 Clara I. Colino
Clara I. Colino José M. Lanao
José M. Lanao Carmen Gutierrez-Millan
Carmen Gutierrez-Millan- 1Area of Pharmacy and Pharmaceutical Technology, Department of Pharmaceutical Sciences, University of Salamanca, Salamanca, Spain
- 2The Institute for Biomedical Research of Salamanca (IBSAL), Salamanca, Spain
Hepatic macrophage populations include different types of cells with plastic properties that can differentiate into diverse phenotypes to modulate their properties in response to different stimuli. They often regulate the activity of other cells and play an important role in many hepatic diseases. In response to those pathological situations, they are activated, releasing cytokines and chemokines; they may attract circulating monocytes and exert functions that can aggravate the symptoms or drive reparation processes. As a result, liver macrophages are potential therapeutic targets that can be oriented toward a variety of aims, with emergent nanotechnology platforms potentially offering new perspectives for macrophage vectorization. Macrophages play an essential role in the final destination of nanoparticles (NPs) in the organism, as they are involved in their uptake and trafficking in vivo. Different types of delivery nanosystems for macrophage recognition and targeting, such as liposomes, solid-lipid, polymeric, or metallic nanoparticles, have been developed. Passive targeting promotes the accumulation of the NPs in the liver due to their anatomical and physiological features. This process is modulated by NP characteristics such as size, charge, and surface modifications. Active targeting approaches with specific ligands may also be used to reach liver macrophages. In order to design new systems, the NP recognition mechanism of macrophages must be understood, taking into account that variations in local microenvironment may change the phenotype of macrophages in a way that will affect the uptake and toxicity of NPs. This kind of information may be applied to diseases where macrophages play a pathogenic role, such as metabolic disorders, infections, or cancer. The kinetics of nanoparticles strongly affects their therapeutic efficacy when administered in vivo. Release kinetics could predict the behavior of nanosystems targeting macrophages and be applied to improve their characteristics. PBPK models have been developed to characterize nanoparticle biodistribution in organs of the reticuloendothelial system (RES) such as liver or spleen. Another controversial issue is the possible toxicity of non-degradable nanoparticles, which in many cases accumulate in high percentages in macrophage clearance organs such as the liver, spleen, and kidney.
Introduction
The use of particles as carriers of therapeutic agents for liver targeting is not a new idea. Hepatic nanoparticle uptake and distribution was initially studied as a drawback to be avoided because it entails a lack of specific selective distribution to other desired targets as well as toxicity and safety concerns. Nevertheless, from another point of view, strategies of selective delivery to different kinds of hepatic cells have also been explored in the search for specific targets of drugs included in nanoparticulated systems, such as hepatocytes, hepatic stellar cells, endothelial cells, and also the Kupffer cells (KC), the resident liver macrophages.
The liver is a very complex organ with many cells that are different in both morphology and functionality but which are nonetheless strongly inter-related. Although there are some other hepatic cells with phagocytic activity, KCs are undoubtedly the main ones responsible for phagocytosis in the liver. Moreover, it is estimated that they constitute 80–90% of all the macrophages present in the body (1).
Kupffer cells are the liver-resident macrophages, considered professional phagocytes to distinguish them from facultative ones. They are the largest mononuclear phagocyte population in the body and constitute approximately 20% of non-parenchymal liver cells. They present functional heterogeneity, likely due to their different origins and intrinsic plasticity (2).
They have an evident role in monitoring the blood entering the zone in order to endocytose debris, degenerated cells and any potentially harmful materials from the gut and circulation stream. Their strategic location at the luminal side of the hepatic sinusoidal endothelium allows them to act as sentinels that capture and process particles. They are involved in antigen presentation and processing and in the modulation of some hepatocyte functions.
Activation of Kupffer cells can induce a series of events to inhibit pathogen replication, recruit other immune cells into the liver, and activate them. On the other hand, interaction derived from infiltration immune cells leads to KC regulation (2). These complex and multiple inter-relationships with other hepatic cells and their concomitant role in immunological processes provide KCs with a wide variety of receptors that can be harnessed for their specific targeting (3, 4).
Due to the involvement of KCs in the evolution of many liver diseases, modulation of their activity may be used for therapeutic ends and nanosystems constitute promising alternatives for achieve this goal. In the present work, we will focus on the proposed nanosystems for targeting hepatic macrophages in order to improve pathological processes in the liver. The role of macrophages in liver diseases, the types of nanovehicles used, their characteristics, and the influence of the phenotype will be revised. Also, pharmacokinetic models for characterizing the biodistribution of NPs in the liver are addressed.
Monocyte–Macrophage System (MPS)
Macrophages have been considered to be a part of different body systems throughout history. Previously considered as reticulo-endothelial system (RES) cells, nowadays, macrophages are generally considered to belong to the monocyte mononuclear phagocyte or monocyte macrophage system (MPS), originally defined as a cell lineage of promonocytes that give rise to monocytes that finally becomes macrophages in tissues. This concept was later reformulated to include dendritic cells (5), and recently also the concept of the MPS has been questioned, based on evidence that tissue resident macrophages may be a separate lineage seeded during embryonic development and capable of self-renewal. The newly proposed nomenclature classifies mononuclear phagocytes according to their ontogeny, location, and/or morphology (6). Regarding the phagocytic cells in liver, the main difference of the newly proposed nomenclature is that macrophages and derived monocyte cells are considered to be separate entities (7).
Macrophages
Macrophages are specialized phagocytic cells strategically distributed in the body and specifically adapted to each tissue once they are settled. They are non-migratory cells that monitor their surrounding environment and process the material they engulf. They also recruit other immune cells, playing an important role in immune defense and homeostatic processes (8). To fulfill their functions, macrophages possess a wide range of sensing molecules specialized according to the tissue in which they are nested in each case.
Due to their central role in homeostasis, inflammation, and immunity, macrophages have arisen as interesting targets for therapeutic intervention (9).
Tissue macrophages are a very heterogeneous group of cells due to their different origins and their adaptation to the local environment. The contributions of embryonic origins and adult bone marrow cells vary depending on the tissues, with even tissue-resident macrophages of prenatal origin deriving from different hematopoietic stem cells.
In view of the new evidence that questions the previous MPS model, it is postulated that there are two groups of macrophages in tissues: one coming from prenatally established populations and a second one originating from infiltrated monocytes that are more related to inflammatory conditions. Figure 1 illustrates the different origins and development processes of tissue-resident macrophages.
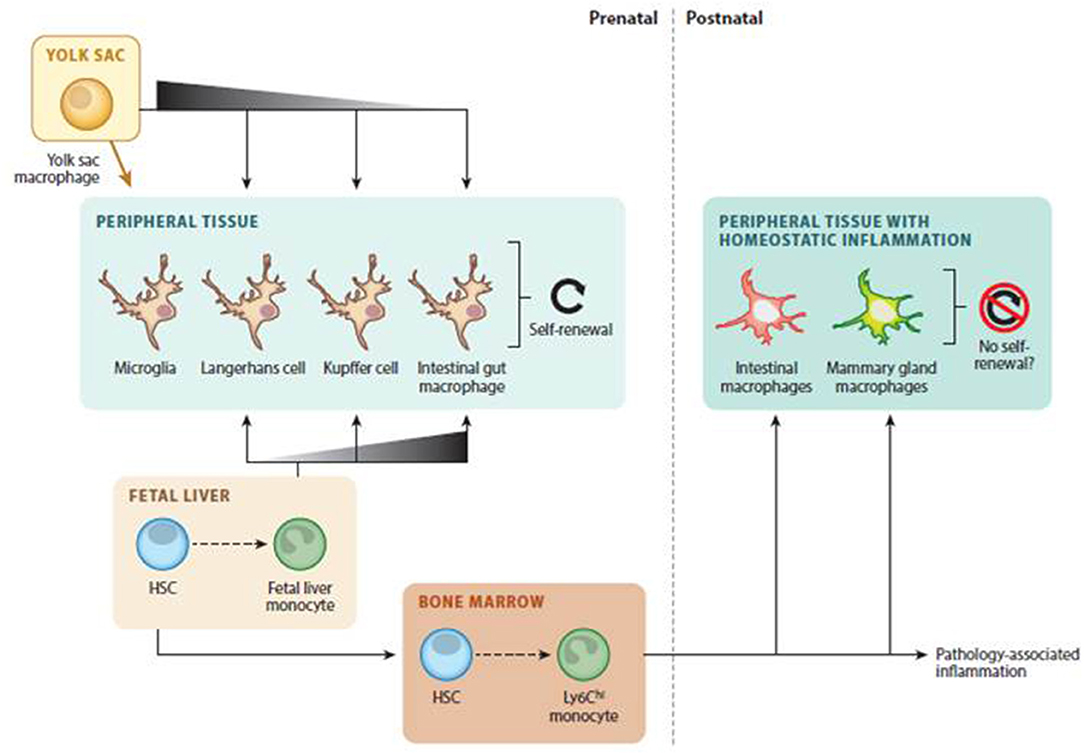
Figure 1. Tissue-resident macrophages development. Adapted with permission (9).
It has been demonstrated in mice that there are also several monocyte-derived tissue macrophage populations whose phenotypes reflect different origins. One such population is made up of macrophages originating from Ly-6C high expressing monocytes (classical monocytes), mainly coming from bone marrow, that express inflammatory chemokine receptors (like CCR2), pattern-recognition receptors, and cytokines. Another population is made up of macrophages derived from Ly-6C low expressing monocytes (non-classical monocytes), mainly coming from the spleen, that present a patrolling behavior and express more scavenging receptors. In the steady state, classical ones can leave the bloodstream and patrol extravascular tissues. They can be converted in some proportion into Ly-6C low expressing monocytes and transport antigens to lymph nodes. Non-classical monocytes patrol the intravascular spaces to clear dying endothelial cells. Under inflammation, classical monocytes differentiate to macrophages that are capable of self-renewal (10).
Despite the differences between mice and humans, genetic expression studies have demonstrated that these two subsets of monocytes are also present in humans, together with another intermediate subset between the classical and the non-classical (11, 12).
With respect to the macrophages that are present in the liver, although traditionally the term hepatic macrophages and Kupffer cells (KC) are used almost interchangeably, modifications to the MPS model mentioned above have also affected this assumption. After injury, heterogeneous hepatic macrophages populations can be observed, such as liver-resident macrophages or KCs and two subsets of bone marrow monocyte-derived macrophages (MoMFs), as well as peritoneal macrophages for subcapsular regions of the liver (13).
Classification
It is well-established in mice that macrophages can undergo two different activation states: M1 or classically activated and M2 or alternatively activated. M1 macrophages can produce pro-inflammatory cytokines and chemokines, high levels of reactive nitrogen and oxygen intermediates. They can also facilitate Th1 response and strong antimicrobial and antineoplastic effect. M2 ones are suppressive, involved in cellular repair and characterized by efficient phagocytic activity and high expression of scavenger, galactose, and mannose-type receptors. M1 and M2 even present different iron, glucose, and amino acid metabolism (2, 4). Although not much information is available regarding human beings, there is evidence to suggest a similar behavior in human macrophages (14). This traditional dichotomic classification seems to be too simplistic in view of recent increases in our knowledge of this area due to new sophisticated characterization techniques. Regarding Kupffer cells, their great flexibility and plasticity allow them to adopt a range of multiple intermediate phenotypes depending on the signals, which can lead to a broad spectrum of activation states (13, 15). Nevertheless, this simple classification into two possible extreme activation states is still used as a reference for KC behavior.
