 Thaís S. Rigoni1
Thaís S. Rigoni1 Natália S. Vellozo1
Natália S. Vellozo1 Mariela Cabral-Piccin1
Mariela Cabral-Piccin1 Laryssa Fabiano-Coelho1
Laryssa Fabiano-Coelho1 Ulisses G. Lopes1
Ulisses G. Lopes1 Alessandra A. Filardy1,2
Alessandra A. Filardy1,2 George A. DosReis1,3
George A. DosReis1,3 Marcela F. Lopes1*
Marcela F. Lopes1*- 1Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Instituto de Microbiologia Paulo de Góes, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 3Instituto Nacional para Pesquisa Translacional em Saúde e Ambiente na Região Amazônica, Conselho Nacional de Desenvolvimento Científico e Tecnológico, Rio de Janeiro, Brazil
Macrophages host Leishmania major infection, which causes cutaneous Leishmaniasis in humans. In the murine model, resistance to infection depends on the host immunity mediated by CD4 T-cell cytokines and macrophages. In association to other stimuli, the Th1 cytokine IFN-γ induces NO-mediated microbial killing by M1/classically-activated macrophages. By contrast, the Th2 cytokine IL-4 promotes M2/alternatively activated macrophages, which express arginase-1 and shelter infection. Other cytokines, such as RANKL, might also participate in the crosstalk between T cells and macrophages to restrict parasite infection. RANKL and its receptor RANK are known to play an essential role in bone remodeling, by inducing osteoclatogenesis. It has also been shown that RANKL stimulates antigen-presenting cells, such as DCs and macrophages, to enhance T cell responses. Here we investigated how RANKL directly modulates the effector macrophage phenotypes and immunity to L. major parasites. We found that inflammatory peritoneal macrophages from B6 mice express RANK and M2 features, such as CD301 (MGL) and CD206 (mannose receptor). Nonetheless, treatment with RANKL or IFN-γ induced macrophage differentiation into more mature F40/80hi macrophages able to produce IL-12 and TNF-α. In parallel, macrophages treated with RANKL, IFN-γ, or RANKL along with IFN-γ progressively downregulated the expression of the M2 hallmarks MGL, arginase-1, and CCL17. Moreover, a synergism between IFN-γ and RANKL enhanced inducible NO synthase (iNOS) expression and NO production by macrophages. These results are consistent with the idea that RANKL helps IFN-γ to induce a M2-like to M1 phenotype shift. Accordingly, concomitant treatment with RANKL and IFN-γ promoted macrophage-mediated immunity to L. major, by inducing NO and ROS-dependent parasite killing. Furthermore, by cooperating with IFN-γ, endogenous RANKL engages CD4 T-cell help toward L. major-infected macrophages to upregulate M1 and Th1 cytokine responses. Therefore, RANKL, in combination with IFN-γ, is a potential local therapeutic tool to improve immune responses in Leishmaniasis, by skewing M2-like into effector M1 macrophages.
Introduction
The Receptor Activator of Nuclear Factor-Kappa B (RANK), a TNF receptor family member, and its ligand RANKL (also known as TRANCE, ODF, and OPGL) are crucial for bone homeostasis, by promoting osteoclast differentiation and activation (1–7). RANK initiates multiple signaling pathways, including NF-κB activation, that lead to gene transcription, iNOS expression, and NO production by osteoclast and macrophages (8–11). Deficiencies in RANK or RANKL cause osteopetrosis owing to abnormal osteoclastogenesis and bone remodeling, but also affect the immune system and lymphoid organogenesis, precluding accurate studies of immunity to pathogens in RANK/RANKL-deficient mice (12–17).
RANKL-RANK axis was first described in the context of immune responses, where T-cell derived RANKL promotes dendritic cell/macrophage function to increase T-cell activation (1, 2, 10, 18–22). Activated T cells and Th1, but not Th2 cells express TRANCE/RANKL (18, 23) and treatment with RANKL stimulates DCs to secrete IL-12 (18, 19). In line with this, a DC therapy with RANKL-treated DCs primed Th1 responses to microbial antigens (20). Conversely, the blockade of RANKL precluded T-cell priming for Th1 responses both in vitro (23) and in vivo (19, 24). RANKL also enhanced APC features and secretion of inflammatory cytokines by bone marrow-derived macrophages (BMDMs), but failed to upregulate IL-12 or high levels of iNOS expression (22, 25). How RANKL modulates macrophage effector functions has not been fully explored.
Leishmania spp. infection causes several clinical forms ranging from localized lesions to disseminated Leishmaniasis, a major health problem in developing countries, by affecting humans and animals. In the experimental cutaneous Leishmaniasis, immunity to Leishmania major infection depends on the Th1/Th2 cytokines produced by CD4 T cells that shape macrophage phenotype, by inducing either classically/M1 or alternatively/M2 activated macrophages (26–32). In resistant mouse strains, Th1 cytokines, such as IFN-γ and TNF-α induce NO production and parasite killing by M1 macrophages. Otherwise, the Th2 cytokine IL-4 increases arginase-1 expression and parasite replication within M2 macrophages in susceptible mice (32–38). Despite efforts and advances, effective vaccination and immunotherapy are not available.
A previous study investigated whether RANKL plays a role as a costimulatory molecule on immune responses to L. major parasites (24). Whereas CD40L-deficient mice are resistant to infection, the blockade of RANKL-RANK interactions precluded IL-12 production by antigen presenting cells and shifted protective Th1 into Th2 responses (24). Nonetheless, it has not been elucidated how RANKL directly affects macrophage effector phenotypes, as well as their ability to fight parasite infection.
Here we investigated the role of RANKL-RANK axis on M1/M2 phenotypes and on macrophage-mediated immunity to L. major. We showed that, whereas peritoneal inflammatory cells express a M2-like phenotype, RANKL has direct effects on macrophages, by promoting the expression of the M1 cytokines TNF-α and IL-12 and by reducing M2-features. Moreover, RANKL synergizes with IFN-γ to increase iNOS expression, NO production, and the control of Leishmania parasites. Furthermore, endogenous RANKL and IFN-γ promote CD4 T-cell help to infected macrophages and upregulate both M1 and Th1 responses to parasite infection.
Materials and Methods
Mice and Parasites
C57BL/6 (B6) and BALB/c mice were obtained from the Oswaldo Cruz Foundation (FIOCRUZ, Rio de Janeiro, Brazil) and maintained in the animal facility at the Federal University of Rio de Janeiro (UFRJ). The animal study was reviewed and approved by the Ethics Committee for Use of Animals at the Federal University of Rio de Janeiro (UFRJ). All experiments were conducted as in the protocol 078/16 (CEUA-UFRJ). L. major LV39 (MRHO/Sv/59/P) parasites were isolated from popliteal lymph nodes of infected BALB/c mice and maintained up to 4 wk at 28°C in Schneider's medium (Sigma-Aldrich, USA), supplemented with 2% sterile human urine, 2 mM of L-glutamine, 10 μg/mL of gentamicin, and 10% fetal bovine serum (FBS, Gibco BRL, South America). For macrophage or mouse infection, Leishmania parasites were cultured until stationary phase at 28°C in Schneider's medium.
Mouse Infection
B6 mice, aging 6–8 weeks, were injected i.p. with 3 ×106 L. major parasites and peritoneal macrophages were collected after 24 h for T-cell/macrophage cocultures. As a source for T cells, B6 mice were infected s.c. (at hind footpads) with 3 ×106 L. major parasites and the spleens were removed at 5 w.p.i.
