 Annika Heimes1
Annika Heimes1 Johanna Brodhagen1
Johanna Brodhagen1 Rosemarie Weikard1
Rosemarie Weikard1 Hans-Martin Seyfert1
Hans-Martin Seyfert1 Doreen Becker1
Doreen Becker1 Marie M. Meyerholz2
Marie M. Meyerholz2 Wolfram Petzl2
Wolfram Petzl2 Holm Zerbe2Martina Hoedemaker3
Holm Zerbe2Martina Hoedemaker3 Laura Rohmeier2,4
Laura Rohmeier2,4 Hans-Joachim Schuberth5
Hans-Joachim Schuberth5 Marion Schmicke6
Marion Schmicke6 Susanne Engelmann7,8
Susanne Engelmann7,8 Christa Kühn1,9*
Christa Kühn1,9*- 1Leibniz Institute for Farm Animal Biology (FBN), Institute of Genome Biology, Dummerstorf, Germany
- 2Clinic for Ruminants with Ambulatory and Herd Health Services, Centre for Clinical Veterinary Medicine, Ludwig-Maximilians-University Munich, Oberschleißheim, Germany
- 3Clinic for Cattle, University of Veterinary Medicine Hanover, Hanover, Germany
- 4Clinic for Swine, Small Ruminants, Forensic Medicine and Ambulatory Service, University of Veterinary Medicine Hanover, Hanover, Germany
- 5Immunology Unit, University of Veterinary Medicine Hanover, Hanover, Germany
- 6Faculty of Natural Sciences III, Martin-Luther Universität Halle-Wittenberg, Halle, Germany
- 7Technical University Braunschweig, Institute for Microbiology, Brunswick, Germany
- 8Helmholtz Centre for Infection Research, Microbial Proteomics, Brunswick, Germany
- 9Agricultural and Environmental Faculty, University Rostock, Rostock, Germany
Mastitis is one of the major risks for public health and animal welfare in the dairy industry. Two of the most important pathogens to cause mastitis in dairy cattle are Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli). While S. aureus generally induces a chronic and subclinical mastitis, E. coli is an important etiological pathogen resulting in an acute and clinical mastitis. The liver plays a central role in both, the metabolic and inflammatory physiology of the dairy cow, which is particularly challenged in the early lactation due to high metabolic and immunological demands. In the current study, we challenged the mammary glands of Holstein cows with S. aureus or E. coli, respectively, mimicking an early lactation infection. We compared the animals' liver transcriptomes with those of untreated controls to investigate the hepatic response of the individuals. Both, S. aureus and E. coli elicited systemic effects on the host after intramammary challenge and seemed to use pathogen-specific targeting strategies to bypass the innate immune system. The most striking result of our study is that we demonstrate for the first time that S. aureus intramammary challenge causes an immune response beyond the original local site of the mastitis. We found that in the peripheral liver tissue defined biological pathways are switched on in a coordinated manner to balance the immune response in the entire organism. TGFB1 signaling plays a crucial role in this context. Important pathways involving actin and integrin, key components of the cytoskeleton, were downregulated in the liver of S. aureus infected cows. In the hepatic transcriptome of E. coli infected cows, important components of the complement system were significantly lower expressed compared to the control cows. Notably, while S. aureus inhibits the cell signaling by Rho GTPases in the liver, E. coli switches the complement system off. Also, metabolic hepatic pathways (e.g., lipid metabolism) are affected after mammary gland challenge, demonstrating that the liver restricts metabolic tasks in favor of the predominant immune response after infection. Our results provide new insights for the infection-induced modifications of the dairy cow's hepatic transcriptome following mastitis.
Introduction
In times of growing consumer awareness, the delivery of high-quality products from healthy animals is a high priority for animal farming and the food industry. One major potential risk for public health and animal welfare in the dairy industry is mastitis, the infection and inflammation of the mammary gland. It is one of the most frequent and costly single-animal diseases in dairy cows (1), raises concerns in human and veterinary medicine due to potentially increasing resistance of pathogens against antimicrobial drugs (2, 3) and, if left untreated, can seriously damage the animal's health (4).
One approach to reduce the incidence of infections in animal production is the breeding of individuals with lower susceptibility to infectious diseases (5). For this purpose, it is important to deeply phenotype the individuals for the target traits when searching for the causal background of the genetic variation. Two of the most important pathogens to cause mastitis in dairy cattle are Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli). While S. aureus generally induces a chronic and subclinical mastitis (6), E. coli is an important etiological pathogen resulting in an acute and clinical mastitis (7). Furthermore, S. aureus is Gram-positive, whereas E. coli is Gram-negative (7). The pathogenesis and infection dynamics of the respective forms of mastitis differ significantly depending on the pathogen type. Gram-negative bacteria release endotoxin or lipopolysaccharide (LPS), a component of their cell wall (6, 7). Many studies consider LPS and its fractions to be a potent factor of the E. coli induced mastitis pathogenesis (7, 8). But it has to be recognized that peripheral LPS and whole pathogen challenge can elicit differential responses (9). Gram-positive bacteria (e.g., S. aureus) rely on completely different virulence factors, e.g., exotoxins (6, 10). Whereas the mammary gland response to these pathogens is increasingly well understood, the consequences for peripheral tissues are less thoroughly investigated.
Thus, in our comprehensive network project, we pursued a holistic approach to mastitis in order to obtain a deeper understanding of the underlying biological networks affected in response to an intramammary challenge with mastitis pathogens at an early stage of lactation associated with particularly high disease incidence. A part of this study had a particular focus on the response of the hepatic transcriptome of cows whose mammary glands were experimentally challenged with live S. aureus or E. coli compared to non-challenged cows. The liver is an important organ as it plays a central role in both, the metabolic and inflammatory physiology of the dairy cow (11). Given the particularly heavy metabolic hepatic workload in early lactation of dairy cows (12), any alteration or impairment of liver function might have detrimental effects on animal health in this critical period. Our hypothesis is that the elucidation of the interaction between metabolism and immune response in the liver of cows might provide new insights into the disease-associated hepatic processes in early lactation. This will open new potential perspectives for the prevention and treatment of mastitis and contribute to the discovery of biomarkers for mastitis incidence.
In our study, we have placed a special focus on the response of the liver transcriptome to S. aureus infection, which is commonly assumed to have no or only rare systemic effects on the host after intramammary infection (13–15). In contrast, E. coli infections are well-known for their systemic effects (7, 13, 16). There are also studies that previously investigated the hepatic transcriptome after experimentally induced E. coli mammary gland infection (11, 17–19). However, to our best knowledge, there are no reports comparing the hepatic transcriptome of cows after an S. aureus mammary gland challenge with that of cows challenged with E. coli and that of non-challenged cows.
Materials and Methods
Animals and Study Design
For our study, we allocated 41 gestating Holstein cows in two experimental groups. Thirty-six of these animals were challenged intramammary with pathogens (either S. aureus or E. coli) in an infection model at the Clinic for Cattle at the University of Veterinary Medicine Hanover, in the following referred to as TiHo. The remaining five animals were brought to the Leibniz Institute for Farm Animal Biology in Dummerstorf, in the following referred to as FBN, to serve as a non-challenged control and to participate in a long-term model. The husbandry and sample collection of both experimental groups were previously described (20–22). For the TiHo cohort, the experiment was approved under the reference number 33.12-42502-04-15/2024 by the Lower Saxony Federal State Office for Consumer Protection and Food Safety. For the FBN cohort, the experiment was approved under the reference number 7221.3-1-055/15 by the responsible authority (LALLF, Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern, Rostock, Germany). Furthermore, this study was submitted to and approved by the ethics committees of the University of Veterinary Medicine Hanover and the Leibniz Institute for Farm Animal Biology, respectively. All ethical evaluations were performed as required by the German Animal Care Law and associated legislative regulations (23).
The challenge scheme for the TiHo cohort followed an established and well-studied model (24, 25). In week 6, at 36 ± 3 days after parturition, 24 healthy animals were challenged with 10,000 CFU (colony forming units) S. aureus 1027 each in both hind quarters of the mammary gland. Twelve animals were challenged with 500 CFU E. coli 1303 in one hind quarter of the mammary gland. Both challenge groups had a control udder quarter infused with sterile sodium chloride solution. The infection doses had been established in previous experiments of our group and had already been applied in a number of studies (13, 25). According to the experience from those studies, the doses were selected to successfully induce a clinical mastitis of S. aureus or E. coli, respectively. A schematic overview of the intramammary challenge scheme is shown in Figure 1. After challenge, body temperature of all animals was monitored by an intravaginal logger.
Figure 1. Scheme of the mammary gland quarters in the pathogen challenge experiment.
Prior to morning milking on day 10 a.p., 2 p.p., 7 p.p., 14 p.p., 21 p.p., and in week 6, blood samples were collected from the Vena jugularis. Serum concentrations of NEFA (non- esterified fatty acids) and BHB (beta-hydroxybutyric acid) were determined using the ABX Pentra 400 (HORIBA, Ltd., Kyoto, Japan). Differences between cohorts (TiHo vs. FBN) were evaluated in a Mixed linear model with fixed effects of day of sampling, cohort, and day of sampling × cohort interaction and a random animal effect with the lme4 package in R (26).
The cows challenged with S. aureus were sacrificed 96 h after challenge, whereas the cows challenged with E. coli were killed 24 h after challenge (captive bolt gun followed by immediate exsanguination). These time points were related to the different peaks of inflammation in the mammary gland after S. aureus or E. coli challenge, according to previous studies (24, 25). The collection of liver tissue samples of the TiHo cohort was carried out during the section of the animals. In the long-term model (FBN cohort), we collected liver samples immediately after captive bolt gun followed by immediate exsanguination at a lactation stage analogous to cows of the TiHo cohort, 6 weeks in their second lactation. The liver tissue samples were dissected from the liver Lobus caudatus, immediately shock frozen in liquid nitrogen, and subsequently stored at −80°C.
Transcriptome Analysis by RNA Sequencing
The frozen liver tissue samples (approximately 30 mg) were grinded using the Precellys 24 tissue homogenizer (peQLab, Erlangen, Germany). Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) via an on-column-purification including a modified DNase I digestion step as described by Weikard et al. (27). Subsequently, total RNA was checked for presence of genomic DNA by PCR (28), and additional DNase I digestion was applied when necessary. RNA concentration and purity were measured on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA). RNA integrity was monitored on the Bioanalyzer 2100 (Agilent Technologies, Böblingen, Germany). For RNA sequencing (RNAseq) library preparation we followed a strand-specific protocol (TruSeq Stranded mRNA LP, Illumina, San Diego, CA) including polyA-selection. Indices were applied for multiplexing during cluster generation and sequencing. The RNAseq libraries were monitored for quality on the Bioanalyzer 2100. Paired end library sequencing (2 × 90 base pairs) was performed on the Illumina HiSeq 2500 system (Illumina, San Diego, CA).
All RNAseq datasets are submitted to the ENA repository (https://www.ebi.ac.uk/ena) and are publicly accessible under project number PRJEB33849, accession numbers ERR3466640 - ERR3466680 at EMBL-EBI.
Bioinformatic Analysis
Demultiplexing was performed with CASAVA v1.8 (Illumina, San Diego, CA). For data processing, we used SAMtools (29) as well as Linux and R-based (30) scripts. Quality of the reads was controlled with FastQC version 0.11.5 (31) and MultiQC version 1.4 (32). We applied Cutadapt version 1.12 (33) to remove adapters and Qualitytrim (34) to eliminate low-quality bases. Read alignment to the bovine reference genome UMD3.1 with Ensembl 87 reference annotation (35) was carried out with Hisat2 version 2.1.0 (36). A guided transcript assembly was performed with StringTie version 1.3.2.d (36). This strategy considered the reference genome annotation and additionally allowed the assembly of reads for transcripts not yet annotated. Using the StringTie (36) merge function, we created a project specific annotation for read counting with featureCounts version 1.5.2 (37). For differential expression analysis we used DESeq2 version 1.18.1 (38). For the analysis, we only included loci with at least four samples with at least 11 reads assigned. For graphical displays of the results, the R packages ggplot2 (39), pheatmap (40), ComplexHeatmap v2.2.0 (41), and EnhancedVolcano (42) were used. To establish clusters of co-expressed genes, we performed a weighted gene co-expression network analysis (43) implemented in the R package WGCNA (44) version 1.68, including all expressed loci from control samples as well as samples from E. coli or S. aureus challenged animals. An adjacency matrix of pairwise correlations between expression levels of all pairs of genes across all samples was generated, reporting the connection strength between gene pairs. For constructing the weighted gene network, we selected a soft thresholding power β as calculated by the picSoft Threshold function. Saturation of the respective scale-free topology index was reached at a value of 7. Hierarchical clustering established network modules. For the module generating function, blockwise Modules, we selected a minimum module size of 30, and a threshold for merging modules of 0.25 while keeping all other parameters at default. We then searched for modules, which displayed a correlation to the challenge effects. To this end, we first calculated the proportion of genes, which showed differential expression for each comparison pair, control versus S. aureus and E. coli challenged animals, respectively. We established a spearman rank correlation between the module eigengene value for each sample and its infection group score (with 0 = control, 1 = S. aureus, 2 = E. coli) assuming no, mild or severe clinical response to pathogen. In our analyses, we furthermore calculated the module membership for each gene and module, which is the correlation between the normalized gene expressions and eigengene value of a sample. We plotted the module membership against the correlation between normalized gene expression and infection identifier (representing a measure of gene trait significance) within modules. For pathway analyses, it is assumed that genes, which are highly interconnected within a network eigengene module, should be involved in closely linked or identical biological pathways. Thus, for those modules showing a high correlation between the module eigengene value for each sample and its infection group score, differentially expressed genes (DEGs) by pathogen comparison were modulewise submitted to Ingenuity Pathway Analysis (IPA, Qiagen, Hilden, Germany) with a threshold for significance of padj < 0.05. In addition, we also tested for enriched KEGG pathways via DAVID (45). In the pathogen-specific analyses, we focused on those modules by pathogen challenge, which were in the highest quartile across all modules regarding the proportion of DEGs.
Finally, IPA pathway analyses were also performed for all DEGs in the comparison between samples from controls and E. coli or S. aureus challenged animals, respectively.
