 Samuel T. Montgomery1
Samuel T. Montgomery1 Dario L. Frey2,3
Dario L. Frey2,3 Marcus A. Mall3,4,5
Marcus A. Mall3,4,5 Stephen M. Stick1,6,7,8
Stephen M. Stick1,6,7,8 Anthony Kicic1,6,7,8,9*, on behalf of the WA Epithelial Research Program (WAERP)6,10 and AREST CF6,7,11,12
Anthony Kicic1,6,7,8,9*, on behalf of the WA Epithelial Research Program (WAERP)6,10 and AREST CF6,7,11,12- 1Faculty of Health and Medical Sciences, School of Biomedical Sciences, The University of Western Australia, Crawley, WA, Australia
- 2Department of Translational Pulmonology, Translational Lung Research Center Heidelberg, University of Heidelberg, Heidelberg, Germany
- 3German Center for Lung Research, Heidelberg, Germany
- 4Department of Pediatric Pulmonology, Immunology and Critical Care Medicine, Charité-Universitätsmedizin Berlin, Berlin, Germany
- 5Berlin Institute of Health, Berlin, Germany
- 6Telethon Kids Institute, The University of Western Australia, Crawley, WA, Australia
- 7Department of Respiratory and Sleep Medicine, Perth Children's Hospital, Nedlands, WA, Australia
- 8Centre for Cell Therapy and Regenerative Medicine, School of Medicine and Pharmacology, The University of Western Australia, Nedlands, WA, Australia
- 9School of Public Health, Curtin University, Bentley, WA, Australia
- 10St John of God Hospital, Subiaco, WA, Australia
- 11Murdoch Children's Research Institute, Melbourne, VIC, Australia
- 12Department of Paediatrics, University of Melbourne, Melbourne, VIC, Australia
Introduction: The responses of cystic fibrosis (CF) airway epithelial cells (AEC) to rhinovirus (RV) infection are likely to contribute to early pathobiology of lung disease with increased neutrophilic inflammation and lower apoptosis reported. Necrosis of AEC resulting in airway inflammation driven by IL-1 signaling is a characteristic finding in CF detectable in airways of young children. Being the most common early-life infection, RV-induced epithelial necrosis may contribute to early neutrophilic inflammation in CF via IL-1 signaling. As little is known about IL-1 and biology of CF lung disease, this study assessed cellular and pro-inflammatory responses of CF and non-CF AEC following RV infection, with the hypothesis that RV infection drives epithelial necrosis and IL-1 driven inflammation.
Methods: Primary AEC obtained from children with (n = 6) and without CF (n = 6) were infected with RV (MOI 3) for 24 h and viable, necrotic and apoptotic events quantified via flow cytometry using a seven-step gating strategy (% total events). IL-1α, IL-1β, IL-1Ra, IL-8, CXCL10, CCL5, IFN-β, IL-28A, IL-28B, and IL-29 were also measured in cell culture supernatants (pg/mL).
Results: RV infection reduced viable events in non-CF AEC (p < 0.05), increased necrotic events in non-CF and CF AEC (p < 0.05) and increased apoptotic events in non-CF AEC (p < 0.05). Infection induced IL-1α and IL-1β production in both phenotypes (p < 0.05) but only correlated with necrosis (IL-1α: r = 0.80; IL-1β: r = 0.77; p < 0.0001) in CF AEC. RV infection also increased IL-1Ra in non-CF and CF AEC (p < 0.05), although significantly more in non-CF AEC (p < 0.05). Finally, infection stimulated IL-8 production in non-CF and CF AEC (p < 0.05) and correlated with IL-1α (r = 0.63 & r = 0.74 respectively; p < 0.0001).
Conclusions: This study found RV infection drives necrotic cell death in CF AEC. Furthermore, RV induced IL-1 strongly correlated with necrotic cell death in these cells. As IL-1R signaling drives airway neutrophilia and mucin production, these observations suggest RV infection early in life may exacerbate inflammation and mucin accumulation driving early CF lung disease. Since IL-1R can be targeted therapeutically with IL-1Ra, these data suggest a new anti-inflammatory therapeutic approach targeting downstream effects of IL-1R signaling to mitigate viral-induced, muco-inflammatory triggers of early lung disease.
Introduction
Cystic Fibrosis (CF) lung disease is progressive, evolves within the first months of life, and is characterized by mucus obstruction and inflammation observable on CT even in the absence of clinical symptoms and often in the absence of detectable respiratory infection (1, 2). Neutrophilic inflammation is a key risk factor for airway disease resulting in bronchiectasis and loss of lung function (3). However, the link between mucus obstruction and airway inflammation has not yet been clearly identified.
Recent evidence from the Australian Respiratory Early Surveillance Team for CF (AREST CF) implicates mucin accumulation as the initial trigger of neutrophilic inflammation in the CF airway (4), and suggests respiratory viral infection may trigger the muco-inflammatory phenotype observed in CF since the heterogeneity of early CF lung disease mirrors the heterogeneity of childhood viral infection (5, 6). Human rhinovirus (RV) appears to be able to manipulate host responses switching from apoptotic to necrotic cell death in airway epithelial cells (AEC) (7, 8). Studies investigating non-bacterial inflammation in the CF airway microenvironment have linked interleukin (IL)-1R signaling driven by IL-1α released from necrotic AEC to neutrophilic inflammation (9, 10). As RV is the most common early life viral infection observed in children with CF (11) and IL-1R signaling has already been detected in the airways of young children with mild disease (12), we hypothesize that resultant neutrophilic inflammation may be driven via this signaling pathway triggered by RV-induced AEC necrosis. However, this proposed mechanism has yet to be investigated.
Given our previous observations of defective responses to RV (8) and IL-1 driven inflammatory responses to necrosis in the pediatric CF airway (12), this study aimed to investigate the direct relationship between RV infection, the type of induced cell death, and IL-1R-driven inflammation in vitro using primary AEC from infants and young children with CF. We obtained primary AEC from young children with and without CF and assessed viable, necrotic and apoptotic events following RV infection utilizing flow cytometry. Using experimental supernatants; IL-1α, IL-1β, IL-1Ra, sIL-1R2, IL-8, CXCL10, CCL5, IFN-β, IL-28A, IL-28B, and IL-29 were measured and subsequently correlated to viable, necrotic and apoptotic responses.
Materials and Methods
Please also refer to the Supplementary Data for full details.