Inflammatory Macrophages
Inflammatory macrophages were obtained 4 days after the i.p. injection of 3% thioglycolate broth. Peritoneal resident macrophages or inflammatory (recruited) macrophages were collected by peritoneal lavage. Inflammatory macrophages were cultured in DMEM (Invitrogen Life Technologies), supplemented with 2 mM glutamine, 5 ×105 M 2-ME, 10 μg/mL gentamicin, 1 mM sodium pyruvate, and 0.1 mM MEM non-essential amino acids (culture medium) plus 10% FBS. Cells were processed and analyzed prior or after culture by flow cytometry and functional assays. Cultures were treated with the following reagents: 0.2–0.5 ng/mL of recombinant IFN-γ (R&D Systems, EUA), 20 ng/mL of recombinant RANKL (R&D Systems, EUA), 200 ng/mL of LPS from Salmonella enterica serovar Typhimurium (Sigma), 10 μM of Bay 11-7082 (Santa Cruz Biotechnology, Dallas, USA) or DMSO (Sigma), 1 mM of N6-(1-imioetil) lysine (L-NIL) from Sigma, 100 μM of deferoxamine (DFO, Sigma) or N-acetyl-L-cysteine (NAC, Sigma). Cultures were maintained up to 3 days at 37°C with 7% of CO2.
Macrophage Infection and Parasite Load
Inflammatory (B6) macrophages were cultured in triplicates at 1.5–3.5 ×105 cells/well in 48-well vessels or at 4 ×105 cells/well on glass coverslips inserted in 24-well plates during 1 h, and then washed. Adherent macrophages were infected for 24 h at a 10:1 parasites/macrophage ratio. Cultures were washed for removal of extracellular parasites and additional non-adherent cells. Infected macrophages were treated with the indicated reagents and maintained in culture medium plus 10% FBS at 37°C and 7% CO2 for 3 days. In cultures established in 48-well vessels, supernatants were replaced by Schneider's medium. Macrophages were further cultured for at least 4 days at 28°C in a BOD incubator (Cienlab) for determination of parasite load. Parasites released in culture supernatants were then counted in a Beckman Coulter (USA) within a range of 3–6 μm, for exclusion of murine cells. For intracellular amastigotes assessment, coverslips were stained with Panotic kit (Laborclin) and parasites were counted within macrophages. Results are expressed as numbers of parasites/100 macrophages.
Flow Cytometry
Peritoneal resident macrophages and inflammatory macrophages were prepared for flow cytometry. For phenotype analyses, inflammatory (B6) macrophages were cultured in triplicates at 2.0–2.5 ×106 cells/well in 24-well plates, treated with the indicated reagents and maintained at 37°C with 7% CO2 during 48 h. Cells were washed in FACS buffer (PBS 0.01% NaN3 plus 2% FBS) and incubated with anti-CD16/CD32 (eBioscience, San Diego, CA, USA) for Fc blocking. We stained cells with allophycocyanin or FITC-labeled anti-F4/80, (eBiosciences, Chicago, IL, USA), Alexa Fluor 488-labeled anti-CD301 (MGL) mAb (AbD Serotec, Kidlington, UK) or control rat IgG2a mAb (R&D Systems, Minneapolis, MN, USA) or with allophycocyanin-labeled anti-CD301 (MGL) mAb or control rat IgG2b mAb (Biolegend, San Diego, CA, USA); PE-labeled anti-RANK mAb (BioLegend), PE-labeled anti-206 (mannose receptor) (Biolegend) or control rat IgG2a mAb (eBioscience). For intracellular staining, we washed, permeabilized, fixed, and stained cells with PE-labeled anti-IL-12p35 or control murine IgG1 mAb (R&D Systems), PE-labeled anti-NOS2 (iNOS) or control rat IgG2a mAb (eBioscience); FITC-labeled anti-Arginase-1 or control sheep IgG mAb (R&D Systems). Cells were acquired with the CellQuest software on a FACSCalibur system (BD Biosciences, San Jose, CA, USA). For analysis, we used the FlowJo software (TreeStar, Ashland, OR, USA). F4/80+ cells were first gated and then evaluated for CD301+ or CD206+ cells, for IL-12p35+ and CD301+ subsets or for iNOS+ and Arginase-1+ subsets, based on the exclusion of background staining with isotype control mAbs.
CD4 T-Cell Purification
Spleens from B6 mice infected at hind footpad with L. major were removed and depleted of red blood cells by Tris-buffered ammonium chloride treatment, followed by nylon wool filtration to obtain enriched T-cell suspensions. Purified CD4 T-cells were obtained by negative selection with a mAb mix, containing anti-CD8, anti-B220, anti-CD11b, and anti-panNK mAbs (BD Biosciences or Ebioscience), and anti-rat IgG magnetic beads (Dynal, Oslo, Norway). Cell suspensions were 85–88% CD4+ cells, as stained for detection of residual CD8 T cells or B cells with anti-CD4, anti-CD5, and anti-CD19 (BD Biosciences) mAbs.
Cocultured Macrophages and CD4 T Cells
For cocultures, peritoneal macrophages from L. major-injected B6 mice were plated in triplicates at 5 ×105 cells/well in 48-well vessels during 1 h and then washed. Adherent macrophages were infected for 7 h at a 10:1 parasites/macrophage ratio. Cultures were washed for removal of extracellular parasites and additional non-adherent (cells/well). Purified CD4 T-cells (5 ×105/well) from infected mice were added to infected macrophages at a 1:1 ratio in 48-well vessels. Cocultures were then treated or not with 10 μg/mL of anti-mouse CD254 (RANKL, Biolegend) or anti-mouse IFN-γ (BD Bioscience) during 48 h. Supernatants were collected for cytokine assays.
Cytokine Assays
Culture supernatants were used for detection of NO (as bellow) and cytokines (IL-12p70, TNF-α, CCL17) by ELISA assay. For that, we used pairs of specific mAbs (R&D Systems, eBioscience or PeproTech), one of which was labeled with biotin, and then developed with streptavidin-alkaline phosphatase (Invitrogen Life Technologies) and p-nitrophenyl phosphate (PNPP substrate, Thermo Scientific Pierce, Waltham, MA, USA), according to manufacturers. Alternatively, Avidin horseradish peroxidase (eBioscience) and 3,3′,5,5′-tetramethylbenzidine (TMB substrate, eBioscience) were used for detection.
Nitric Oxide
Production of NO was determined indirectly by the quantification of nitrites. Culture supernatants were mixed with Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, 2% H3PO4; Sigma) in a 1:1 ratio. A standard curve with known concentrations of sodium nitrite (NaNO2) was used and the results were expressed as nitrites (μM). The optical density was determined at 540 nm on a plate spectrophotometer (Versa Max, Molecular Devices).
Statistics
Results are expressed as the mean of technical replicates per treatment and S.E.M in figures. For parasite load, data were transformed to log of parasites per mL for statistical analysis. Data were analyzed by unpaired Student's two-tailed t-test, by using the GraphPad Prism (v. 6.0). Otherwise, results were analyzed by one-way ANOVA, followed by the indicated post-tests. The numbers of independent repeat experiments and significant differences are indicated in the figure legends.