Results
Statistics and Distribution of Samples
RNAseq generated a total of 4.5 billion reads corresponding to an average of 110 million reads per liver sample. Ninety-eight percent of reads mapped at least once to the bovine reference genome UMD3.1 (35).
A multidimensional scaling (MDS) plot represents the distribution of hepatic transcriptome data from cows having received an intramammary challenge with S. aureus and E. coli and non-challenged control samples along PC (principal component) 1 and PC2 (Supplementary Image 1). The results clearly demonstrate that there is an essential clustering of animals with respect to the pathogen challenge, but also a substantial variation of animals within the pathogen groups, particularly for the E. coli challenge. Single E. coli challenged animals cluster with S. aureus challenged individuals, while four animals challenged with S. aureus form an outgroup for PC1. The control group cows cluster together, but they are not distinct from the majority of S. aureus challenged cows. Similar results were obtained, when cluster analyses between samples were conducted (Figure 2). The cluster tree is essentially divided into two branches equivalent to E. coli or S. aureus/control animals. However, the E. coli branch of the cluster tree contains three animals challenged with S. aureus. The five control animals formed a subgroup within the branch of essentially S. aureus challenged animals. This is in line with data on serum NEFA and BHB. In the sixth lactation week (immediately before the challenge) there was no significant difference between individuals in the TiHo and FBN cohort [NEFA: TiHo: 388 μmol/l ± 44.1 (s.e.) FBN: 342 μmol/l ± 107.9 (s.e.), p = 0.69; BHB: TiHo: 0.457 mmol/l ± 0.078 (s.e.), FBN: 0.592 mmol/l ± 0.190 (s.e.), p = 0.51].
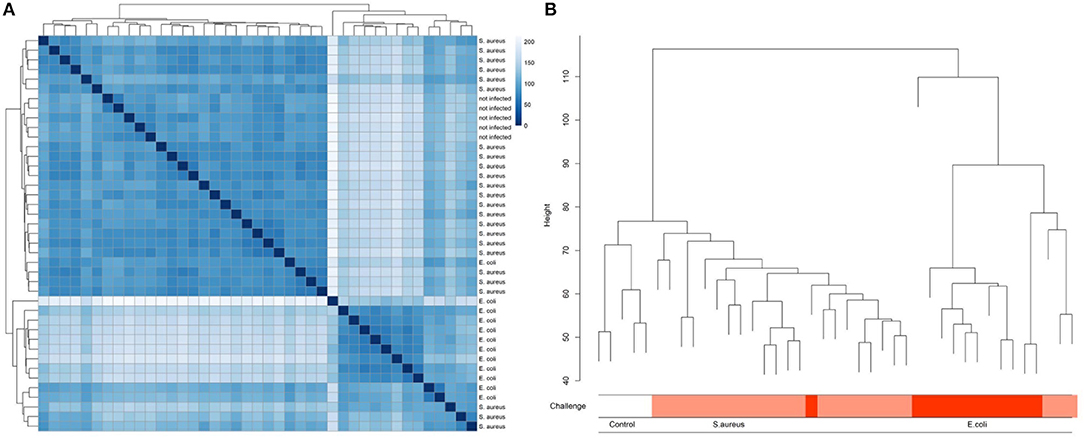
Figure 2. Heat map (A) and clustering (B) of liver transcriptome expression data from cows after experimental intramammary challenge with S. aureus and E. coli and non-challenged control cows.
Differential Expression Analysis
S. aureus Challenge
S. aureus mammary gland infection is usually considered to be a subclinical inflammation with local pathological manifestation. However, the body temperature course data after the pathogen challenge monitored by the intravaginal logger indicated a clinical response to the challenge as demonstrated by a maximum mean body temperature exceeding 40.0°C in 83% of the S. aureus challenged animals during the course of challenge. This clinical pathogen response is supported by the hepatic transcriptome analysis, which revealed a total of 3,672 genes differently expressed between liver samples of S. aureus challenged cows compared to the control group (padj < 0.05, 2,021 lower and 1,651 higher expressed in S. aureus, see also Supplementary Table 1). Of these loci, 2,137 were annotated genes (58%). Several of the loci with very low padj values and high log2 Fold Changes are yet unannotated in the bovine genome and merit further investigation. More than 55% of the DEGs displayed a lower expression after S. aureus challenge compared to untreated controls. Although the number of DEGs in the liver in response to intramammary S. aureus challenge is substantial, it is lower than observed for the E. coli challenge. Comparison of the volcano plots from S. aureus vs. E. coli challenge (Figure 3) demonstrated the substantially stronger effect of E. coli compared to S. aureus challenge, particularly regarding strongly upregulated genes.
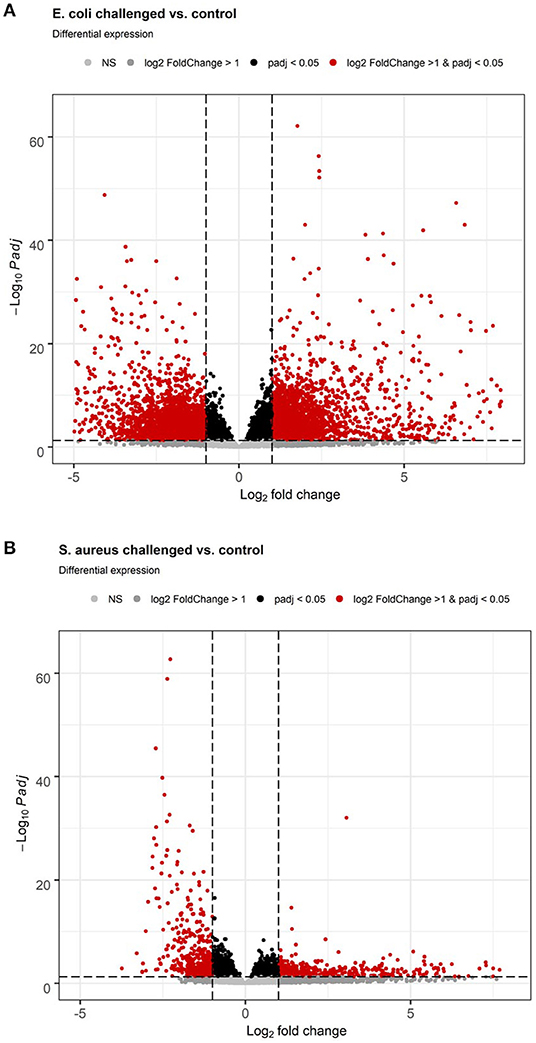
Figure 3. Volcano plot of differential gene expression analysis of the hepatic transcriptome between individuals with intramammary pathogen challenge and controls. (A) E. coli vs. control. (B) S. aureus vs. control.
E. coli Challenge
E. coli mammary infection usually elicits a systemic inflammatory response. Correspondingly, all challenged cows developed fever (inner body temperature > 39.5°C) in the time period 24 h after challenge. The average maximum temperature of the animals was 41.8°C (22). This strong systemic response was also reflected in the liver transcriptome. When comparing the liver transcriptome of cows challenged with E. coli with that of the non-challenged control group, 10,648 loci were significantly differentially expressed (padj < 0.05, see Supplementary Table 2, Figure 3). Thus, more than 50% of all loci passing the threshold for being included in the differential expression analysis (20,723) were differentially expressed between the E. coli challenged cows and the control group. Of these significantly DEGs, 6,720 were annotated genes (63%). There was a tendency for more DEGs with higher expression in E. coli challenged animals than with lower expression compared to untreated controls (3,548 higher, 3,172 lower expressed; Figure 3).
Module Analysis Searching for Clusters of Genes Associated With Pathogen Challenge
Data Analysis Across Pathogen Challenge Groups
To identify further mechanisms in the hepatic pathogen-specific response to intramammary infection, we performed a module-based cluster analysis across all samples. The respective WGCNA analysis yielded 31 distinct modules (Supplementary Table 3). We found 15 modules, which were significantly correlated with the infection category (0 = control, 1 = S. aureus, 2 = E. coli, Table 1). Six of those modules showed a negative and nine modules a positive correlation: Within the modules with significant correlation to the infection identifier E. coli eight modules were in the upper quartile across all modules regarding the proportion of DEGs (Table 1). For the infection identifier S. aureus, four significantly correlated modules were in the upper quartile across all modules regarding the proportion of DEGs. However, in contrast to the E. coli infection identifier, where all significantly correlated modules in the upper quartile regarding the proportion of DEGs were also significantly correlated to the infection identifier, half of the modules in the upper quartile did not show this pattern for the S. aureus category. Instead, for the S. aureus identifier, four modules without significant correlation to the infection category score were in the upper quartile regarding the proportion of DEGs. This applied particularly to the modules “yellow” and “grey60”. It has to be considered that three of those modules only harbored a limited number of annotated genes.
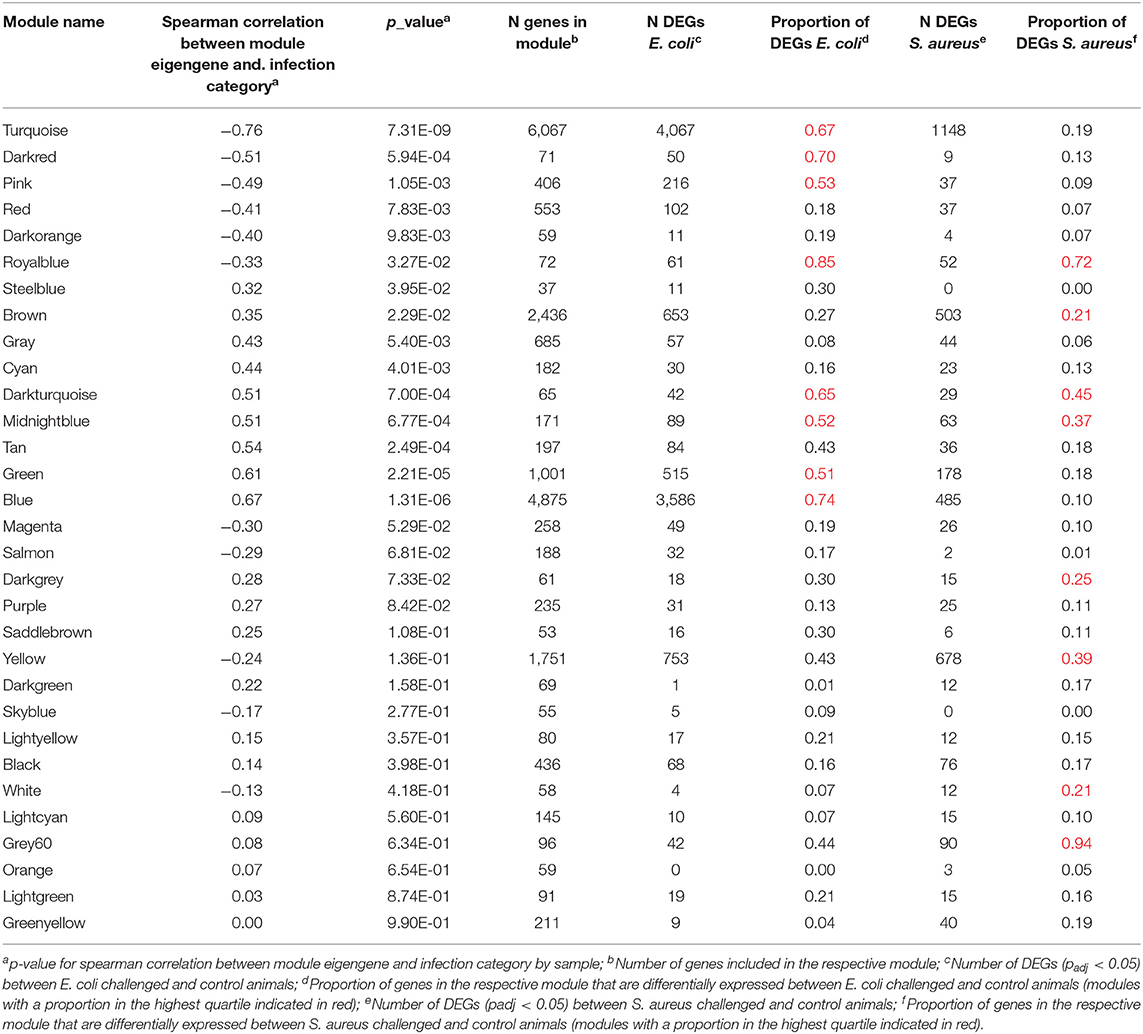
Table 1. Module characteristics after WGCNA analysis of the hepatic transcriptomes including individuals challenged intramammary with E. coli or S. aureus and unchallenged controls.
For each module and each gene, we then plotted the module membership measure (a parameter for how well a gene fits into the assigned module) against the correlation between normalized gene expression and infection category (representing a measure of gene-trait relationship significance). While modules with a significant correlation to the infection category showed a very clear linear relationship of those two parameters (see Figures 4C,D), we did not observe this pattern for uncorrelated modules (Figures 4A,B).