Differences not only in activation but also in origin have a great impact on the different subsets that can be defined for liver macrophages. The ontogeny and maintenance of resident hepatic macrophages have been the objects of many studies by important research groups, which have demonstrated that, besides the existence of self-renewal processes from prenatally settled local precursor cells, Kupffer cell populations are maintained by the infiltration of circulating bone-derived monocytes that differentiate into Kupffer cells in the liver (16, 17). Murine models have provided evidence that MoMF can contribute to regenerating the resident liver macrophage population when KC are massively depleted (18).
On the other hand, even some studies with models of sterile liver injury have shown phagocytes with an expression of the transcription factor GATA-6, suggesting macrophage infiltration from the peritoneal cavity (19).
Role of Kupffer Cells in Hepatic Diseases: Cytokines and Chemokines
Due to their physiological functions, Kupffer cells are involved in local cell communication and homeostasis maintenance. The prominent role of KCs in immune processes, particle engulfment, antigen presentation, and the attraction and stimulation of T cells is well-known. They recruit other immune cells in the liver and release mediators to initiate response in other liver cells (2, 13).
Regarding other types of hepatic macrophages, it is known that bone marrow-derived macrophages participate in liver repair and regeneration but functional differences with Kupffer cells have not been clearly established. Following their activation, they produce cytokines that trigger a cascade of responses in other cells. If we accept the classical M1/M2 classification, it can be said that M1 KCs release proinflammatory cytokines, including tumor necrosis factor (TNF)-a, IL-6 and IL-1β, while M2 KCs release IL-4, IL-10, IL-13, and transforming growth factor-β. Consequently, the balance between these two phenotypes can ultimately lead to many different effects such as liver damage and wound repair (20).
The complex roles and expression of different KC phenotypes can lead to both protective and harmful responses (2, 21), and since it is required that their activation be precise, timely and localized, any KC dysregulation can lead to significant pathology (15). However, the results of some studies and hypotheses regarding these opposing effects are disputed. In some cases, KC depletion can be beneficial because of the reduction of inflammation or fibrosis but, on the other hand, the suppression of KC's role against pathogen invasion can clearly be harmful. Further studies must be done in order to conclude if KC depletion could prevent or exacerbate liver damage.
Thus, liver macrophages play an essential role in many pathogenic stages such as acute liver injury, fatty liver disease, fibrosis, cirrhosis, and liver tumors (22), constituting potential therapeutic targets for liver disease treatments. However, the pathogenesis of the disease treated and the phenotype of the macrophages targeted are points to consider when targeting macrophages (23).
In response to liver injuries such as alcoholic liver and non-alcoholic fatty liver disease (NAFLD) diseases, Kupffer cells are activated and a polarization to an M1-like phenotype is promoted in resident as well as monocyte-derived macrophages (24, 25). When injury ends, there is a switch to M2 restorative macrophages that release anti-inflammatory cytokines, regenerative growth factors, and matrix degrading metalloproteinase (MMP) expression, which promote tissue repair (26).
Macrophage polarization also has an important role in the growth and development of tumoral tissues. Macrophages in tumor tissues (TAMs) are mostly M2-like cells that produce tumor-promoting cytokines and growth factors that promote tumor expansion, angiogenesis, metastasis, and immune cell evasion. TAMs also contribute to drug resistance (27, 28).
Targeting Liver Macrophages With NPs
Delivery Systems for Macrophages Recognition and Targeting
Liver macrophages are specialized in the internalization of foreign nanoparticles, playing an essential role in its destination in the organism, since they are involved in their uptake and trafficking in vivo. Therapeutic capacity and clearance mechanisms in clinically relevant nanomedicines have been linked to macrophage activity. However, due to their pathophysiological roles in diseases (29), liver macrophages are also potential therapeutic targets for a variety of aims, from cell activation to monocyte recruitment or macrophage differentiation (30). Emerging nanotechnology systems may offer new perspectives for macrophage vectorization. After intravenous administration, nanoparticles are opsonized in the bloodstream before being phagocytized by macrophages and accumulated in the RES organs. This passive targeting promotes the accumulation of the NPs in the liver, a process that increases within tumors due to the EPR (enhanced permeability and retention) effect (31).
Kupffer cells internalize NPs through multiple scavenger, toll-like, mannose, and Fc receptors (23). The mechanisms involved are macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and additional endocytotic pathways (1, 32, 33). Clathrin-mediated endocytosis has been pointed out as responsible for the internalization of size ranges of approximately 100–350 nm, while caveolin-mediated mechanism is responsible for the endocytosis of 20–100 nm particles (34–36). Macropynocitosis allows for large volume extracellular internalization of 0.5–5 μm nanosystems (23). The internalization process is then modulated by the size of the NPs as well as other characteristics such as charge. Larger nanoparticles generally show more efficient hepatic uptake: a diameter >200 nm is preferred for liver deposition (1, 37–39). Charged NPs, especially those with a positive charge, are taken up to a greater extent than those with a neutral charge (23, 39). Shape also has a great impact on NP uptake and elongated NPs are taken up less by macrophages than spherical ones (40). However, cylindrical silica NPs showed the highest accumulation in the liver compared to other shapes (41). Moreover, surface hydrophobic NPs tend to be opsonized by proteins that make them attractive to the phagocytic cells of the MPS (42–44).
Therefore, the success of therapeutic strategies targeting liver macrophages involves the use of different NP types with appropriate properties to allow them to be preferentially taken up by the liver. Incorporation of ligands to the surface of NPs increases specificity through active targeting (21, 31).
Liposomes
Liposomes are biodegradable vesicles with an aqueous core and a phospholipidic membrane that can carry hydrophilic as well as hydrophobic compounds. They have the advantage of being both biocompatible and biodegradable, whereas their instability is one of their main drawbacks. Macrophages, especially KCs, readily phagocytose circulating liposomes (45), causing them to accumulate in the liver (46). The liposomes proposed for the vectorization to hepatic macrophages have mainly sizes of ~100 nm due to restrictions on parenteral formulations. This passive targeting has been exploited for the administration of anti-infective drugs that have an effect on liver macrophages, some examples of which are shown in Table 1. For instance, commercial liposomal formulations of amphotericin B (Ambisome) allow the drug to accumulate in spleen and liver macrophages that constitute a reservoir of Leishmania, reducing the drug's nephrotoxicity (54, 55). Besides, the encapsulation of vancomycin in liposomes improves its poor penetration into cells, allowing its targeting to Kupffer cells. In a mouse model, such formulations reduced the intracellular Methicillin-resistant Staphylococcus aureus (MRSA) reservoir where the bacteria can survive and proliferate, significantly increasing the survival rate of infected mice over those treated with the drug solution (48, 49).
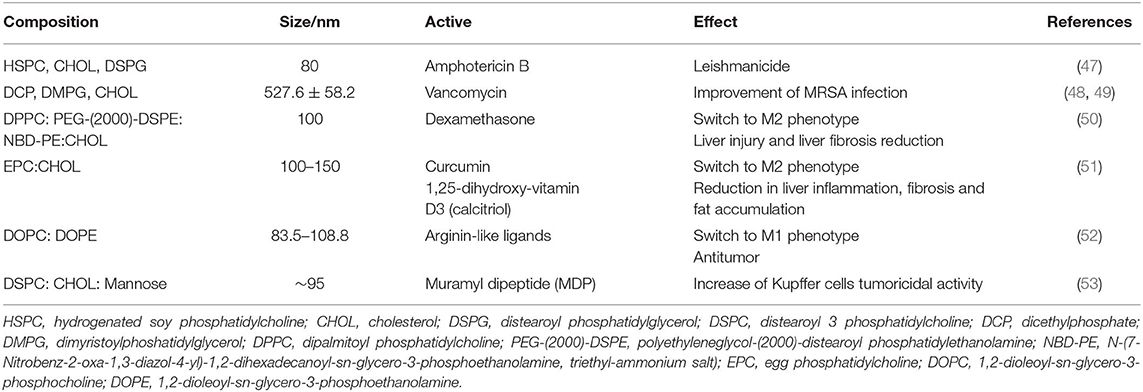
Table 1. Characteristics of some liposomes proposed for liver macrophages targeting.
Liposomes are also suitable vehicles for the targeting of anti-inflammatory compounds such as dexamethasone, curcumin, or calcitriol to the liver. They show improved results over the free drug in the treatment of acute and chronic liver disease models in mice. Pharmacokinetic studies show the preferential uptake of the liposomes by the liver although they accumulate not only in Kupffer cells but also in monocytes, infiltrating macrophages and, to a lesser extent, T cells. Liposomes also induce a repolarization of macrophages to a regulatory phenotype (50, 51).
Pathological conditions may influence the liposomes behavior, and recently, changes in spatial distribution and a decrease in the liposomes uptake by macrophages has been described in liver fibrosis (56).
Liposomes can be decorated for active vectorization with surface modifiers, such as mannose that has been proposed for the treatment of liver tumors in order to increase liposome uptake by macrophages via receptor-mediated endocytosis. The increase in mannose ligand concentration leads to a higher accumulation percentage in the livers of mice. Also, active targeting of an immunomodulator to liver by mannose-decorated liposomes resulted in more effective inhibition of metastasis than when delivered by liposomes without mannose (53, 57).
Substances attached to liposome surface may produce other effects. Some arginine-like ligands may switch macrophages to the M1 phenotype in order to achieve an antitumor effect. In a study with a library of this kind of ligands, nitroarginine, and acetylglutamine DOPE:DOPC liposomes were the most effective for the redirection of macrophage phenotypes (52).
In summary, preferentially uptake of liposomes by liver macrophages make them suitable vehicles for the vectorization of anti-inflammatory, anti-infective or other drugs for the treatment of liver diseases. When associated with anti-inflammatory compounds, they are able to promote the macrophages' regulatory state and reduce the dose for the treatment of acute and chronic liver injury. The decoration of the surface of liposomes with arginine-like and mannose ligands may be useful for anti-tumor treatments.
Lipoplexes
Inhibition of regulatory pathways triggered by macrophages via the use of gene therapies is another strategy for the treatment of macrophage-associated diseases. Lipid-based NPs are the most successful non-viral vehicles for targeting RNAi to Kupffer cells and can reach a high efficiency of transfection. Although they are sometimes referred to as liposomes, lipoplexes are usually different in both structure and composition. They are based on cationic lipids that are able to both bind and condense negatively charged iRNA through electrostatic interactions and to deliver the payload into the cytoplasm of target cells (58). They also incorporate neutral lipids in order to attenuate the toxicity of cationic lipids. Besides cell selectivity, the success of these kinds of therapies depends on transfection efficacy. Lipoplexes are engulfed by Kupffer cells through macropinocytosis and clathrin-mediated mechanisms after IV administration, but RNAi escape from endosomes is a rate-limiting step for these therapies (59). Proper lipid design allows RNAi to reach the cellular cytoplasm where it exerts its action. In this way, cationic lipid C12-200 eposide would prevent RNAi lysosomal degradation (58) through a micropinocytosis internalization mechanism as macropinosomes do not follow the endosomic degradation pathway (23). It was applied to inhibit the PD-1/PD-L1 pathway that contributes to the persistence of viral liver infections. Lipid NPs of 70–80 nm size were efficiently internalized (66.5%) and expressed by Kupffer cells after in vivo IV administration in viral-infected mice. This led to an enhanced antiviral effect. This promising antiviral immunotherapy may be applicable to vaccine development, to treat diverse viral liver infections and other diseases such as hepatocarcinome (60).
