) for 15 h or with MHV-68 (MOI 10) for 48 h, BMDCs were co-incubated with CD8+ T cells (A,B) or CD4+ T cells (C–E). CD8+ T cells specific for (A) ORF6487 or (B) ORF61524 epitopes were co-incubated for 4 h; CD4+ T cells specific for (C) ORF6593, (D) ORF61343, or (E) ORF61691 epitopes for 15 h. BMDCs pulsed with the cognate MHC-class-I (A,B) and -II-restricted peptides (C–E) at a concentration of 1 or 1,000 ng/ml, respectively, were used as positive controls: (A) ORF6-487, (B) ORF61−524, (C) ORF6-593, (D) ORF61−343, and (E) ORF61−691. BMDCs pulsed with irrelevant peptide (ctrl peptide) as well as uninfected or MVA-F6-infected BMDCs served as negative or infection controls, respectively. Cytokine production [IFNγ (A–E), TNFα (A,B)] was determined by ICS and FACS analysis. Data are mean ± SEM of n = 4 mice per group, pooled from two independent experiments.
Protection From Early Latency but Not From Established Latency by Prophylactic RecMVA Vaccines After MHV-68 Challenge
Following acute infection via the intranasal route, MHV-68 scatter from lungs to secondary lymphoid organs where persistent infection is established in latently infected B-lymphocytes, dendritic cells and macrophages (10). Splenomegaly is a marker for latency as a result of increased B and T cell numbers which peaks at day 14 post infection (24, 49). Accordingly, we determined the ability of recMVA vaccines to protect from viral latency by quantifying the latent viral load and the reactivation capacity of latent viruses (50). We vaccinated mice i.p. followed by a homologous boost vaccination at day 5. Four weeks after the boost, mice were challenged i.n. with MHV-68 (Figure 5A). At day 17 or day 45 post challenge, corresponding to the early or established latent phase of infection, respectively, protection was evaluated.
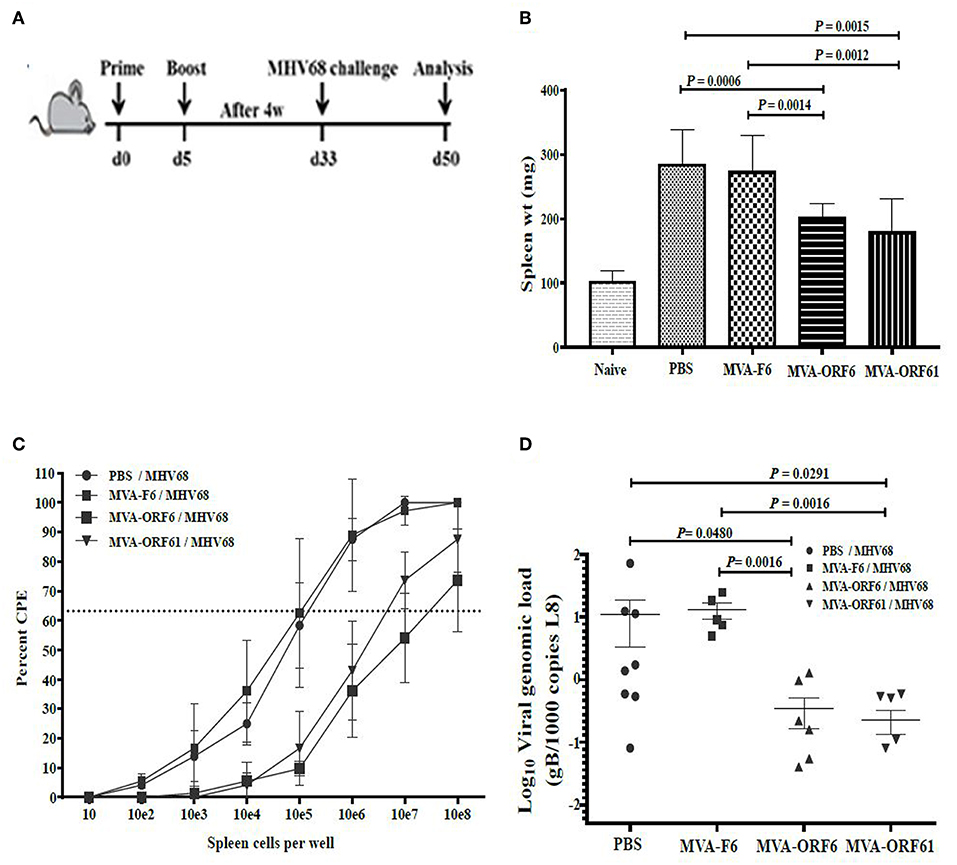
Figure 5. Protection from MHV-68 early latency. (A) C57BL/6 mice were i.p. prime/boost vaccinated using 1 × 108 IU/200 μl MVA-ORF6, MVA-ORF61, MVA-F6 (as infection control) or PBS (as mock control). Four weeks later, mice were challenged i.n. by using 5 × 104 PFU/20 μl of MHV-68. At 17 days post challenge, (B) spleen weights, and (C) latent viral load by ex vivo reactivation or (D) by Real Time PCR were determined. (B) Mean spleen weight ± SEM of n = 8–10 mice per group, pooled from three independent experiments. Age-matched mice which received no treatment at all (no vaccination, no challenge) served as control littermates (naïve, spleen weight ± SEM of n = 4). (C,D) Single splenocytes suspensions from day 17 after MHV-68 challenge were used (C) to determine the latent viral load by ex vivo reactivation of splenocytes or (D) for isolation of DNA to measure latent viral copy load by Real Time PCR analysis. (C) Mean frequency of reactivation replication of latent virus ± SEM from n = 6–9 mice per group, pooled from three independent experiments. The horizontal dotted line specifies the point of 63.2% Poisson distribution, set by the non-linear regression that was used to calculate the frequency within the cells. For calculation of significance, frequencies of reactivation events were statistically analyzed by paired t-test over all cell dilutions. The statistical difference of MVA-ORF6 immunized group to PBS is P = 0.008 and to MVA-F6 P = 0.005. The statistical difference of MVA-ORF61 immunized group to PBS is P = 0.009 and to MVA-F6 P = 0.007. (D) Mean viral genomic load in the spleen from n = 5–8 mice per group, pooled from two independent experiments is indicated by horizontal bars. Each symbol represents an individual mouse. Statistical significance (P).