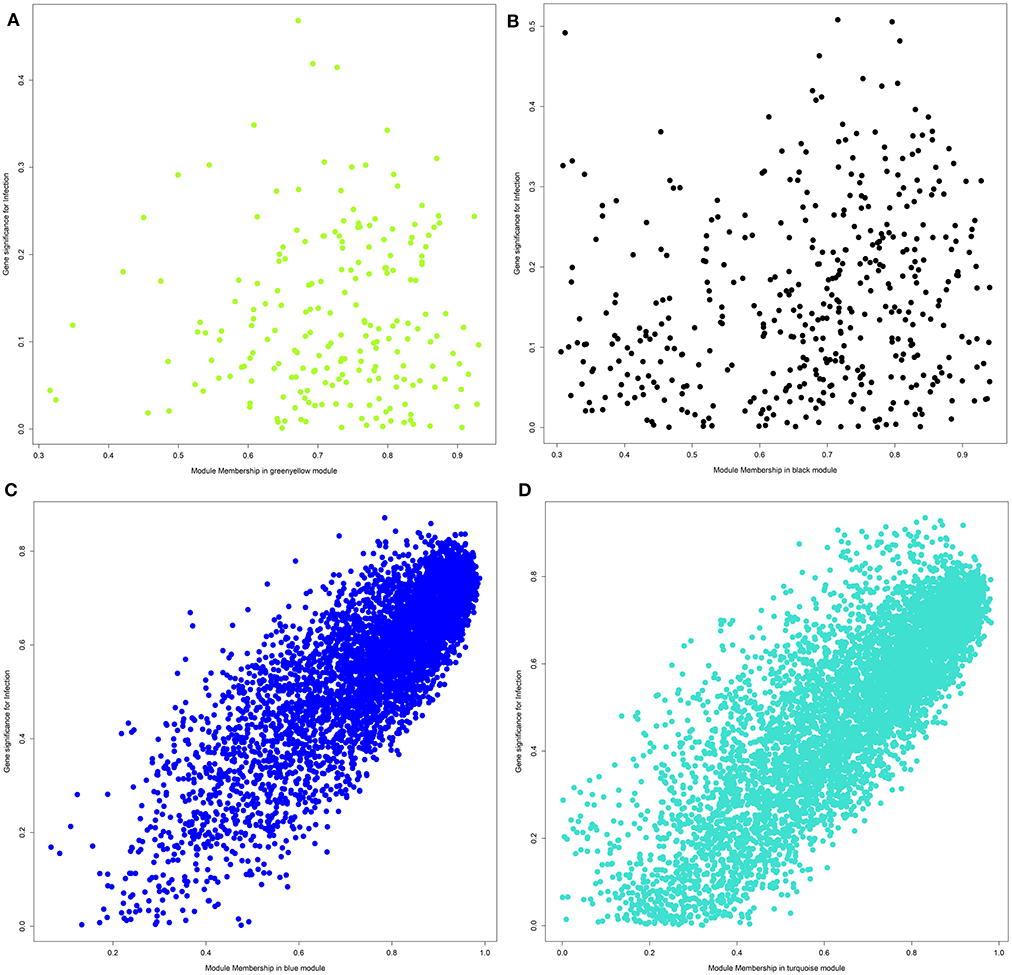
Figure 4. Plot of module membership against the correlation between normalized gene expression and infection category for each gene within a module. (A) Module greenyellow; (B) module black; (C) module blue; (D) module turquoise.
Data Analysis Within Pathogen Challenge Groups
For subsequent pathway analyses, we focused on those modules within the upper quartile across all modules regarding the proportion of DEGs and also on those with a significant correlation to the infection identifier. There were a number of modules with a significant correlation to the infection identifier, which demonstrated pathogen-challenge specific differences in the proportion of DEGs (Table 1). These modules were further analyzed within pathogen challenge groups by clustering analyses via heatmaps (Supplementary Images 2, 3) and were investigated for pathways and biological function GO terms enriched by DEGs.
E. coli Challenge
For E. coli challenge, pathway enrichment analysis highlighted the modules turquoise, darkred, pink, green and blue (Supplementary Image 2, Tables 1, 2, Supplementary Tables 4–6). These modules showed a clustering of samples for infection status and had a number of biological pathways and functions enriched with DEGs. The module turquoise by far harbored the largest number of genes in our dataset (Table 1) and showed pathways enriched with DEGs involved in Fatty Acid Beta Oxidation and Acute Phase response (Table 2, Supplementary Table 4). A particularly strong inactivation or activation according to the calculated z-scores was predicted for these two pathways. Accordingly, all members of the IPA Fatty Acid Beta Oxidation pathway were downregulated. DAVID enrichment analysis further revealed many KEGG pathways and biological function GO terms for fat and amino acid metabolism enriched with DEGs from this module (e.g., Valine, leucine, and isoleucine degradation, Fatty acid metabolism or PPAR signaling pathway, Supplementary Table 6). This fits in with the observation that several transcription factors known to be activators of fatty acid metabolism (e.g., PPARA, PPARG, PPARD) were found in the IPA analyses as upstream regulators with a predicted inhibition action status (Supplementary Table 5). Taken together, the data indicate that genes in this module are substantially involved in the inactivation of fatty acid metabolism and amino acid degradation highlighting the impaired metabolic function of the liver after intramammary E. coli challenge. The module is of particular interest, because it links the lipid metabolism with the immune response. The pathway Acute Phase response, one of the major tools of innate immune response is significantly enriched with DEGs from this pathway. Its positive z-score indicates its activation. The nuclear receptor HNF4A that, for example, regulates the metabolism in the liver, was the most prominent transcription factor among the upstream regulators and was predicted to be strongly inactivated (Supplementary Table 5). This is consistent with its known role of being involved in both the metabolic and the immune-related functions of the hepatocytes. In contrast, the effects described for DEGs in the turquoise module were not observed for S. aureus challenge, and this module was not in the highest quartile regarding the proportion of DEGs for S. aureus.
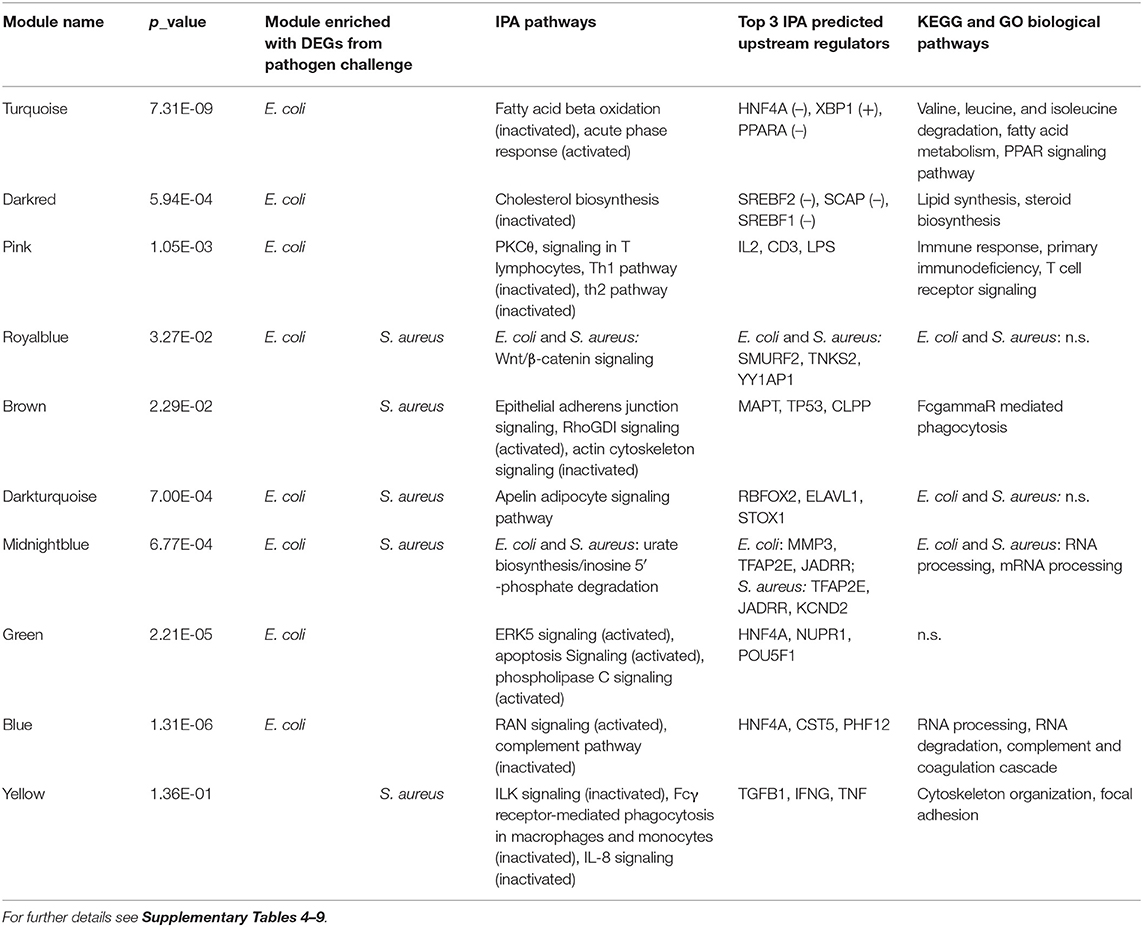
Table 2. Summary of ingenuity and DAVID enrichment analysis with DEGs from WGCNA modules within pathogen challenge groups.
The darkred module contained only 71 genes (Table 1, Supplementary Table 4) but also displayed a particular cluster of genes involved in metabolism. The dominant feature in the enrichment analysis of this module was the Cholesterol Biosynthesis pathway, which was observed as an inactivated pathway in IPA and was supported by corresponding biological function GO terms and KEGG pathway data from DAVID analysis (Table 2, Supplementary Tables 4, 6). Predicted upstream regulators for DEGs in this module were, e.g., SREBF1 and SREBF2, which were assigned an inactivated status (Supplementary Table 5). They are known to be important for cholesterol homeostasis by regulating the transcription of sterol-regulated genes, and in our study, they were highly significantly (padj. = 0.0003 and 0.0002 for SREBF1 and SREBF2, respectively) lower expressed in E. coli challenged animals compared to controls. However, they themselves were not assigned to this module, but to the turquoise module.
In contrast to the turquoise and darkred modules, the pink module comprises DEGs, which were predominantly enriched in pathways and biological function GO terms associated with immune response, namely PKCθ Signaling in T Lymphocytes and Th1 and Th2 pathways (Table 2, Supplementary Tables 4, 6). Interestingly, almost all pathways for this module showed an inactivation status, this was most evident for the Th1 pathway. Suppressed hepatic response to E. coli challenge in the mammary gland is consistent with the prediction that upstream regulators with anti-inflammatory and attenuating cytokine signaling role like IL10 or SOCS1 are activated in the liver, and immune response stimulating factors like IL2 and IFNG are inactivated (Supplementary Table 5). Thus, this module provides a first indication on the inhibition of at least some sections of the hepatic immune response to intramammary E. coli challenge.
The module blue was another large module with >4,800 associated genes (Table 1), of which 2,386 were DEGs in the E. coli challenge (Table 2). In this module, we found the pathways and biological function GO terms related to protein and RNA processing significantly enriched with DEGs (Supplementary Tables 4, 6). Interestingly, also the Complement pathway, a central component of the innate immune response, was highlighted, because it had the lowest z-score of all enriched pathways indicating that this pathway is inactivated in E. coli challenged animals.
With 1,001 assigned genes in total, the module green (Table 1) was also a major module in size and provided also a conclusive clustering of E. coli challenged and control animals (Supplementary Image 2). However, despite >500 DEGs, only 14 IPA pathways and no KEGG pathway or biological function GO terms showed any significant enrichment with DEGs (Table 2, Supplementary Table 4). Thus, the specific function of the DEGs in this module is inconclusive, although, e.g., the ERK signaling pathway displayed enrichment.
S. aureus Challenge
For S. aureus challenge, the modules brown, yellow, royalblue, and grey60 were of particular interest, because they had a proportion of DEGs within the highest quartile across all modules and/or showed a clustering of samples for infection status (Supplementary Image 3, Tables 1, 2). In addition, except royalblue, they were not highly enriched with DEGs in the E. coli challenge compared to all other modules (Table 2).
The module brown is the largest module, which comprised a high proportion of DEGs for S. aureus challenge compared to all modules (Tables 1, 2). It showed an enrichment of DEGs for the IPA Epithelial Adherens Junction, Rho GDI signaling and Actin Cytoskeleton Signaling pathways (Supplementary Table 7). While for the Rho GDI signaling pathway activation was predicted, Actin Cytoskeleton signaling showed a highly negative z-score indicating inactivation. The DAVID analysis added further confirmation that DEGs in this module are involved in immune-related regulation of the actin cytoskeleton (Supplementary Table 9). The KEGG pathway FcγR mediated phagocytosis was significantly enriched with DEGs from this module. In the process of bacterial phagocytosis, FcγR crosslinking is known to induce changes in the actin cytoskeleton. Pathway enrichment analogous to the brown module was not observed in the E. coli challenge associated modules (Table 2).
For the yellow module, enrichment analyses with DEGs from the S. aureus challenge highlighted the ILK signaling, Fcγ Receptor mediated Phagocytosis in Macrophages and Monocytes, and IL8 signaling within the total of 118 enriched IPA pathways (Supplementary Table 7). All three had a negative z-score indicating inactivation of these pathways after S. aureus challenge. The major upstream regulators predicted for the DEGs in this module (TGFB1, IFNG, TNF, Supplementary Table 8) provide a direct link between the indicated pathways and an immune response to the pathogen.
Further modules, which display a clustering of samples by infection status are royalblue and grey60 (Supplementary Image 3). Yet, pathway analysis did not provide meaningful data for these modules due to a low number of annotated DEGs (Tables 1, 2). Nevertheless, particularly the module grey60 merits future further analysis due to a substantial number of genes with highly significant differential expression and high log2 FoldChange. Further attempts for improved functional annotation will be conducted, e.g., within the global initiative Functional Annotation of Animal Genomes (FAANG, www.faang.org).
Pathway Enrichment Analysis of All DEGs After Intramammary S. aureus Challenge
When interpreting the results, it has to be considered that we have monitored the response of the animals in one tissue while the direct pathogen contact took place in another tissue. Thus, regulatory signals might not be directly measured at gene expression level in the liver, e.g., if the regulatory signal is a cytokine produced by blood monocytes. This is different to in vitro challenges, where only those signaling pathways that are active within the respective cell are monitored by expression analysis. Furthermore, we decided to analyze the DEGs across all modules within a pathogen challenge, because the analysis of genes by cluster has the limitation that a gene can only be member of a single cluster, but is involved in multiple pathways. This is demonstrated e.g., by SREB1 and SREB2, which were predicted as upstream regulators of DEGs after E. coli challenge in the module darkred. However, they themselves were assigned to another module (turquoise). Thus, we complemented the cluster-based data analysis with pathway analyses across all DEGs with pathogen challenge, E. coli or S. aureus.