Study Population and Establishment of Primary Cell Culture
This study was approved by the relevant institutional Human Ethics Committees with written consent obtained from parents or guardians. This study included samples from six clinically stable infants and children with CF (mean age 2.9 ± 1.8 years old; Table 1) participating in the AREST CF early surveillance program (2), and samples from six children without CF (mean age 3.8 ± 1.9 years old; Table 1) recruited upon admission to hospital for elective non-respiratory related surgery. Cystic fibrosis transmembrane conductance regulator (CFTR) genotype was determined as part of newborn screening (Table 1). Current bacterial infection in CF samples was determined as part of standard clinical practice using gold-standard microbiological screening, with previous infection the presence of a bacterial infection at any previous visit. Prior wheeze was determined by parent-reported wheeze in the three-months prior to recruitment. Children without CF had no respiratory symptoms observed at time of recruitment. Samples were attained by brushing of the tracheal mucosa of children with a single-sheathed nylon bronchial cytology brush as previously described (8, 13). After collection, primary AEC cultures were established as previously described (14).
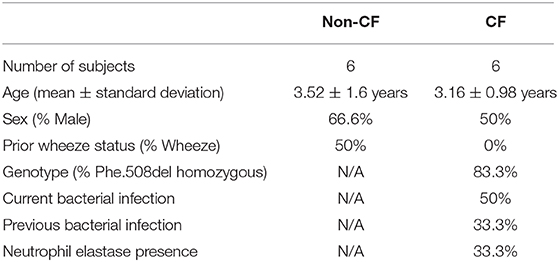
Table 1. Demographics of the study population.
Human RV Infection
Human rhinovirus 1b (RV1b) was propagated as previously described (15). To simulate an acute RV infection in vitro, primary AEC were infected with ~2.95 × 105 TCID50/mL. To ensure responses were due to actively replicating virus, controls were exposed to an UV-inactivated RV1b at the same TCID50 as previously described (16). After 24- and 48-h cells were collected for analysis via flow cytometry and supernatant collected for cytokine measurement. As the peak concentration of RV viral load following infection is observed 24 h post-infection (17, 18), this timepoint was chosen for analysis. Viral load was assessed via qPCR as previously described (19). Infection with RV1b induced typical viral cytokine production from both non-CF and CF AEC (Table S1). Data from 48 h of RV1b infection is presented in the Supplementary Data.
Flow Cytometry
A flow cytometry methodology to measure cell death and disassembly was adapted for use with AEC (20). Briefly, primary cells were detached from culture surfaces via gentle trypsinization, combined with cells obtained from supernatant following centrifugation, and resuspended at a concentration of 106 cells/mL in annexin binding buffer (ThermoFisher Scientific, Scoresby, VIC, Australia). Tubes containing 100 μL of cell suspension were stained for 15 min with 100 μL of Annexin V/AlexaFluor488 (ThermoFisher Scientific, Scoresby, VIC, Australia) (1:40 v/v) and TO-PRO-3 (10 μM final concentration) (ThermoFisher Scientific, Scoresby, VIC, Australia) in annexin binding buffer and flow cytometry performed via a FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). A total of 20,000 events were recorded during acquisition for each sample. Analysis was performed using FlowJo software v10.4 (FlowJo LLC, Ashland, OR, USA) using a seven-step gating strategy to separate events into viable, necrotic, A5+ apoptotic, A5- apoptotic, apoptotic bodies and cellular debris as previously described (20) (Figure S1). Cutoffs used for positive forward scatter (FSC) and side scatter (SSC) were 50 k. Events were grouped into “viable”, “necrotic”, and “apoptotic” for further analysis. Data are presented as percentage of total events (% total).
Cytokine Measurement
Interleukin (IL)-1α, IL-1β, and interferon- beta (IFN-β) protein production was determined using commercially available AlphaLISA kits (Perkin Elmer, Waltham, MA, USA) in cell-free culture supernatant. Similarly, IL-8 (BD Biosciences, San Diego, CA, USA), IL-1 receptor antagonist (IL-1Ra), soluble IL-1 receptor 2 (sIL-1R2), C-X-C motif chemokine 10 (CXCL10), Chemokine (C-C motif) ligand 5 (CCL5), IL-28A, IL-28B, and IL-29 protein production (R&D Systems, Minneapolis, MN, USA) were all determined using commercially available ELISA kits performed according to manufacturer's instructions. Samples below the detection range were arbitrarily reported as half the lower limit and included in the analysis with all other samples as previously described (21).
Statistical Analysis
Data were analyzed using GraphPad Prism v7.04 (GraphPad Software, La Jolla, CA, USA). Data were natural log transformed where appropriate. Comparisons between paired data were performed using Wilcoxon matched pairs signed rank test and Friedman's test with Dunn's multiple comparisons test presented as mean ± standard deviation. Comparisons between unpaired data were performed using Mann-Whitney tests presented as mean ± standard deviation. Associations between flow cytometry events and cytokines measured were assessed using Spearman's rank-order correlations. A two tailed P value < 0.05 was considered statistically significant.
Results
Demographic data for the study populations are summarized in Table 1. Sex and age were similar between cohorts, with most children with CF homozygous for the p.Phe508del mutation. Infection with RV1b resulted in increased rhinovirus load measured via qPCR compared to UV-inactivated RV1b (31.3 ± 29.8 copy #/ng RNA vs. 2.37 × 107 ± 1.46 × 107 copy #/ng RNA; p < 0.05), increased typical pro-inflammatory viral cytokines CXCL10 and CCL5 (Figure S2), and type I and III interferon responses (Figures S3, S4).
Rhinovirus Infection Increases Necrosis but Not Apoptosis in CF AEC
To determine the cellular response to rhinovirus infection, we measured viable, necrotic, and apoptotic events in non-CF (n = 6) and CF (n = 6) AEC (Figure 1). Infection with RV1b resulted in reduced viable events in non-CF AEC (57.6 ± 9.8% vs. 35.4 ± 9.8%; p < 0.05) and CF AEC (65.1 ± 17.5% vs. 49.8 ± 19.4%; p < 0.05) (Figure 1A), and significantly elevated necrotic events in non-CF AEC (8.7 ± 2.3% vs. 12.6 ± 4.9%; p < 0.05) and CF AEC (8.5 ± 1.8% vs. 11.8 ± 3.5%; p < 0.05) (Figure 1B). RV1b infection significantly increased apoptotic events in non-CF AEC (28.8 ± 8.5% vs. 36.9 ± 6.1%; p < 0.05), however, this was not observed for CF AEC (23.5 ± 16.6% vs. 26.2 ± 11.9%) (Figure 1C). Similarly, infection with RV1b for 48 h decreased viable events, increased necrotic events, and increased apoptotic events in both non-CF and CF AEC (Figure S5).