Results
RANK Expression and Inflammatory Macrophage Phenotype
To investigate the role of RANKL-RANK axis in macrophage-mediated immunity, we first determined RANK expression on F4/80int-inflammatory vs. F4/80hi-peritoneal (resident) macrophages from B6 mice (Figure 1A). After i.p. thioglycolate injection, about 20–30% of recruited (inflammatory) macrophages expressed RANK at higher levels compared with resident macrophages (Figure 1A). In unstimulated cultures, however, inflammatory macrophages upregulated both F4/80 and RANK expression (Figure 1A, lower panels). Next, we determined the effector phenotype of inflammatory (B6) macrophages (Figure 1 and Supplementary Figure 1). About 80% of elicited-peritoneal macrophages expressed the M2 marker MGL (CD301) and variable percentages of other M2 features (39), such as the mannose receptor (MR; CD206) (Figures 1B,C), the IL-4Rα subunit (CD124) (Supplementary Figure 1A), and arginase-1, but not the M1 hallmarks IL-12p35 (Supplementary Figure 1B) and iNOS (not shown). Moreover, a subset of CD206hi macrophages expressed IL-10 (Supplementary Figure 1D). Thus, inflammatory peritoneal macrophages seem to express a M2-like phenotype.
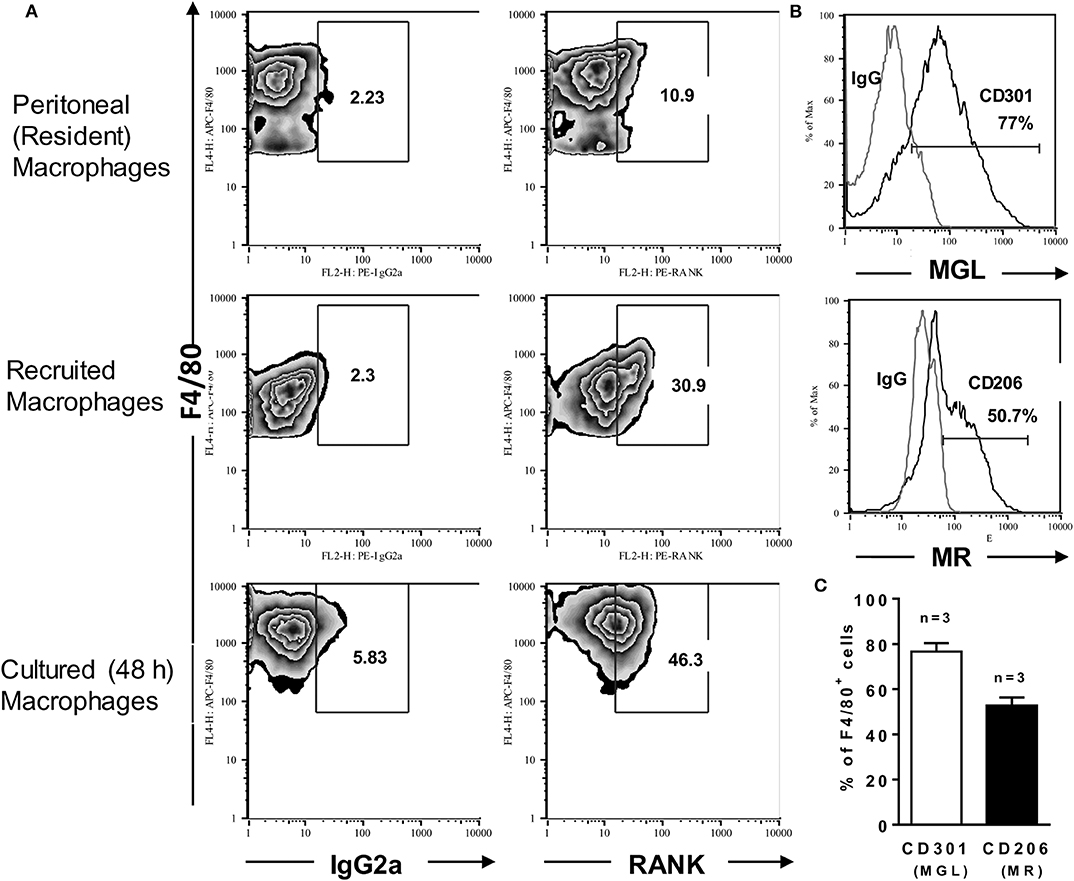
Figure 1. Inflammatory macrophages express RANK and a M2-like phenotype. Peritoneal (resident) macrophages and thioglycolate-elicited (inflammatory) macrophages from B6 mice were analyzed for the expression of RANK or for the M2 hallmarks CD301 (MGL) and CD206 (MR). (A) Peritoneal cells were stained with anti-F4/80 and anti-RANK or control IgG2a. Where indicated, inflammatory macrophages were cultured in medium only during 48 h before staining. (B,C) F4/80+ inflammatory macrophages were also stained with anti-CD301 or control IgG2b and anti-CD206 or control IgG2a, as indicated. In (C), results are expressed as means and S.E.M. (n = 3 mice). Data are representative of 3 independent experiments.
RANKL Upregulates F4/80 Expression by Macrophages
Since RANKL and IFN-γ have been used together to improve the expression of the costimulatory molecule CD86 in BMDMs (22), we first investigated whether RANKL and/or IFN-γ would also affect the expression of APC features (Supplementary Figure 2) in B6 inflammatory macrophages, which also expressed the MHC-class II I-Ab molecule (not shown). Here we observed that, compared with unstimulated macrophages, treatment with either RANKL and/or IFN-γ induced more mature macrophages, as assessed by higher F4/80 expression (Supplementary Figure 2, upper panels). We also found that untreated macrophages expressed the costimulatory CD80, CD86, and the coinhibitory PD-L1 molecules (Supplementary Figure 2A). Moreover, independent of treatment, macrophages remained positive for both costimulatory and coinhibitory molecules, although each treatment with RANKL and/or IFN-γ have differentially modulated the expression of CD80, CD86, and PD-L1 (Supplementary Figure 2B). It should be noticed, however, that macrophages treated with RANKL along with IFN-γ expressed the lowest levels of costimulatory molecules associated with the highest levels of the coinhibitory PDL-1 (Supplementary Figure 2B).
RANKL Cooperates With IFN-γ to Induce Effector Macrophages
Next, we addressed the functional phenotypes of RANKL-treated macrophages. First, we observed that inflammatory macrophages from both L. major-susceptible BALB/c and resistant B6 mice expressed increased NO production only after treatment with both RANKL and IFN-γ (Figure 2A). The synergistic effect of suboptimal doses of IFN-γ (40) and RANKL (9) on NO production was comparable to treatment with LPS and IFN-γ (Figure 2A, right panel). In all the subsequent studies we employed only inflammatory macrophages from the B6 (Th1) mouse strain, which develops IFN-γ-dependent macrophage responses to L. major infection (41). We found that treatment with either RANKL or IFN-γ affected cytokine secretion by macrophages, by inducing TNF-α and IL-12p70, whereas RANKL reduced the M2 chemokine CCL17 only in the presence of IFN-γ (Figure 2B). Therefore, RANKL modulates macrophage function, either directly, by inducing the secretion of proinflammatory cytokines or in combination with IFN-γ to increase NO production and reduce a M2 chemokine. To address the role of NF-κB signaling in macrophage responses to RANKL, we used the IKK signaling inhibitor Bay 11-7082. Interestingly, IKK signaling inhibition only partially reduced TNF-α and IL-12 responses to different treatments, but completely blocked NO production in macrophages treated with IFN-γ or RANKL along with IFN-γ (Figure 2C). These results indicate that RANKL and/or IFN-γ differentially regulate gene transcription, as previously discussed (10, 38, 42), as well as that each M1-functional (IL-12, TNF-α, NO) response expresses variable degrees of NF-κB signaling dependence (Figure 2C).