The Ingenuity Pathway Analysis (IPA) revealed a substantial number of 124 significantly enriched canonical pathways in the liver transcriptome in response to intramammary S. aureus challenge (see Supplementary Table 10). Analogous to the modulewise pathway analyses, the main hepatic processes affected by S. aureus challenge are (i) Epithelial Adherens Junctions and Actin Cytoskeleton Signaling, (ii) ILK Signaling and Integrin Signaling, and (iii) RhoGDI Signaling (Table 3).
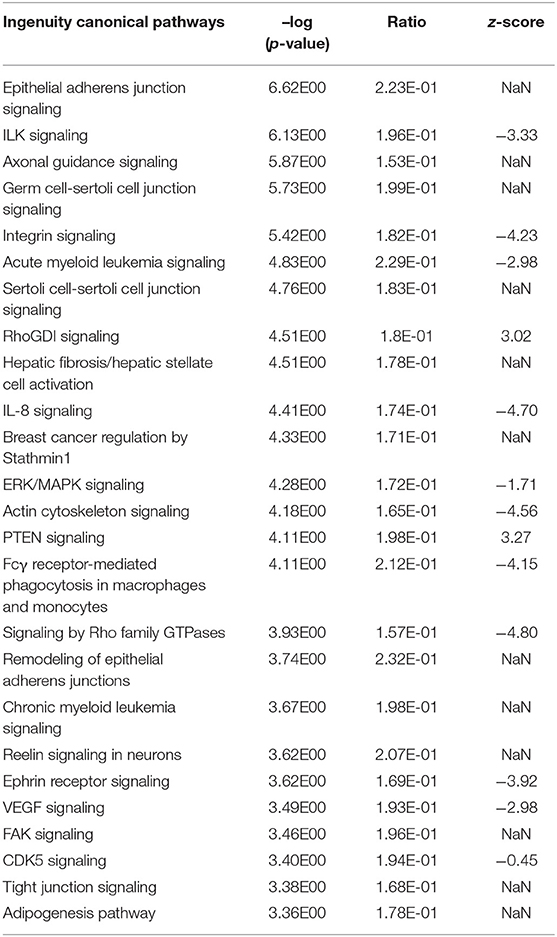
Table 3. Top 25 significantly enriched Ingenuity canonical pathways in the hepatic transcriptome comparing S. aureus challenged to non-challenged cows (NaN, no activation score).
It was striking that numerous significantly enriched pathways were involved in the organization and maintenance of the cytoskeleton and/or intracellular or cell to cell signaling (e.g., Epithelial Adherens Junction Signaling, ILK Signaling, Integrin Signaling, RhoGDI Signaling, ERK/MAPK Signaling, Actin Cytoskeleton Signaling, Signaling by Rho family GTPases, Remodeling of Epithelial Adherens Junctions, FAK Signaling, and Tight Junction Signaling). All pathways with an indicated activation status in the Top 25 list of significantly enriched Ingenuity canonical pathways were inactivated except for RhoGDI Signaling and PTEN Signaling (Table 3).
Epithelial Adherens Junctions and Actin Cytoskeleton Signaling
Analogous to the S. aureus associated module brown, Epithelial Adherens Junction Signaling was the most significantly affected pathway (see Table 3, Supplementary Image 4) in the liver of S. aureus challenged cows, which is associated with the also modified pathways Remodeling of Epithelial Adherens Junctions and Tight Junction Signaling. All but one of the 33 significantly DEGs allocated in this pathway were lower expressed in response to intramammary S. aureus challenge compared to control animals. Epithelial adherens junctions, also known as the cadherin/catenin complex, are the most important regulators of the actin cytoskeleton (46, 47), which alongside the tight junctions, function as principle mediators of cell-cell adhesion. The Actin Cytoskeleton Signaling itself had a negative z-score (−4.56) in our S. aureus challenge study, which reflects its substantial inactivation in infected animals (see Table 3, Figure 5). This pathway plays an important role in dynamic processes such as cell motility, axon guidance, cytokinesis, and phagocytosis. Its inactivation is consistent with the analogous negative z-scores also for ILK Signaling and Integrin Signaling (see below), which we had also observed for the S. aureus associated module yellow.
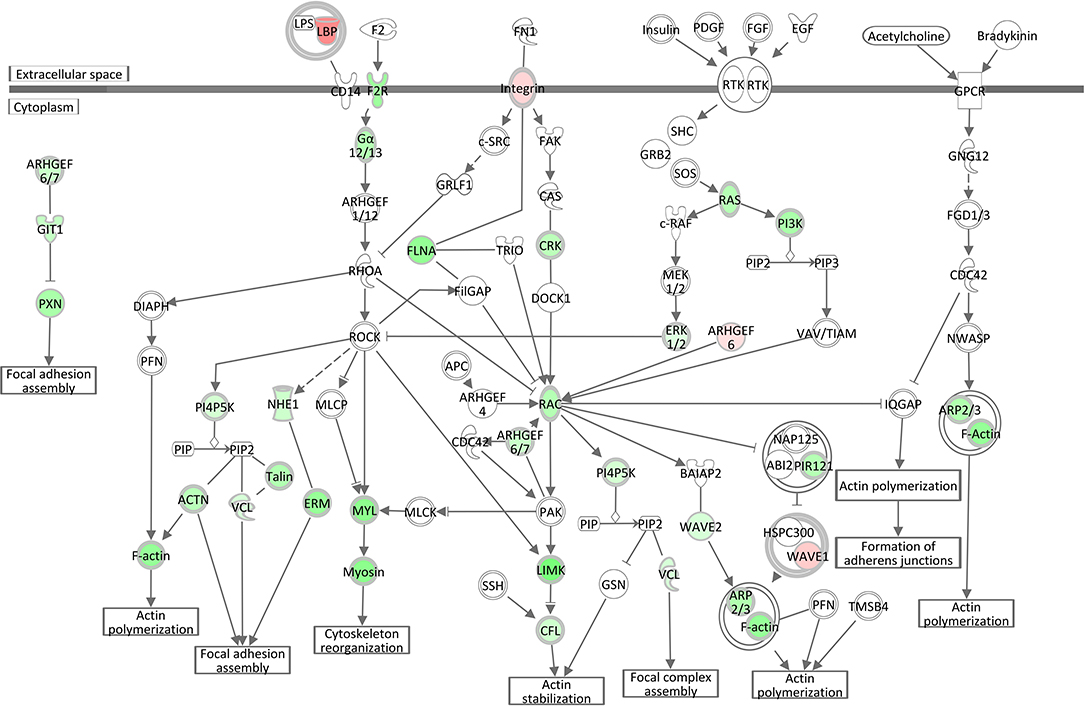
Figure 5. Gene expression in the actin cytoskeleton signaling pathway for the hepatic transcriptome of S. aureus challenged animals compared to the control liver transcriptome: green = lower expression and red = higher expression in S. aureus challenged animals compared to untreated control (adapted from IPA, Qiagen).
ILK Signaling and Integrin Signaling
The negative z-scores (−3.33) for the ILK (integrin-linked kinase) Signaling pathway and for the Integrin Signaling pathway (−4.23) show that both pathways were clearly inactivated in the liver of cows intramammary challenged with S. aureus (Table 3, Figure 6, Supplementary Image 5). The ILK protein is responsible for the conjunctions between integrins and the actin cytoskeleton (48), providing the link between the two pathway complexes. As indicated in Figure 6, ILK signaling mediated by beta integrin is central to a cascade of downstream processes including cell adhesion and cytoskeletal reorganization. Correspondingly to ILK Signaling, the Integrin Signaling pathway (Supplementary Image 5) displays significant enrichment of DEGs. While alpha integrins show a higher expression in the liver of S. aureus challenged cows compared to controls, beta integrin expression displays an opposite direction and is obviously a down-regulating driver for the inactivated ILK and Integrin signaling cascades.
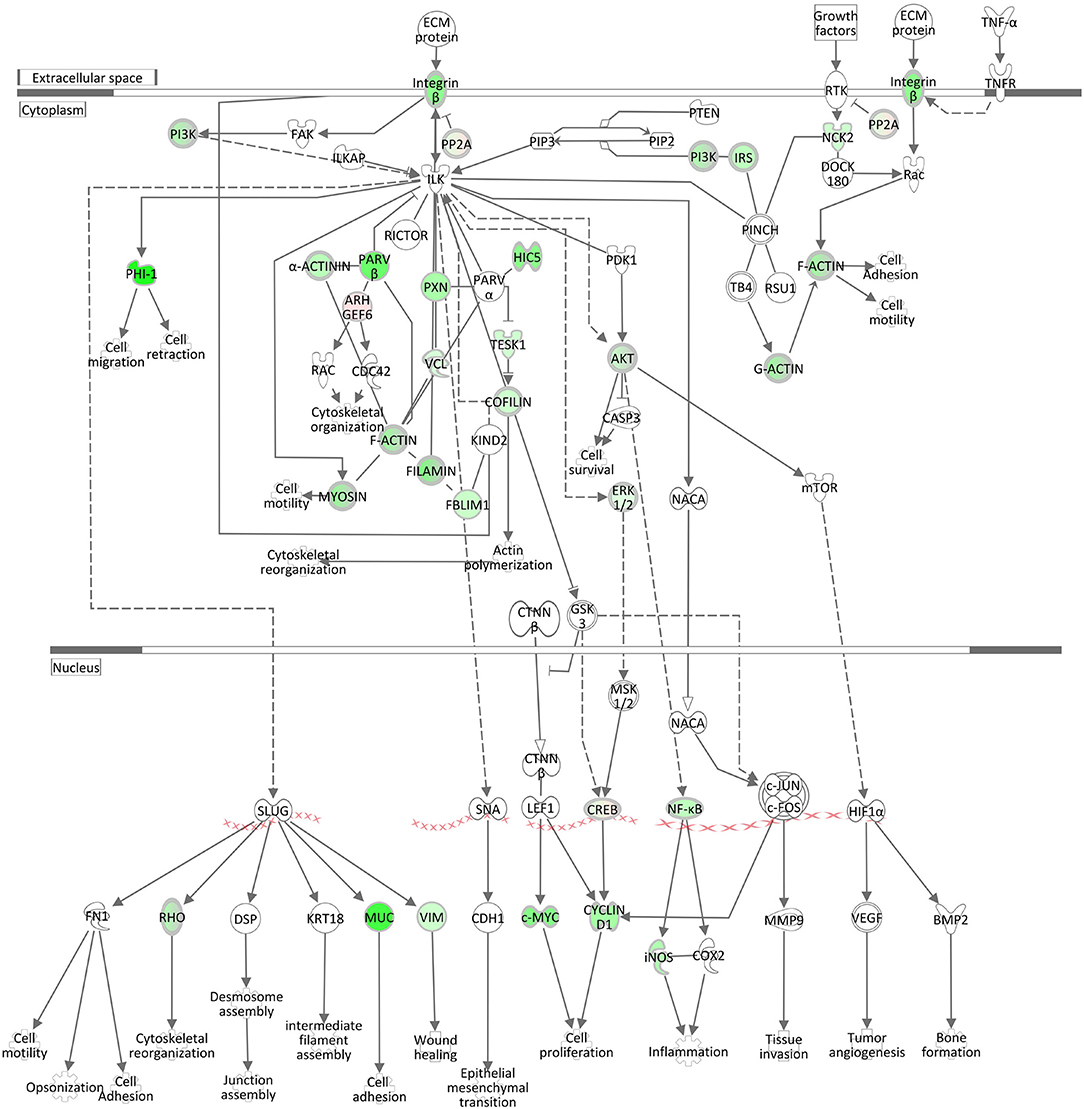
Figure 6. Gene expression of the ILK Signaling pathway in the hepatic transcriptome of S. aureus challenged animals compared to the control liver transcriptome: green = lower expression and red = higher expression in S. aureus challenged animals compared to untreated control (adapted from IPA, Qiagen).
Signaling by Rho Family GTPases and RhoGDI Signaling
Rho family GTPases are known to function as molecular switches that regulate a variety of biological processes, including cell proliferation, apoptosis, differentiation, migration, cytoskeletal reorganization, and membrane trafficking. The pathway Signaling by Rho Family GTPases was assigned a z-score of −4.8, the lowest activation score when comparing the hepatic transcriptome of S. aureus challenged cows to the control group (Table 3, Supplementary Table 10). This pathway was also enriched for DEGs in the S. aureus associated module brown. The low z-score indicates that genes involved in this pathway were considerably downregulated in response to intramammary challenge. Of the total 254 molecules included in this IPA pathway, 40 could be found as significantly differentially expressed in our analysis. Except for three genes, all displayed a downregulated gene expression level in response to S. aureus challenge (see also Supplementary Table 11). As presented in Figure 7, this pathway is associated with a variety of cytoskeletal processes affecting rearrangements of the plasma-membrane-associated actin cytoskeleton, cellular proliferation, and cytokinesis in liver tissue in response to the intramammary S. aureus challenge. This is consistent with the findings regarding the downregulation of the pathways Epithelial Adherens Junction Signaling, ILK Signaling, Integrin Signaling, and Actin Cytoskeleton Signaling (see above) but also with the upregulation of the RhoGDI Signaling pathway, which was assigned a positive z-score of 3.02. The RhoGDI (Rho GDP-dissociation inhibitor) is described as an inhibitor of Rho family GTPases activity by hampering the dissociation of GDP, preventing the subsequent binding of GTP (49). Thus, the activation of this network is plausible in the context of our analysis.