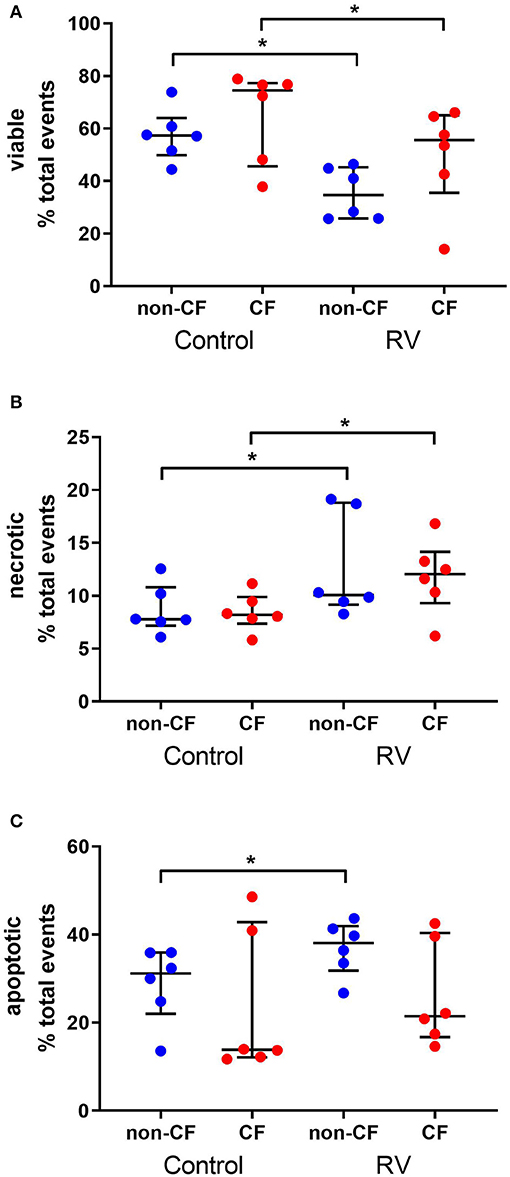
Figure 1. Rhinovirus infection of non-CF and CF AEC decreases viable events, increases necrotic events, and increases apoptotic events in non-CF AEC only. Non-CF (n = 6) and CF (n = 6) AEC infected with RV1b for 24 h were assessed for changes in viable (A), necrotic (B), and apoptotic (C) events measured via flow cytometry. Infection with RV1b for 24 h resulted in (A) decreased viable events in non-CF and CF AEC compared to controls, (B) increased necrotic events in non-CF and CF AEC compared to controls, and (C) increased apoptotic events in non-CF AEC compared to controls. *p < 0.05.
IL-1α and IL-1β Are Increased in Supernatant and Correlate With Cell Death Following Rhinovirus Infection
We next investigated the role of IL-1 signaling in the inflammatory response following rhinovirus-induced cell death in vitro by measuring IL-1α and IL-1β protein following RV1b infection and correlated these with viable, necrotic, and apoptotic events in non-CF and CF AEC (Figure 2). Infection with RV1b increased IL-1α in non-CF (61.6 ± 31.7 pg/mL vs. 511 ± 252 pg/mL; p < 0.05) and CF AEC supernatant compared to controls (46.2 ± 32.7 pg/mL vs. 236 ± 93.1 pg/mL; p < 0.05) (Figure 2A). IL-1α was higher in supernatant from non-CF AEC when compared to CF AEC (p < 0.05). Similarly, IL-1β protein was significantly elevated post infection in both non-CF (4.4 ± 2.3 pg/mL vs. 20.9 ± 9.9 pg/mL; p < 0.05) and CF AEC (3.9 ± 3.6 pg/mL vs. 24.2 ± 18.7 pg/mL; p < 0.05) (Figure 2B) supernatant when compared to controls. Upon analysis, IL-1α was found to be negatively correlated with viable events measured in non-CF AEC only (r = −0.63, p < 0.0001), positively correlated with necrotic events measured in CF AEC (r = 0.80, p < 0.0001), as well as apoptotic events measured in non-CF (r = 0.47, p = 0.0011) (Figures 3A–C). Similarly, IL-1β was negatively correlated with viable events measured in non-CF (r = −0.47, p = 0.0029), strongly positively correlated with necrotic events measured in CF AEC (r = 0.77, p < 0.0001). A weak correlation was also observed between IL-1β and apoptotic events measured in non-CF AEC only (r = 0.37, p < 0.05) (Figures 3D–F). Infection with RV1b for 48 h produced similar responses, with increased IL-1α and IL-1β following infection (Figure S6) significantly associated with necrotic events only in CF AEC, but with apoptotic events in non-CF and CF AEC (Figure S7).
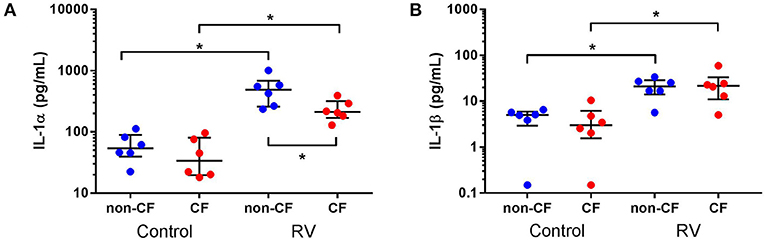
Figure 2. IL-1α and IL-1β is increased in supernatant from non-CF and CF AEC following rhinovirus infection. Supernatant from non-CF (n = 6) and CF (n = 6) AEC infected with RV1b at for 24 h was assessed for levels of IL-1α and IL-1β protein. Infection with RV1b for 24 h resulted in (A) increased IL-1α from non-CF and CF AEC compared to control, with higher levels in non-CF supernatant compared to CF supernatant post-infection, and (B) increased IL-1β from non-CF and CF AEC compared to controls. *p < 0.05.
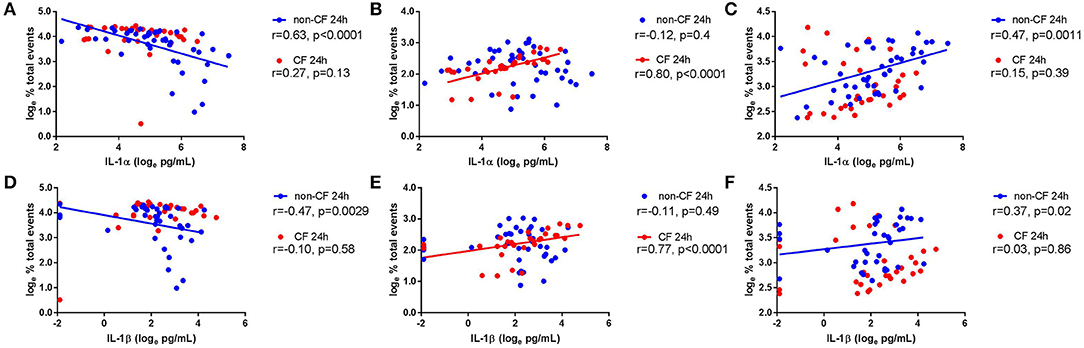
Figure 3. IL-1α and IL-1β in supernatant are associated with necrotic events in CF AEC but not non-CF AEC following 24 h of rhinovirus infection. IL-1α and IL-1β protein in supernatant from non-CF (n = 44) and CF (n = 32) AEC following RV1b infection for 24 h were assessed for correlations with the corresponding changes in viable, necrotic and apoptotic events measured via flow cytometry. IL-1α protein in supernatant was (A) significantly correlated with decreased viable events in non-CF AEC but not CF AEC, (B) significantly correlated with increased necrotic events in CF AEC but not non-CF AEC, and (C) significantly correlated with increased apoptotic events in non-CF AEC but not CF AEC. Similarly, IL-1β protein in supernatant was (D) significantly correlated with decreased viable events in non-CF AEC but not CF AEC, (E) significantly correlated with increased necrotic events in CF AEC but not non-CF AEC, and (F) significantly correlated with increased apoptotic events in non-CF AEC but not CF AEC.