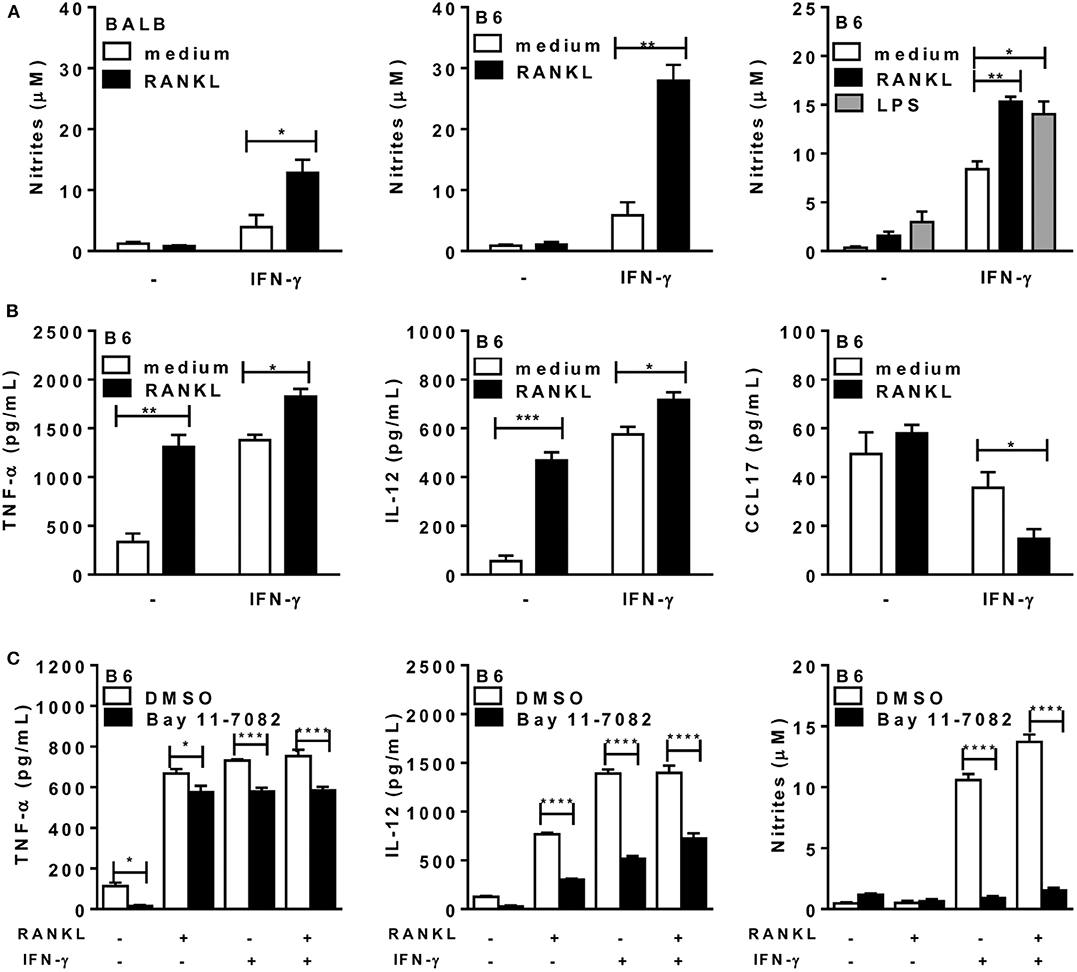
Figure 2. RANKL and IFN-γ increase cytokine and NO production by macrophages. Inflammatory macrophages from B6 or BALB/c mice were cultured in triplicates with medium or RANKL in the presence or absence of IFN-γ during 48 h. Some cultures also received the IKK-signaling inhibitor Bay 11-7082 or control diluent (DMSO). (A,C) Culture supernatants were tested for NO production by the Griess method. (B,C) Culture supernatants were assayed for TNF-α, IL-12p70, and CCL17 by ELISA. Results are expressed as means and S.E.M. For (A,B), significant differences between macrophages treated with or without RANKL were analyzed by t-test. Data are representative of 3 independent experiments. In (A, right panel) LPS (200 ng/mL) was used as a positive control and the results were analyzed by one-way ANOVA, followed by Tukey post-test. In (C), data were analyzed by one-way ANOVA, followed by Bonferroni post-test. Significant differences were indicated for *P <0.05, **P <0.01, ***P <0.001, and ****P <0.0001.
RANKL and IFN-γ Promote a M2-Like to M1 Phenotype Shift
To investigate the effects of RANKL on M1/M2 phenotypes of B6 inflammatory macrophages, we analyzed the expression of MGL (as a M2 marker) and intracellular IL-12p35 (as a M1 marker) (Figure 3), as well as the expression of iNOS and arginase-1 (Figure 4) enzymes, which control L-arginine metabolism (33). At steady state, unstimulated F4/80+ macrophages expressed a M2-like (MGL+) phenotype, but not IL-12p35 (Figures 3A,B). Conversely, treatment with RANKL reduced MGL and increased IL-12p35 expression (Figures 3A,B). More interesting, RANKL suppressed arginase-1 and cooperated with IFN-γ to increase iNOS expression at higher levels (Figures 4A,B). These findings might explain how RANKL and IFN-γ synergize to increase NO production by macrophages (Figure 2A). Next, we performed parallel cytometric analyses of multiple M1/M2 hallmarks, as well as secreted cytokines (Supplementary Figures 3, 4). In unstimulated cultures, at least 80% of macrophages expressed MGL (but not iNOS) (Supplementary Figure 4A) and different percentages of the M2 markers CD206, CD124, and arginase-1 (Supplementary Figure 3). The M2 hallmarks and the secretion of CCL17 chemokine were progressively downregulated by RANKL, IFN-γ, and RANKL along with IFN-γ (Supplementary Figures 3, 4). By contrast, iNOS expression and IL-12 production were upregulated in treated macrophages (Supplementary Figures 4A,C), indicating a M2-like to M1 shift. Moreover, we observed a subset of double positive macrophages, which expressed both MGL and iNOS in cultures treated with RANKL (Supplementary Figure 4B). Therefore, in the presence of RANKL, M2-like macrophages can give rise to M1-intermediate cells (with lower iNOS expression), whereas cooperation with IFN-γ is required to induce a bona fide M1 phenotype with higher iNOS expression and reduced M2 features.