At day 17, MVA-ORF6- and MVA-ORF61-pre-immunized mice showed a highly significant reduction in spleen weight (Figure 5B) and a decrease in latent virus as quantified by the number of reactivating splenocytes (Figure 5C) as compared to the control groups (MVA-F6 or PBS). In MVA-F6 or PBS immunized control groups, the frequency of splenocytes allowing for MHV-68 reactivation from latency was 1 in 5,565 cells and 1 in 5,964 cells, respectively. The amount of reactivating splenocytes was 1 in 64,609 cells MVA-ORF61 and 1 in 117,065 cells for MVA-ORF6 immunized animals. Likewise compared to the control groups, MVA-ORF6 or MVA-ORF61 vaccination resulted in a significant decrease of the splenic latent viral genomic load as quantified by real-time PCR (Figure 5D). These results were corroborated by the IFNγ producing CD8+ T cell frequencies in MVA-ORF6 or MVA-ORF61 vaccinated mice after MHV-68 challenge which were significantly higher (Figure 6A). Interestingly, the amount of IFNγ producing CD4+CD62L- T cells were significantly lower in these two vaccination groups as compared to the controls (MVA-F6 or PBS) (Figure 6B). Since these activated IFNγ producing ORF6- and ORF61-specific CD4+ T cells were also significantly reduced in absolute numbers, we suggest that the decrease was not due to a shift in the relative distribution but rather indicates the consumption of these T cell specificities as effectors for direct killing of virus infected cells (Supplementary Figure 3). There were no obvious differences regarding antigen-specific TNFα production in CD8+ T cells (Figure 6C) or IL-2 production in CD4+ T cells (Figure 6D, Supplementary Figure 3) underlining the importance of IFNγ for protection in early latency.
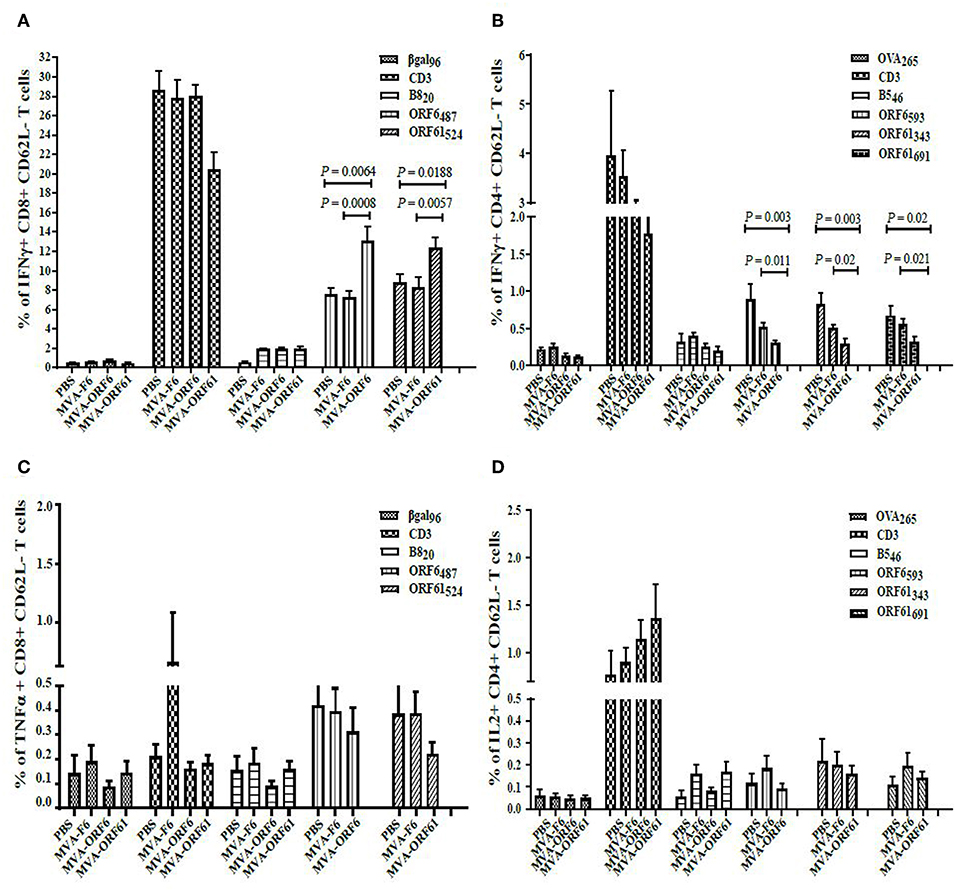
Figure 6. CD8+ and CD4+ T cell responses in recMVA-prime/boost vaccinated mice 17 days after MHV-68 challenge. (A) Detection of activated (CD62L−) IFNγ- or (C) TNFα-producing ORF6- and ORF61-specific CD8+ T cells (ICS followed by FACS analysis). Splenocytes were stimulated with peptide epitopes ORF6487/ORF61524, βgal499 (negative control) or B820 (vaccination control). (B) Detection of activated (CD62L-) IFNγ- or (D) IL2-producing ORF6- and ORF61-specific CD4+ T cells. Splenocytes were stimulated with peptide epitopes ORF6593, ORF61343-, ORF61691, OVA265 (negative control) or B546 (vaccination control). (A–D) CD3 indicates T cell stimulation using anti-CD3 antibodies as non-antigen-specific positive control. The statistical significance was calculated using an un-paired t-test. Data shown are mean ± SEM of n = 11 mice for each group, pooled from three independent experiments.
Surprisingly, both vaccines failed to confer long-term reduction in latency, since we could not detect a sustained decrease in spleen weight (Figure 7A) and viral genomic load (Figure 7B) at day 45 post challenge as compared to the controls. Interestingly, the T cell analysis showed a significantly higher IFNγ-producing CD8+ T cell response directed against ORF6 and ORF61 as compared to control vaccinated animals (Figure 7C), while CD4+ T cell frequencies for both, ORF6 and ORF61, were comparable in all groups (Figure 7D). In addition, MVA-ORF6 vaccinated mice contained significantly more ORF6-specific TNFα-producing CD8+ T cells compared to the other vaccine groups (Figure 7C, lower panel) which corroborated the data obtained for IFNγ. In contrast, ORF61-specific TNFα-production was comparable between vaccine groups (Figure 7C, lower panel). Interestingly, we failed to detect ORF6- or ORF61-specific IL-2 production in all CD4+ T cells at day 45 (Figure 7D, lower panel) which may indicate that the low CD4+ T cell response seen in long-term latency might be due to lack of proliferation or even exhaustion. Thus, although present in high numbers, antigen-specific CD8+ T cells failed to control latency at later time points.