However, cationic lipids are used as vehicles for RNAi forming lipoplexes. A high transfection efficacy in Kupffer cells can be achieved with proper lipid selection, showing promising results in immunotherapy.
Inorganic Nanoparticles
Inorganic NPs are a broad group of metallic and non-metallic nanomaterials. Some of them are non-biodegradable, which constitutes a pitfall for their use. However, they possess excellent properties such as small size, high surface area, and easy functionalization and may induce per se responses in macrophages with different therapeutic applications as shown in Table 2.

Table 2. Some examples of inorganic nanoparticles for liver macrophage targeting.
Inorganic NPs have been used in the diagnostic and treatment of liver fibrosis and recently this topic has been reviewed in depth (65). For instance, the reduction of inflammatory macrophage activity caused by ceriumoxide NPs has been proposed to prevent hepatic dysfunction in septic rats. The NPs attenuate the expression of a number of different inflammatory macrophage mediators that are associated with sepsis, improving rat survival (61). This downregulation of Kupffer cell activity was also reported for gold nanoparticles (GNPs) in two rat liver-injury models causing antioxidant and antifibrotic effects (62).
Inflammatory diseases may also be treated by the switch of macrophages from an inflammatory (“M1”) to an anti-inflammatory (“M2”) phenotype. Carbohydrates are able to induce phenotypic changes promoting one or other activation state depending on their physical and chemical characteristics (66). As an example, glucomannan carbohydrate-decorated silicon oxide nanoparticles promote M2 polarization in macrophages by inducing clustering of mannose receptors (MR) on the cell surface. Although this was assayed in a murine inflammatory bowel disease model, it may be applied to other inflammatory diseases (63).
Conversely, induction of an immune response was the aim of calcium phosphate polyetilenimine/SiO2 nanoparticles used as carriers of a Toll-like-3 ligand. The NPs targeted the liver with 30–40% NP-positive cells when administered intravenously to mice and could be applied to vaccination (67).
Another group of inorganic NPs with applications in liver macrophage vectorization is superparamagnetic iron oxide nanoparticles (SPIONs), which are promising nanomaterials as diagnostic, iron supplement, and drug carrier agents. Surface modifications can render a high biocompatibility (64). They are phagocytized by macrophages and induce a pro-inflammatory response (68–70) through the activation of the Toll-like receptor 4 (71). Recently, they have been proposed for the reeducation of M2 tumor-associated macrophages to an antitumor M1 state in cancer treatment. This effect was studied for carboxymethyldextran-coated iron oxide NPs (Ferumoxytol®), which are approved by the FDA for the treatment of iron deficiency and other clinical uses. In a mouse in vivo model, these NPs inhibited tumor growth and prevented metastasis development. This activity was associated with the increase of M1 macrophages that may have been promoted by iron overload (72, 73). The uptake mechanism of carboxy-dextran coated SPIONs by human macrophages is a clathrin-mediated and scavenger receptor endocytosis although macropinocytosis may also contribute to internalization (74). The recognition of SPIONs by macrophages depends on the particle size and surface modifier, and it is better for positively charged particles and for a size of ~60 nm of size, although such SPIONs show cytotoxicity (71, 74, 75).
The efficient SPION uptake by macrophages allows its use for labeling macrophages in a cellular therapy for the treatment of liver cirrhosis. The labeled cells were tracked in vivo by magnetic resonance and no effects on phagocytic activity or cell viability were observed (75). AuNPs have also been used to this end and 50 nm was proposed as the optimal size for labeling without toxicity concerns for both NPs types (76).
In summary, inorganic nanoparticles are able to stimulate and also inhibit the activity of macrophages. Moreover, they can promote a switch to a specific macrophage state. The desired effect depends on the disease to be treated. The interaction of SPIONs with macrophages has been well-characterized and a mathematical model for the prediction of NPs uptake has even been developed (74). However, further insights are needed to clarify the relationship between the characteristics of NPs and their therapeutic and toxic effects.
Polymeric NPs
Polymeric NPs are colloid systems made of natural or synthetic polymers. They consist of a matrix in which the drug is homogeneously distributed or may be structured in a nucleus and a polymeric shell (nanocapsules) (54). They display great versatility. A fundamental feature of these nanosystems is their biodegradability, which is essential for intravenous administration.
Polymers of polylactic–glycolic acids (PLGA) are the most used for drug delivery as they are compatible and biodegradable compounds, and are excipients approved by the FDA. NPs of these polyesthers show excellent properties as drug carriers for liver macrophages vectorization (77) in order to improve efficacy or reduce side effects. PLGA NPs have been proposed as carriers of an inhibitor of the spleen tyrosine kinase SYK. This enzyme is overexpressed in M1 macrophages and shows a positive correlation with the pathogenesis of NASH and alcoholic hepatitis in patients. Although the bare inhibitor was more effective in vitro, its incorporation to 160 nm PLGA nanoparticles improved its intrahepatic delivery and therapeutic efficacy in vivo. This ameliorated fibrosis, inflammation, and steatosis in mice after IV administration (Figure 2) (78).
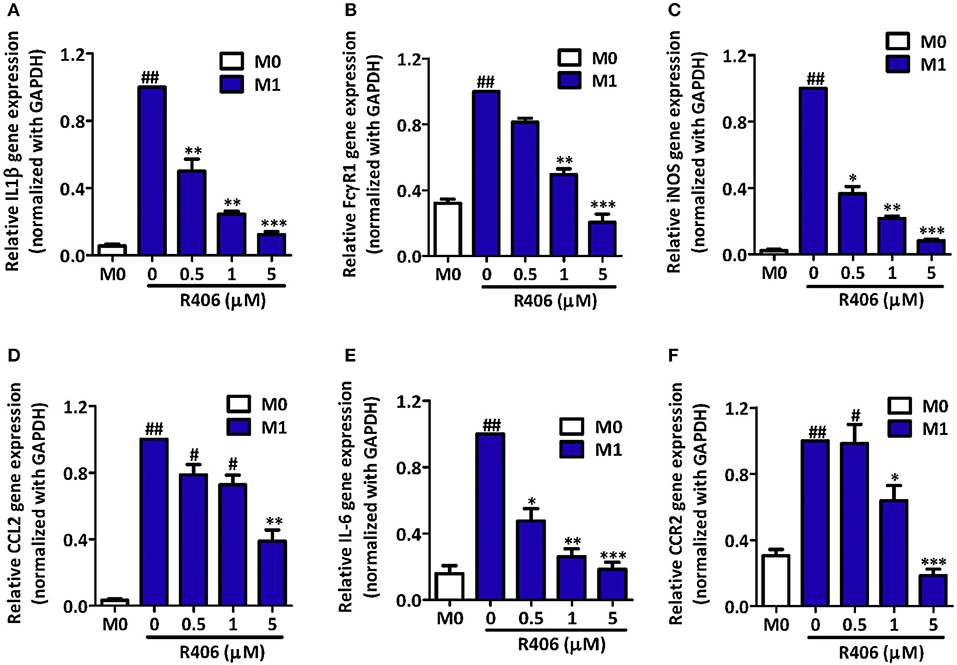
Figure 2. Inhibition of M1-specific differentiation and inflammatory markers by R406 in RAW macrophages. Gene expression of M1 markers IL-1β (A); FcγR1 (B); iNOS (C); CCL2 (D); IL-6 (E); and CCR2 (F) in RAW 264.7 cells after incubation with medium alone (M0) or M1 stimulus with R406 (0, 0.5, 1, and 5 μM). Expression values for the respective genes in untreated M1 macrophages were set at 1.0 to calculate the relative gene expression. Data are presented as mean + SEM. #p < 0.05, ##p < 0.01 denotes significance versus control M0 macrophages. *p < 0.05, **p < 0.01, and ***p < 0.001 denotes significance versus M1-differentiated macrophages. Reproduced with permission (78).
The objective pursued for rosiglitazone NP incorporation was to reduce their serious side effects such as the increased risk of fatal cardiac arrhythmia. Rosiglitazone vectorization to macrophages using 200 nm PLGA/polyvinylic acid (PVA) nanospheres allows for its selective delivery to circulating monocytes and Kupffer cells, reducing obesity-related inflammatory reaction in the white adipose tissue and liver and mitigating undesired effects (79).
Cationic polymers have been proposed as carriers of genetic material to reduce the expression of proteins related to liver diseases. This was the objective of RNAi for protein NOGOB encapsulation in [poly(amine-co-ester) (PACE) terpolymers] NPs. NOGOB promotes M1 polarization, stimulating the progression of alcoholic liver disease and liver fibrosis. The in vivo spleen administration of the NPs with a size of 240–300 nm allowed up to 60% Nogo-B protein suppression (80). This is a high transfection efficacy although alternative administration routes adequate for clinical use should be assayed. High transfection efficacy was also achieved by PLAcore/PVAshell NPs loaded with PEI-CD98 siRNA. The objective was the downregulation of CD98, a factor that is overexpressed in the livers of non alcoholic fatty liver disease (NAFLD) patients playing as a key inducer in this disease. The IV administration of CD98siRNA NPs with a size of 273.1 ± 19.3 nm effectively targeted liver hepatocytes and Kupffer cells, leading to a significant decrease of major proinflammatory cytokines and markers of NAFLD (81).
Chitosan, a biodegradable, positively charged polymer of natural origin is also widely used as a vehicle of RNAi. Quaternary chitosan NPs were designed for vectorization to macrophages of the RNAi of proinflammatory cytokine tumor necrosis factor TNFα. The NPs with sizes between 210 and 279 nm and zeta potentials from 14 to 22 mV achieved a high cellular uptake efficiency near 100% by RAW-274-7 macrophages. NPs cross-linking with TPP reduced their size and zeta potential, resulting in better transfection abilities (82). Polyethylenimine (PEI) is another cationic polymer with high transfection efficacy due to the proton–sponge phenomenon. In order to increase the chitosan's ability to transfect and reduce the PEI cytotoxicity, both components are mixed. NPs of sizes from 150 to 200 nm with both polymers achieved high macrophages transfection in vitro with non-cell toxicity (83). Functionalization of chitosan and other polymers, such as dendrimers with mannose, allows for active vectorization with a better selectivity for macrophage vectorization of RNAi and drugs (84–86).
The applications of polymeric NPs extend to HIV infections, where macrophages play a central role as virus reservoirs. Kutscher et al. designed GLU-decorated chitosan (CS) shell and polylactic-co-glycolic acid (PLGA) core nanoparticles (GLU-CS-PLGA) that recognize special receptors expressed in infected macrophages for the delivery of the antiretroviral drug nevirapine (87). Also, polymeric NPs modified with folic acid may target atazanavir/ritonavir to activated macrophages that overexpress folate receptor at an elevated level (88).
Polyesthers and chitosan are the polymers most often used in order to target drugs and RNAi to liver macrophages. To this end, NPs sizes of 150 to 300 nm have been prepared. Decoration with mannose and other ligands allow NPs to be selectively uptaken.