IL-1Ra Is Increased in Supernatant Following Rhinovirus Infection
Since we observed differential responses in IL-1 signaling, we next assessed IL-1R regulatory protein expression, namely IL-1Ra and sIL-1R2, by non-CF and CF AEC following infection with RV1b (Figure 4). Rhinovirus infection resulted in increased IL-1Ra production from non-CF (1368.2 ± 205.6 pg/mL vs. 8149.0 ± 3013.1 pg/mL; p < 0.05) and CF AEC (1930.4 ± 870.4 pg/mL vs. 5334.1 ± 1425.4 pg/mL; p < 0.05) compared to control, with significantly higher IL-1Ra observed in non-CF AEC after infection compared to CF AEC (p < 0.05) (Figure 4A). There was no difference in sIL-1R2 protein production between non-CF or CF AEC, however, sIL-1R2 was significantly induced after infection in CF AEC when compared to non-CF AEC (16.5 ± 2.1 pg/mL vs. 49.8 ± 38.1 pg/mL; p < 0.05) (Figure 4B). Similarly, infection with RV1b for 48 h increased IL-1Ra but not sIL-1R2 production in both non-CF and CF AEC (Figure S8).
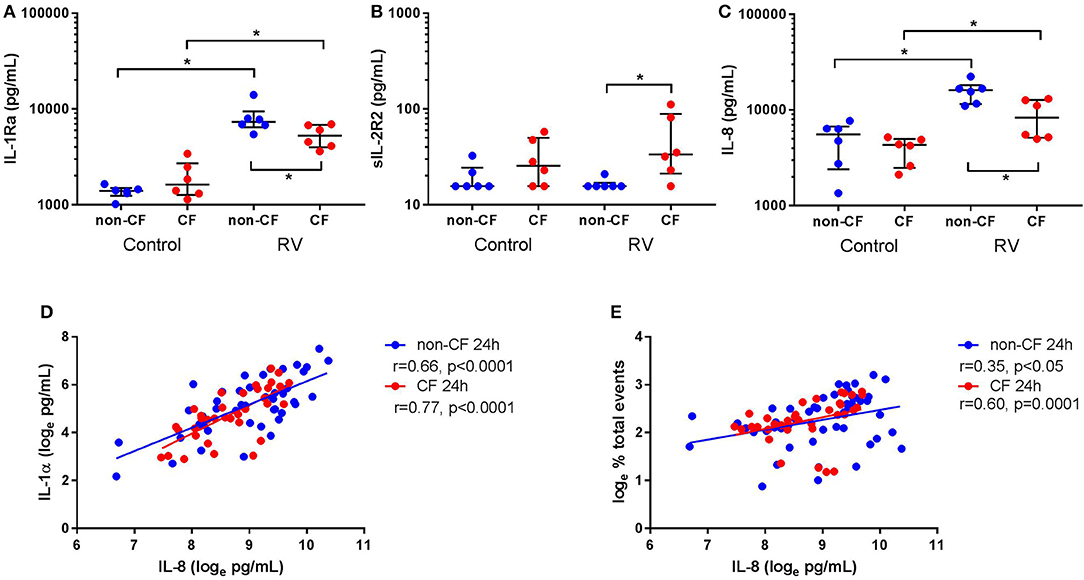
Figure 4. Rhinovirus infection increases IL-1Ra and IL-8 signaling in supernatant from non-CF and CF AEC. Supernatant from non-CF (n = 6) and CF (n = 6) AEC infected with RV1b for 24 h was assessed for levels of IL-1Ra, sIL-1R2, IL-8 protein, and correlations between IL-8 and IL-1α protein or necrotic events measured via flow cytometry. Infection with RV1b for 24 h resulted in (A) increased IL-1Ra from non-CF and CF AEC compared to control, with significantly higher IL-1Ra in supernatant from non-CF AEC compared to CF AEC, (B) no change in sIL-1R2 from non-CF and CF AEC compared to control, but significantly higher sIL-1R2 after infection in supernatant from CF AEC compared to non-CF AEC, and (C) increased IL-8 in supernatant from non-CF and CF AEC compared to control, with significantly higher IL-8 in supernatant from non-CF AEC compared to CF AEC which (D) significantly correlated with IL-1α levels in supernatant from non-CF and CF AEC and (E) significantly correlated with increased necrotic events in both non-CF and CF AEC. *p < 0.05.
IL-8 Is Increased in Supernatant and Associated With IL-1α and Necrotic Events Following Rhinovirus Infection
We next measured inflammation downstream of IL-1R activation by measuring levels of the main neutrophil chemoattractant, IL-8, by non-CF and CF AEC following RV1b infection. Viral infection resulted in a significant increase in IL-8 protein by both non-CF (4890.5 ± 2426.7 pg/mL vs. 15656.4 ± 4102.1 pg/mL; p < 0.05) and CF AEC (3915.3 ± 1262.1 pg/mL vs. 8762.8 ± 3919.0 pg/mL; p < 0.01) compared to relevant controls (Figure 4C), with significantly higher IL-8 produced by non-CF AEC compared to CF AEC (p < 0.05). After infection for 48 h, IL-8 was significantly increased in non-CF and CF AEC (Figure S9A). When analyzed for associations with IL-1 signaling and cell death, IL-8 was positively correlated with necrotic events in non-CF and CF AEC (r = 0.35, p < 0.05 and r = 0.60, p = 0.0001 respectively) (Figure 4D), and IL-1α in non-CF and CF AEC (r = 0.63 & r = 0.74 respectively; p < 0.0001) (Figure 4E). Similar responses were observed following 48 h of infection, with significant associations between IL-8 and IL-1α and necrotic events in non-CF and CF AEC (Figures S9B,C).
Discussion
Our previous work demonstrated a defective response of CF AEC to RV infection (8), and an inflammatory response to epithelial necrosis in CF driven by IL-1R signaling (9) that is already detectable in the airways of infants and children with CF in the absence of bacterial infection (12). In the current study, we add to these earlier findings by conducting a series of in vitro experiments on AEC from children with and without CF focusing on the response of the epithelium to RV infection. Utilizing flow cytometry we observed increased necrosis in CF AEC associated with IL-1R signaling, but increased apoptosis in non-CF AEC associated with IL-1R signaling. When we assessed the IL-1 receptor antagonist IL-1Ra, we found that RV induced IL-Ra production in both phenotypes however this was significantly higher in non-CF AEC. This corresponded with increased IL-8 following RV infection that was significantly higher in non-CF AEC. Furthermore, production of IL-8 was associated with IL-1α and epithelial necrosis in non-CF and CF AEC.