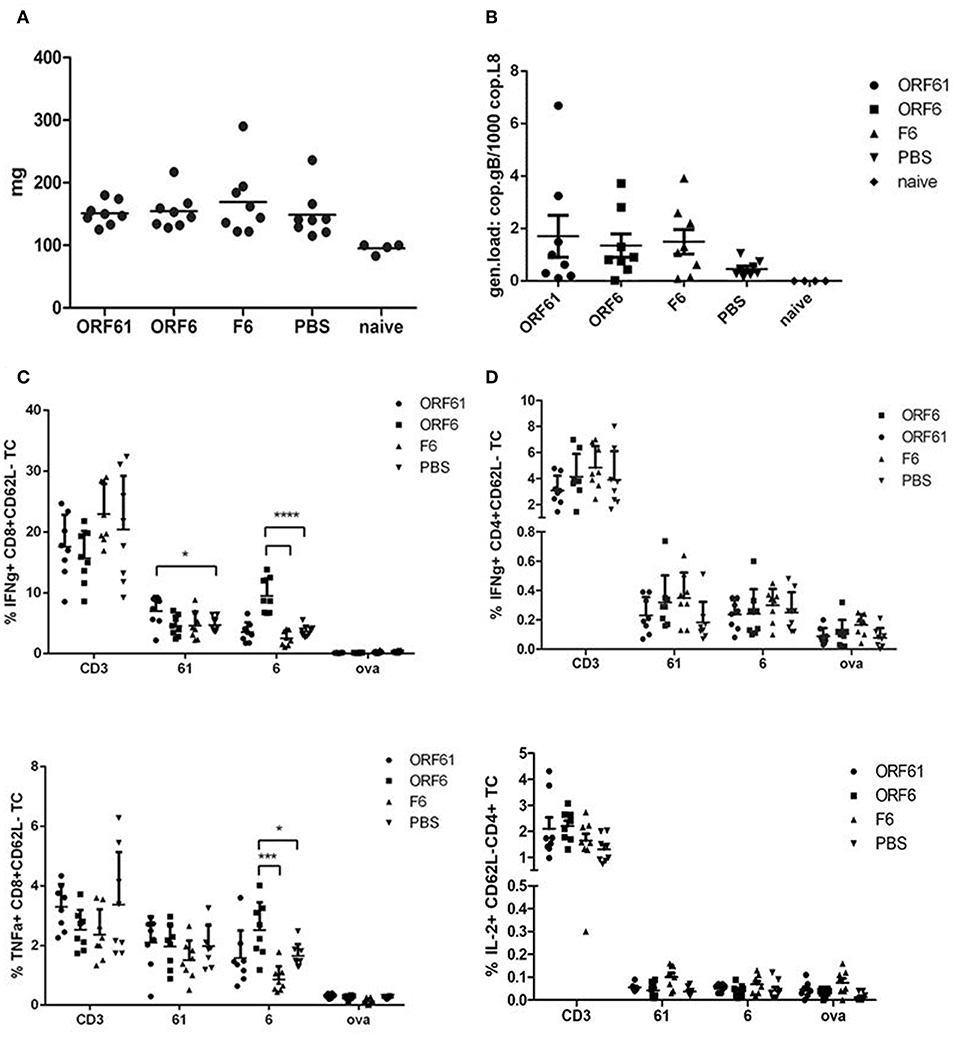
Figure 7. Vaccination with MVA-ORF6 or MVA-ORF61 fails to protect from MHV-68 latency at day 45. C57BL/6 mice were i.p. prime/boost vaccinated using 1 × 108 IU/200 μl MVA-ORF6, MVA-ORF61, MVA-F6 (as infection control) or PBS (as mock control). Four weeks later, mice were challenged i.n. with 5 × 104 PFU/20 μl of MHV-68. Mice which received no treatment at all (no vaccination, no challenge) served as control littermates (naïve). At 45 days post challenge, (A) whole spleen weights or (B) latent viral copy load by Real Time PCR of DNA isolated from single splenocyte suspensions were determined. (A) Mean spleen weight (horizontal bars) of n = 4–8 mice per group pooled from two independent experiments. (B) Mean viral genomic load in spleen from n = 4–8 mice per group pooled from two independent experiments as indicated by horizontal bars. (C) CD8+ and (D) CD4+ T cell responses in recMVA-prime/boost vaccinated mice 45 days after MHV-68 challenge. (C) Detection of activated (CD62L-) IFNγ- (upper panel) or TNFα-producing (lower panel) ORF6- and ORF61-specific CD8+ T cells (ICS followed by FACS analysis). Splenocytes were stimulated with peptide epitopes ORF6487, ORF61524, or OVA257 (negative control). (D) Detection of activated (CD62L-) IFNγ- (upper panel) or IL-2-producing (lower panel) ORF6- and ORF61-specific CD4+ T cells. Splenocytes were stimulated with peptide epitopes ORF6593, ORF61343, or OVA265 (negative control). (C,D) CD3 indicates T cell stimulation using anti-CD3 antibody as non-antigen-specific positive control. The statistical significance was calculated using an un-paired t-test. Data shown are mean ± SEM of n = 8 mice for each group, pooled from two independent experiments. Statistical significance (P); *P ≤ 0.05; ****P ≤ 0.0001. (A–D) Each symbol represents an individual mouse.
Loss of Protection in Late Latency at Day 45 Is Not Driven by T Cell Exhaustion
As mentioned above, T cell exhaustion especially targeting CD4+ T cells during the course of MHV-68 infection could be a reason for the transient and timely limited protective capacity of recMVA vaccines. We therefore monitored the CD8+ and CD4+ T cell responses at day 17 and day 45 post challenge for the expression of characteristic markers of T cell exhaustion such as the inhibitory receptors PD-1 and CTLA-4 as well as the transcription factors Eomes and T-bet (Figure 8). Interestingly, the expression of exhaustion marker PD-1 was highly significantly reduced in CD4+ and CD8+ T cells in all groups at day 45 compared to day 17 (Figure 8A). The expression of CTLA-4 did not change between day 45 and day 17 in CD8+ T cells and was significantly reduced in CD4+ T cells for all groups apart from the MVA-ORF6-vaccinated group which showed comparable expression of CTLA-4 at days 45 and 17 (Figure 8B). In addition, PD-1/T-bet double positive cells most likely representing early exhausted T cells as well as PD-1/Eomes double positive cells indicating most likely terminally exhausted T cells were significantly diminished in CD4+ as well as CD8+ T cells in all corresponding groups at day 45 compared to day17 (Figure 8C). Of note, T cell frequencies were considerably low and close to detection limit for all markers except PD-1 at day 45. Since exhaustion markers were not increased in any of the groups by day 45, but in contrast strongly decreased at day 45 with the exception of CTLA-4 in CD8+ T cells, we conclude that T cell exhaustion did not contribute to the loss of protection by recMVA vaccines in established latency at day 45.