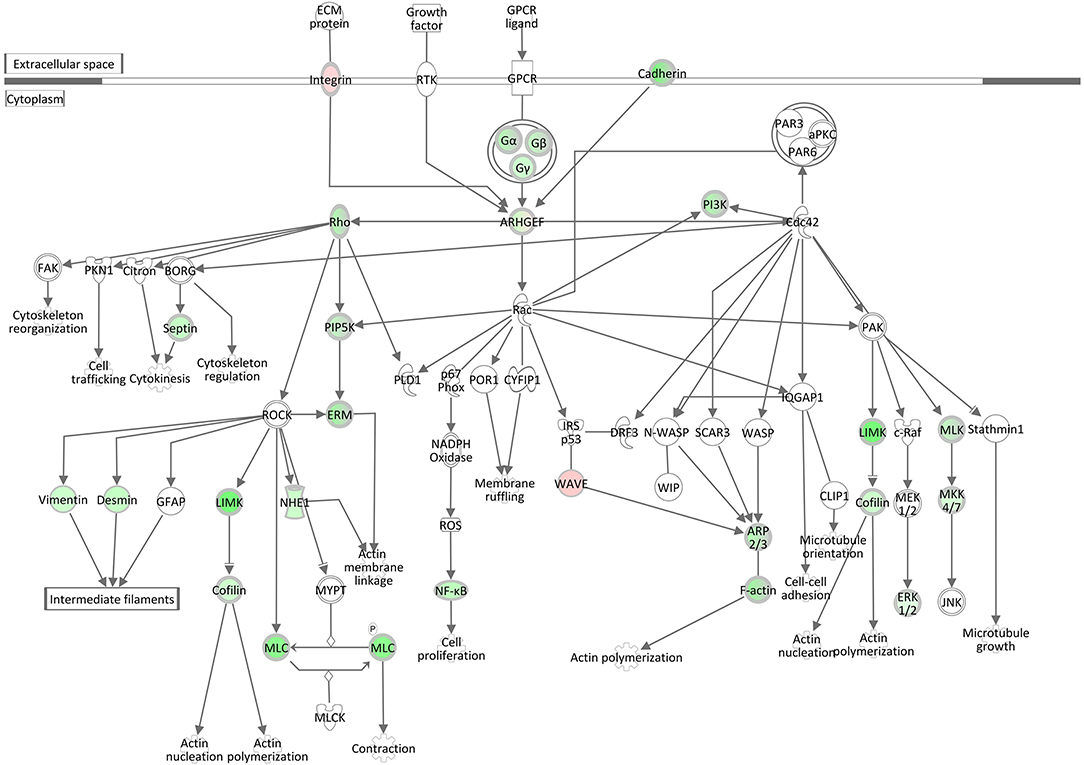
Figure 7. Gene expression of the signaling by rho family GTPases pathway in the hepatic transcriptome of S. aureus challenged animals compared to the control liver transcriptome: green = lower expression in S. aureus challenged animals compared to untreated control; red = higher expression in S. aureus challenged animals compared to untreated control (adapted from IPA, Qiagen).
In summary, the results from the pathway analysis within the S. aureus associated modules as well as across all genes point toward a key role of actin cytoskeleton processes in the liver after intramammary S. aureus challenge.
Analysis of Potential Drivers and Upstream Regulators in the Liver in Response to Intramammary S. aureus Challenge
Pathway analysis had highlighted β integrin at the intersection of the enriched pathways after S. aureus challenge (see above). To obtain information on the further transmission of the initial signals, the potential upstream regulators were monitored, which had been predicted for the DEGs by IPA. In response to S. aureus challenge, miRNAs were the most prominent upstream regulators in the hepatic transcriptome with the highest positive activation z-scores (Supplementary Table 12). Within the top 15 predicted upstream regulators with the lowest (negative) activation z-score is TGFB1. Because TGFB1 is known for its function in regulating immune responses and involvement in mastitis (13), it is a striking regulatory candidate. It was also by far the most significant upstream regulator predicted for DEGs in the S. aureus-associated module yellow. At transcription level, TGFB1 showed a highly significant lower expression in S. aureus infected cows compared to control cows (Supplementary Table 1), which supports its predicted regulatory function.
Pathway Enrichment Analysis of All DEGs After Intramammary E. coli Challenge
Ingenuity pathway analysis across all DEGs in the E. coli challenge revealed a slightly higher number (148) of significantly enriched canonical pathways in the liver transcriptome (Supplementary Table 10) compared to S. aureus challenge (see above). However, the main pathways identified as enriched after E. coli intramammary challenge differed from those obtained from the analysis of S. aureus challenged animals. This is in line with the differences detected in the modulewise data analysis. Key hepatic processes affected by E. coli challenge and attributed the highest activation scores, are (i) RXR Activation (and associated pathways) together with lipid metabolism (Adipogenesis, Fatty Acid Beta Oxidation), and (ii) Acute Phase Response and the Complement System (Table 4, Supplementary Table 10).
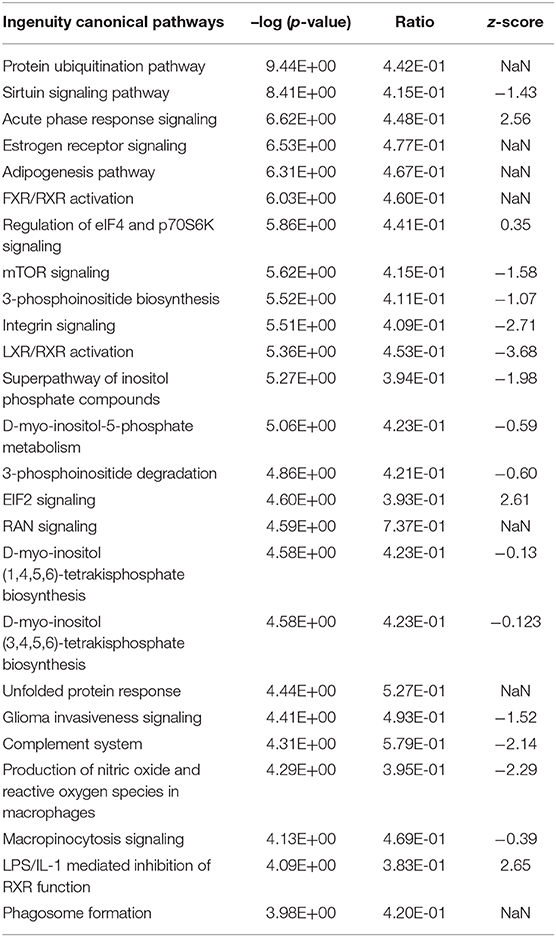
Table 4. Top 25 significantly enriched Ingenuity canonical pathways in the hepatic transcriptome comparing E. coli challenged to non-challenged cows (NaN, no activation score).
RXR Activation and Fat Metabolism
Modulewise analysis had indicated a major effect of E. coli challenge on lipid metabolism demonstrated by the most significantly enriched pathways for DEGs in the dominating module turquoise. When including all DEGs in the E. coli challenge in the enrichment analysis, the LXR/RXR Activation pathway was significantly enriched and had one of the lowest negative z-scores (z-score = −3.68), indicating a substantial downregulation of this pathway in E. coli challenged vs. control animals. This is in agreement with the positive activation score for the pathway LPS/IL-1 mediated Inhibition of RXR Function (z-score = 2.65). As presented in Supplementary Image 6, a downregulation of the key receptor RXRα gene expression has far-reaching consequences for genes involved in lipid and cholesterol metabolism, bile acid homeostasis, and organic anion transport. Coordinated interplay of the nuclear receptors LXR and RXR is known for sensitive fine-tuning of the balance and maintenance of the body fat and cholesterol homeostasis (50). This fits the observation of an enrichment of the pathways Adipogenesis and Fatty Acid beta Oxidation in response to E. coli intramammary challenge also detected for the E. coli challenge associated module turquoise. Furthermore, due to the effects on the pathway LPS/IL-1 mediated Inhibition of RXR Function, a link is also provided for an immune response to the intramammary pathogen challenge also seen in the turquoise module.
Decreased Expression of Key Genes in Bovine Energy Metabolism
In addition to canonical pathways associated with fat metabolism, we also observed a significantly differential expression of particular genes involved in biochemical pathways associated with energy metabolism (Supplementary Table 13, Supplementary Images 7, 8). This conclusive, coordinated accumulation of DEGs had not been detected by IPA analysis, but it resulted in highly significantly enriched KEGG pathways Citrate Cycle (padj < 0.40E-16) and Glycolysis/Gluconeogenesis (p < 0.5E-19). All genes encoding key enzymes in glycolysis/gluconeogenesis showed a lower transcription in the cows with intramammary E. coli challenge compared to controls (e.g., PCK1, PCK2, ALDOB, FBP1, GPI, HK1, G6PC, see Supplementary Image 7). In addition, important genes involved in mitochondrial energy metabolism (e.g., PCCA, PCCB, CPT1A, CPT1B, PDK1, PDK2, PDK4) and particularly in the citrate cycle (e.g., IDH1, ACO1, SDHA, SDHB, FH, see Supplementary Image 8) were significantly differentially expressed.
Acute Phase Response Signaling and Complement System
Acute Phase Response Signaling, an essential component within the innate immune responses, was within the top significantly enriched Ingenuity canonical pathways and also had one of the highest activation scores in the hepatic transcriptome comparing E. coli challenged to non-challenged cows (z-score = 2.56). Figure 8 displays the very coordinated activation of this pathway in response to intramammary E. coli challenge with an increased expression of key cytokine receptor genes (TNFR, IL1R1, OSMR, GP130).
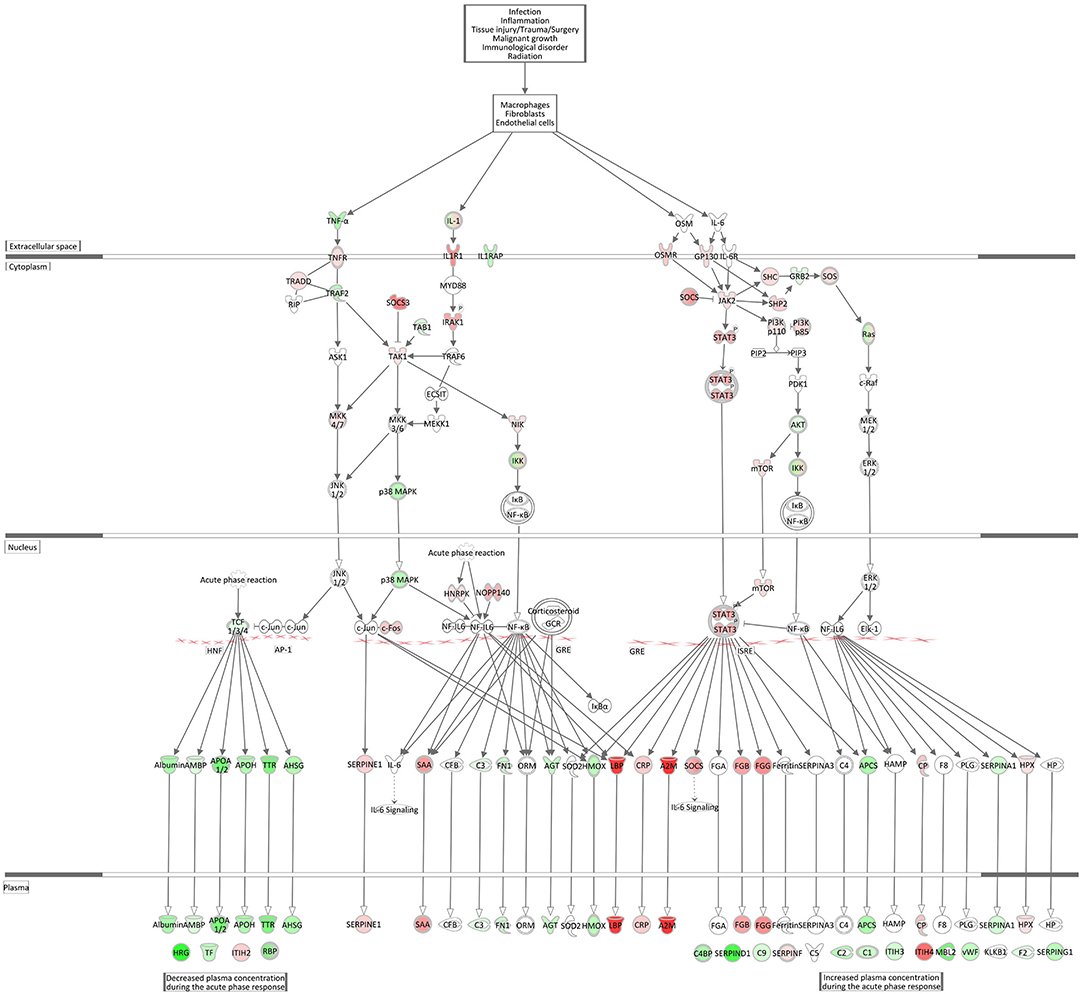
Figure 8. Gene expression of the acute phase response signaling in the E. coli challenged liver transcriptome compared to control hepatic transcriptome: green = lower expression and red = higher expression in E. coli challenged animals compared to untreated control (adapted from IPA, Qiagen).
The Complement System pathway, which is as well an integral part of the innate immune system acting as bridge between innate and acquired immunity, was also highly significantly enriched with significantly DEGs after E. coli challenge. However, surprisingly and in contrast to the acute phase response pathway it displayed a low negative z-score indicating its strong and coordinated inactivation in E. coli challenged animals (z-score = −2.14). As shown in Figure 9, the vast majority of complement factors were significantly transcriptionally downregulated in the livers of E. coli challenged cows compared to the control, while in contrast some inhibitors of the complement system (C1QBP, DAF/CD55, MCP/CD46) were correspondingly upregulated in the challenged animals.