This study provides several novel insights into the mechanisms surrounding pro-inflammatory responses and cell death following RV infection in the CF airway. Our data shows RV infection directly increases necrotic events in both non-CF and CF AEC supporting previous data where rhinovirus protease 3C increased necrosis in nasal AEC (7). The lack of apoptosis in CF AEC supports previous work in our laboratory where dampened apoptosis was observed following RV infection (8). This study supports data suggesting RV infection drives lytic cell death (7), potentially responsible for the increased viral load observed in CF (8, 22).
Delayed apoptosis was also observed in CF AEC following RV infection in this study. Defective apoptotic responses have been observed in AEC and neutrophils in CF (8, 23, 24), we hypothesize reported accumulation of apoptotic cells in the CF airway may be suggestive evidence of defective efferocytosis (25, 26). Cleavage of the phosphatidylserine receptor by neutrophil elastase specifically disrupts phagocytosis of apoptotic cells (26, 27) and as free neutrophil elastase is increased in the CF airway (28, 29), it may explain the reduced apoptotic response and defective efferocytosis observed in the CF airway. Additionally, as suggested by the data in this study, a delayed apoptotic response following RV infection of AEC may also contribute to the defective apoptosis and increased viral load observed in CF (8, 22). The study by Vandivier et al. also found evidence of secondary necrosis following delayed apoptosis, potentially further exacerbating inflammation in the airway via release of DAMPs such as IL-1 signaling (26). As phagocytosis of apoptotic cells can induce anti-inflammatory cytokine production (30, 31), impaired clearance of apoptotic cells may have an additive effect on airway inflammation via reduced anti-inflammatory capacity.
Neutrophilic inflammation is a key risk factor for airway disease resulting in bronchiectasis and loss of lung function (3) which is observed in the absence of detectable bacterial infection (1, 2, 10). It is therefore important to elucidate triggers of early inflammation prior to bacterial colonization of the CF airway. As IL-1R signaling has been investigated as a key pathway driving neutrophilic and eosinophilic inflammation in the airway (9, 12, 32, 33), we next investigated IL-1α and IL-1β signaling following RV infection of AEC. As IL-1α is constitutively active, it can be released directly from necrotic cells in the airway epithelium (9) or actively secreted following activation of the NLRP3 inflammasome and caspase-1 (34, 35) which is required for IL-1β cleavage and release. Activation of the NLRP3 inflammasome has been reported following RV infection resulting from calcium flux resulting from RV ion channel protein 2B activity (36), potassium efflux from lytic cell death such as necrosis or pyroptosis (37), and dysregulated sodium transport due to ENaC upregulation (38). It has also been observed in other inflammatory respiratory diseases with RV associated exacerbations as a hallmark of disease like asthma or COPD, where viral-induced cell death likely contributes to morbidity (36, 39, 40). In this study, we found increased IL-1α and IL-1β alongside increased necrotic cell death suggesting NLRP3 activation could potentially exacerbate the inflammatory cascade following RV infection. This finding supports previously reported data that both IL-1α and IL-1β are released from AEC following RV infection and implicated active secretion via NLRP3 activation (41, 42). Additionally, IL-1α and IL-1β in supernatants of airway mucopurulent secretions have been shown to regulate both MUC5B and MUC5AC through IL-1R (43–45). Release of IL-1α is primarily through AEC while IL-1β in the CF lung is mainly released from macrophages and interstitial mononuclear cells (46, 47), potentially explaining the differences between IL-1α and IL-1β levels observed in this study when compared to levels reported in other studies in ex-vivo samples (43). This data suggests IL-1α and IL-1β observed following RV-A infection may exacerbate mucus hyperconcentration and obstruction evident in the CF airway (4, 43).
Furthermore, we found IL-1α and IL-1β significantly correlated with necrotic events in CF AEC only, while IL-1α and IL-1β correlated with apoptotic events in non-CF AEC only. Studies utilizing the β-ENaC murine model of CF-like lung disease have observed the presence of mucus obstruction and airway neutrophilia in germ-free conditions (48, 49), with “sterile” inflammation in the CF airway triggered by IL-1α released from necrotic AEC (9, 50). IL-1α is measurable in the airways of young children with CF with mild lung disease and associated with structural lung disease measured via CT in the absence of detectable bacterial infection, suggesting a role for IL-1α in the inflammatory cascade in the CF airway environment in the absence of detectable bacterial infection (12). The current study observed levels of IL-1α higher than measured in BALf in young children with CF, suggesting clinically relevant amounts of IL-1α are released from AEC following RV infection. There was higher IL-1α detected in non-CF AEC compared to CF AEC in response to RV infection suggesting IL-1α release from CF AEC occurs predominantly via necrotic cell death post-infection, and release from non-CF AEC via apoptotic cell death. Additionally, IL-1α is associated with viability of non-CF AEC, suggesting overall cell death had a greater effect on IL-1α release in non-CF AEC. Apoptotic cell death is considered immunologically silent due to efficient phagocytosis (51), however, in an in vitro monoculture there is a lack of clearance which results in secondary necrosis and cellular breakdown (52). While epithelial cells can self-phagocytize to reduce inflammatory consequences (53, 54), clearance of apoptotic cells relies on professional phagocytes like macrophages (55) and failure leads to release of immunostimulatory danger associated molecular patterns such as IL-1α (56). Secondary necrosis of AEC in vitro may potentially explain the differences in IL-1α detected between phenotypes, likely due to the observed and reported lack of apoptosis in CF AEC following RV infection (8). Defective apoptosis due to cleavage of apoptotic signaling receptors by neutrophil elastase and manipulation of phagocytic ability by Pseudomonas aeruginosa in monocytes has been reported in CF (26, 57). As IL-1α is increased in the CF airway during bacterial infection (12), we hypothesize defective apoptotic signaling and efferocytosis may play a role in IL-1R-activated neutrophilic inflammation in the CF airway before and after bacterial colonization of the CF airway.
Several recent studies have shown the potential for anti-inflammatory therapy by blocking of IL-1R via genetic deletion of the receptor or pharmacological inhibition via IL-1Ra to inhibit IL-8 expression and neutrophilic inflammation (9, 43). Deletion of IL-1R and IL-1Ra treatment in the βENaC-transgenic mouse significantly reduced IL-1β, neutrophils present in the airway and levels of keratinocyte chemoattractant—a murine IL-8 ortholog (9). This finding was also observed in primary AEC grown at air-liquid interface after stimulation with supernatants of airway mucopurulent secretions, with IL-1Ra treatment reducing IL-8 mRNA (43). The present study found increased IL-1Ra following RV infection in both non-CF and CF AEC, although IL-1Ra was higher in non-CF AEC when compared to CF AEC. This did not correspond with a reduction in IL-8 signaling likely as a result of the amount measured being dramatically lower than the therapeutic concentrations used in other studies (9, 43). RV infection increased IL-8 in both non-CF and CF AEC, however it was significantly higher in non-CF AEC post-infection. This contrasts with previous data by Sutanto et al. which demonstrated significantly higher IL-8 from CF AEC post-RV infection (8). However, differences in the viral titer used for infection and shorter timepoint may have contributed toward the differences in the observed findings.