Other Nanosystems
Exosomes are phospholipidic nanoparticles of endosomal origin that are secreted by cells. They display similar advantages to synthetic nanoparticles, but they usually show higher biocompatibility and physiological activity. Like the majority of nano-sized vesicles, they also accumulate in the liver and may target Kupffer cells. Exosomes derived from mesenchymal stem cells reduce the levels of proinflammatory factors in murine macrophages in vitro with a decrease in biochemical and histological damage (89). These effects were also observed in an in vivo experimental lethal hepatic injury mouse model. The beneficial effect of these vesicles on reducing mortality implies modulation of the inflammatory response and activation of protective mechanisms to limit cell death (90).
Mesenchymal stem cell exosomes are then new types of nanovehicles with anti-inflammatory and protective effect in liver injury that constitute promising strategies for liver disease treatments.
Impact of Macrophage Phenotype in NPs Uptake and Toxicity
Nanosystems that specifically target and deliver therapeutics to polarized macrophages are of interest due to the role that they play in liver diseases. In order to improve the design of those drug delivery systems, the interaction of nanoparticles with macrophages of different phenotypes must be understood, which is why it has been the subject of several studies. Increased NP sequestration by M1 phenotypes has been reported as they are involved in biological processing of foreign materials. Thus, incorporation of phagocytosis promoters in lipid-latex nanoparticles allowed them to target inflammatory M1 macrophages (91). Also, a higher sequestration by inflammatory phenotype was found in vitro and in vivo for spherical silica nanoparticles. This was attributed to the silanol terminal groups that would attach to the receptors of anionic groups that are overexpressed in M1 macrophages (92).
However, M2 macrophages show increased expression of mannose and galactose receptors (93). This makes it possible to target specifically anti-inflammatory phenotypes with mannose–decorated nanoparticles (94). Pluronic and chitosan-based NPs of 150 to 265 nm, decorated with mannose, with positive and negative zeta potentials, respectively, can selectively target M2 macrophages to treat inflammatory diseases and HIV infections. The charge of the NPs and their degree of internalization are dependent on mannose density (84, 94).
Recently, a comparative study on gold nanoparticle uptake by human monocyte-derived macrophages of different phenotypic polarization showed, in general, higher internalization of gold nanoparticles by M2-polarized human macrophages in comparison with the M1-polarized cells. The extent of the uptake was positively correlated with the expression of M2 markers CD163 and CD206. Further investigation in human Kupffer cells showed comparable internalization of nanoparticles by those unstimulated Kupffer cells with a mixed M1/M2 phenotype and the M2-polarized cells, both of which ingested more nanoparticles than the M1-polarized cells did (28).
In short, various types of nanosystems have been designed for targeting polarized macrophages, although further research is necessary to define the NP characteristics necessary for promoting preferential M1 or M2 uptake.
Hepatic Biodistribution of Nanoparticles
The kinetics of nanoparticles when administered in vivo strongly affects their therapeutic efficacy. Nanoparticular systems are recognized by macrophages of the mononuclear phagocyte system and tend to accumulate mainly in organs such as the liver, the spleen, or the lungs (95). The selective distribution of nanoparticular systems in these types of organs facilitates their use for the diagnosis and treatment of different types of pathologies.
Experimental studies conducted with cell lines and animals have brought about a clarification of the mechanisms of penetration into macrophages and hepatocytes using chitosan nanoparticles (96).
Figure 3 shows the arrangement of chitosan nanoparticles in Kupffer cells, hepatocytes, and in whole animals.
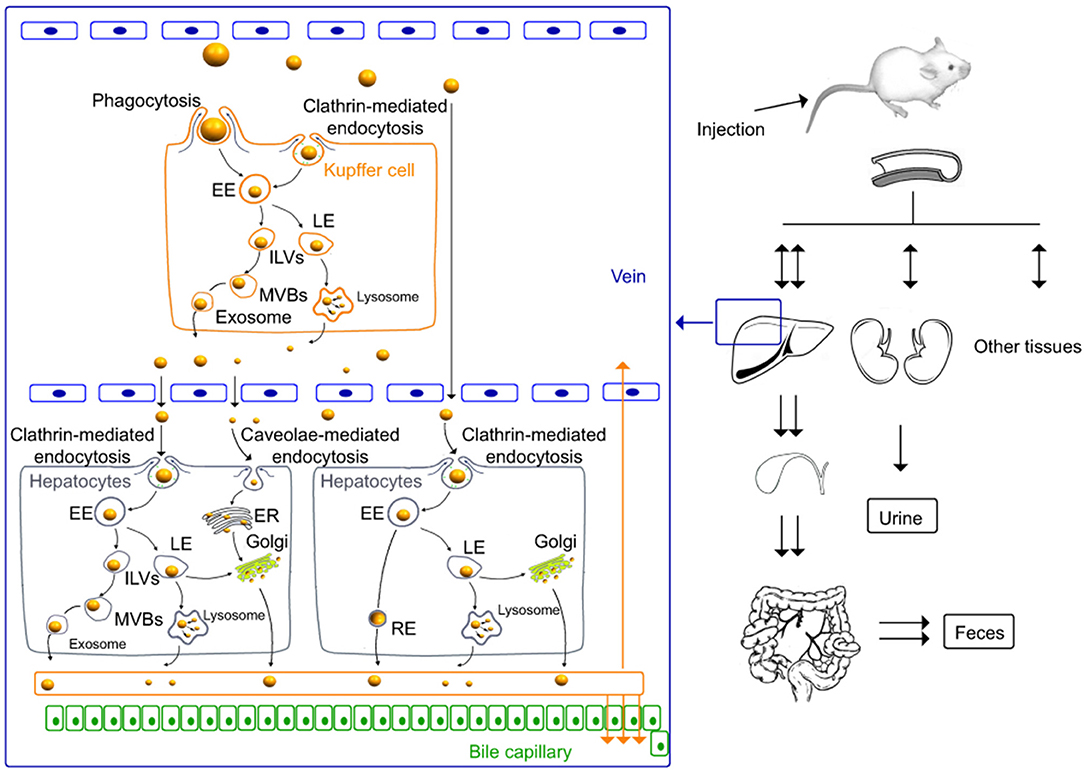
Figure 3. In vivo distribution and elimination of chitosan nanoparticles in Kupffer cells and rat hepatocytes (96). International Journal of Nanomedicine. Reproduced with permission from Dove Medical Press Ltd.
As shown in Figure 3, in vivo studies in mice demonstrate that, at the level of Kupffer cells, the cellular uptake of chitosan nanoparticles is produced via mechanisms of phagocytosis and clathrin- and caveolin-mediated endocytosis and their release through the lysosomal and multivesicular pathways. In addition, nanoparticles penetrate intracellularly into hepatocytes. The renal and hepatobiliary excretion pathways constitute the main routes of elimination in vivo, observing a slow elimination with a nanoparticle half-life of >60 days (96). In addition, in vitro studies conducted with murine macrophage cell lines show that chitosan nanoparticle uptake was clathrin-mediated endocytosis as a primary mechanism and also via phagocytosis as a secondary mechanism. After internalization, a large proportion of the nanoparticles may be excreted from the cells by lysosome-mediated and multivesicular body-mediated exocytosis (97).
Short-term biodistribution of silica nanoparticles in mice demonstrates their high accumulation in organs of the reticulo-endothelial system such as liver and spleen. At the same time, the animals in the experiment showed a clear increase in the number of hepatic macrophages over time. Aggregates of macrophages or microgranulomes increased between 6 and 24 h after silica nanoparticle administration. Clearance of silica nanoparticles from the liver appears to be slower than from the spleen, probably due to hepatic processing and biliary excretion (98).
Other authors also describe the retention of silica nanoparticles and the development of fibrosis in rat livers up to 60 days after IV administration (99).
Iron oxide nanoparticles are a type of system increasingly used for magnetic resonance imaging in diagnostic techniques (100). The administration of polyacrylic acid-coated iron oxide nanoparticles is associated with a selective distribution in the liver that produces proinflammatory activation and liver toxicity in mice. A high accumulation of iron was also observed by macrophages phagocytosis in the periportal zone of the hepatic acinus of the liver and in the splenic red pulp of the spleen which demonstrates the specific uptake of this type of nanoparticles by the monocyte–macrophage system (101).
Nanosystems are currently being combined with cell-based platforms with interesting biodistribution properties and with different therapeutic objectives.
As an example, plasmonic gold nanostars (GNS) were incorporated into immune system cells such as dendritic cells or macrophages obtained from bone marrow in order to investigate the biodistribution of these types of cells in a murine lymphoma model (102).
Figure 4 shows biodistribution in organs and tissues at different times in mice after IV administration of GNS-labeled macrophage cells. The quantification of gold in the different tissues was performed using an ICP-MS technique (102).
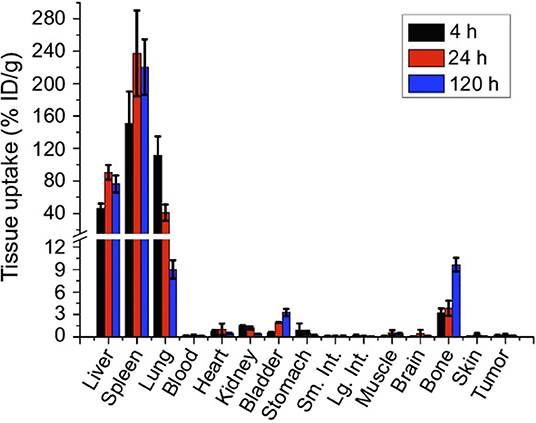
Figure 4. Biodistribution of GNS-labeled macrophage cells at different times after IV administration in mice (102). International Journal of Nanomedicine. Reproduced with permission from Dove Medical Press Ltd.
As shown in Figure 4, after IV administration of GNS-labeled macrophage cells, a specific distribution of this cell-based nanosystem preferably in the spleen, liver, and lung was observed, indicating that these organs had high macrophage cell accumulation. This type of cell-based delivery system presents interesting applications for cell-tracking studies (102).
Stabilin (-1 and -2) are specific receptors for the cellular uptake of different substances such as antisense oligonucleotides in the liver through clathrin-mediated endocytosis (103). Recent studies have been conducted to determine the mechanisms involved in the uptake of nanoparticles by hepatic macrophages. For this reason, an embryonic zebrafish model has been used to evaluate the interaction of nanoparticles with macrophages and endothelial cells using liposomes as nanoparticles. This research has demonstrated the role of the stabilin-2 receptor in the uptake of nanoparticles by endothelial cells. The nanoparticle uptake proved to be independent of the type of material and the functional properties of the nanoparticles but was influenced by the surface charge of the nanoparticle. In addition, the interaction between endothelial cells and nanoparticles can be blocked by competitive inhibitors of stabilin-2 such as dextran sulfate (104).
In another work about the mechanisms involved in the anti-inflammatory activity and in the recognition of crystals and nanomaterials by macrophages, cell-surface receptors or membrane cholesterol have been described as mechanisms involved in crystal nanoparticle recognition although other phagocytosis mechanisms are still unknown (105).