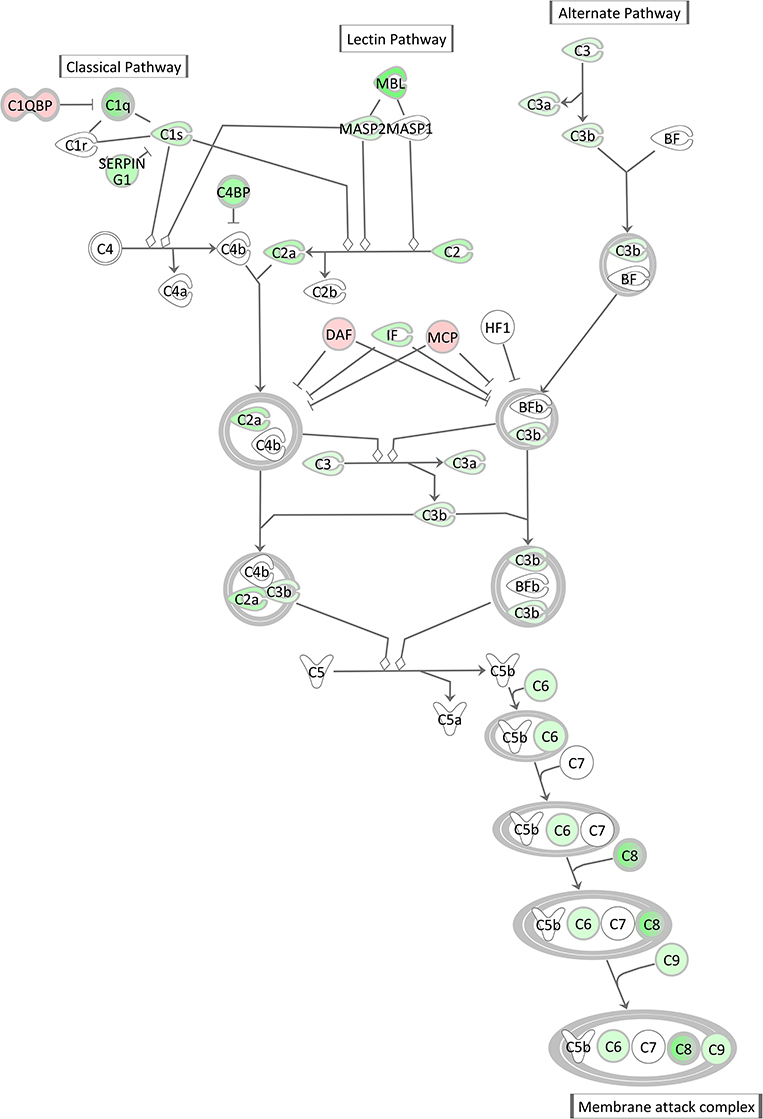
Figure 9. Gene expression of the complement system in the hepatic transcriptome of E. coli challenged animals compared to the control liver transcriptome: green = lower expression and red = higher expression in E. coli challenged animals compared to untreated control (adapted from IPA, Qiagen).
Analysis of Potential Upstream Regulators in the Liver in Response to Intramammary E. coli Challenge
The IPA analysis predicted a large number of upstream regulators in response to intramammary E. coli challenge (see Supplementary Table 12). Most prominently, HNF4, known as a major transcription factor in hepatocytes, stood out with extremely high significance (p < 8.06E-83) and a high negative activation score. Indication on inactivation was obtained also for other known regulators of liver (lipid) metabolism (SREBF2, SREBF1, PPARG). This has already been demonstrated by the results from the module turquoise. This module comprised the largest number of genes and showed a strong correlation of module membership of genes and gene-infection association (see Figure 4). For the DEGs in this module, the enrichment analysis showed a strong association to amino acid and fatty acid metabolism and identified HNF4A as upstream regulator with a strongly predicted inactivation. The predicted inactivation of those factors was confirmed by a significantly differential expression of the respective genes themselves. Analogously, the positive activation score of XBP1 and ERN1, which were on top of the list of genes with a predicted positive activation score, were also supported by a higher expression in the E. coli challenged animals compared to the controls. The XBP1 protein is a transcription factor that regulates the expression of genes important to the proper functioning of the immune system and in the cellular stress response (51). ERN1 (IRE1a) functions as a sensor of unfolded proteins in the endoplasmic reticulum and triggers an intracellular signaling pathway termed the unfolded protein response.
Discussion
In summary, the results from the module-based expression analyses are very congruent to those obtained across the entire dataset. Our intramammary gland challenge experiment with two divergent mastitis pathogens revealed three key findings:
(i) Although S. aureus mastitis is commonly categorized as a local inflammation, we demonstrated that the liver is substantially affected via challenge of the udder displaying in part similar, in part opposite signatures of infection as mammary gland epithelial cells.
(ii) S. aureus and E. coli dictate a pathogen-specific signature in the hepatic transcriptome after intramammary challenge.
(iii) Both pathogens initiate activating and deactivating effects on the innate immune response of the liver after intramammary challenge.
Intramammary S. aureus-Induced Challenge Triggers a Massive Hepatic Response
The liver is known to be central for both, metabolism and (innate) immune responses. Mammary gland challenge and infection by S. aureus is commonly considered a local, non-generalized process. Previous S. aureus challenge experiments with an analogous model had indicated no or only a moderate systemic host response (52) and no data were provided regarding liver involvement in this process. However, the fact that during the course of infection, the animals displayed an increase of body temperature with an average peak of >40.5°C (22) demonstrates that a clinical response to the intramammary S. aureus challenge had occurred. This is also documented by a massive change in liver function observed in the hepatic gene expression analysis. In our study, the biological pathway analysis of the hepatic transcriptome response to S. aureus intramammary challenge revealed that numerous significantly enriched pathways were affected such as the actin cytoskeleton and cell signaling (e.g., Epithelial Adherens Junction Signaling, ILK Signaling, Integrin Signaling, RhoGDI Signaling, ERK/MAPK Signaling, Actin Cytoskeleton Signaling, Signaling by Rho family GTPases, Remodeling of Epithelial Adherens Junctions, see Table 3, Supplementary Table 7). This indicates that multiple hepatic functions are affected by intramammary S. aureus challenge including lipid metabolism, innate immune response, citrate cycle, and gluconeogenesis. Although the pathogen challenge took place in the mammary gland, these crucial response effects to the invading pathogen were noted in the peripheral liver tissue.
Our study provides evidence that intramammary S. aureus challenge negatively affected hepatic pathways directly involved in combating the invading pathogen. This is already visible from the volcano plot of DEGs (Figure 3). The plot demonstrated a dominance of those particularly strongly DEGs (log2 Fold Change) that are lower expressed in pathogen challenged than in control animals compared to genes that are upregulated. For example, the Fcγ Receptor-Mediated Phagocytosis in Macrophages and Monocytes pathway (CD32) as well as the important cytokine/chemokine pathway IL8 Signaling (Table 3) were downregulated due to S. aureus challenge. The activation of Fcγ receptors favors the antibody-mediated phagocytosis of pathogens by macrophages or neutrophilic granulocytes, which is an essential part of the immune defense (53, 54). The downregulation of these hepatic processes suggest that such antibacterial mechanisms are attenuated in response to intramammary S. aureus challenge.
Furthermore, the pathway analysis revealed that Integrin Signaling had one of the lowest negative activation scores of all significantly enriched pathways, implying that this pathway was very substantially downregulated in hepatic tissue due to intramammary S. aureus challenge. Interestingly, β integrin genes showed a lower expression in the liver of S. aureus challenged cows compared to controls, in contrast to alpha integrin genes. In line with Integrin Signaling being downregulated in response to S. aureus challenge we also observed a downregulation of closely associated biological pathways, such as ILK Signaling and Actin Cytoskeleton signaling. Since ILK is at the center of cell-matrix adhesion and interacts with the cytoplasmic tail of beta integrins and couples it with the actin cytoskeleton, ILK signaling appears to be a central link between the affected pathways for innate immune responses against pathogenic invasion.
A further partner integrated into the biological and molecular crosstalk in response to intramammary S. aureus challenge is the pathway of Epithelial Adherens Junctions, which are crucial regulators of the actin cytoskeleton to maintain tissue homeostasis in critical cell processes and challenges that include tissue barrier function.
The central S. aureus-induced hepatic host response appears to be elicited by Actin Cytoskeleton signaling. The actin cytoskeleton is a dynamic structure necessary for cell and tissue organization, also comprising the epithelial barriers; thus, dynamic remodeling of the actin cytoskeleton is included as an essential component of a variety of cellular processes. For successful infection and replication, many pathogens are known to hijack the cytoskeleton using effector proteins introduced into the host cytosol by specialized secretion systems (55). This is associated with processes involving actin and actin-binding proteins in spatiotemporal actin polymerization/depolymerization processes. The rearrangement of the cytoskeleton and adhesion complexes linked to cell migration appears to be a major host structural component that is obviously manipulated by extracellular pathogens to defend against the infection. Interestingly, enhanced rather than downregulated actin signaling and actin-cytoskeleton rearrangement was recently described to occur after short term (3 h) mammary infection with S. aureus and was suggested to be involved in internalization of S. aureus 1027 into mammary epithelial cells (56).
S. aureus and E. coli Elicit a Pathogen-Specific Signature in the Hepatic Transcriptome After Intramammary Challenge
The pathogen-specific signatures were initially demonstrated by modules of genes associated predominantly to a specific pathogen challenge. Subsequent pathway analyses revealed that particular biological processes are affected depending on the pathogen type, S. aureus or E. coli. Intramammary S. aureus challenge displayed that specific pathways predominantly connected to the cellular actin cytoskeleton were compromised in the liver transcriptome, e.g., ILK Signaling or Actin Cytoskeleton Signaling. The RhoGDI signaling, signaling by Rho family GTPases and ERK/MAPK signaling also fit into this cellular and molecular crosstalk in response to intramammary S. aureus challenge. Both, Rho family GTPases (including Rac, CDC42, and Rho) and classical extracellular activated kinase ERK1/2 MAP kinases have been identified as key integrators triggering cytoskeleton rearrangements. Signals from chemokine receptors and integrin molecules that coordinate these processes are transmitted to Rho family GTPases and passed on to cytoskeletal target proteins, which leads to the initiation of numerous processes including cell migration, morphogenesis, cytokinesis, and endo/exocytosis (57). Examples of DEGs in our study from these pathways with effects on the actin cytoskeleton are MYL9, PIEZO1, FLNA, RALBP1, RIN3, MYOC1C, and CDH5 (58–64). All these genes were significantly lower expressed in the livers of S. aureus challenged animals. Simultaneously, genes included in RhoGDI signaling, the inhibitor of Rho family GTPases and binding partner that can tightly control Rho GTPases, are substantially higher expressed in S. aureus challenged compared to control cows. This again supports our hypothesis that there is a central effect of intramammary S. aureus infection on the hepatocyte actin cytoskeleton.
Remarkably, this pathway was significantly enriched only in the liver transcriptome in response to intramammary S. aureus challenge, but not after the E. coli challenge, which is in line with previous data in the mammary gland (56). This provides evidence that this pathway is specifically modulated during S. aureus challenge. It has been known that several S. aureus virulence factors can affect Rho GTPase activity in humans (65–69). Some of these factors such as SCIN, CHIP, and EDIN A-C are human specific and not encoded by the bovine isolate S. aureus 1027 used in this study (10). For others like the superantigen like proteins Ssl5, Ssl7, and Ssl10, the respective coding sequences have been detected in the genome of S. aureus 1027 (10). Both Ssl5 and Ssl10 interfere with Rho GTPase activity by binding G protein-coupled receptors (GPCRs) (70, 71). Besides, Ssl5 increased binding of chemokines to cells independent of chemokine receptors through their common glycosaminoglycan-binding site (70). For Ssl7 it has been shown that it targets C5 and thereby inhibits complement-mediated cell lysis possibly by blocking C5b-9 formation (72). The precise role of these proteins during interaction of S. aureus with the bovine host and in development of bovine mastitis, however, has still to be clarified.
While for S. aureus, the particular pathogen component responsible for the immune response is yet unclear, this is different for E. coli. For E. coli, LPS is known to be the key pathogen associated molecular pattern that is predominantly responsible for the inflammatory response to pathogen challenge (73). This is also confirmed by LPS being one of the most significantly predicted upstream regulators of DEGs in the module turquoise associated with E. coli challenge. Initiated by LPS, an entire cascade of LXR/RXR mediated signaling seems to be elicited (Supplementary Image 6).
Intramammary E. coli challenge also triggered a significant hepatic transcriptome response, but this was different to challenge by S. aureus. The function of LXR/RXR was significantly downregulated in challenged animals. This adaptation of the hepatic transcriptome seems to be pathogen-specific for the E. coli challenge. Retinoid X receptors (RXRs) are class II nuclear receptors; this subgroup includes inter alia liver X receptors (LXRs) (74). While LXRβ is expressed ubiquitously, LXRα is liver associated (75). LXRs are also activated by altered forms of cholesterol (76) and are fundamental for the transcriptional control of lipid metabolism in the liver (74, 76). The module darkred harboring DEGs enriched for the Cholesterol Biosynthesis pathway provided data supporting effects on cholesterol synthesis in the liver by intramammary E. coli challenge.
In recent years, several reports (74–76) shed light on the interactions between RXRs and the immune system, and especially on the acute phase response to infection (APR) (74). The APR is the first line of defense of the innate immune system induced by e.g., infections, injuries, or neoplasia (77) and is one of the most prominent pathways being upregulated in the liver after intramammary lipopolysaccharide (LPS) challenge (11, 18). Thus, the activated APR signaling observed in our hepatic transcriptome analysis in response to E. coli challenge is in accordance with data from the literature. Moreover, FGG, FGB, and A2M genes involved in APR signaling, were significantly upregulated in our analysis of DEGs.
In addition to its immunological effects, the APR has negative metabolic consequences: In lipid metabolism the APR leads to increased lipolysis, decreased fatty acid oxidation, and inhibition of bile acid synthesis (78). The RXRs seem to be a link between immunological and metabolic adaptations after infection. E. coli LPS as well as proinflammatory cytokines TNFalpha and IL1beta decreased the abundance of RXR proteins in the liver of hamsters (74). In a bovine cell model, Wang et al. (75) found that a synthetic LXR agonist decreased the synthesis of inflammatory cytokines in bovine mammary epithelial cells (bMECs) after challenge with E. coli LPS. Moreover, using transcriptome analysis of liver and mammary gland tissue samples following intramammary E. coli challenge, Moyes et al. (11) found that an increase in the expression of APR genes was accompanied by a decrease of key metabolic enzymes, including those of the lipid metabolism. Analogous data were obtained in our study indicating impaired gluconeogenesis and mitochondrial energy metabolism (Supplementary Table 13, Supplementary Images 7, 8), which are important metabolic processes in the lactating dairy cow. Respective differences in gene expression for S. aureus challenged cows were not observed: neither e.g., PCK1, HK1, IDH1, nor CPTA displayed significant differences between challenged and non-challenged cows.
These studies in the literature and our results from the hepatic transcriptome analysis consistently show that particularly E. coli mastitis is not only to be considered at the immunological level, but also in the context of metabolic changes. This is especially essential in case of severe clinical E. coli mastitis. Particularly the early lactation is a sensitive time period for mastitis, a time in which the liver is eventually confronted with the double burden of (bacterial) infection and increased metabolic demands for milk production. The aspect of an increased metabolic challenge of the animal due to pathogen-specific adaptions of the hepatic transcriptome might be a new approach for the treatment and prevention of the disease via direct interventions not only directly at the mammary gland, but also at systemic level.