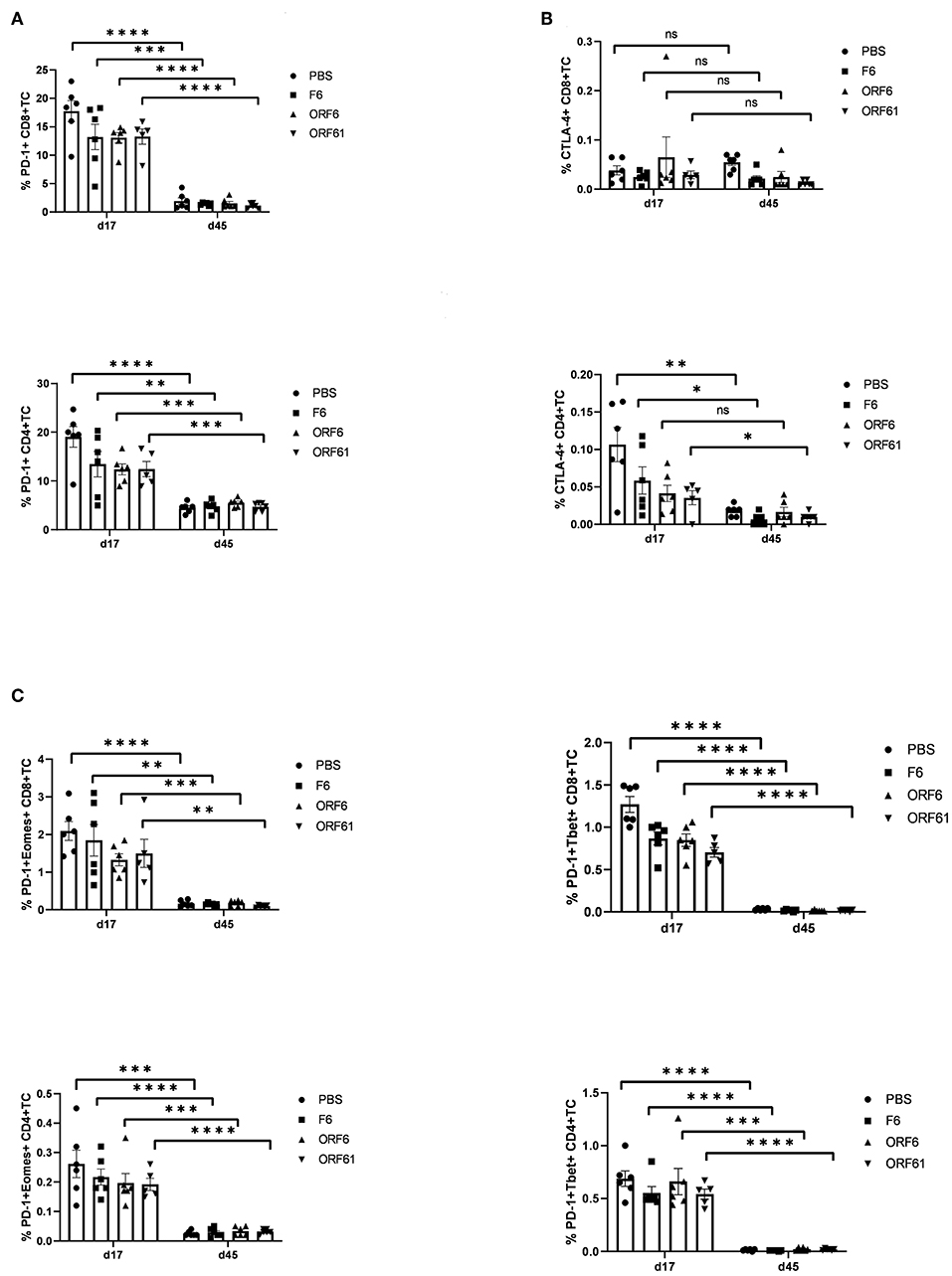
Figure 8. Lack of protection by recMVA vaccines in long-term latency at day 45 post challenge is not due to T cell exhaustion. C57BL/6 mice were i.p prime/boost vaccinated with 1 × 108 IU/200 μl MVA-ORF6, MVA-ORF61, MVA-F6 (as infection control) or PBS (as mock control). Four weeks later, mice were challenged i.n. with 5 × 104 PFU/20 μl MHV-68. On day 17 (d17) or day 45 (d45) post challenge, splenocytes were prepared and the expression of exhaustion markers such as inhibitory receptors PD-1 (A) and CTLA-4 (B) or transcription factors such as Eomes or T-bet within PD-1-positive T cells (C) determined for CD8+ (upper panels) and CD4+ T cells (lower panels) using specific antibodies (FACS analysis). The statistical significance was calculated using an un-paired t-test. Data shown are mean ± SEM of n = 5–6 mice for each group, pooled from two independent experiments. Statistical significance (P); *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; n.s., Non-significant.
Discussion
Recombinant vaccines based on MVA expressing MHV-68 antigens have been constructed employing an MVA-BAC recombination system based on the en passant technique which has been described as a comparatively fast and easy method (35). In favor of possible vaccine application, we applied a self-excisable MVA-BAC in which the BAC cassette was subsequently removed from recMVA genomes by site specific recombination (41). BAC-less MVA-ORF6 and MVA-ORF61 efficiently expressed ther recombinant target genes and displayed standard growth kinetics in vitro.
As T cells are pivotal to control γHV infections, we monitored the T cell response after immunization with recMVA and investigated the efficacy of MHV-68-infected target cells to stimulate CD8+ and CD4+ T cells in vitro, as an indication of direct antigen processing and presentation. Nevertheless, direct as well as cross-priming contribute to MVA-induced CD8+ T cell responses (51–53). After a single vaccination with MVA-ORF6 or MVA-ORF61 we observed strong but subdominant target antigen-specific effector CD8+ T cell responses during the acute phase of infection (8 dpi) (43, 54–56). As expected, in the memory phase of infection CD8+ T cell frequencies contracted to about 10% of the initial maximum (Supplementary Figures 1A,C). ORF6 and−61 antigens were expressed under control of the modified PH5 early/late promoter resulting in enhanced transgene expression (57). In addition, both MVA-ORF6 and MVA-ORF61 vaccines induced effector CD4+ T cell responses upon primary vaccination, but only ORF6-specific CD4+ T cells were clearly detectable in the memory phase (35 dpi) (Supplementary Figures 1B,D). Of note, next to ORF61-specific memory CD4+ T cells, MVA-ORF61-induced B546-specific memory CD4+ T cell responses were hardly detectable as well, implicating that these CD4+ T cells primarily differentiated into effector T cells (CD62Llow) after immunization leaving low memory CD4+ T cell responses (58). RecMVA has been often used as vector for boost vaccinations (59–61) and allows for short-term prime-boost immunization strategies (43). Likewise, our data indicate that MVA-ORF6− and -ORF61 efficiently expanded CD4+ and CD8+ T cells following this short-term prime-boost regimen even at low to undetectable numbers of pre-existing memory CD4+ T cells.