Pathological conditions may also influence NPs uptake. Liver fibrosis profoundly changes the myeloid compartment in the liver, with decreasing numbers of Kupffer cells and increasing numbers of MoMF. With the aim of investigating the changes in the targeting properties of different nanosystems in hepatic fibrosis, Ergen et al. studied the biodistribution of three intravenously injected carrier material, i.e., 10 nm poly(N-(2-hydroxypropyl)methacrylamide) polymers, 100-nm PEGylated liposomes, and 2,000 nm poly(butyl cyanoacrylate) microbubbles, in two fibrosis mice models. They found a decreased uptake of polymers and microbubbles by almost all myeloid cells of the fibrotic liver. However, liposomes had an overall higher targeting efficiency for endothelial and myeloid cells, which remained high even in fibrotic livers with around 60% carrier positive cells in healthy livers and after induction of liver fibrosis, although with a low specificity for the various cell populations. In all cases, Kupffer cells and monocyte-derived macrophages were the cells with increased percentage of carrier-positive cells (56).
Pharmacokinetic Models
Different pharmacokinetic models such as compartmental and especially physiologically based pharmacokinetic (PBPK) models have been developed to characterize the disposition of drugs in different organs and tissues, and especially in the liver, when they are administered in different types of nanoparticles (98, 106–112).
Classic pharmacokinetic models, such as the two-compartmental model, have been proposed to characterize the accumulation in the reticulo-endothelial system and in the livers of mice, as well as the elimination of superparamagnetic iron oxide nanoparticles (SPIONs). The model allows binding to Kupffer cells and extrahepatic clearance of nanoparticles to be characterized using dynamic magnetic resonance imaging (MRI) as seen in Figure 5 (106). The constants Kin and Kout describe the kinetics of nanoparticles associated with and dissociated from the macrophage, and the constant Ke describes the elimination of nanoparticles from the blood compartment by the extrahepatic RES (97).
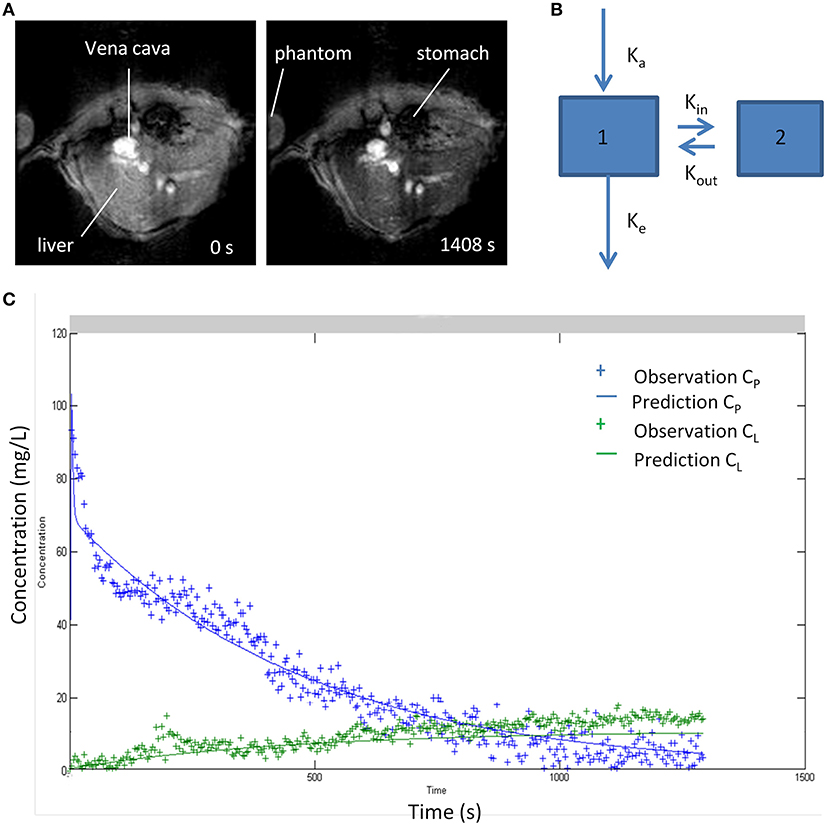
Figure 5. (A) MR images of liver before and after 20 min of intravenous injection of iron oxide nanoparticles (B) Two-compartment kinetic model used to characterize blood pharmacokinetics and distribution in liver tissue of superparamagnetic iron oxide nanoparticle (SPIO). Compartments 1 and 2 represent blood and liver. (C) Pharmacokinetic profiles of nanoparticles in blood and liver fitted to the two-compartment model (106). Reproduced with permission.
The kinetic parameters of distribution in the liver Kin and Kout are related to Kupffer cell numbers, allowing the function of the reticulo-endothelial system to be evaluated in different situations and presents therapeutic applications in liver disease, allowing the chronic liver injury to be evaluated, taking into account that the macrophages are integrated in different stages of the inflammatory process (106).
Physiologically Based Pharmacokinetic (PBPK) Models
PBPK models have been developed to characterize nanoparticle biodistribution in organs of the reticuloendothelial system (RES) such as the liver or the spleen.
PBPK models constitute an interesting strategy for modeling and simulation that allow the kinetic behavior of drugs in animals and humans to be predicted with a physiological basis. These models are based on grouping the body into different compartments that are assimilated to different organs and tissues. These compartments are defined by the volume of tissue and the blood flow that irrigates it. The mass balance of the drug throughout the body is defined through first-order differential equations. The distribution of the drug in each of the tissues can be perfusion-limited or diffusion-limited (112–114). Perfusion rate-limited kinetics occurs when blood flow limits tissue distribution. In the steady state, the concentration of drug in the tissue is in balance with the concentration of drug in the blood through a specific partition coefficient for each tissue. This type of distribution occurs in lipid molecules that easily penetrate the tissue. Permeability rate-limited kinetics occurs when permeability across the membrane constitutes the limiting process of intratisular distribution and occurs mostly in more polar or hydrophilic molecules (112, 113).
Specific PBPK models that consider the specific disposition properties of nanoparticles have been developed to characterize the biodistribution and elimination of nanoparticulate systems in the whole body. This model assumes the existence of two PBPK submodels: one to characterize the disposition of the nanoparticles and another to characterize the disposition of the free drug as shown in Figure 6. This model predicts a greater accumulation of nanoparticles in the liver, spleen, and lungs due to the vascular structure of these tissues and the uptake of nanoparticles by the mononuclear phagocytic system. On the other hand, this type of model has some limitations, given that certain processes, such as aggregation or degradation of nanoparticles, among others, can change the properties of nanoparticles and their arrangement in different organs and tissue. In addition, this type of model allows the interaction between the drug, the nanoparticle, and the complex physiology of the organism to be modeled and simulated (112).
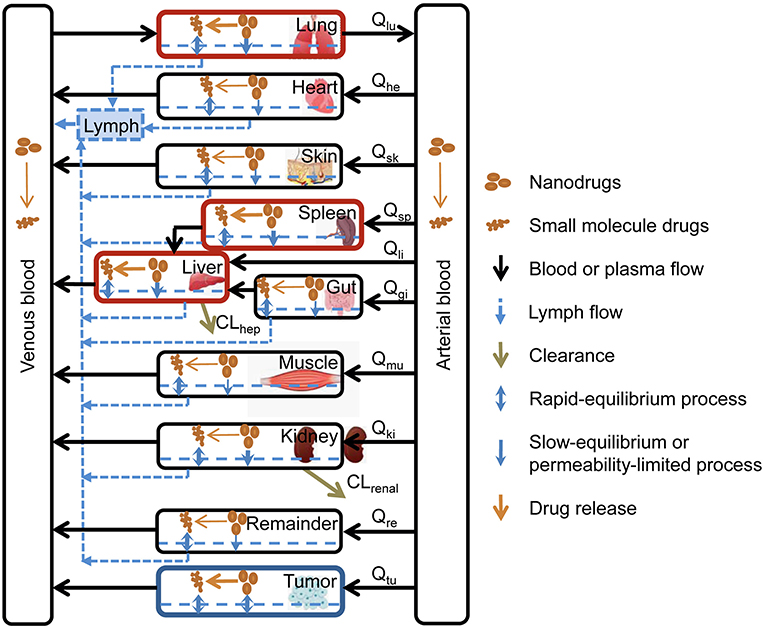
Figure 6. Specific PBPK model to characterize the biodistribution of nanoparticles (112). Reproduced with permission.
Some of these PBPK models have been designed to characterize the role of macrophages in drug tissue concentrations by combining the data from clinical studies with in vitro data. The model allows the concentration of the antibiotic moxifloxacin in tissues from biopsies to be predicted, including those from interstitial fluid, intracellular fluid, vascular space, and macrophages. The study showed that macrophages contribute to the accumulation of the drug in tissues from biopsies (109).
This same type of PBPK model has also been used to predict behavior in liver diseases such as liver cirrhosis. This model allows changes in the plasma concentrations of different drugs, such as alfentanil or lidocaine among others associated with Child-Pugh class A, B, and C liver cirrhosis, to be predicted (115).
Previously, general PBPK models that allow the distribution of drugs incorporated in different types of nanoparticles to be simulated have been described (111). The model was tested with different kinds of nanoparticles with differences in drug dose, size, charge, shape, or surface properties. This model also considers saturable phagocytosis of the nanoparticles. This model is based on another previously published model to characterize the biodistribution of PAA-PEG (116).
This model considers 10 anatomical compartments and each compartment is divided into three subcompartments that represent blood, tissue, and phagocytic cells.
Bigger nanoparticles are better recognized by the macrophages than smaller ones. On the other hand, cationic nanoparticles are better recognized by macrophages than anionic or neutral ones (112, 117). Based on this model, when nanoparticles are injected into the blood, they are captured by phagocytic cells of organs of the reticulo-endothelial system (RES) such as the liver and the spleen depending on their electric charge, size, and agglomeration state, among other factors (111).
In the field of toxicokinetics, PBPK models have been applied to characterize the biodistribution of silver nanoparticles compared to ionic silver. The PBPK model predicts a higher accumulation of silver in the liver as silver nanoparticles in comparison with ionic silver (118).
Nanoparticle Toxicity
The toxicity of nanosystems is an important issue that may limit its applicability. Once in the organism, the processing and final fate of nanoparticles is dependent on their composition.
Liposomes are biodegradable phospholipidic-based nanosystems, and once administered, they are degraded by serum proteins in the blood circulation or by intracellular lipases. The degradation products of liposomes are their constituent lipid molecules that can be further metabolized by the body. This also holds for other lipids forming solid lipid nanoparticles. However, positively charged lipids show limited compatibility as they may induce cytokine activation and cellular toxicity with apoptosis (119).
Polymeric nanoparticles are modified in the organism, giving rise to constituent monomeric units or modified polymer chains. For biodegradable polymers, degradation products are smaller than the renal molecular weight cutoff size and can follow renal elimination (120, 121). For non-biodegradable components, the larger molecules may be cleared by hepatobiliary or the mononuclear phagocyte system, but their biotransformation in hepatocytes and macrophages may cause toxicity (122, 123). As their lipidic counterparts, cationic polymers such as PEI show high cellular toxicity (1).