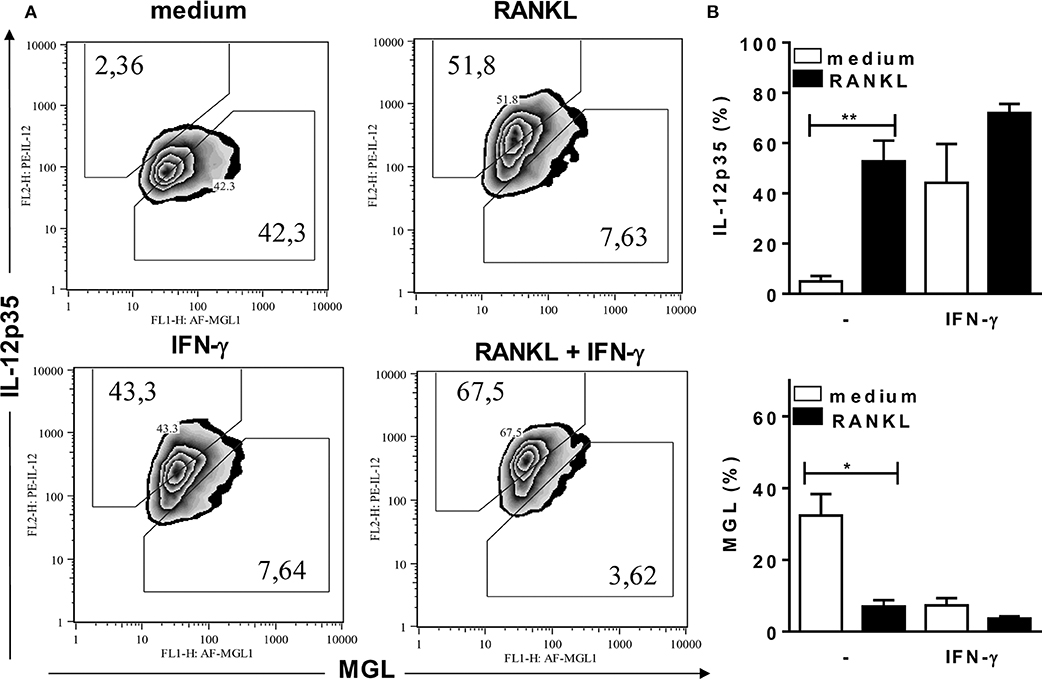
Figure 3. RANKL induces a M2-like to M1 phenotype shift. Inflammatory (B6) macrophages were cultured in triplicates with medium or RANKL in the presence or absence of IFN-γ. After 48 h, cells were harvested and stained with anti-F4/80, anti-CD301 (MGL), anti-IL-12p35, or control mAbs. (A) Plots depict F4/80+ cells as evaluated for CD301 (MGL) and intracellular IL-12p35 expression. (B) Results are expressed as means and S.E.M. Significant differences between macrophages treated with or without RANKL were analyzed by t-test and indicated for *P <0.05, and **P <0.01. Data are representative of 2 independent experiments.
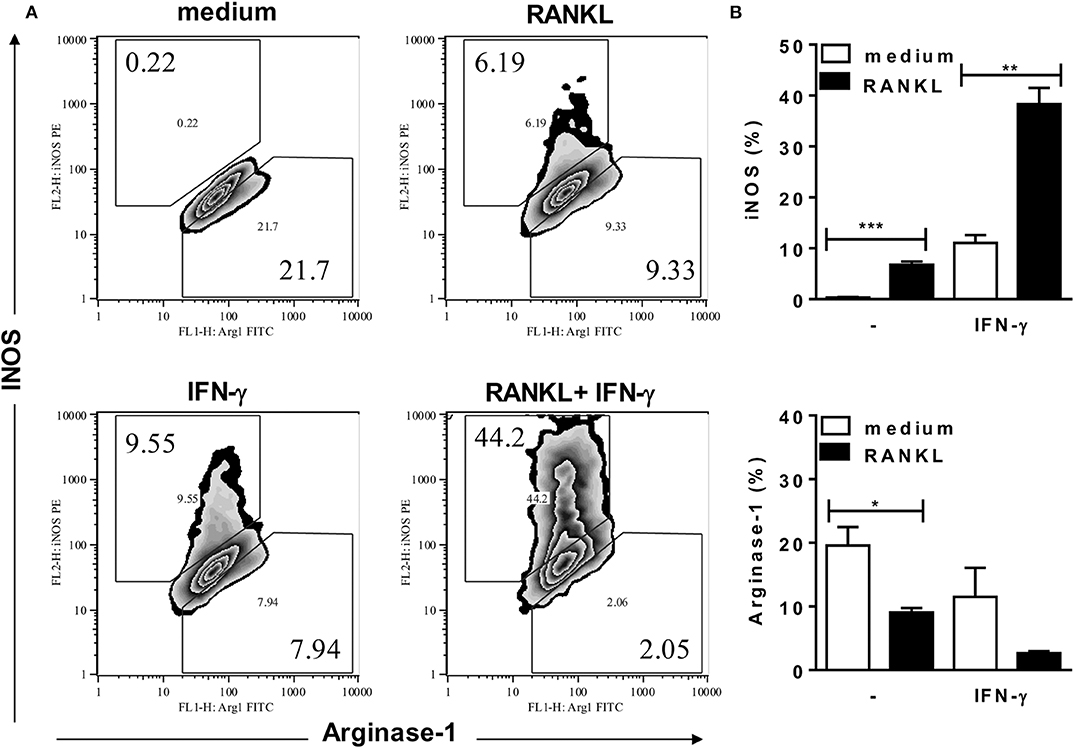
Figure 4. RANKL requires IFN-γ to induce high iNOS expression. Inflammatory (B6) macrophages were cultured in triplicates with medium or RANKL in the presence or absence of IFN-γ. After 48 h, cells were harvested and stained with anti-F4/80, anti-iNOS, anti-arginase-1 or control mAbs. (A) Plots depict F4/80+ cells as evaluated for intracellular iNOS and arginase-1 expression. (B) Results are expressed as means and S.E.M. Significant differences between macrophages treated with or without RANKL were analyzed by t-test and indicated for *P <0.05, **P <0.01, and ***P <0.001. Data are representative of 3 independent experiments.
RANKL and IFN-γ Help Macrophages to Control Infection
Next, we infected macrophages with L. major to test whether RANKL could affect macrophage-mediated immunity for parasite control (Figure 5). Although treatment with RANKL alone was not able to reduce parasite infection, it did enhance L. major control by IFN-γ-treated macrophages (Figure 5). Moreover, iNOS inhibition completely blocked parasite killing by macrophages treated with RANKL plus IFN-γ (Figures 5A,C). Nonetheless, the use of ROS inhibitors DFO (Figures 5B,C) or NAC (not shown) also blocked the control of L. major parasites, although NO production has actually increased in these conditions (Figure 5D). These results indicate that, although necessary, NO was not sufficient to control parasite infection by effector M1 macrophages generated in the presence of RANKL and IFN-γ.