Furthermore, the endogenous processing and presentation of viral antigens by infected target cells was analyzed by using antigen-specific CD8+/CD4+ T cell lines which were generated from recMVA-vaccinated mice. These effector memory-like T cells were used as in vitro readout system (37, 43). MHV-68 ORF6 which encodes for a single-stranded DNA binding protein, seemed to be efficiently processed and both, MHC-I- and -II restricted epitopes, presented as ORF6-specific CD8+ and CD4+ T cells were activated upon exposure to MVA-ORF6 or MHV-68-infected target cells. After infection of target cells with recMVA or MHV-68 encoding ORF61 a ribonuleotide-reductase large subunit protein, CD8+ T cells were specifically activated, while CD4 T cells were less responsive (recMVA) or completely failed to respond (MHV-68). Interestingly, EBV-BPLF1 is known to interact with its large subunit with ribonucleotide-reductase and removes its ubiquitin chain, thereby decreasing the activity of ribonucleotide-reductase in vitro, while MHV-68 ribonucleotide-reductase has been shown to colocalize with subnuclear structures named PML-nuclear bodies (promyelocytic leukemia protein) (62). We propose the possibility that processing of ORF61 as an antigen encoding MHC-II-restricted epitopes might be decreased in vitro due to the MHV-68 deubiquitinase enzyme (DUB) encoded by ORF64 similar as in the case of EBV. Further studies are required to elucidate the processing of the ORF61 antigen.
Our data indicate that recMVA vaccines induce MHV-68–specific long-term memory T cell responses after primary as well as boost vaccination. Previously, post exposure vaccinations with recombinant replication-competent vaccinia viruses (VACV-ORF6487 and VACV-Orf61524) have shown a transient viral replication control in lungs but had no impact on latency establishment (28). Therefore, we were interested in the protective efficacy of recMVA on the establishment of MHV-68 latency at early (day 17) and late time points (day 45) in a prophylactic vaccination setting. At day 17 in early latency, the spleen weights and splenic viral copy loads were significantly reduced in both groups of recMVA-immunized mice. However, in established latency at day 45, both vaccination groups were comparable to the controls and the protective capacity was lost. CD8+ T cells are involved in resolving splenomegaly which is important in control of long-term latent infection, while the manifestation of splenomegaly is driven by CD4+ T cells depending on MHV-68-infected B-cells in the spleen (63–67). We observed that both rec-ORF6-MVA and rec-ORF61-MVA vaccines maintained strong effector CD8+ T cell responses for both time points tested in the latent phase of MHV-68 infection. Those antigen specific CD8+ T cells were able to proliferate efficiently in response to a challenge with MHV-68 encoding the cognate epitopes. In contrast, ORF6- and ORF61-specific effector CD4+ T cells were significantly reduced in frequency and absolute numbers as compared to mice without pre-existing immunity to ORF6 or ORF61 in early latency, while they were indistinguishable from the controls at day 45.
CD8+ T cells are not sufficient to prevent lytic or persistent MHV-68 infection, however they contribute to CD4+ T cells which mainly control the latent infection (67). These CD4+ T cells have effector functions which are mediated by IFNγ production and/or cytotoxicity. Our data corroborate this view, since rec-MVA induced strong effector CD4+ T cells producing IFNγ which may have been consumed to control the latent infection. MHV-68 shows persistent infection in IFNγ−/− or IFNγ receptor−/− mice, indicating a role of IFNγ in limiting the lytic infection or inhibiting reactivation from latent infection (68, 69). The loss of ORF6 and ORF61 specific CD4+ T cells in recMVA-vaccinated groups after challenge supports this effector function and correlates with decreased spleen weight as a marker for splenomegaly (63, 70) as well as the reduced latent virus reservoir in these mice early at day 17. However, these CD4+ T cells were unable to control long-term latency and were present at comparable frequency as in control groups, possibly because they were consumed and/or unable to proliferate. The latter is supported by the loss of IL-2 production in these cells at day 45 supporting the view that particularly CD4+ T cells may be required for long-term infection control (27, 71). T cell exhaustion as described for some chronic infections (72) could be a reason for the restricted protective capacity of recMVA vaccines. However, a comparative T cell analysis for exhaustion markers such as the inhibitory receptors PD-1 and CTLA-4 or the transcription factors Eomes and T-bet at day 17 and day 45 excluded a role of T cell exhaustion at day 45. The CD4+ T cell frequencies expressing PD-1 or CTLA-4 were significantly decreased in all corresponding groups at day 45 compared to day 17, with the exception of the MVA-ORF6 group. Similarly, we found a significant decrease of CD4+ and CD8+ T cell frequencies coexpressing exaustion markers such as PD-1 and T-bet (early exhaustion) or PD-1 and Eomes (terminal exhaustion) (72–74) in corresponding groups at day 45 compared to day 17. We therefore conclude that the lack of protection by recMVA vaccines in established latency in this model was not mediated by T cell exhaustion.
Up to now exclusively live-attenuated γHV e.g., latency-deficient mutants were able to partially protect from latency (18, 75–78). A more recent study demonstrated that the protection mediated by latency-deficient mutants does not require immunity to any of the known latency genes in MHV-68. This suggests that lytic access to the latency reservoir is indeed a viable target for control (79). The prime/boost vaccination regimen based on recMVA expressing ORF6 and ORF61 had a profound impact on the quality and quantity of MHV-68-specific T cell responses. The vaccines proved to be protective in early latency by limiting latent viral infection. The failure to protect from long-term latency offers now the chance and basis to test advanced strategies to further enhance the CD4+ T cell response such as heterologous prime/boost regimens by combining antibody-targeted vaccines or DNA-, protein- or viral vector-based (e.g., Adeno, VSV) vaccines for priming and MVA for boosting. These approaches have been shown to significantly increase target antigen-specific T cell responses and enhance their efficacy in preclinical models as well as in clinical studies (80, 81). Alternatively, direct targeting of ORF6 or-61 antigens to the MHC class II processing pathway, e.g., by fusion to the MHC class II-associated invariant chain Ii, may be an option. A recent study provided evidence that combined intranasal/intramuscular vaccination of mice using a heterologous adenovirus-based prime/boost protocol reduced the risk of latency establishment after intranasal MHV-68 challenge. Interestingly, exclusively CD8+ T cells were targeted by the polytope vaccine and it was hypothesized that their redirection to the mucosal viral entry site abrogated the infection before latency was established (82). Alternatively, tissue-resident memory cells may have been induced and efficiently controlled the initial viral challenge load at the mucosa. The recMVA evaluated here seem promising model candidates to test immunotherapeutic approaches for long-term latent virus control even in immunocompromised hosts, e.g., by simultaneous vaccination with both constructs or in combination with adoptive transfer of antigen-specific T cells. Our findings have important implication for the use and the future design of vaccine based immunotherapy against gammaherpesvirus infections.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was carried out in accordance with the recommendations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocol was approved by the North Rhine-Westphalia State Agency for Nature, Environment and Consumer Protection (LANUV), Germany (Permit Numbers: A116/12 and G237/14).