The non-biodegradability of some inorganic NPs is a factor that leads to liver toxicity (124) and limits their applicability in humans. When NPs are not decomposed by the phagocytosis process, they will remain within the cell and be sequestered in the spleen and liver for long periods of time (122, 125–127). Once the nanoparticle-filled phagocyte dies, those nanoparticles are taken up again by other phagocytes of the same organ, resulting in a similar total amount of nanoparticles accumulated (128). NPs accumulate in the liver and especially in Kupffer cells (129) and could cause fibrosis and other histological tissue changes. They produce oxidative stress that in turn modulates the autophagic process in the liver, disrupting liver metabolism and homeostasis (65). This hepatic oxidative damage has been reported in both in vitro and in vivo models for AuNPs, SiO2NPs, and AgNPs, with differences among the NP compositions (130–134). However, long-term effects need to be further characterized (65).
Tunable properties of NPs, such as shape, surface charge, and size, may affect their toxicity (135, 136). For instance, cerium oxide (CeO2) NPs with rod-like shape showed higher and dose-dependently enhanced macrophage cytotoxicity responses with respect to cubic/octahedral NPs (137). The surface charge is also an important parameter that affects NP clearance rate. One study with mesoporous silica NPs (MSNs) showed that positively charged NPs were rapidly excreted from the liver into the gastrointestinal tract while negatively charged NPs remained sequestered in the liver (138). Also, the degradation of silica NPs to silicic acid for their renal excretion depends on the characteristics of the NPs, such as porosity, size, and surface chemistry (139–141).
Toxicity also depends greatly on the dose of the NPs, with cytotoxicity reported over certain doses for inorganic nanoparticles such as cerium oxide and gold NPs (61, 142, 143).
Therefore, tailoring the characteristics of NPs and controlling the doses of inorganic NPs used could overcome the toxicity problems in the liver and allow for their clinical application (65).
Biodegradable SPIONs or SiNPs offer a safer alternative. For instance, SPION administration is well-tolerated, and long-term in vivo biodistribution studies have shown that they can be transformed to non-superparamagnetic iron forms and eliminated with no signs of toxicity (64). However, in another study, toxicity of SPIONs and ultrasmall superparamagnetic iron oxide nanoparticles (USPIO) in human macrophages has been described (144).
Conclusions
Different types of NPs such as liposomes, solid lipid nanoparticles, inorganic NPs, or exosomes have been proposed for targeting liver macrophages in order to treat liver diseases. They are used as delivery systems but may also induce changes in macrophage phenotypes with influence in the progression of the illness. Lipids and polymeric NPs as vectors of RNAi may exert therapeutic effects by inhibiting regulatory pathways triggered by macrophages. Moreover, the interaction of NPs with macrophages depends on their phenotype, although this issue must be studied in more depth. The physiological-based pharmacokinetic models have been mainly used to describe and simulate NPs destination in the organism. Although strategies to vectorize liver macrophages with NPs are promising, compatibility issues, especially long-term toxicity, are drawbacks for certain systems, especially non-biodegradable ones.
Author Contributions
All authors have equally contributed to the manuscript conception and preparation. JL and CC elaborated an abstract and an index and then all the three authors organized a well-balanced distribution of the different items. For their part of the work, the responsible person looked for the references, redacted the text and selected the images. CG-M was responsible of references database organization. CG-M and CC adapted the format to the journal template. JL, CG-M, and CC revised the whole content and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. (2016) 240:332–48. doi: 10.1016/j.jconrel.2016.01.020
2. Ma Y, Yang M, He Z, Wei Q, Li J. The biological function of kupffer cells in liver disease. In: Ghosh A, editor. Biology of Myelomonocytic Cells. London: IntechOpen (2017).
3. Meijer DK, Molema G. Targeting of drugs to the liver. Semin Liver Dis. (1995) 15:202–56. doi: 10.1055/s-2007-1007278
4. Li P, He K, Li J, Liu Z, Gong J. The role of Kupffer cells in hepatic diseases. Mol Immunol. (2017) 85:222–9. doi: 10.1016/j.molimm.2017.02.018
5. Yona S, Gordon S. From the reticuloendothelial to mononuclear phagocyte system—the unaccounted years. Front Immunol. (2015) 6:328. doi: 10.3389/fimmu.2015.00328
6. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
7. Hume DA, Irvine KM, Pridans C. The mononuclear phagocyte system: the relationship between monocytes and macrophages. Trends Immunol. (2019) 40:98–112. doi: 10.1016/j.it.2018.11.007
8. Mildner A, Yona S, Jung S. A close encounter of the third kind: monocyte-derived cells. Adv Immunol. (2013) 120:69–103. doi: 10.1016/B978-0-12-417028-5.00003-X
9. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
10. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
11. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
12. Schmidl C, Renner K, Peter K, Eder R, Lassmann T, Balwierz PJ, et al. Transcription and enhancer profiling in human monocyte subsets. Blood. (2014) 123:e90–9. doi: 10.1182/blood-2013-02-484188
13. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. (2019) 3:730–43. doi: 10.1002/hep4.1356
14. Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. (2013) 121:e57–69. doi: 10.1182/blood-2012-06-436212
15. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. (2013) 3:785–97. doi: 10.1002/cphy.c120026
16. Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. (2007) 110:4077–85. doi: 10.1182/blood-2007-02-073841
17. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
18. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
19. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell. (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
20. Sato K, Hall C, Glaser S, Francis H, Meng F, Alpini G. Pathogenesis of Kupffer cells in cholestatic liver injury. Am J Pathol. (2016) 186:2238–47. doi: 10.1016/j.ajpath.2016.06.003
21. Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-targeted therapeutics for metabolic disease. Trends Pharmacol Sci. (2018) 39:536–46. doi: 10.1016/j.tips.2018.03.001
22. Ritz T, Krenkel O, Tacke F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol. (2018) 330:175–82. doi: 10.1016/j.cellimm.2017.12.007
23. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today. (2015) 10:487–510. doi: 10.1016/j.nantod.2015.06.006
24. Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes. (2010) 59:916–25. doi: 10.2337/db09-1403
25. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. (2018) 67:1270–83. doi: 10.1002/hep.29544
26. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
27. He H, Ghosh S, Yang H. Nanomedicines for dysfunctional macrophage-associated diseases. J Control Release. (2017) 247:106–26. doi: 10.1016/j.jconrel.2016.12.032
28. MacParland SA, Tsoi KM, Ouyang B, Ma XZ, Manuel J, Fawaz A, et al. Phenotype determines nanoparticle uptake by human macrophages from liver and blood. ACS Nano. (2017) 11:2428–43. doi: 10.1021/acsnano.6b06245
29. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. (2010) 31:3657–66. doi: 10.1016/j.biomaterials.2010.01.065
30. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
31. Li L, Wang H, Ong ZY, Xu K, Ee PLR, Zheng S, et al. Polymer- and lipid-based nanoparticle therapeutics for the treatment of liver diseases. Nano Today. (2010) 5:296–312. doi: 10.1016/j.nantod.2010.06.007
32. Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. (2007) 2:469–78. doi: 10.1038/nnano.2007.223
33. Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. (2011) 32:3435–46. doi: 10.1016/j.biomaterials.2011.01.021
34. Parton RG. Caveolae meet endosomes: a stable relationship? Dev Cell. (2004) 7:458–60. doi: 10.1016/j.devcel.2004.09.009
35. Pelkmans L, Burli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. (2004) 118:767–80. doi: 10.1016/j.cell.2004.09.003
36. Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci USA. (2008) 105:11613–8. doi: 10.1073/pnas.0801763105
37. Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine. (2016) 11:673–92. doi: 10.2217/nnm.16.5
38. Kulkarni SA, Feng SS. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. (2013) 30:2512–22. doi: 10.1007/s11095-012-0958-3
39. Faraji AH, Wipf P. Nanoparticles in cellular drug delivery. Bioorg Med Chem. (2009) 17:2950–62. doi: 10.1016/j.bmc.2009.02.043
40. Arnida, Janat-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur J Pharm Biopharm. (2011) 77:417–23. doi: 10.1016/j.ejpb.2010.11.010
41. Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. (2010) 141:320–7. doi: 10.1016/j.jconrel.2009.10.014
42. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. (2013) 9:1521–32. doi: 10.1002/smll.201201390
43. Esmaeili F, Ghahremani MH, Esmaeili B, Khoshayand MR, Atyabi F, Dinarvand R. PLGA nanoparticles of different surface properties: preparation and evaluation of their body distribution. Int J Pharm. (2008) 349:249–55. doi: 10.1016/j.ijpharm.2007.07.038
44. Hassan R, Tammam SN, Safy SE, Abdel-Halim M, Asimakopoulou A, Weiskirchen R, et al. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur J Pharm Biopharm. (2019) 134:96–106. doi: 10.1016/j.ejpb.2018.11.018
45. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Reports. (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
46. Samuelsson E, Shen H, Blanco E, Ferrari M, Wolfram J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids Surf B Biointerfaces. (2017) 158:356–62. doi: 10.1016/j.colsurfb.2017.07.014
47. Stone NR, Bicanic T, Salim R, Hope W. Liposomal amphotericin B (AmBisome((R))): a review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs. (2016) 76:485–500. doi: 10.1007/s40265-016-0538-7
48. Sande L, Sanchez M, Montes J, Wolf AJ, Morgan MA, Omri A, et al. Liposomal encapsulation of vancomycin improves killing of methicillin-resistant Staphylococcus aureus in a murine infection model. J Antimicrob Chemother. (2012) 67:2191–4. doi: 10.1093/jac/dks212