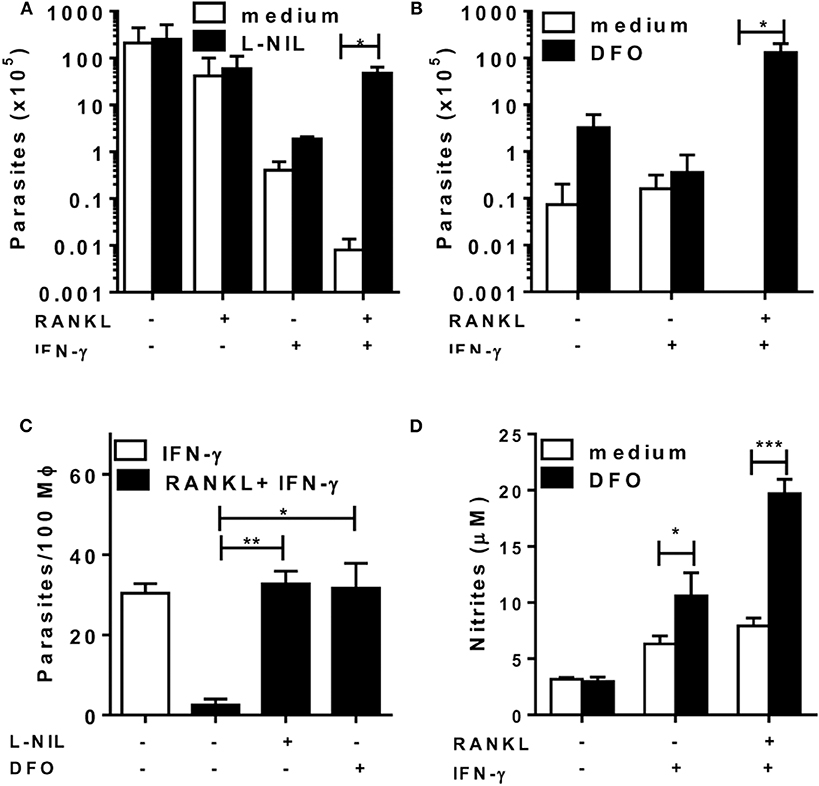
Figure 5. RANKL and IFN-γ induce L. major-parasite control. Inflammatory (B6) macrophages were infected with L. major (1:10) during 24 h and then treated with RANKL, IFN-γ, L-NIL (1 mM) or DFO (100 μM) during 72 h. (A–C) Parasitic burden was determined as (A,B) released promastigotes (parasites/mL) or (C) by counting amastigotes within macrophages. (D) Culture supernatants were tested for NO production by the Griess method. Results are expressed as means and S.E.M. Significant differences between macrophages treated without and with L-NIL or DFO were analyzed by t-test and indicated for *P <0.05, **P <0.01, and ***P <0.001. Data are representative of 2 independent experiments.
RANKL and IFN-γ Cooperate in CD4 T-Cell Help to Macrophages
Finally, we addressed the role of paracrine/surface RANKL and IFN-γ in the crosstalk between T cells and macrophages at the effector phase of immune responses, by using neutralizing mAbs against RANKL and IFN-γ. For that, we purified CD4 T-cells from L. major-infected mice, which were cocultured with peritoneal macrophages from mice injected with L. major. Macrophages were infected in vitro in order to present antigens to CD4 T cells and upregulate IL-12, IFN-γ, and TNF-α responses (Figure 6). Treatment with anti-RANKL reduced both IL-12 and IFN-γ, but not TNF-α in cocultures, whereas anti-IFN-γ reduced IL-12 secretion and completely blocked the production of TNF-α (Figures 6A–C). These results are consistent with the idea that endogenous (T-cell derived) RANKL and IFN-γ might help macrophages to produce IL-12 and upregulate Th1 responses.
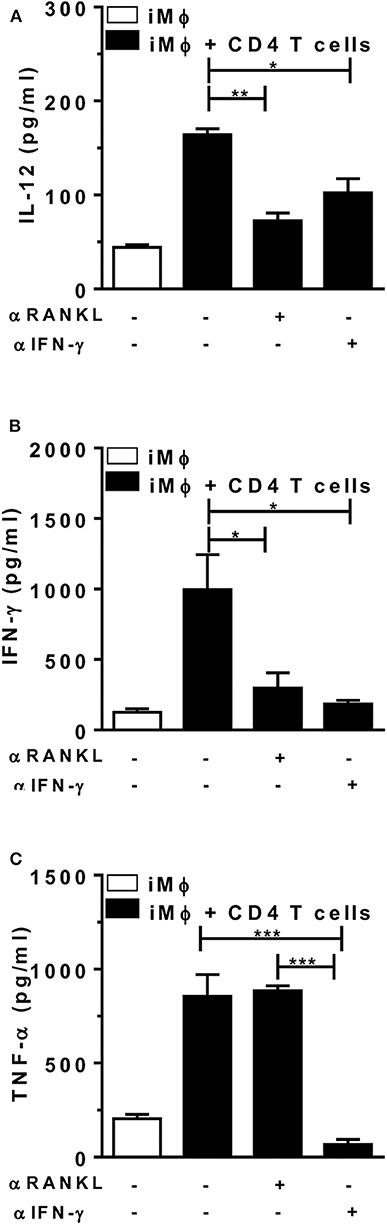
Figure 6. Endogenous RANKL is involved in CD4 T-cell help to macrophages. Peritoneal (B6) macrophages (from mice injected i.p. with L. major) were infected in vitro and then cultured alone or with CD4 T cells from L. major-infected mice (at 5 w.p.i.). Cultures were treated or not with anti-RANKL or anti-IFN-γ-neutralizing antibodies. (A-C) After 48 h, culture supernatants were collected and assayed for (A) IL-12p70, (B) IFN-γ, and (C) TNF-α by ELISA. Results are expressed as means and S.E.M. Results from cocultures were analyzed by one-way ANOVA, followed by Tukey post-test. Significant differences were indicated for *P <0.05, **P <0.01, ***P <0.001.
Discussion
RANKL might play a costimulatory role in the immune responses mediated by APCs and T cells during L. major infection, as previously suggested (24). Now, we expand this view, by proposing that RANKL works in concert with IFN-γ to improve macrophage effector mechanisms for macrophage-mediated immunity and inflammatory responses. Whereas, inflammatory macrophages from B6 mice express RANK and a M2-like phenotype, RANKL induces their differentiation into F4/80hi-mature macrophages, able to secrete inflammatory cytokines. Moreover, RANKL cooperates with IFN-γ to skew M2-like into M1 macrophages, which express iNOS, NO production, and parasite killing, as discussed below.
RANKL-RANK axis is known to play a major role in osteoclastogenesis. In combination with other cytokines, RANKL promotes osteoclast maturation, but it also induces regulatory mechanisms, such as NO production by osteoclasts to maintain bone homeostasis (9). In certain conditions, RANKL might also induce iNOS expression and NO production by macrophages (9, 11). Moreover, the presence of RANKL in the vicinity of iNOS-expressing macrophages in rat bones indicates that, in addition to the role in osteoclastogenesis, RANKL might promote osteogenesis, by inducing M1 macrophages (25). Conversely, depending on the tissue environment and cell origin, RANKL might induce M2 macrophages (43), as well as either IL-12- or IL-10-producing DCs (44), and play tolerogenic/immunoregulatory effects in vivo. These examples highlight how RANKL affects different immune responses in a context/cell-dependent fashion, and that specific features of each model/disease should be considered to design studies to address the role of RANKL on immune cells.
It has been reported that RANKL has direct effects on DCs, by inducing IL-12 secretion and APC function, both relevant for T-cell activation and vaccine development (18, 20). Similarly, whereas B6 inflammatory macrophages expressed a M2-like phenotype, we found that RANKL directly induced IL-12 and TNF-α responses. Moreover, we show, for the first time, that RANKL favors M1 phenotype, by inducing IL-12, but suppressing MGL, arginase-1, and other M2 features. By contrast, although RANKL (at suboptimal doses) may have a limited effect on iNOS expression (22, 25) and NO production, it does act in synergism with IFN-γ to induce iNOS/NO by macrophages. It is reasonable to propose that a higher threshold for potential harmful NO responses demands either multiple signaling pathways initiated by both RANKL and IFN-γ, including NF-κB activation [our data and Guerrini and Takayanagi (10)], as well as epigenetic and post-transcriptional mechanisms induced by IFN-γ to upregulate iNOS expression (38).