Author Contributions
BS, ST, KT, HA, and ID conceived and designed the experiments. BS, ST, and HA performed the experiments. BS, ST, and ID analyzed the data. BS and ID wrote the paper.
Funding
This work was supported by the DFG [grant number GRK 1949], the DAAD and MOI II Graduate School.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank B. Steer and R. Tao for expert technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02984/full#supplementary-material
References
1. Odumade OA, Hogquist KA, Balfour HH Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev. (2011) 24:193–209. doi: 10.1128/CMR.00044-10
2. Pellet C, Kerob D, Dupuy A, Carmagnat MV, Mourah S, Podgorniak MP, et al. Kaposi's sarcoma-associated herpesvirus viremia is associated with the progression of classic and endemic Kaposi's sarcoma. J Invest Dermatol. (2006) 126:621–7. doi: 10.1038/sj.jid.5700083
3. Cesarman E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. (2011) 305:163–74. doi: 10.1016/j.canlet.2011.03.003
4. Brambilla L, Boneschi V, Berti E, Corbellino M, Parravicini C. HHV8 cell-associated viraemia and clinical presentation of Mediterranean Kaposi's sarcoma. Lancet. (1996) 347:1338. doi: 10.1016/S0140-6736(96)90989-7
5. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. (1995) 332:1186–91. doi: 10.1056/NEJM199505043321802
6. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. (1994) 266:1865–9. doi: 10.1126/science.7997879
7. Kieff E, Rickinson AB. Epstein-Barr virus and its replication. In: Fields BN, Knipe DM, Howley P, et al., editors. Fields Virology. Philadelphia, PA: Lippincott–Raven (2001). p. 2511–75.
8. Levy R, Miller RA. Therapy of lymphoma directed at idiotypes. J Natl Cancer Inst Monogr. (1990) 10, 61–68.
9. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. (1995) 86:1276–80. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276
10. Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. (2000) 165:1074–81. doi: 10.4049/jimmunol.165.2.1074
11. Sixbey JW, Vesterinen EH, Nedrud JG, Raab-Traub N, Walton LA, Pagano JS. Replication of Epstein-Barr virus in human epithelial cells infected in vitro. Nature. (1983) 306:480–3. doi: 10.1038/306480a0
12. Probert M, Epstein MA. Morphological transformation in vitro of human embryo fibroblasts by Epstein-Barr virus: preliminary observations. Science. (1972) 175:202–3.
13. Virgin HW IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, et al. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. (1997) 71:5894–904.
14. Forrest JC, Krug LT, Speck SH. Murine gammaherpesvirus 68 infection of mice: a small animal model for characterizing basic aspects of gammaherpesvirus pathogenesis. In: Damania B, Pipas JM, editors. DNA Tumor Viruses. New York, NY: Springer (2009). p. 735–75. doi: 10.1007/978-0-387-68945-6_28
15. Liu L, Usherwood EJ, Blackman MA, Woodland DL. T-cell vaccination alters the course of murine herpesvirus 68 infection and the establishment of viral latency in mice. J Virol. (1999) 73:9849–57.
16. Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, et al. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol. (2003) 77:7696–701. doi: 10.1128/JVI.77.13.7696-7701.2003
17. Hoegh-Petersen M, Thomsen AR, Christensen JP, Holst PJ. Mucosal immunization with recombinant adenoviral vectors expressing murine gammaherpesvirus-68 genes M2 and M3 can reduce latent viral load. Vaccine. (2009) 27:6723–30. doi: 10.1016/j.vaccine.2009.08.104
18. Jia Q, Freeman ML, Yager EJ, McHardy I, Tong L, Martinez-Guzman D, et al. Induction of protective immunity against murine gammaherpesvirus 68 infection in the absence of viral latency. J Virol. (2010) 84:2453–65. doi: 10.1128/JVI.01543-09
19. Obar JJ, Donovan DC, Crist SG, Silvia O, Stewart JP, Usherwood EJ. T-cell responses to the M3 immune evasion protein of murid gammaherpesvirus 68 are partially protective and induced with lytic antigen kinetics. J Virol. (2004) 78:10829–32. doi: 10.1128/JVI.78.19.10829-10832.2004
20. Usherwood EJ, Ward KA, Blackman MA, Stewart JP, Woodland DL. Latent antigen vaccination in a model gammaherpesvirus infection. J Virol. (2001) 75:8283–8. doi: 10.1128/JVI.75.17.8283-8288.2001
21. May JS, Stevenson PG. Vaccination with murid herpesvirus-4 glycoprotein B reduces viral lytic replication but does not induce detectable virion neutralization. J Gen Virol. (2010) 91(Pt 10):2542–52. doi: 10.1099/vir.0.023085-0
22. Stewart JP, Micali N, Usherwood EJ, Bonina L, Nash AA. Murine gamma-herpesvirus 68 glycoprotein 150 protects against virus-induced mononucleosis: a model system for gamma-herpesvirus vaccination. Vaccine. (1999) 17:152–7. doi: 10.1016/S0264-410X(98)00190-X
23. Freeman ML, Lanzer KG, Cookenham T, Peters B, Sidney J, Wu T-T, et al. Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J Virol. (2010) 84:2881–92. doi: 10.1128/JVI.02229-09
24. Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J Gen Virol. (1992) 73 (Pt 9):2347–56. doi: 10.1099/0022-1317-73-9-2347
25. Stevenson PG, Belz GT, Altman JD, Doherty PC. Changing patterns of dominance in the CD8+ T cell response during acute and persistent murine gamma-herpesvirus infection. Eur J Immunol. (1999) 29:1059–67.