49. Surewaard BG, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, et al. Correction: identification and treatment of the Staphylococcus aureus reservoir in vivo. J Exp Med. (2016) 213:3087. doi: 10.1084/jem.2016033411032016c
50. Bartneck M, Scheyda KM, Warzecha KT, Rizzo LY, Hittatiya K, Luedde T, et al. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials. (2015) 37:367–82. doi: 10.1016/j.biomaterials.2014.10.030
51. Maradana MR, Yekollu SK, Zeng B, Ellis J, Clouston A, Miller G, et al. Immunomodulatory liposomes targeting liver macrophages arrest progression of non-alcoholic steatohepatitis. Metabolism. (2018) 78:80–94. doi: 10.1016/j.metabol.2017.09.002
52. Bygd HC, Ma L, Bratlie KM. Physicochemical properties of liposomal modifiers that shift macrophage phenotype. Mater Sci Eng C Mater Biol Appl. (2017) 79:237–44. doi: 10.1016/j.msec.2017.05.032
53. Opanasopit P, Sakai M, Nishikawa M, Kawakami S, Yamashita F, Hashida M. Inhibition of liver metastasis by targeting of immunomodulators using mannosylated liposome carriers. J Control Release. (2002) 80:283–94. doi: 10.1016/S0168-3659(02)00006-8
54. Pei Y, Yeo Y. Drug delivery to macrophages: challenges and opportunities. J Control Release. (2016) 240:202–11. doi: 10.1016/j.jconrel.2015.12.014
55. Torrado JJ, Espada R, Ballesteros MP, Torrado-Santiago S. Amphotericin B formulations and drug targeting. J Pharm Sci. (2008) 97:2405–25. doi: 10.1002/jps.21179
56. Ergen C, Niemietz PM, Heymann F, Baues M, Gremse F, Pola R, et al. Liver fibrosis affects the targeting properties of drug delivery systems to macrophage subsets in vivo. Biomaterials. (2019) 206:49–60. doi: 10.1016/j.biomaterials.2019.03.025
57. Hagimori M, Chinda Y, Suga T, Yamanami K, Kato N, Inamine T, et al. Synthesis of high functionality and quality mannose-grafted lipids to produce macrophage-targeted liposomes. Eur J Pharm Sci. (2018) 123:153–61. doi: 10.1016/j.ejps.2018.07.036
58. Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. (2010) 107:1864–9. doi: 10.1073/pnas.0910603106
59. Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. (2013) 31:638–46. doi: 10.1038/nbt.2612
60. Dolina JS, Sung SS, Novobrantseva TI, Nguyen TM, Hahn YS. Lipidoid nanoparticles containing PD-L1 siRNA delivered in vivo enter Kupffer cells and enhance NK and CD8(+) T cell-mediated hepatic antiviral immunity. Mol Ther Nucleic Acids. (2013) 2:e72. doi: 10.1038/mtna.2012.63
61. Selvaraj V, Nepal N, Rogers S, Manne ND, Arvapalli R, Rice KM, et al. Inhibition of MAP kinase/NF-kB mediated signaling and attenuation of lipopolysaccharide induced severe sepsis by cerium oxide nanoparticles. Biomaterials. (2015) 59:160–71. doi: 10.1016/j.biomaterials.2015.04.025
62. de Carvalho TG, Garcia VB, de Araujo AA, da Silva Gasparotto LH, Silva H, Guerra GCB, et al. Spherical neutral gold nanoparticles improve anti-inflammatory response, oxidative stress and fibrosis in alcohol-methamphetamine-induced liver injury in rats. Int J Pharm. (2018) 548:1–14. doi: 10.1016/j.ijpharm.2018.06.008
63. Gan J, Dou Y, Li Y, Wang Z, Wang L, Liu S, et al. Producing anti-inflammatory macrophages by nanoparticle-triggered clustering of mannose receptors. Biomaterials. (2018) 178:95–108. doi: 10.1016/j.biomaterials.2018.06.015
64. Rojas JM, Sanz-Ortega L, Mulens-Arias V, Gutierrez L, Perez-Yague S, Barber DF. Superparamagnetic iron oxide nanoparticle uptake alters M2 macrophage phenotype, iron metabolism, migration and invasion. Nanomedicine. (2016) 12:1127–38. doi: 10.1016/j.nano.2015.11.020
65. Tee JK, Peng F, Ho HK. Effects of inorganic nanoparticles on liver fibrosis: optimizing a double-edged sword for therapeutics. Biochem Pharmacol. (2019) 160:24–33. doi: 10.1016/j.bcp.2018.12.003
66. Lundahl MLE, Scanlan EM, Lavelle EC. Therapeutic potential of carbohydrates as regulators of macrophage activation. Biochem Pharmacol. (2017) 146:23–41. doi: 10.1016/j.bcp.2017.09.003
67. Sokolova V, Shi Z, Huang S, Du Y, Kopp M, Frede A, et al. Delivery of the TLR ligand poly(I:C) to liver cells in vitro and in vivo by calcium phosphate nanoparticles leads to a pronounced immunostimulation. Acta Biomater. (2017) 64:401–10. doi: 10.1016/j.actbio.2017.09.037
68. Mulens-Arias V, Rojas JM, Perez-Yague S, Morales MP, Barber DF. Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials. (2015) 52:494–506. doi: 10.1016/j.biomaterials.2015.02.068
69. Laskar A, Eilertsen J, Li W, Yuan XM. SPION primes THP1 derived M2 macrophages toward M1-like macrophages. Biochem Biophys Res Commun. (2013) 441:737–42. doi: 10.1016/j.bbrc.2013.10.115
70. Kodali V, Littke MH, Tilton SC, Teeguarden JG, Shi L, Frevert CW, et al. Dysregulation of macrophage activation profiles by engineered nanoparticles. ACS Nano. (2013) 7:6997–7010. doi: 10.1021/nn402145t
71. Jin R, Liu L, Zhu W, Li D, Yang L, Duan J, et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials. (2019) 203:23–30. doi: 10.1016/j.biomaterials.2019.02.026
72. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumor growth by inducing pro-inflammatory macrophage polarization in tumor tissues. Nat Nanotechnol. (2016) 11:986–94. doi: 10.1038/nnano.2016.168
73. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. (2011) 121:985–97. doi: 10.1172/JCI44490
74. Lunov O, Zablotskii V, Syrovets T, Rocker C, Tron K, Nienhaus GU, et al. Modeling receptor-mediated endocytosis of polymer-functionalized iron oxide nanoparticles by human macrophages. Biomaterials. (2011) 32:547–55. doi: 10.1016/j.biomaterials.2010.08.111
75. Sharkey J, Starkey Lewis PJ, Barrow M, Alwahsh SM, Noble J, Livingstone E, et al. Functionalized superparamagnetic iron oxide nanoparticles provide highly efficient iron-labeling in macrophages for magnetic resonance-based detection in vivo. Cytotherapy. (2017) 19:555–69. doi: 10.1016/j.jcyt.2017.01.003
76. Sun X, Gamal M, Nold P, Said A, Chakraborty I, Pelaz B, et al. Tracking stem cells and macrophages with gold and iron oxide nanoparticles – the choice of the best suited particles. Appl Mater Today. (2019) 15:267–79. doi: 10.1016/j.apmt.2018.12.006
77. Park JK, Utsumi T, Seo YE, Deng Y, Satoh A, Saltzman WM, et al. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine. (2016) 12:1365–74. doi: 10.1016/j.nano.2016.01.013
78. Kurniawan DW, Jajoriya AK, Dhawan G, Mishra D, Argemi J, Bataller R, et al. Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis. J Control Release. (2018) 288:227–38. doi: 10.1016/j.jconrel.2018.09.004
79. Di Mascolo D, C JL, Aryal S, Ramirez MR, Wang J, Candeloro P, et al. Rosiglitazone-loaded nanospheres for modulating macrophage-specific inflammation in obesity. J Control Release. (2013) 170:460–8. doi: 10.1016/j.jconrel.2013.06.012
80. Cui J, Piotrowski-Daspit AS, Zhang J, Shao M, Bracaglia LG, Utsumi T, et al. Poly(amine-co-ester) nanoparticles for effective Nogo-B knockdown in the liver. J Control Release. (2019) 304:259–67. doi: 10.1016/j.jconrel.2019.04.044
81. Canup BS, Song H, Le Ngo V, Meng X, Denning TL, Garg P, et al. CD98 siRNA-loaded nanoparticles decrease hepatic steatosis in mice. Dig Liver Dis. (2017) 49:188–96. doi: 10.1016/j.dld.2016.11.008
82. Xiao B, Ma P, Ma L, Chen Q, Si X, Walter L, et al. Effects of tripolyphosphate on cellular uptake and RNA interference efficiency of chitosan-based nanoparticles in Raw 264.7 macrophages. J Colloid Interface Sci. (2017) 490:520–8. doi: 10.1016/j.jcis.2016.11.088
83. Iranpur Mobarakeh V, Modarressi MH, Rahimi P, Bolhassani A, Arefian E, Atyabi F, et al. Optimization of chitosan nanoparticles as an anti-HIV siRNA delivery vehicle. Int J Biol Macromol. (2019) 129:305–15. doi: 10.1016/j.ijbiomac.2019.02.036
84. Chu S, Tang C, Yin C. Effects of mannose density on in vitro and in vivo cellular uptake and RNAi efficiency of polymeric nanoparticles. Biomaterials. (2015) 52:229–39. doi: 10.1016/j.biomaterials.2015.02.044
85. Jain K, Verma AK, Mishra PR, Jain NK. Surface-engineered dendrimeric nanoconjugates for macrophage-targeted delivery of amphotericin B: formulation development and in vitro and in vivo evaluation. Antimicrob Agents Chemother. (2015) 59:2479–87. doi: 10.1128/AAC.04213-14
86. Xiao B, Laroui H, Ayyadurai S, Viennois E, Charania MA, Zhang Y, et al. Mannosylated bioreducible nanoparticle-mediated macrophage-specific TNF-alpha RNA interference for IBD therapy. Biomaterials. (2013) 34:7471–82. doi: 10.1016/j.biomaterials.2013.06.008
87. Xavier RJ, Podolsky DK. Unraveling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
88. Puligujja P, McMillan J, Kendrick L, Li T, Balkundi S, Smith N, et al. Macrophage folate receptor-targeted antiretroviral therapy facilitates drug entry, retention, antiretroviral activities, and biodistribution for reduction of human immunodeficiency virus infections. Nanomedicine. (2013) 9:1263–73. doi: 10.1016/j.nano.2013.05.003
89. Jiang L, Zhang S, Hu H, Yang J, Wang X, Ma Y, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate acute liver failure by reducing the activity of the NLRP3 inflammasome in macrophages. Biochem Biophys Res Commun. (2019) 508:735–41. doi: 10.1016/j.bbrc.2018.11.189
90. Haga H, Yan IK, Takahashi K, Matsuda A, Patel T. Extracellular vesicles from bone marrow-derived mesenchymal stem cells improve survival from lethal hepatic failure in mice. Stem Cells Transl Med. (2017) 6:1262–72. doi: 10.1002/sctm.16-0226
91. Bagalkot V, Badgeley MA, Kampfrath T, Deiuliis JA, Rajagopalan S, Maiseyeu A. Hybrid nanoparticles improve targeting to inflammatory macrophages through phagocytic signals. J Control Release. (2015) 217:243–55. doi: 10.1016/j.jconrel.2015.09.027
92. Herd HL, Bartlett KT, Gustafson JA, McGill LD, Ghandehari H. Macrophage silica nanoparticle response is phenotypically dependent. Biomaterials. (2015) 53:574–82. doi: 10.1016/j.biomaterials.2015.02.070
93. Shrivastava R, Shukla N. Attributes of alternatively activated (M2) macrophages. Life Sci. (2019) 224:222–31. doi: 10.1016/j.lfs.2019.03.062
94. Hatami E, Mu Y, Shields DN, Chauhan SC, Kumar S, Cory TJ, et al. Mannose-decorated hybrid nanoparticles for enhanced macrophage targeting. Biochem Biophys Rep. (2019) 17:197–207. doi: 10.1016/j.bbrep.2019.01.007
95. Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. (2012) 134:2139–47. doi: 10.1021/ja2084338
96. Jiang LQ, Wang TY, Wang Y, Wang ZY, Bai YT. Co-disposition of chitosan nanoparticles by multi types of hepatic cells and their subsequent biological elimination: the mechanism and kinetic studies at the cellular and animal levels. Int J Nanomedicine. (2019) 14:6035–60. doi: 10.2147/IJN.S208496