Here we show that RANKL and IFN-γ promote the differentiation of M1 macrophages, which might be able to activate IFN-γ-producing Th1 cells to further upregulate M1 effector mechanisms. In contrast to Park et al. (22), we found that treatment with RANKL and IFN-γ upregulated the coinhibitory PD-L1 molecule and reduced macrophage expression of costimulatory features required for T-cell priming. It is likely that the differentiation into effector M1 macrophages is followed by reduced ability to prime naïve T cells along with increased regulatory activity on effector PD-1+ T cells, potentially deleterious to protect against Leishmania infection (45). In order to understand the role of RANKL and IFN-γ at the effector phase of Th1 and M1 responses, we investigated the crosstalk between CD4 T cells and macrophages from L. major-infected mice, by using anti-RANKL and anti-IFN-γ to neutralize endogenously produced cytokines. Whereas, both RANKL and IFN-γ concurred for IL-12 responses, RANKL was also required for IFN-γ production by T cells. Here, we highlighted a previously unexplored role for RANKL and IFN-γ in the interplay between Th1 cells and infected macrophages, by upregulating each other cytokine responses to parasite infection. Altogether, these results are relevant for both T-cell and macrophage-mediated immunity to L. major infection and expand previous findings on the role of RANKL as a costimulatory molecule that stimulate APC-T cell crosstalk in vivo (24). In a model, where CD40L-deficient mice were resistant to L. major, the blockade of RANKL induced susceptibility to infection, by reducing Th1 responses (24). Based on our findings, we suggest that in addition to possible defective APC/T-cell responses, mice lacking RANKL/RANK axis might have limited M1 effector mechanisms for parasite killing, such as iNOS expression and NO production.
We further addressed the role of RANKL on the potential effector mechanisms for macrophage-mediated immunity toward L. major parasites. We show that RANKL and IFN-γ cooperate to help parasite killing by B6 inflammatory macrophages in a NO-dependent fashion. More interesting, whereas NO plays a role as an effector molecule, ROS-related mechanisms might also concur for parasite killing by macrophages stimulated with RANKL and IFN-γ. Accordingly, L. major infection is able to induce ROS production by macrophages [data not shown and Goncalves et al. (46)]. Moreover, inhibition of peroxidase, by using DFO (or NAC), suppressed parasite killing even in the presence of increased NO production. These findings are consistent with the idea that both NO and ROS are necessary to kill L. major, through a peroxinitrite-dependent mechanism (47). Nonetheless, in contrast to these direct effects of NAC/DFO on infected B6 macrophages stimulated with RANKL and IFN-γ, the use of NAC to treat susceptible BALB/c mice reduced both lesions and parasite burden, most likely by increasing Th1 and M1-cytokine responses to L. major infection (48).
It has been reported that RANKL can act as an adjuvant in vaccines, by enhancing T-cell activation and Th1 responses induced by DCs (20, 49). Interestingly, a DNA vaccine containing RANKL gene stimulated antigen-specific T-cell response to Trypanosoma cruzi infection (49). Here we show that RANKL was able to act on inflammatory immature cells to induce more mature macrophages that express F4/80 at higher level. Moreover, RANKL costimulated the effector arm of Th1 responses, by potentiating the M1 effector mechanisms and parasite killing by infected macrophages. It is worth investigating whether RANKL could counteract a maturation deficit, which underlies defective macrophage immunity to Leishmania infection in susceptible mice (50–53). According to this idea, we found that RANKL and IFN-γ also enhance NO production in susceptible BALB/c macrophages. Hence, RANKL, in combination with IFN-γ, has potential use for local therapy, as previously suggested for IL-12 (54), as well as for therapeutic vaccines, reducing the risk of systemically side effects on bone homeostasis. Altogether, our results indicate that RANKL produced by activated T cells might costimulate T-cell-macrophage crosstalk: 1- to upregulate IL-12 and induction of Th1 responses; 2- to promote M1-mediated immunity in concert with IFN-γ. Therefore, RANKL might help protective Th1-mediated immune responses to Leishmania infection, by directly acting on macrophages to shift M2-like into effector M1 macrophages.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee for Use of Animals at the Federal University of Rio de Janeiro (UFRJ).
Author Contributions
TR performed and designed experiments, analyzed data, and co-wrote the manuscript. NV, MC-P, LF-C, and AF performed cell culture, cytokine, and flow cytometry assays. UL analyzed and discussed data. GD designed the research, supervised the experiments, and analyzed data. ML designed the research, supervised the experiments, analyzed data, and wrote the manuscript. TR, NV, MC-P, LF-C, UL, and AF approved the final version of the manuscript.
Funding
This work was supported by the Brazilian National Research Council (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq), the Rio de Janeiro State Science Foundation (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, FAPERJ), and the National Institute of Science and Technology (INCT-INPeTAm/CNPq/MCT). ML, UL, and AF are research fellows at CNPq, Brazil. We also received fellowships from CNPq (TR), FAPERJ (AF and MC-P), and the American Association of Immunologists (NV and ML).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank George Alexandre DosReis (in memoriam) for his major scientific and advisor contribution to this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00886/full#supplementary-material
References
1. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. (1997) 390:175–9. doi: 10.1038/36593
2. Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. (1997) 186:2075–80. doi: 10.1084/jem.186.12.2075
3. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. (1998) 93:165–76. doi: 10.1016/s0092–8674(00)81569-x
4. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. (1998) 95:3597–602. doi: 10.1073/pnas.95.7.3597
5. Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. (1998) 253:395–400. doi: 10.1006/bbrc.1998.9788
6. Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. (1999) 96:3540–5. doi: 10.1073/pnas.96.7.3540
7. Nagy V, Penninger JM. The RANKL-RANK story. Gerontology. (2015) 61:534–42. doi: 10.1159/000371845
8. Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. (2002) 277:44347–56. doi: 10.1074/jbc.M202009200
9. Zheng H, Yu X, Collin-Osdoby P, Osdoby P. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem. (2006) 281:15809–20. doi: 10.1074/jbc.M513225200
10. Guerrini MM, Takayanagi H. The immune system, bone and RANKL. Arch Biochem Biophys. (2014) 561:118–23. doi: 10.1016/j.abb.2014.06.003
11. Takada Y, Aggarwal BB. Evidence that genetic deletion of the TNF receptor p60 or p80 in macrophages modulates RANKL-induced signaling. Blood. (2004) 104:4113–21. doi: 10.1182/blood-2004–04-1607
12. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
13. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. (1999) 13:2412–24. doi: 10.1101/gad.13.18.2412
14. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. (2011) 17:1231–4. doi: 10.1038/nm.2452
15. Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. (2007) 39:960–2. doi: 10.1038/ng2076
16. Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. (2008) 83:64–76. doi: 10.1016/j.ajhg.2008.06.015
17. Pangrazio A, Cassani B, Guerrini MM, Crockett JC, Marrella V, Zammataro L, et al. RANK-dependent autosomal recessive osteopetrosis: characterization of five new cases with novel mutations. J Bone Miner Res. (2012) 27:342–51. doi: 10.1002/jbmr.559
18. Josien R, Wong BR, Li HL, Steinman RM, Choi Y. TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J Immunol. (1999) 162:2562–8.
19. Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y. TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med. (1999) 189:1025–31. doi: 10.1084/jem.189.7.1025
20. Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, et al. TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J Exp Med. (2000) 191:495–502. doi: 10.1084/jem.191.3.495
21. Cremer I, Dieu-Nosjean MC, Marechal S, Dezutter-Dambuyant C, Goddard S, Adams D, et al. Long-lived immature dendritic cells mediated by TRANCE-RANK interaction. Blood. (2002) 100:3646–55. doi: 10.1182/blood-2002–01-0312
22. Park HJ, Park OJ, Shin J. Receptor activator of NF-kappaB ligand enhances the activity of macrophages as antigen presenting cells. Exp Mol Med. (2005) 37:524–32. doi: 10.1038/emm.2005.65
23. Chen NJ, Huang MW, Hsieh SL. Enhanced secretion of IFN-gamma by activated Th1 cells occurs via reverse signaling through TNF-related activation-induced cytokine. J Immunol. (2001) 166:270–6. doi: 10.4049/jimmunol.166.1.270
24. Padigel UM, Kim N, Choi Y, Farrell JP. TRANCE-RANK costimulation is required for IL-12 production and the initiation of a Th1-type response to Leishmania major infection in CD40L-deficient mice. J Immunol. (2003) 171:5437–41. doi: 10.4049/jimmunol.171.10.5437
25. Huang R, Wang X, Zhou Y, Xiao Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Res. (2017) 5:17019. doi: 10.1038/boneres.2017.19
26. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. (2002) 2:845–58. doi: 10.1038/nri933
27. McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev. (2004) 201:206–24. doi: 10.1111/j.0105–2896.2004.00190.x
28. Alexander J, Bryson K. T helper (h)1/Th2 and Leishmania: paradox rather than paradigm. Immunol Lett. (2005) 99:17–23. doi: 10.1016/j.imlet.2005.01.009
29. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol. (2011) 9:604–15. doi: 10.1038/nrmicro2608
30. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol. (2016) 16:581–92. doi: 10.1038/nri.2016.72
31. Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. (2018) 215:357–75. doi: 10.1084/jem.20171389
32. Tomiotto-Pellissier F, Bortoleti B, Assolini JP, Goncalves MD, Carloto ACM, Miranda-Sapla MM, et al. Macrophage polarization in leishmaniasis: broadening horizons. Front Immunol. (2018) 9:2529. doi: 10.3389/fimmu.2018.02529
33. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
34. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/s1471-4906(02)02302-5
35. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
36. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
37. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
38. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. (2020) 15:123–147. doi: 10.1146/annurev-pathmechdis-012418–012718
39. Allman WR, Dey R, Liu L, Siddiqui S, Coleman AS, Bhattacharya P, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci USA. (2015) 112:E4094–103. doi: 10.1073/pnas.1421580112
40. Vila-del Sol V, Diaz-Munoz MD, Fresno M. Requirement of tumor necrosis factor alpha and nuclear factor-kappaB in the induction by IFN-gamma of inducible nitric oxide synthase in macrophages. J Leukocyte Biol. (2007) 81:272–83. doi: 10.1189/jlb.0905529
41. Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, et al. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol. (2010) 184:877–85. doi: 10.4049/jimmunol.0902346
42. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Direct Autoimmun. (2010) 11:27–60. doi: 10.1159/000289196
43. Meng YH, Zhou WJ, Jin LP, Liu LB, Chang KK, Mei J, et al. RANKL-mediated harmonious dialogue between fetus and mother guarantees smooth gestation by inducing decidual M2 macrophage polarization. Cell Death Dis. (2017) 8:e3105. doi: 10.1038/cddis.2017.505
44. Williamson E, Bilsborough JM, Viney JL. Regulation of mucosal dendritic cell function by receptor activator of NF-kappa B (RANK)/RANK ligand interactions: impact on tolerance induction. J Immunol. (2002) 169:3606–12. doi: 10.4049/jimmunol.169.7.3606
45. da Fonseca-Martins AM, Ramos TD, Pratti JES, Firmino-Cruz L, Gomes DCO, Soong L, et al. Immunotherapy using anti-PD-1 and anti-PD-L1 in Leishmania amazonensis-infected BALB/c mice reduce parasite load. Sci Rep. (2019) 9:20275. doi: 10.1038/s41598–019-56336–8
46. Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of Leishmania major infection. J Exp Med. (2011) 208:1253–65. doi: 10.1084/jem.20101751
47. Linares E, Giorgio S, Mortara RA, Santos CX, Yamada AT, Augusto O. Role of peroxynitrite in macrophage microbicidal mechanisms in vivo revealed by protein nitration and hydroxylation. Free Radical Biol Med. (2001) 30:1234–42. doi: 10.1016/s0891–5849(01)00516–0
48. Rocha-Vieira E, Ferreira E, Vianna P, De Faria DR, Gaze ST, Dutra WO, et al. Histopathological outcome of Leishmania major-infected BALB/c mice is improved by oral treatment with N-acetyl-l-cysteine. Immunology. (2003) 108:401–8. doi: 10.1046/j.1365–2567.2003.01582.x
49. Miyahira Y, Akiba H, Katae M, Kubota K, Kobayashi S, Takeuchi T, et al. Cutting edge: a potent adjuvant effect of ligand to receptor activator of NF-kappa B gene for inducing antigen-specific CD8+ T cell response by DNA and viral vector vaccination. J Immunol. (2003) 171:6344–8. doi: 10.4049/jimmunol.171.12.6344
50. Beil WJ, Meinardus-Hager G, Neugebauer DC, Sorg C. Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leukocyte Biol. (1992) 52:135–42.
51. Sunderkotter C, Kunz M, Steinbrink K, Meinardus-Hager G, Goebeler M, Bildau H, et al. Resistance of mice to experimental leishmaniasis is associated with more rapid appearance of mature macrophages in vitro and in vivo. J Immunol. (1993) 151:4891–901.
52. Steinbrink K, Schonlau F, Rescher U, Henseleit U, Vogel T, Sorg C, et al. Ineffective elimination of Leishmania major by inflammatory (MRP14-positive) subtype of monocytic cells. Immunobiology. (2000) 202:442–59. doi: 10.1016/s0171-2985(00)80103-5
53. Vellozo NS, Pereira-Marques ST, Cabral-Piccin MP, Filardy AA, Ribeiro-Gomes FL, Rigoni TS, et al. All-Trans retinoic acid promotes an M1- to M2-phenotype shift and inhibits macrophage-mediated immunity to leishmania major. Front Immunol. (2017) 8:1560. doi: 10.3389/fimmu.2017.01560
Keywords: classically-activated macrophage, leishmaniasis, M1 and M2 macrophages, nitric oxide, ODF, OPGL, RANKL, TRANCE
Citation: Rigoni TS, Vellozo NS, Cabral-Piccin M, Fabiano-Coelho L, Lopes UG, Filardy AA, DosReis GA and Lopes MF (2020) RANK Ligand Helps Immunity to Leishmania major by Skewing M2-Like Into M1 Macrophages. Front. Immunol. 11:886. doi: 10.3389/fimmu.2020.00886
Received: 20 December 2019; Accepted: 16 April 2020;
Published: 12 May 2020.
Edited by:
Heinrich Korner, University of Tasmania, AustraliaReviewed by:
Walderez Ornelas Dutra, Federal University of Minas Gerais, BrazilHira Nakhasi, Center for Biologics Evaluation and Research (FDA), United States
Copyright © 2020 Rigoni, Vellozo, Cabral-Piccin, Fabiano-Coelho, Lopes, Filardy, DosReis and Lopes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela F. Lopes, bWFyY2VsYWxAYmlvZi51ZnJqLmJy