Both Pathogens Initiate Activating and Deactivating Effects on the Innate Immune Response in the Liver After Intramammary Challenge
There was a remarkable effect of E. coli challenge on the hepatic expression of the complement system genes. This comprised a lower expression of direct components of the complement system and a higher expression of genes encoding inhibitors of the complement system (C1QBP, DAF/CD55, MCP/CD46) in the challenged animals. Similar to the APR, the complement system is a powerful weapon of the innate immune response to fight against invading pathogens (79). However, our transcriptional analysis of livers after intramammary E. coli challenge revealed that the complement system [particularly its C1q (e.g., antigen-specific) governed branch] is incapable to react accordingly to the pathogen challenge while the APR seems to be activated, although with some balancing processes.
Günther et al. (80) challenged four first lactation Holstein cows with E. coli 1303 (the same strain as used in our experiment) and analyzed mRNA from udder tissue on a microarray platform. They also found a significant transcriptional downregulation of some components of the complement system, especially the first component of the classical pathway (C1 complex) and components in the membrane attack complex (MAC). Interestingly, Günther et al. also found complement inhibitors, such as DAF (decay-accelerating factor 1) and C1QBP (complement C1q-binding protein), to be increased in infected mammary gland tissue. Our data indicate that this effect is also reflected in the liver, the main tissue synthesizing complement system components. Based on these data, we suggest that while intramammary E. coli challenge triggers transcriptional processes of the APR in the liver, it also induces in parallel an active shut-down of the complement system already at transcript level.
The E. coli 1303 strain used in our study was studied by Leimbach et al. (81, 82), who reported that this strain does not carry many known virulence factors, which are thought to be advantageous to a mastitis pathogen. However, Abreu and Barbosa (79) reviewed various mechanisms and strategies of human pathogenic E. coli strains that might be used to either passively or even actively inhibit and overcome the damaging attacks of the complement system. The shutdown of the complement system as observed in our study drastically weakens the innate immune response and partially disarms the infected organism. This might be an important aspect of the pathogenesis of E. coli induced mastitis and might be responsible for the severe course of the disease. Alternatively, the shutdown of the complement system could represent an, albeit dangerous, regulatory mechanism to counterbalance the massive activation of the APR and its subsequent consequences.
The balanced activation of the immune system in the liver is also well reflected by alterations at transcriptional level of genes involved in different parts of the APR. While TNFalpha and IL1beta showed a lower expression in the hepatic transcriptome of intramammary E. coli challenged cows compared to control cows, the respective receptor genes (TNFR and IL1R1) displayed an inverse ratio of expression levels. In addition, while obviously major acute phase protein genes (AM2, LBP) were upregulated via STAT3/STAT3A in the course of challenge, TCF1/3/4 was significantly lower expressed with correspondingly lower expression of genes encoding albumin (ALB), transferrin (TF), or histidine rich protein (HRG), a peptide with antimicrobial activity against E. coli (83).
In our pathway enrichment analysis, TGFB1 has been prioritized as potential regulator serving for the transmission of initial signals of e.g., β integrin and FcγR to the hepatic transcriptome response to intramammary S. aureus challenge. This suggests that TGFB1 could be a potential signaling driver of alterations in the hepatic transcriptome of pathogen challenged cows. Cross talk between TGFB1 and integrins is well described (84). TGFB1 is the most important factor among the numerous cytokines and growth factors with various effects on immune cells (85). It prevents the immune system from attacking the body's own cells (85) by suppression of the immune system. The TGFB1 gene expression has been reported to be involved in regulating immune response associated with experimentally induced S. aureus mastitis (13). In our study, its strong candidacy as key regulator of the immunomodulatory response to intramammary S. aureus challenge in the liver transcriptome is supported by its highly significant lower hepatic gene expression level in S. aureus infected cows compared to controls and by its highly significant predicted role as an inactivated upstream regulator.
In the hepatic transcriptome of intramammary E. coli infected cows, TGFB1 expression levels were also significantly downregulated compared to the untreated controls. The inactivation of this immunosuppressor is also in line with the canonical pathway analysis of E. coli infected vs. controls, where the TGF beta Signaling pathway had a high positive z-score (3.02), indicating a considerable activation of this pathway in the livers of E. coli infected animals. Thus, the lower hepatic immunoinhibitor gene expression in response to S. aureus or E. coli intramammary challenge has initiated an activation of the immune response via upregulation of expression levels of target genes acting downstream in this pathway (e.g., SMAD1, SMA5). This supports a well and tightly orchestrated regulation of the immune response beyond the local site of bacterial infection.
Our experiment has proved that the liver is affected from an intramammary pathogen challenge with both S. aureus and E. coli, respectively. We found that both pathogens can attack the immune system in an important metabolic organ that is located at a considerable distance from the site of the animal's direct pathogen contact. Thus, only antibiotic therapy of cows suffering from mastitis might not be sufficient. Immunostimulant drugs could favor the course of the disease but would have to be used in a pathogen-specific manner. Especially for E. coli mastitis, the influences of the infection on the liver metabolism should not be neglected. Particularly in the early lactation, the cow's energy demand for milk production can hardly be covered by the energy consumed with the feed (12). An infection, which also leads to adaptations of important metabolic pathways in the liver (lipid metabolism, glycolysis/gluconeogenesis, mitochondrial energy metabolism), further increases this energy deficit. Therefore, energy supplementation (e.g., glucose) could be beneficial for cows suffering from E. coli mastitis.
Potential Impact of Early Lactation Environmental Effects
Differences in the environment between the infection and control groups could have introduced a potential bias possibly impacting the interpretation of our study. Thus, we compared initially the metabolic status of the challenge groups and the control group prior to the intramammary pathogen challenge, which might have resulted in environmentally, but not challenge-induced hepatic differences between the groups. However, neither blood serum NEFA nor BHB provided indication for a significantly different metabolic status of the different groups. Furthermore, an overlay of the results from the differential expression analysis added further proof that the vast majority of expression differences were due to pathogen challenge. To this end, we also compared the Top 25 significantly enriched canonical pathways for two separate input gene lists in E. coli and S. aureus challenge (Supplementary Tables 14, 15, Supplementary Image 9): List 1 comprises genes differentially expressed when comparing the E. coli group vs. control but not differentially expressed in S. aureus vs. control and list 2 vice versa. If environment indeed had been the main driver of the observed hepatic transcriptome effects, few and similar pathways should have resulted from the two lists. However, the comparison of the pathways enriched in the analysis of E. coli or S. aureus challenged animals, respectively, vs. control showed distinct differences in affected canonical pathways or their activation status (Supplementary Image 9). In addition to our cluster-based module analysis, this provides further evidence that indeed pathogen response and not e.g., divergent environmentally induced metabolic status had driven the transcriptomic differences between pathogen challenge group and control.
Conclusions
Both, S. aureus and E. coli elicited systemic effects on the host after intramammary challenge and seem to use pathogen-specific targeting strategies to bypass the innate immune system: While S. aureus inhibits the cell signaling via integrin, FcγR and Rho GTPases in the liver, E. coli switches the complement system off. Also metabolic hepatic pathways (e.g., lipid metabolism or gluconeogenesis and citrate cycle) are affected after mammary gland challenge, demonstrating that the liver reduces metabolic tasks in favor of the predominant immune response after infection. Therefore, a revised, pathogen-specific treatment regime for mastitis, which goes beyond mere antibiotic therapy, might be beneficial for the course of the infection.
The most striking result of our study is that we demonstrate for the first time that S. aureus udder challenge causes an immune response beyond the original local site of the mastitis. We found that in the peripheral liver tissue defined biological pathways are switched on/off in a coordinated manner to balance the immune response in the entire organism. TGFB1 signaling plays a crucial role in this context.
Data Availability Statement
RNA-Seq datasets are submitted to the ENA repository (https://www.ebi.ac.uk/ena, Project number PRJEB33849, accession numbers ERR3466640 - ERR3466680) at EMBL-EBI.
Ethics Statement
The animal study was reviewed and approved by the FBN cohort, the experiment was conducted under the reference number 7221.3-1-055/15 with the approval by the responsible authority (LALLF, Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern, Rostock, Germany). For the TiHo cohort, the experiment was performed under the reference number 33.12-42502-04-15/2024 by the Lower Saxony Federal State Office for Consumer Protection and Food Safety. This study was submitted to and approved by the ethics committees of the Leibniz Institute for Farm Animal Biology and the University of Veterinary Medicine Hanover, respectively. All ethical evaluations were performed as required by the German Animal Care law and associated legislative regulations (23).
Author Contributions
RW, H-MS, WP, HZ, MH, H-JS, MS, SE, and CK designed research. AH, JB, MM, and LR performed research. AH, RW, DB, and CK analyzed data and wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by Funds of the German Government's Special Purpose Fund held at Landwirtschaftliche Rentenbank. The funding body had no role in the design of the study, collection, analysis, and interpretation of data nor in writing the manuscript. Furthermore, the funders had no role in the decision to publish the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the laboratory staff at the FBN Dummerstorf as well as the Ph.D. students of the ChronMast project at the TiHo Hanover. We also thank the Förderverein Bioökonomieforschung for continuous support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00715/full#supplementary-material
References
1. Halasa T, Huijps K, Osteras O, Hogeveen H. Economic effects of bovine mastitis and mastitis management: a review. Vet Q. (2007) 29:18–31. doi: 10.1080/01652176.2007.9695224
2. Roesch M, Perreten V, Doherr MG, Schaeren W, Schällibaum M, Blum JW. Comparison of antibiotic resistance of udder pathogens in dairy cows kept on organic and on conventional farms. J Dairy Sci. (2006) 89:989–97. doi: 10.3168/jds.S0022-0302(06)72164-6
3. Shea KM. Antibiotic resistance: what is the impact of agricultural uses of antibiotics on children's health? Pediatrics. (2003) 112:253–8.