There are number of unique strengths to the current study. Firstly, primary AEC from pediatric patients were used for experiments in this study, as most immortalized cell lines that are commonly used in CF research are derived from adult donors and may not accurately recapitulate phenotypic differences observed following RV infection in primary AEC isolated from the pediatric airway (8). Secondly, primary cell cultures were passaged before use in this study to distance in vitro cultures from the inflammatory environment from which they were isolated to minimize any pro-inflammatory influences from the in vivo airway milieu (58). While using freshly isolated AEC for in vitro studies may more accurately recapitulate the environment in the CF airway, it could obfuscate mild and virus-specific inflammatory responses. Finally, the use of a more robust flow cytometry methodology that captures events related to apoptotic cell disassembly to analyze cell death allows us to have greater confidence in data generated (20), as conventional methodologies utilizing propidium iodide staining are suggested to have a false positive rate of up to 40% (59).
For this study, we used a submerged monolayer culture model that doesn't fully represent the physiological features of a differentiated respiratory epithelium (60). However, as the basal cells are epithelial progenitors, they are likely to represent intrinsic properties of the respiratory epithelium. Additionally, since viral replication and pro-inflammatory responses are elevated in air-liquid interface compared to monolayer culture (61) subtle phenotypic and mechanistic differences might be more easily identified in an air-liquid interface system. Rhinovirus species affect viral replication and inflammatory responses differently (17, 62), thus the implications of the findings from this study are limited to RV-A infections. However, RV-A has been reported as the most common strain present in adults with CF and associated with more severe clinical outcomes (63). We used a laboratory strain of RV-A (RV1b) that has been reported to induce cytotoxicity more readily than community-derived strains (8, 64, 65) and therefore future work will focus on corroborating the findings of this study using community RV strains of various serotype in order to determine if all RV induce inflammation via IL-1 signaling (66).
In summary, we have demonstrated that RV-A infection of non-CF and CF AEC drives necrotic cell death specifically associated with IL-1α and IL-1β in CF AEC. Viral infection also drove increased IL-8 release associated with necrotic cell death, implicating necrotic cell death following RV infection as a trigger of IL-1R-mediated neutrophilic inflammation in the CF airway. Collectively, these results suggest a role for RV infection as a trigger of IL-1R-driven neutrophilic inflammation in the early life CF airway (Figure 5). Mucin accumulation and hyperconcentration has been identified as the earliest trigger of cystic fibrosis lung disease (4), and linked to IL-1 signaling in vitro (43) creating a positive feedback cycle capable of inducing neutrophilic inflammation in the absence of bacterial infection. Previous studies have highlighted the potential translation of IL-1Ra as a novel anti-inflammatory therapy in CF (9, 12, 67, 68), with the aim to prevent further mucus obstruction and viral-induced, muco-inflammatory triggers of early lung disease in young CF children.
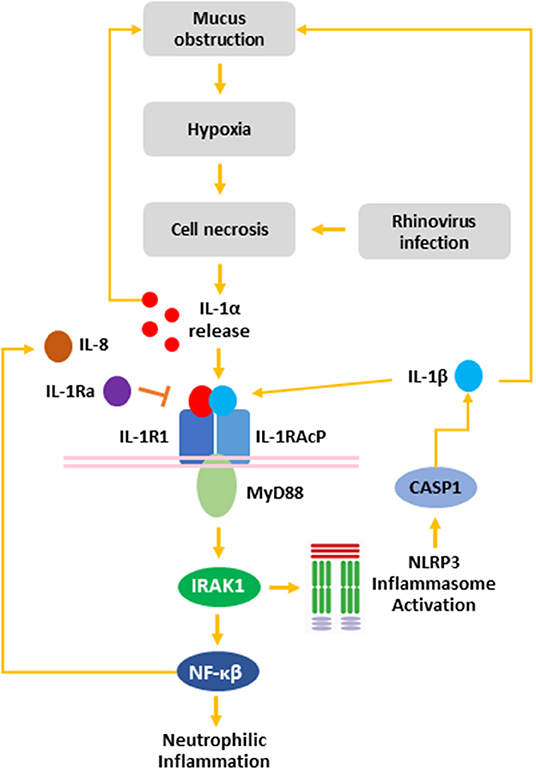
Figure 5. The role of rhinovirus infection in the IL-1 inflammatory response in the CF airway. Mucus obstruction in the CF airway leads to hypoxia of the airway epithelium and epithelial necrosis. Additionally, rhinovirus infection induced necrosis of AEC resulting in release of IL-1α from necrotic cells. Binding of IL-1α to IL-1R recruits MyD88 to the IL-1R:IL-1RAcP complex. Activation of MyD88 leads to IRAK1 activation, which activates the NLRP3 inflammasome leading to CASP1 activation and IL-1β secretion. Additionally, IRAK1 activates NF-κβ, which induces IL-8 release leading to neutrophilic airway inflammation. Both IL-1α and IL-1β induce mucin secretion, which leads to further mucus obstruction creating a positive feedback loop capable of exacerbating CF airway disease. IL-1R activation can be blocked by IL-1Ra to inhibit signaling downstream of IL-1R. IL-1α, interleukin-1 alpha; IL-1β, interleukin-1 beta; IL-1R1, interleukin-1 receptor 1; IL-1Ra, interleukin-1 receptor antagonist IL-1RAcP, interleukin-1 receptor accessory protein; IL-8, interleukin-8; IRAK, interleukin-1 receptor-activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NFκB, nuclear factor kappa beta; NLRP3, nod-like receptor protein 3.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by The University of Western Australia Human Research Ethics Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
SM, MM, SS, and AK contributed conception and design of the study. SM and DF acquired the data and performed the statistical analysis. SM, DF, MM, SS, and AK contributed to data analysis and interpretation. SM wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the US Cystic Fibrosis Foundation, Cystic Fibrosis Australia, Cystic Fibrosis Western Australia, and the German Federal Ministry of Education and Research (82DZL004A1). SS is a NHMRC Practitioner Fellow. AK is a Rothwell Family Fellow.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the subjects and families for their generous contributions to the AREST CF program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00596/full#supplementary-material