26. Coleman HM, de Lima B, Morton V, Stevenson PG. Murine gammaherpesvirus 68 lacking thymidine kinase shows severe attenuation of lytic cycle replication in vivo but still establishes latency. J Virol. (2003) 77:2410–7. doi: 10.1128/JVI.77.4.2410-2417.2003
27. Blackman MA, Flano E, Usherwood E, Woodland DL. Murine gamma-herpesvirus-68: a mouse model for infectious mononucleosis? Mol Med Tdy. (2000) 6:488–90. doi: 10.1016/S1357-4310(00)01813-X
28. Stevenson PG, Belz GT, Castrucci MR, Altman JD, Doherty PC. A gamma-herpesvirus sneaks through a CD8 (+) T cell response primed to a lytic-phase epitope. Proc Natl Acad Sci USA. (1999) 96:9281–6. doi: 10.1073/pnas.96.16.9281
29. Liu L, Flano E, Usherwood EJ, Surman S, Blackman MA, Woodland DL. Lytic cycle T cell epitopes are expressed in two distinct phases during MHV-68 infection. J Immunol. (1999) 163:868–74.
30. Obar JJ, Fuse S, Leung EK, Bellfy SC, Usherwood EJ. Gammaherpesvirus persistence alters key CD8 T-cell memory characteristics and enhances antiviral protection. J Virol. (2006) 80:8303–15. doi: 10.1128/JVI.00237-06
31. Freeman ML, Burkum CE, Cookenham T, Roberts AD, Lanzer KG, Huston GE, et al. CD4 T cells specific for a latency-associated γ-herpesvirus epitope are polyfunctional and cytotoxic. J Immunol. (2014) 193:5827–34. doi: 10.4049/jimmunol.1302060
32. Altenburg AF, Kreijtz JH, de Vries RD, Song F, Fux R, Rimmelzwaan GF, et al. Modified vaccinia virus ankara (MVA) as production platform for vaccines against influenza and other viral respiratory diseases. Viruses. (2014) 6:2735–61. doi: 10.3390/v6072735
33. Gilbert SC. Clinical development of Modified Vaccinia virus Ankara vaccines. Vaccine. (2013) 31:4241–6. doi: 10.1016/j.vaccine.2013.03.020
34. Gomez CE, Perdiguero B, Garcia-Arriaza J, Esteban M. Clinical applications of attenuated MVA poxvirus strain. Expert Rev Vaccines. (2013) 12:1395–416. doi: 10.1586/14760584.2013.845531
35. Cottingham MG, Gilbert SC. Rapid generation of markerless recombinant MVA vaccines by en passant recombineering of a self-excising bacterial artificial chromosome. J Virol Methods. (2010) 168:233–6. doi: 10.1016/j.jviromet.2010.04.012
36. Tischer BK, Smith GA, Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol. (2010) 634:421–30. doi: 10.1007/978-1-60761-652-8_30
37. Thiele F, Tao S, Zhang Y, Muschaweckh A, Zollmann T, Protzer U, et al. Modified vaccinia virus Ankara-infected dendritic cells present CD4+ T-cell epitopes by endogenous major histocompatibility complex class II presentation pathways. J Virol. (2015) 89:2698–709. doi: 10.1128/JVI.03244-14
38. Adler H, Messerle M, Wagner M, Koszinowski UH. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol. (2000) 74:6964–74. doi: 10.1128/JVI.74.15.6964-6974.2000
39. Staib C, Drexler I, Sutter G. Construction and isolation of recombinant MVA. Methods Mol Biol. (2004) 269:77–100. doi: 10.1385/1-59259-789-0:077
40. Tischer BK, Kaufer BB. Viral bacterial artificial chromosomes: generation, mutagenesis, and removal of mini-F sequences. J Biomed Biotechnol. (2012) 2012:472537. doi: 10.1155/2012/472537
41. Cottingham MG, Andersen RF, Spencer AJ, Saurya S, Furze J, Hill AV, et al. Recombination-mediated genetic engineering of a bacterial artificial chromosome clone of modified vaccinia virus Ankara (MVA). PLoS ONE. (2008) 3:e1638. doi: 10.1371/journal.pone.0001638
42. Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol. (1998) 79 (Pt 2):347–52. doi: 10.1099/0022-1317-79-2-347
43. Kastenmuller W, Gasteiger G, Gronau JH, Baier R, Ljapoci R, Busch DH, et al. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J Exp Med. (2007) 204:2187–98. doi: 10.1084/jem.20070489
44. Flach B, Steer B, Thakur NN, Haas J, Adler H. The M10 locus of murine gammaherpesvirus 68 contributes to both the lytic and the latent phases of infection. J Virol. (2009) 83:8163–72. doi: 10.1128/JVI.00629-09
45. Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol. (2007) 81:13200–8. doi: 10.1128/JVI.01148-07
46. Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. (2006) 40:191–7. doi: 10.2144/000112096
47. Cottingham MG, Carroll MW. Recombinant MVA vaccines: dispelling the myths. Vaccine. (2013) 31:4247–51. doi: 10.1016/j.vaccine.2013.03.021
48. Baur K, Brinkmann K, Schweneker M, Patzold J, Meisinger-Henschel C, Hermann J, et al. Immediate-early expression of a recombinant antigen by modified vaccinia virus ankara breaks the immunodominance of strong vector-specific B8R antigen in acute and memory CD8 T-cell responses. J Virol. (2010) 84:8743–52. doi: 10.1128/JVI.00604-10
49. Flano E, Kim IJ, Moore J, Woodland DL, Blackman MA. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J Immunol. (2003) 170:3828–34. doi: 10.4049/jimmunol.170.7.3828
50. Usherwood EJ, Stewart JP, Robertson K, Allen DJ, Nash AA. Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J Gen Virol. (1996) 77 (Pt 11):2819–25. doi: 10.1099/0022-1317-77-11-2819
51. Gasteiger G, Kastenmuller W, Ljapoci R, Sutter G, Drexler I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J Virol. (2007) 81:11925–36. doi: 10.1128/JVI.00903-07
52. Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell. (2015) 162:1322–37. doi: 10.1016/j.cell.2015.08.004
53. Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8(+) T cells orchestrate pDC-XCR1(+) dendritic cell spatial and functional cooperativity to optimize priming. Immunity. (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
54. Croft NP, Smith SA, Wong YC, Tan CT, Dudek NL, Flesch IE, et al. Kinetics of antigen expression and epitope presentation during virus infection. PLoS Pathog. (2013) 9:e1003129. doi: 10.1371/journal.ppat.1003129
55. Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, et al. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med. (2005) 201:95–104. doi: 10.1084/jem.20041912
56. Moutaftsi M, Tscharke DC, Vaughan K, Koelle DM, Stern L, Calvo-Calle M, et al. Uncovering the interplay between CD8, CD4 and antibody responses to complex pathogens. Future Microbiol. (2010) 5:221–39. doi: 10.2217/fmb.09.110
57. Becker PD, Norder M, Weissmann S, Ljapoci R, Erfle V, Drexler I, et al. Gene expression driven by a strong viral promoter in MVA increases vaccination efficiency by enhancing antibody responses and unmasking CD8(+) T cell epitopes. Vaccines. (2014) 2:581–600. doi: 10.3390/vaccines2030581
58. MacLeod MK, Clambey ET, Kappler JW, Marrack P. CD4 memory T cells: what are they and what can they do? Semin Immunol. (2009) 21:53–61. doi: 10.1016/j.smim.2009.02.006
59. Ewer K, Rampling T, Venkatraman N, Bowyer G, Wright D, Lambe T, et al. A monovalent chimpanzee adenovirus ebola vaccine boosted with MVA. N Engl J Med. (2016) 374:1635–46. doi: 10.1056/NEJMoa1411627
60. Joachim A, Nilsson C, Aboud S, Bakari M, Lyamuya EF, Robb ML, et al. Potent functional antibody responses elicited by HIV-I DNA priming and boosting with heterologous HIV-1 recombinant MVA in healthy Tanzanian adults. PLoS ONE. (2015) 10:e0118486. doi: 10.1371/journal.pone.0118486
61. Reyes-Sandoval A, Berthoud T, Alder N, Siani L, Gilbert SC, Nicosia A, et al. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect Immun. (2010) 78:145–53. doi: 10.1128/IAI.00740-09
62. Full F, Hahn AS, Grosskopf AK, Ensser A. Gammaherpesviral tegument proteins, PML-nuclear bodies and the ubiquitin-proteasome system. Viruses. (2017) 9:308. doi: 10.3390/v9100308
63. Ehtisham S, Sunil-Chandra NP, Nash AA. Pathogenesis of murine gammaherpesvirus infection in mice deficient in CD4 and CD8 T cells. J Virol. (1993) 67:5247–52.