4. De Vliegher S, Fox LK, Piepers S, McDougall S, Barkema HW. Invited review: mastitis in dairy heifers: nature of the disease, potential impact, prevention, and control. J Dairy Sci. (2012) 95:1025–40. doi: 10.3168/jds.2010-4074
5. Martin P, Barkema HW, Brito LF, Narayana SG, Miglior F. Symposium review: novel strategies to genetically improve mastitis resistance in dairy cattle. J Dairy Sci. (2018) 101:2724–36. doi: 10.3168/jds.2017-13554
6. Keane OM. Symposium review: intramammary infections-major pathogens and strain-associated complexity. J Dairy Sci. (2019) 102:4713–26. doi: 10.3168/jds.2018-15326
7. Burvenich C, Van Merris V, Mehrzad J, Diez-Fraile A, Duchateau L. Severity of E-coli mastitis is mainly determined by cow factors. Vet Res. (2003) 34:521–64. doi: 10.1051/vetres:2003023
8. Vedrine M, Berthault C, Leroux C, Reperant-Ferter M, Gitton C, Barbey S, et al. Sensing of Escherichia coli and LPS by mammary epithelial cells is modulated by O-antigen chain and CD14. PLoS ONE. (2018) 13:e0202664. doi: 10.1371/journal.pone.0202664
9. Schwarz JM, Bilbo SD. LPS elicits a much larger and broader inflammatory response than Escherichia coli infection within the hippocampus of neonatal rats. Neurosci Lett. (2011) 497:110–5. doi: 10.1016/j.neulet.2011.04.042
10. Wolf C, Kusch H, Monecke S, Albrecht D, Holtfreter S, von Eiff C, et al. Genomic and proteomic characterization of Staphylococcus aureus mastitis isolates of bovine origin. Proteomics. (2011) 11:2491–502. doi: 10.1002/pmic.201000698
11. Moyes KM, Sørensen P, Bionaz M. The impact of intramammary Escherichia coli challenge on liver and mammary transcriptome and cross-talk in dairy cows during early lactation using RNAseq. PLoS ONE. (2016) 11:e0157480. doi: 10.1371/journal.pone.0157480
12. Weber C, Hametner C, Tuchscherer A, Losand B, Kanitz E, Otten W, et al. Variation in fat mobilization during early lactation differently affects feed intake, body condition, and lipid and glucose metabolism in high-yielding dairy cows. J Dairy Sci. (2013) 96:165–80. doi: 10.3168/jds.2012-5574
13. Jensen K, Gunther J, Talbot R, Petzl W, Zerbe H, Schuberth HJ, et al. Escherichia coli-and Staphylococcus aureus-induced mastitis differentially modulate transcriptional responses in neighbouring uninfected bovine mammary gland quarters. BMC Genomics. (2013) 14:36. doi: 10.1186/1471-2164-14-36
14. Schukken YH, Gunther J, Fitzpatrick J, Fontaine MC, Goetze L, Holst O, et al. Host-response patterns of intramammary infections in dairy cows. Vet Immunol Immunopathol. (2011) 144:270–89. doi: 10.1016/j.vetimm.2011.08.022
15. Song M, He Y, Zhou H, Zhang Y, Li X, Yu Y. Combined analysis of DNA methylome and transcriptome reveal novel candidate genes with susceptibility to bovine Staphylococcus aureus subclinical mastitis. Sci Rep. (2016) 6:29390. doi: 10.1038/srep29390
16. Mitterhuemer S, Petzl W, Krebs S, Mehne D, Klanner A, Wolf E. Escherichia coli infection induces distinct local and systemic transcriptome responses in the mammary gland. BMC Genomics. (2010) 11:138. doi: 10.1186/1471-2164-11-138
17. Chang G, Petzl W, Vanselow J, Gunther J, Shen X, Seyfert HM. Epigenetic mechanisms contribute to enhanced expression of immune response genes in the liver of cows after experimentally induced Escherichia coli mastitis. Vet J. (2015) 203:339–41. doi: 10.1016/j.tvjl.2014.12.023
18. Jiang L, Sørensen P, Røntved C, Vels L, Ingvartsen KL. Gene expression profiling of liver from dairy cows treated intra-mammary with lipopolysaccharide. BMC Genomics. (2008) 9:443. doi: 10.1186/1471-2164-9-443
19. Jorgensen HB, Buitenhuis B, Rontved CM, Jiang L, Ingvartsen KL, Sorensen P. Transcriptional profiling of the bovine hepatic response to experimentally induced E. coli Mastitis Physiol Genomics. (2012) 44:595–606. doi: 10.1152/physiolgenomics.00084.2011
20. Heimes A, Brodhagen J, Weikard R, Hammon HM, Meyerholz MM, Petzl W, et al. Characterization of functional traits with focus on udder health in heifers with divergent paternally inherited haplotypes on BTA18. BMC Vet Res. (2019) 15:241. doi: 10.1186/s12917-019-1988-4
21. Meyerholz MM, Rohmeier L, Eickhoff T, Hülsebusch A, Jander S, Linden M, et al. Genetic selection for bovine chromosome 18 haplotypes associated with divergent somatic cell score affects postpartum reproductive and metabolic performance. J Dairy Sci. (2019) 102:9983–94. doi: 10.3168/jds.2018-16171
22. Rohmeier L, Petzl W, Koy M, Eickhoff T, Hülsebusch A, Jander S, et al. In vivo model to study the impact of genetic variation on clinical outcome of mastitis in uniparous dairy cows. BMC Vet Res. (2020) 16:33. doi: 10.1186/s12917-020-2251-8
23. Tierschutzgesetz. Amended by Announcement of 18-05-2006 I 1206, 1313; last amended by Article 101 G v 20-11-2019 I 1626. (1972). Available online at: https://www.gesetze-im-internet.de/tierschg/BJNR012770972.html
24. Petzl W, Gunther J, Pfister T, Sauter-Louis C, Goetze L, von Aulock S, et al. Lipopolysaccharide pretreatment of the udder protects against experimental Escherichia coli mastitis. Innate Immun. (2012) 18:467–77. doi: 10.1177/1753425911422407
25. Petzl W, Zerbe H, Gunther J, Yang W, Seyfert HM, Nurnberg G, et al. Escherichia coli, but not Staphylococcus aureus triggers an early increased expression of factors contributing to the innate immune defense in the udder of the cow. Vet Res. (2008) 39:18. doi: 10.1051/vetres:2007057
26. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. (2015) 67:1. doi: 10.18637/jss.v067.i01
27. Weikard R, Goldammer T, Brunner RM, Kuehn C. Tissue-specific mRNA expression patterns reveal a coordinated metabolic response associated with genetic selection for milk production in cows. Physiol Genomics. (2012) 44:728–39. doi: 10.1152/physiolgenomics.00007.2012
28. Weikard R, Goldammer T, Eberlein A, Kuehn C. Novel transcripts discovered by mining genomic DNA from defined regions of bovine chromosome 6. BMC Genomics. (2009) 10:186. doi: 10.1186/1471-2164-10-186
29. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence alignment/map format and SAMtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
30. Beever JE, Fisher SR, Guerin G, Lewin HA. Mapping of eight human chromosome 1 orthologs to cattle chromosomes 3 and 16. Mamm Genome. (1997) 8:533–6. doi: 10.1007/s003359900493
31. FastQ. Version 0.11.5, released 2016-03-08. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
32. MultiQC. Version 1.4, released 2018-01-11. Available online at: http://multiqc.info/
33. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. (2011) 17:10–2. doi: 10.14806/ej.17.1.200
34. Qualitytrim. Version 1.6.0, release 2015-01-05. Available online at: https://bitbucket.org/arobinson/qualitytrim
35. Bovine, Genome assembly UMD3,.1. Ensembl annotation version 87, released 2016-12-08. Available online at: ftp://ftp.ensembl.org/././pub/release-87/fasta/bos_taurus/dna/
36. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringtie and ballgown. Nat Protoc. (2016) 11:1650–67. doi: 10.1038/nprot.2016.095
37. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
38. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
40. Kolde R. Pheatmap: Pretty Heatmaps. Version 1.0.12. (2019). Available online at: https://CRAN.R-project.org/package=pheatmap
41. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. (2016) 32:2847–9. doi: 10.1093/bioinformatics/btw313
42. Blighe K, Rana S, Lewis M. EnhancedVolcano: Publication-Ready Volcano Plots With Enhanced Colouring and Labeling. Version 1.4.0. (2019). Available online at: https://github.com/kevinblighe/EnhancedVolcano
43. Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. (2012) 46:1–17. doi: 10.18637/jss.v046.i11
44. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. (2008) 9:559. doi: 10.1186/1471-2105-9-559
45. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. (2009) 4:44–57. doi: 10.1038/nprot.2008.211
46. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. (2008) 1778:660–9. doi: 10.1016/j.bbamem.2007.07.012
47. McEwen AE, Escobar DE, Gottardi CJ. Signaling from the adherens junction. Subcell Biochem. (2012) 60:171–96. doi: 10.1007/978-94-007-4186-7_8
48. Yen C-F, Wang H-S, Lee C-L, Liao S-K. Roles of integrin-linked kinase in cell signaling and its perspectives as a therapeutic target. Gynecol Minim Invasive Ther. (2014) 3:67–72. doi: 10.1016/j.gmit.2014.06.002
49. Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. (2005) 390:1–9. doi: 10.1042/BJ20050104
50. Murthy S, Born E, Mathur SN, Field FJ. LXR/RXR activation enhances basolateral efflux of cholesterol in CaCo2 cells. J Lipid Res. (2002) 43:1054–64. doi: 10.1194/jlr.M100358-JLR200
51. Yoshida H, Nadanaka S, Sato R, Mori K. XBP1 is critical to protect cells from endoplasmic reticulum stress: evidence from site-2 protease-deficient Chinese hamster ovary cells. Cell Struct Funct. (2006) 31:117–25. doi: 10.1247/csf.06016
52. Bannerman DD, Paape MJ, Lee J-W, Zhao X, Hope JC, Rainard P. Escherichia coli and Staphylococcus aureus elicit differential innate immune responses following intramammary infection. Clin Diagn Lab Immunol. (2004) 11:463–72. doi: 10.1128/CDLI.11.3.463-472.2004
53. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. (1999) 17:593–623. doi: 10.1146/annurev.immunol.17.1.593
54. Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, et al. Fcγ receptor–mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J Exp Med. (2000) 191:669–82. doi: 10.1084/jem.191.4.669
55. Colonne PM, Winchell CG, Voth DE. Hijacking host cell highways: manipulation of the host actin cytoskeleton by obligate intracellular bacterial pathogens. Front Cell Infect Microbiol. (2016) 6:107. doi: 10.3389/fcimb.2016.00107
56. Gunther J, Petzl W, Bauer I, Ponsuksili S, Zerbe H, Schuberth HJ, et al. Differentiating Staphylococcus aureus from Escherichia coli mastitis: S. aureus triggers unbalanced immune-dampening and host cell invasion immediately after udder infection. Sci Rep. (2017) 7:4811. doi: 10.1038/s41598-017-05107-4
57. Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells. (2010) 29:311–25. doi: 10.1007/s10059-010-0053-8
58. Arif E, Sharma P, Solanki A, Mallik L, Rathore YS, Twal WO, et al. Structural analysis of the Myo1c and Neph1 complex provides insight into the intracellular movement of Neph1. Mol Cell Biol. (2016) 36:1639–54. doi: 10.1128/MCB.00020-16
59. Hu J, Lu J, Goyal A, Wong T, Lian G, Zhang J, et al. Opposing FlnA and FlnB interactions regulate RhoA activation in guiding dynamic actin stress fiber formation and cell spreading. Hum Mol Genet. (2017) 26:1294–304. doi: 10.1093/hmg/ddx047
60. Janson C, Kasahara N, Prendergast GC, Colicelli J. RIN3 is a negative regulator of mast cell responses to SCF. PLoS ONE. (2012) 7:e49615. doi: 10.1371/journal.pone.0049615
61. Neel NF, Rossman KL, Martin TD, Hayes TK, Yeh JJ, Der CJ. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol Cell Biol. (2012) 32:1374–86. doi: 10.1128/MCB.06291-11
62. Nourse JL, Pathak MM. How cells channel their stress: interplay between piezo1 and the cytoskeleton. Semin Cell Dev Biol. (2017) 71:3–12. doi: 10.1016/j.semcdb.2017.06.018
63. Schubert C, Pryds A, Zeng S, Xie Y, Freund KB, Spaide RF, et al. Cadherin 5 is regulated by corticosteroids and associated with central serous chorioretinopathy. Hum Mutat. (2014) 35:859–67. doi: 10.1002/humu.22551
64. Sharili AS, Kenny FN, Vartiainen MK, Connelly JT. Nuclear actin modulates cell motility via transcriptional regulation of adhesive and cytoskeletal genes. Sci Rep. (2016) 6:33893. doi: 10.1038/srep33893
65. Aktories K. Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Microbiol. (2011) 9:487–98. doi: 10.1038/nrmicro2592
66. Bestebroer J, De Haas CJ, Van Strijp JA. How microorganisms avoid phagocyte attraction. FEMS Microbiol Rev. (2010) 34:395–414. doi: 10.1111/j.1574-6976.2009.00202.x
67. Boquet P, Lemichez E. Bacterial virulence factors targeting Rho GTPases: parasitism or symbiosis? Trends Cell Biol. (2003) 13:238–46. doi: 10.1016/S0962-8924(03)00037-0
68. De Jong NWM, van Kessel KPM, van Strijp JAG. Immune evasion by Staphylococcus aureus. Microbiol Spectr. (2019) 7:GPP3-0061-2019. doi: 10.1128/microbiolspec.GPP3-0061-2019
69. Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. (2005) 3:948–58. doi: 10.1038/nrmicro1289
70. Bestebroer J, van Kessel KP, Azouagh H, Walenkamp AM, Boer IG, Romijn RA, et al. Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood. (2009) 113:328–37. doi: 10.1182/blood-2008-04-153882
71. Walenkamp AME, Boer IGJ, Bestebroer J, Rozeveld D, Timmer-Bosscha H, Hemrika W, et al. Staphylococcal superantigen-like 10 inhibits CXCL12-induced human tumor cell migration. Neoplasia. (2009) 11:333–44. doi: 10.1593/neo.81508
72. Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol. (2005) 174:2926–33. doi: 10.4049/jimmunol.174.5.2926
73. Petzl W, Zerbe H, Gunther J, Seyfert HM, Hussen J, Schuberth HJ. Pathogen-specific responses in the bovine udder. Models and immunoprophylactic concepts. Res Vet Sci. (2018) 116:55–61. doi: 10.1016/j.rvsc.2017.12.012
74. Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem. (2000) 275:16390–9. doi: 10.1074/jbc.M000953200
75. Wang J, Xiao C, Wei Z, Wang Y, Zhang X, Fu Y. Activation of liver X receptors inhibit LPS-induced inflammatory response in primary bovine mammary epithelial cells. Vet Immunol Immunopathol. (2018) 197:87–92. doi: 10.1016/j.vetimm.2018.02.002
76. A-González N, Castrillo A. Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim Biophys Acta Mol Basis Dis. (2011) 1812:982–94. doi: 10.1016/j.bbadis.2010.12.015
77. Gruys E, Toussaint MJM, Niewold TA, Koopmans SJ. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci. (2005) 6:1045–56. doi: 10.1631/jzus.2005.B1045
78. Hardardottir I, Grunfeld C, Feingold KR. Effects of endotoxin on lipid metabolism. Biochem Soc Trans. (1995) 23:1013–8. doi: 10.1042/bst0231013
79. Abreu AG, Barbosa AS. How Escherichia coli circumvent complement-mediated killing. Front Immunol. (2017) 8:452. doi: 10.3389/fimmu.2017.00452
80. Gunther J, Koczan D, Yang W, Nurnberg G, Repsilber D, Schuberth HJ, et al. Assessment of the immune capacity of mammary epithelial cells: comparison with mammary tissue after challenge with Escherichia coli. Vet Res. (2009) 40:31. doi: 10.1051/vetres/2009014
81. Leimbach A, Poehlein A, Vollmers J, Görlich D, Daniel R, Dobrindt U. No evidence for a bovine mastitis Escherichia coli pathotype. BMC Genomics. (2017) 18:359. doi: 10.1186/s12864-017-3739-x
82. Leimbach A, Poehlein A, Witten A, Wellnitz O, Shpigel N, Petzl W, et al. Whole-genome draft sequences of six commensal fecal and six mastitis-associated Escherichia coli strains of bovine origin. Genome Announc. (2016) 4:e00753–16. doi: 10.1128/genomeA.00753-16
83. Rydengard V, Olsson AK, Morgelin M, Schmidtchen A. Histidine-rich glycoprotein exerts antibacterial activity. FEBS J. (2007) 274:377–89. doi: 10.1111/j.1742-4658.2006.05586.x
84. Munger JS, Sheppard D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. (2011) 3:a005017. doi: 10.1101/cshperspect.a005017
Keywords: mastitis, bovine, S. aureus, E. coli, transcriptome, liver
Citation: Heimes A, Brodhagen J, Weikard R, Seyfert H-M, Becker D, Meyerholz MM, Petzl W, Zerbe H, Hoedemaker M, Rohmeier L, Schuberth H-J, Schmicke M, Engelmann S and Kühn C (2020) Hepatic Transcriptome Analysis Identifies Divergent Pathogen-Specific Targeting-Strategies to Modulate the Innate Immune System in Response to Intramammary Infection. Front. Immunol. 11:715. doi: 10.3389/fimmu.2020.00715
Received: 05 August 2019; Accepted: 30 March 2020;
Published: 29 April 2020.
Edited by:
Robert Braidwood Sim, University of Oxford, United KingdomReviewed by:
Joan Clària, Hospital Clínic de Barcelona, SpainDan Anthony Mitchell, University of Warwick, United Kingdom
Copyright © 2020 Heimes, Brodhagen, Weikard, Seyfert, Becker, Meyerholz, Petzl, Zerbe, Hoedemaker, Rohmeier, Schuberth, Schmicke, Engelmann and Kühn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christa Kühn, a3VlaG5AZmJuLWR1bW1lcnN0b3JmLmRl