97. Jiang LQ, Wang TY, Webster TJ, Duan HJ, Qiu JY, Zhao ZM, et al. Intracellular disposition of chitosan nanoparticles in macrophages: intracellular uptake, exocytosis, and intercellular transport. Int J Nanomedicine. (2017) 12:6383–98. doi: 10.2147/IJN.S142060
98. Waegeneers N, Brasseur A, Van Doren E, Van der Heyden S, Serreyn PJ, Pussemier L, et al. Short-term biodistribution and clearance of intravenously administered silica nanoparticles. Toxicol Rep. (2018) 5:632–8. doi: 10.1016/j.toxrep.2018.05.004
99. Zhuravskii S, Yukina G, Kulikova O, Panevin A, Tomson V, Korolev D, et al. Mast cell accumulation precedes tissue fibrosis induced by intravenously administered amorphous silica nanoparticles. Toxicol Mech Methods. (2016) 26:260–9. doi: 10.3109/15376516.2016.1169341
100. Farrell BT, Hamilton BE, Dósa E, Rimely E, Nasseri M, Gahramanov S, et al. Using iron oxide nanoparticles to diagnose CNS inflammatory diseases and PCNSL. Neurology. (2013) 81:256–63. doi: 10.1212/WNL.0b013e31829bfd8f
101. Couto D, Freitas M, Costa VM, Chisté RC, Almeida A, Lopez-Quintela MA, et al. Biodistribution of polyacrylic acid-coated iron oxide nanoparticles is associated with proinflammatory activation and liver toxicity. J Appl Toxicol. (2016) 36:1321–31. doi: 10.1002/jat.3323
102. Liu Y, Huang W, Xiong C, Huang Y, Chen BJ, Racioppi L, et al. Biodistribution and sensitive tracking of immune cells with plasmonic gold nanostars. Int J Nanomedicine. (2019) 14:3403–11. doi: 10.2147/IJN.S192189
103. Miller CM, Donner AJ, Blank EE, Egger AW, Kellar BM, Ostergaard ME, et al. Stabilin-1 and stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res. (2016) 44:2782–94. doi: 10.1093/nar/gkw112
104. Campbell F, Bos FL, Sieber S, Arias-Alpizar G, Koch BE, Huwyler J, et al. Directing nanoparticle biodistribution through evasion and exploitation of stab2-dependent nanoparticle uptake. ACS Nano. (2018) 12:2138–50. doi: 10.1021/acsnano.7b06995
105. Nakayama M. Macrophage recognition of crystals and nanoparticles. Front Immunol. (2018) 9:103. doi: 10.3389/fimmu.2018.00103
106. Liu T, Choi H, Zhou R, Chen IW. Quantitative evaluation of the reticuloendothelial system function with dynamic MRI. PLoS ONE. (2014) 9:e103576. doi: 10.1371/journal.pone.0103576
107. Sweeney LM, MacCalman L, Haber LT, Kuempel ED, Tran CL. Bayesian evaluation of a physiologically-based pharmacokinetic (PBPK) model of long-term kinetics of metal nanoparticles in rats. Regul Toxicol Pharmacol. (2015) 73:151–63. doi: 10.1016/j.yrtph.2015.06.019
108. Cheng YH, Riviere JE, Monteiro-Riviere NA, Lin Z. Probabilistic risk assessment of gold nanoparticles after intravenous administration by integrating in vitro and in vivo toxicity with physiologically based pharmacokinetic modeling. Nanotoxicology. (2018) 12:453–69. doi: 10.1080/17435390.2018.1459922
109. Edginton AN, Ahr G, Willmann S, Stass H. Defining the role of macrophages in local moxifloxacin tissue concentrations using biopsy data and whole-body physiologically based pharmacokinetic modeling. Clin Pharmacokinet. (2009) 48:181–7. doi: 10.2165/00003088-200948030-00004
110. Aborig M, Malik PRV, Nambiar S, Chelle P, Darko J, Mutsaers A, et al. Biodistribution and physiologically-based pharmacokinetic modeling of gold nanoparticles in mice with interspecies extrapolation. Pharmaceutics. (2019) 11:E179. doi: 10.3390/pharmaceutics11040179
111. Carlander U, Li D, Jolliet O, Emond C, Johanson G. Toward a general physiologically-based pharmacokinetic model for intravenously injected nanoparticles. Int J Nanomedicine. (2016) 11:625–40. doi: 10.2147/IJN.S94370
112. Yuan D, He H, Wu Y, Fan J, Cao Y. Physiologically based pharmacokinetic modeling of nanoparticles. J Pharm Sci. (2019) 108:58–72. doi: 10.1016/j.xphs.2018.10.037
113. Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet Syst Pharmacol. (2013) 2:e63. doi: 10.1038/psp.2013.41
114. Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. (2016) 6:430–40. doi: 10.1016/j.apsb.2016.04.004
115. Edginton AN, Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet. (2008) 47:743–52. doi: 10.2165/00003088-200847110-00005
116. Li M, Al-Jamal KT, Kostarelos K, Reineke J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano. (2010) 4:6303–17. doi: 10.1021/nn1018818
117. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumors. Nat Rev Mater. (2016) 1:16014. doi: 10.1038/natrevmats.2016.14
118. Bachler G, von Goetz N, Hungerbuhler K. A physiologically based pharmacokinetic model for ionic silver and silver nanoparticles. Int J Nanomedicine. (2013) 8:3365–82. doi: 10.2147/IJN.S46624
119. Epstein-Barash H, Gutman D, Markovsky E, Mishan-Eisenberg G, Koroukhov N, Szebeni J, et al. Physicochemical parameters affecting liposomal bisphosphonates bioactivity for restenosis therapy: internalization, cell inhibition, activation of cytokines and complement, and mechanism of cell death. J Control Release. (2010) 146:182–95. doi: 10.1016/j.jconrel.2010.03.011
120. Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, et al. Renal clearance of quantum dots. Nat Biotechnol. (2007) 25:1165–70. doi: 10.1038/nbt1340
121. Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. (2012) 41:2545–61. doi: 10.1039/c2cs15327k
122. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. (2008) 3:703–17. doi: 10.2217/17435889.3.5.703
123. Zamboni WC, Szebeni J, Kozlov SV, Lucas AT, Piscitelli JA, Dobrovolskaia MA. Animal models for analysis of immunological responses to nanomaterials: challenges and considerations. Adv Drug Deliv Rev. (2018) 136–137:82–96. doi: 10.1016/j.addr.2018.09.012
124. Cornu R, Beduneau A, Martin H. Influence of nanoparticles on liver tissue and hepatic functions: a review. Toxicology. (2020) 430:152344. doi: 10.1016/j.tox.2019.152344
125. Balasubramanian SK, Jittiwat J, Manikandan J, Ong CN, Yu LE, Ong WY. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials. (2010) 31:2034–42. doi: 10.1016/j.biomaterials.2009.11.079
126. Karmali PP, Simberg D. Interactions of nanoparticles with plasma proteins: implication on clearance and toxicity of drug delivery systems. Expert Opin Drug Deliv. (2011) 8:343–57. doi: 10.1517/17425247.2011.554818
127. Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. (2012) 14:282–95. doi: 10.1208/s12248-012-9339-4
128. Sadauskas E, Danscher G, Stoltenberg M, Vogel U, Larsen A, Wallin H. Protracted elimination of gold nanoparticles from mouse liver. Nanomedicine. (2009) 5:162–9. doi: 10.1016/j.nano.2008.11.002
129. Fraga S, Brandao A, Soares ME, Morais T, Duarte JA, Pereira L, et al. Short- and long-term distribution and toxicity of gold nanoparticles in the rat after a single-dose intravenous administration. Nanomedicine. (2014) 10:1757–66. doi: 10.1016/j.nano.2014.06.005
130. Abdelhalim MAK, Moussa SAA, Qaid HAY. The protective role of quercetin and arginine on gold nanoparticles induced hepatotoxicity in rats. Int J Nanomedicine. (2018) 13:2821–5. doi: 10.2147/IJN.S160995
131. Yu Y, Duan J, Li Y, Jing L, Yang M, Wang J, et al. Silica nanoparticles induce liver fibrosis via TGF-beta1/Smad3 pathway in ICR mice. Int J Nanomedicine. (2017) 12:6045–57. doi: 10.2147/IJN.S132304
132. Piao MJ, Kang KA, Lee IK, Kim HS, Kim S, Choi JY, et al. Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol Lett. (2011) 201:92–100. doi: 10.1016/j.toxlet.2010.12.010
133. Ramadi KB, Mohamed YA, Al-Sbiei A, Almarzooqi S, Bashir G, Al Dhanhani A, et al. Acute systemic exposure to silver-based nanoparticles induces hepatotoxicity and NLRP3-dependent inflammation. Nanotoxicology. (2016) 10:1061–74. doi: 10.3109/17435390.2016.1163743
134. Gaiser BK, Hirn S, Kermanizadeh A, Kanase N, Fytianos K, Wenk A, et al. Effects of silver nanoparticles on the liver and hepatocytes in vitro. Toxicol Sci. (2013) 131:537–47. doi: 10.1093/toxsci/kfs306
135. Hu Q, Zhao F, Fan M, He C, Yang X, Huang Z, et al. The influence of titanium dioxide nanoparticles on their cellular response to macrophage cells. Comp Biochem Physiol C Toxicol Pharmacol. (2019) 223:42–52. doi: 10.1016/j.cbpc.2019.05.006
136. Mohammadpour R, Yazdimamaghani M, Cheney DL, Jedrzkiewicz J, Ghandehari H. Subchronic toxicity of silica nanoparticles as a function of size and porosity. J Control Release. (2019) 304:216–32. doi: 10.1016/j.jconrel.2019.04.041
137. Forest V, Leclerc L, Hochepied JF, Trouve A, Sarry G, Pourchez J. Impact of cerium oxide nanoparticles shape on their in vitro cellular toxicity. Toxicol In Vitro. (2017) 38:136–41. doi: 10.1016/j.tiv.2016.09.022
138. Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JA, et al. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials. (2010) 31:5564–74. doi: 10.1016/j.biomaterials.2010.03.048
139. King EJ, McGeorge M. The biochemistry of silicic acid: the solution and excretion of silica. Biochem J. (1938) 32:426–33. doi: 10.1042/bj0320426
140. Yu T, Hubbard D, Ray A, Ghandehari H. in vivo biodistribution and pharmacokinetics of silica nanoparticles as a function of geometry, porosity, and surface characteristics. J Control Release. (2012) 163:46–54. doi: 10.1016/j.jconrel.2012.05.046
141. He Q, Zhang Z, Gao Y, Shi J, Li Y. Intracellular localization and cytotoxicity of spherical mesoporous silica nano- and microparticles. Small. (2009) 5:2722–9. doi: 10.1002/smll.200900923
142. de Araujo RFJ, de Araujo AA, Pessoa JB, Freire Neto FP, da Silva GR, Leitao Oliveira AL, et al. Anti-inflammatory, analgesic and anti-tumor properties of gold nanoparticles. Pharmacol Rep. (2017) 69:119–29. doi: 10.1016/j.pharep.2016.09.017
143. Zhang N, Chen H, Liu AY, Shen JJ, Shah V, Zhang C, et al. Gold conjugate-based liposomes with hybrid cluster bomb structure for liver cancer therapy. Biomaterials. (2016) 74:280–91. doi: 10.1016/j.biomaterials.2015.10.004
Keywords: hepatic macrophages, nanoparticles, drug delivery, biodistribution, Kupffer cells, toxicity
Citation: Colino CI, Lanao JM and Gutierrez-Millan C (2020) Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 11:218. doi: 10.3389/fimmu.2020.00218
Received: 31 October 2019; Accepted: 27 January 2020;
Published: 04 March 2020.
Edited by:
Ruchi Bansal, University of Twente, NetherlandsReviewed by:
Florent Ginhoux, Singapore Immunology Network (Agency for Science, Technology and Research), SingaporeMatthias Bartneck, University Hospital RWTH Aachen, Germany
Copyright © 2020 Colino, Lanao and Gutierrez-Millan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Lanao, am1sYW5hb0B1c2FsLmVz