64. Usherwood EJ, Ross AJ, Allen DJ, Nash AA. Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J Gen Virol. (1996) 77 (Pt 4):627–30. doi: 10.1099/0022-1317-77-4-627
65. Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HI. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. (1996) 70:6775–80.
66. Cardin RD, Brooks JW, Sarawar SR, Doherty PC. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J Exp Med. (1996) 184:863–71. doi: 10.1084/jem.184.3.863
67. Belz GT, Stevenson PG, Castrucci MR, Altman JD, Doherty PC. Postexposure vaccination massively increases the prevalence of gamma-herpesvirus-specific CD8+ T cells but confers minimal survival advantage on CD4-deficient mice. Proc Natl Acad Sci USA. (2000) 97:2725–30. doi: 10.1073/pnas.040575197
68. Dutia BM, Clarke CJ, Allen DJ, Nash AA. Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J Virol. (1997) 71:4278–83.
69. Weck KE, Dal Canto AJ, Gould JD, O'Guin AK, Roth KA, Saffitz JE, et al. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus-induced vascular disease. Nat Med. (1997) 3:1346–53. doi: 10.1038/nm1297-1346
70. Nash AA, Dutia BM, Stewart JP, Davison AJ. Natural history of murine gamma-herpesvirus infection. Philos Trans R Soc Lond B Biol Sci. (2001) 356:569–79. doi: 10.1098/rstb.2000.0779
71. Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. (2011) 29:351–97. doi: 10.1146/annurev-immunol-072710-081639
72. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
73. Pauken KE, Wherry EJ. SnapShot: T cell exhaustion. Cell. (2015) 163:1038. doi: 10.1016/j.cell.2015.10.054
74. Saeidi A, Zandi K, Cheok KK, Saeidi H, Wong WF, Lee CYQ, et al. T-cell exhaustion in chronic infections: reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol. (2018) 9:2569. doi: 10.3389/fimmu.2018.02569
75. Boname JM, Coleman HM, May JS, Stevenson PG. Protection against wild-type murine gammaherpesvirus-68 latency by a latency-deficient mutant. J Gen Virol. (2004) 85(Pt 1):131–5. doi: 10.1099/vir.0.19592-0
76. Fowler P, Efstathiou S. Vaccine potential of a murine gammaherpesvirus-68 mutant deficient for ORF73. J Gen Virol. (2004) 85(Pt 3):609–13. doi: 10.1099/vir.0.19760-0
77. Rickabaugh TM, Brown HJ, Martinez-Guzman D, Wu TT, Tong L, Yu F, et al. Generation of a latency-deficient gammaherpesvirus that is protective against secondary infection. J Virol. (2004) 78:9215–23. doi: 10.1128/JVI.78.17.9215-9223.2004
78. Tibbetts SA, McClellan JS, Gangappa S, Speck SH, Virgin HW. Effective vaccination against long-term gammaherpesvirus latency. J Virol. (2003) 77:2522–9. doi: 10.1128/JVI.77.4.2522-2529.2003
79. Lawler C, Simas JP, Stevenson PG. Vaccine protection against Murid Herpesvirus-4 is maintained when the priming virus lacks known latency genes. Immunol Cell Biol. (2019). doi: 10.1111/imcb.12299. [Epub ahead of print].
80. Ruhl J, Citterio C, Engelmann C, Haigh T, Dzionek A, Dreyer J, et al. Heterologous prime-boost vaccination protects against EBV antigen-expressing lymphomas. J Clin Invest. (2019) 129:2071–87. doi: 10.1172/JCI125364
81. Kardani K, Bolhassani A, Shahbazi S. Prime-boost vaccine strategy against viral infections: mechanisms and benefits. Vaccine. (2016) 34:413–23. doi: 10.1016/j.vaccine.2015.11.062
Keywords: vaccinia virus, MVA, T cell response, viral vector vaccine, MHV-68, gammaherpesvirus
Citation: Samreen B, Tao S, Tischer K, Adler H and Drexler I (2019) ORF6 and ORF61 Expressing MVA Vaccines Impair Early but Not Late Latency in Murine Gammaherpesvirus MHV-68 Infection. Front. Immunol. 10:2984. doi: 10.3389/fimmu.2019.02984
Received: 13 June 2019; Accepted: 05 December 2019;
Published: 18 December 2019.
Edited by:
Pedro A. Reche, Complutense University of Madrid, SpainReviewed by:
Luka Cicin-Sain, Helmholtz Center for Infection Research, GermanyWayne Robert Thomas, University of Western Australia, Australia
Francisco Sobrino Castello, Severo Ochoa Molecular Biology Center (CSIC-UAM), Spain
Copyright © 2019 Samreen, Tao, Tischer, Adler and Drexler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingo Drexler, aW5nby5kcmV4bGVyQG1lZC51bmktZHVlc3NlbGRvcmYuZGU=
†These authors have contributed equally to this work