References
1. Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. (2009) 180:146–52. doi: 10.1164/rccm.200901-0069OC
2. Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. (2009) 155:623–8 e1. doi: 10.1016/j.jpeds.2009.05.005
3. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. (2013) 368:1963–70. doi: 10.1056/NEJMoa1301725
4. Esther CR, Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med. (2019) 11:eaav3488. doi: 10.1126/scitranslmed.aav3488
5. Meyer KC, Sharma A. Regional variability of lung inflammation in cystic fibrosis. Am J Respir Crit Care Med. (1997) 156:1536–40. doi: 10.1164/ajrccm.156.5.9701098
6. Willner D, Haynes MR, Furlan M, Hanson N, Kirby B, Lim YW, et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. Am J Respir Cell Mol Biol. (2012) 46:127–31. doi: 10.1165/rcmb.2011-0253OC
7. Lötzerich M, Roulin PS, Boucke K, Witte R, Georgiev O, Greber UF. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. (2018) 9:272. doi: 10.1038/s41419-018-0306-6
8. Sutanto EN, Kicic A, Foo CJ, Stevens PT, Mullane D, Knight DA, et al. Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: effects of nonviral and viral stimulation. Am J Respir Cell Mol Biol. (2011) 44:761–7. doi: 10.1165/rcmb.2010-0368OC
9. Fritzsching B, Zhou-Suckow Z, Trojanek JB, Schubert SC, Schatterny J, Hirtz S, et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am J Respir Crit Care Med. (2015) 191:902–13. doi: 10.1164/rccm.201409-1610OC
10. Balazs A, Mall MA. Mucus obstruction and inflammation in early cystic fibrosis lung disease: emerging role of the IL-1 signaling pathway. Pediatr Pulmonol. (2019) 54(Suppl. 3):S5–S12. doi: 10.1002/ppul.24462
11. Deschamp AR, Hatch JE, Slaven JE, Gebregziabher N, Storch G, Hall GL, et al. Early respiratory viral infections in infants with cystic fibrosis. J Cyst Fibros. (2019) 18:844–50. doi: 10.1016/j.jcf.2019.02.004
12. Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, et al. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros. (2018) 17:715–22. doi: 10.1016/j.jcf.2018.05.006
13. Lane C, Burgess S, Kicic A, Knight D, Stick S. The use of non-bronchoscopic brushings to study the paediatric airway. Respir Res. (2005) 6:53–. doi: 10.1186/1465-9921-6-53
14. Martinovich KM, Iosifidis T, Buckley AG, Looi K, Ling K-M, Sutanto EN, et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci Rep. (2017) 7:17971. doi: 10.1038/s41598-017-17952-4
15. Lee W-M, Chen Y, Wang W, Mosser A. Growth of human rhinovirus in H1-HeLa cell suspension culture and purification of virions. In: Jans DA, Ghildyal R, editors. Rhinoviruses: Methods and Protocols. New York, NY: Springer New York (2015). p. 49–61. doi: 10.1007/978-1-4939-1571-2_5
16. Kicic A, Stevens PT, Sutanto EN, Kicic-Starcevich E, Ling KM, Looi K, et al. Impaired airway epithelial cell responses from children with asthma to rhinoviral infection. Clin Exp Allergy. (2016) 46:1441–55. doi: 10.1111/cea.12767
17. Nakagome K, Bochkov YA, Ashraf S, Brockman-Schneider RA, Evans MD, Pasic TR, et al. Effects of rhinovirus species on viral replication and cytokine production. J Allergy Clin Immunol. (2014) 134:332–41. doi: 10.1016/j.jaci.2014.01.029
18. Sykes A, Macintyre J, Edwards MR, del Rosario A, Haas J, Gielen V, et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. (2014) 69:240. doi: 10.1136/thoraxjnl-2012-202909
19. Bochkov YA, Palmenberg AC, Lee W-M, Rathe JA, Amineva SP, Sun X, et al. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat Med. (2011) 17:627–32. doi: 10.1038/nm.2358
20. Jiang L, Tixeira R, Caruso S, Atkin-Smith GK, Baxter AA, Paone S, et al. Monitoring the progression of cell death and the disassembly of dying cells by flow cytometry. Nat Protoc. (2016) 11:655. doi: 10.1038/nprot.2016.028
21. Garratt LW, Sutanto EN, Ling KM, Looi K, Iosifidis T, Martinovich KM, et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur Respir J. (2015) 46:384–94. doi: 10.1183/09031936.00212114
22. Dauletbaev N, Das M, Cammisano M, Chen H, Singh S, Kooi C, et al. Rhinovirus load is high despite preserved interferon-β response in cystic fibrosis bronchial epithelial cells. PLoS ONE. (2015) 10:e0143129–e. doi: 10.1371/journal.pone.0143129
23. Gray RD, Hardisty G, Regan KH, Smith M, Robb CT, Duffin R, et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax. (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
24. Moriceau S, Lenoir G, Witko-Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun. (2010) 2:260–6. doi: 10.1159/000295791
25. Maiuri L, Raia V, De Marco G, Coletta S, de Ritis G, Londei M, et al. DNA fragmentation is a feature of cystic fibrosis epithelial cells: a disease with inappropriate apoptosis? FEBS Lett. (1997) 408:225–31. doi: 10.1016/S0014-5793(97)00347-5
26. Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, et al. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. (2002) 109:661–70. doi: 10.1172/JCI0213572
27. Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. (2001) 276:1071–7. doi: 10.1074/jbc.M003649200
28. Dittrich AS, Kühbandner I, Gehrig S, Rickert-Zacharias V, Twigg M, Wege S, et al. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur Respir J. (2018) 51:1701910. doi: 10.1183/13993003.01910-2017
29. Birrer P, McElvaney NG, Rüdeberg A, Sommer CW, Liechti-Gallati S, Kraemer R, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. J Cystic Fibr. (1994) 150:207–13. doi: 10.1164/ajrccm.150.1.7912987
30. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. (1998) 101:890–8. doi: 10.1172/JCI1112
31. McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J Immunol. (1999) 163:6164–72.
32. Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. (2007) 13:851–6. doi: 10.1038/nm1603
33. Brown R, Paulsen M, Schmidt S, Schatterny J, Frank A, Hirtz S, et al. Lack of IL-1 receptor signaling reduces spontaneous airway eosinophilia in juvenile mice with muco-obstructive lung disease. Am J Respir Cell Mol Biol. (2019) 62:300–9. doi: 10.1165/rcmb.2018-0359OC
34. Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, et al. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci USA. (2011) 108:18055–60. doi: 10.1073/pnas.1109176108
35. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. (2009) 106:20388–93. doi: 10.1073/pnas.0908698106
36. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol. (2013) 49:923–34. doi: 10.1165/rcmb.2013-0032OC
37. da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. (2019) 10:346. doi: 10.1038/s41419-019-1579-0
38. Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. eLife. (2019) 8:e49248. doi: 10.7554/eLife.49248
39. Han M, Bentley JK, Rajput C, Lei J, Ishikawa T, Jarman CR, et al. Inflammasome activation is required for human rhinovirus-induced airway inflammation in naive and allergen-sensitized mice. Mucosal Immunol. (2019) 12:958–68. doi: 10.1038/s41385-019-0172-2
40. Radzikowska U, Eljaszewicz A, Wawrzyniak P, Dreher A, Globinska A, Smolinska S, et al. Rhinovirus triggers increased inflammasome activation in human bronchial epithelium in asthma. Eur Respir J. (2017) 50(suppl 61):PA996. doi: 10.1183/1393003.congress-2017.PA996
41. Piper SC, Ferguson J, Kay L, Parker LC, Sabroe I, Sleeman MA, et al. The role of interleukin-1 and interleukin-18 in pro-inflammatory and anti-viral responses to rhinovirus in primary bronchial epithelial cells. PLoS ONE. (2013) 8:e63365. doi: 10.1371/journal.pone.0063365
42. Shi L, Manthei DM, Guadarrama AG, Lenertz LY, Denlinger LC. Rhinovirus-induced IL-1β release from bronchial epithelial cells is independent of functional P2X7. Am J Respir Cell Mol Biol. (2012) 47:363–71. doi: 10.1165/rcmb.2011-0267OC
43. Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest. (2019) 129:10. doi: 10.1172/JCI125669
44. Abdullah LH, Coakley R, Webster MJ, Zhu Y, Tarran R, Radicioni G, et al. Mucin production and hydration responses to mucopurulent materials in normal versus cystic fibrosis airway epithelia. Am J Respir Crit Care Med. (2018) 197:481–91. doi: 10.1164/rccm.201706-1139OC
45. Gray T, Coakley R, Hirsh A, Thornton D, Kirkham S, Koo JS, et al. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L320–30. doi: 10.1152/ajplung.00440.2002
46. Tang A, Sharma A, Jen R, Hirschfeld AF, Chilvers MA, Lavoie PM, et al. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS ONE. (2012) 7:e37689. doi: 10.1371/journal.pone.0037689
47. Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ Res. (2014) 115:875–83. doi: 10.1161/CIRCRESAHA.115.304437
48. Livraghi-Butrico A, Kelly EJ, Klem ER, Dang H, Wolfgang MC, Boucher RC, et al. Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal Immunol. (2012) 5:397–408. doi: 10.1038/mi.2012.17
49. Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. (2004) 10:487–93. doi: 10.1038/nm1028
50. Montgomery ST, Mall MA, Kicic A, Stick SM. Hypoxia and sterile inflammation in cystic fibrosis airways: mechanisms and potential therapies. Eur Respir J. (2017) 49(1). doi: 10.1183/13993003.00903-2016
51. Szondy Z, Sarang Z, Kiss B, Garabuczi É, Köröskényi K. Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front Immunol. (2017) 8:909. doi: 10.3389/fimmu.2017.00909
52. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. (2010) 584:4491–9. doi: 10.1016/j.febslet.2010.10.046
53. Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville MC, et al. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. (2005) 12:107–14. doi: 10.1038/sj.cdd.4401517
54. Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. (2013) 493:547–51. doi: 10.1038/nature11714
55. Gordon S, Plüddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immuno. (2018) 9:127. doi: 10.3389/fimmu.2018.00127
56. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. (2017) 22:1189–204. doi: 10.1007/s10495-017-1413-z
57. Bianchi SM, Prince LR, McPhillips K, Allen L, Marriott HM, Taylor GW, et al. Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med. (2008) 177:35–43. doi: 10.1164/rccm.200612-1804OC
58. Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O'Neal W, et al. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem. (2005) 280:17798–806. doi: 10.1074/jbc.M410618200
59. Rieger AM, Hall BE, Luong LT, Schang LM, Barreda DR. Conventional apoptosis assays using propidium iodide generate a significant number of false positives that prevent accurate assessment of cell death. J Immunol Methods. (2010) 358:81–92. doi: 10.1016/j.jim.2010.03.019
60. Schögler A, Blank F, Brügger M, Beyeler S, Tschanz SA, Regamey N, et al. Characterization of pediatric cystic fibrosis airway epithelial cell cultures at the air-liquid interface obtained by non-invasive nasal cytology brush sampling. Respir Res. (2017) 18:215. doi: 10.1186/s12931-017-0706-7
61. Ito K, Huang S, Constant S. Comparison of respiratory virus infection between human nasal epithelial cell monolayer and air-liquid interface 3D culture. Eur Respir J. (2011) 38(Suppl. 55):p3506.
62. Rajan D, McCracken CE, Kopleman HB, Kyu SY, Lee FE-H, Lu X, et al. Human rhinovirus induced cytokine/chemokine responses in human airway epithelial and immune cells. PLoS ONE. (2014) 9:e114322. doi: 10.1371/journal.pone.0114322
63. Flight WG, Bright-Thomas RJ, Tilston P, Mutton KJ, Guiver M, Morris J, et al. Incidence and clinical impact of respiratory viruses in adults with cystic fibrosis. Thorax. (2014) 69:247. doi: 10.1136/thoraxjnl-2013-204000
64. Bossios A, Psarras S, Gourgiotis D, Skevaki CL, Constantopoulos AG, Saxoni-Papageorgiou P, et al. Rhinovirus infection induces cytotoxicity and delays wound healing in bronchial epithelial cells. Respir Res. (2005) 6:114. doi: 10.1186/1465-9921-6-114
65. Wark PAB, Grissell T, Davies B, See H, Gibson PG. Diversity in the bronchial epithelial cell response to infection with different rhinovirus strains. Respirology. (2009) 14:180–6. doi: 10.1111/j.1440-1843.2009.01480.x
66. de Almeida MB, Zerbinati RM, Tateno AF, Oliveira CM, Romão RM, Rodrigues JC, et al. Rhinovirus C and respiratory exacerbations in children with cystic fibrosis. Emerg Infect Dis. (2010) 16:996–9. doi: 10.3201/eid1606.100063
67. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun. (2016) 7:10791. doi: 10.1038/ncomms10791
Keywords: cystic fibrosis, airway epithelium, rhinovirus, interleukin-1, necrosis
Citation: Montgomery ST, Frey DL, Mall MA, Stick SM and Kicic A (2020) Rhinovirus Infection Is Associated With Airway Epithelial Cell Necrosis and Inflammation via Interleukin-1 in Young Children With Cystic Fibrosis. Front. Immunol. 11:596. doi: 10.3389/fimmu.2020.00596
Received: 27 November 2019; Accepted: 13 March 2020;
Published: 09 April 2020.
Edited by:
Christian Herr, Saarland University Hospital, GermanyReviewed by:
Léna Royston, Geneva University Hospitals (HUG), SwitzerlandJakob Usemann, University Children's Hospital Zurich, Switzerland
Copyright © 2020 Montgomery, Frey, Mall, Stick and Kicic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Kicic, YW50aG9ueS5raWNpY0B0ZWxldGhvbmtpZHMub3JnLmF1