 Dahee Shim
Dahee Shim Hagyu Kim
Hagyu Kim Sung Jae Shin
Sung Jae Shin- 1Department of Microbiology, Institute for Immunology and Immunological Diseases, Brain Korea 21 Program for Leading Universities and Students (PLUS) Project for Medical Science, Yonsei University College of Medicine, Seoul, South Korea
- 2Department of Life Science, Research Institute for Natural Sciences, College of Natural Sciences, Hanyang University, Seoul, South Korea
Tuberculosis (TB) is a leading cause of death worldwide following infection with Mycobacterium tuberculosis (Mtb), with 1.5 million deaths from this disease reported in 2018. Once the bacilli are inhaled, alveolar and interstitial macrophages become infected with Mtb and differentiate into lipid-laden foamy macrophages leading to lung inflammation. Thus, the presence of lipid-laden foamy macrophages is the hallmark of TB granuloma; these Mtb-infected foamy macrophages are the major niche for Mtb survival. The fate of TB pathogenesis is likely determined by the altered function of Mtb-infected macrophages, which initiate and mediate TB-related lung inflammation. As Mtb-infected foamy macrophages play central roles in the pathogenesis of Mtb, they may be important in the development of host-directed therapy against TB. Here, we summarize and discuss the current understanding of the alterations in alveolar and interstitial macrophages in the regulation of Mtb infection-induced immune responses. Metabolic reprogramming of lipid-laden foamy macrophages following Mtb infection or virulence factors are also summarized. Furthermore, we review the therapeutic interventions targeting immune responses and metabolic pathways, from in vitro, in vivo, and clinical studies. This review will further our understanding of the Mtb-infected foamy macrophages, which are both the major Mtb niche and therapeutic targets against TB.
Introduction
Tuberculosis (TB) is a chronic inflammatory disease caused by a Mycobacterium tuberculosis (Mtb) infection (1). When the Mtb bacilli become inhaled into alveoli, the bacilli are phagocytosed by alveolar macrophages in the lung (2). Phagocytosed Mtb uses various approaches to avoid host defense mechanisms, such as inhibition of phagosome maturation, expression of virulence-associated factors, inhibition of phagolysosomal fusion, and protection from reactive oxidative radicals (3, 4). Following infection with Mtb, alveolar macrophages migrate to the interstitium and induce inflammatory responses, resulting in the extravasation of dendritic cells, neutrophils, natural killer cells, T cells, and B cells (2). These infiltrated immune cells surround the infected alveolar macrophages, which are reservoirs of Mtb, to construct TB granulomas (5–7). Thus, understanding the fate of alveolar macrophages at the initial infectious phase is critical for preventing TB pathogenesis.
During the construction of TB granulomas, Mtb-infected macrophages accumulate lipid bodies in their cytosolic area, differentiating into foamy macrophages, which are hallmarks of TB lesions (8, 9). The accumulated bubble-like lipid bodies contain cholesteryl esters and triglycerides (10). Mtb-infected foamy macrophages play central roles in granuloma development, maintenance, and infection dissemination (9). In advanced granulomas, the core region is characterized by caseous necrosis, which further leads to the formation of a lipid-rich environment (6, 8, 9, 11). In granulomas, Mtb can grow and persist in foamy macrophages and the necrotic core (7). When foamy macrophages leave the original granuloma, a secondary granuloma is established, promoting dissemination (11). As the primary niche for Mtb, determining the features of Mtb-infected foamy macrophages is essential for investigating and controlling TB pathogenesis. This review describes the current understanding of foamy macrophages infected with Mtb.
Macrophages
Differentiation and General Features of Lung Macrophages
Macrophages are well-classified by their ontogeny (12–14). The functions and phenotypes of macrophages are influenced by their developmental origins and locations. Macrophages are generally formed as three major precursors: embryonic yolk sac precursor, fetal liver precursors, and bone-marrow derived blood monocytes (13, 14). In the steady state lungs, macrophages consist of alveolar macrophages and interstitial macrophages (15). The alveolar macrophages originate from fetal liver macrophages and fetal monocytes under the control of granulocyte/macrophage colony-stimulating factor (GM-CSF), peroxisome proliferator-activated receptors-gamma (PPAR-γ), and the lung microenvironment (16–19). To maintain homeostatic regulation, alveolar macrophages have a unique enhancer repertoire, including Spi-C and Car4, which are induced by the macrophage lineage-determining factor PU.1 in the lung-specific microenvironment (20, 21). A recent study demonstrated that mammalian target of rapamycin (mTOR) signaling is also required for the self-renewing ability of alveolar macrophages, accompanied by a distinctive metabolic signature based on the expression of sterol regulatory binding protein (SREBP) target genes (22). While undergoing tissue imprinting in the lungs, alveolar macrophages generally express immunosuppressive genes, including tumor growth factor-β (TGF-β) and interleukin-10 (IL-10) (17, 23). Alveolar macrophages have unique phenotypic features; they are loosely adherent, round-shaped cells expressing high levels of cluster of differentiation 206 (CD206), which detects microbial carbohydrates and high levels of scavenger receptors, such as macrophage scavenger receptor class A and macrophage receptor with collagenous structure (23). Notably, alveolar macrophages also express high levels of CD11c and CD170; moreover, their expression of F4/80 and CR3 is low or absent compared with that in other parts of the lung or peripheral tissue-resident macrophages (17).
In contrast, interstitial macrophages originate from primitive yolk sac macrophages and bone marrow monocytes (15). These cells are smaller and have monocyte-like morphology, which is characterized by a high nuclear/cytoplasm ratio with cytosolic vacuoles compared to alveolar macrophages (24). Interstitial macrophages express high levels of CD11b, CD64, F4/80, and the proto-oncogene tyrosine-protein kinase MER (25). Following invasion by microorganisms, interstitial macrophages play a role in the second-line defense via their phagocytosis and antigen presentation abilities (26). These interstitial macrophages have diverse immune responses depending on the activation stimulus, explained by the concepts of M1 and M2 macrophages. The activated spectrums and concepts for the M1 and M2 macrophages are further described below.
General Functions and Metabolic Programing of M1 and M2 Macrophages and Their Distinct Roles in Lung Inflammation by Mtb Infection
Overall, when macrophages are infected with bacteria or viruses, they elicit pro-inflammatory responses by releasing anti-microbial proteins and cytokines, such as complement proteins, tumor necrosis factor α, IL-1β, IL-6, IL-12, and IL-23 (27–29). These pro-inflammatory macrophages can activate endothelial cells in the blood vessels to support extravasation of other immune cells into inflamed areas (28, 29). Macrophages also trigger T cell responses through their antigen presenting abilities via major histocompatibility complex II molecules (30). In vitro, macrophages can be classically activated by lipopolysaccharide or interferon γ (IFN-γ) to mimic bacterial infection or pro-inflammatory activation, respectively. These classically activated macrophages are named as M1 macrophages (31). From the perspective of immunometabolism, M1 macrophages are well-known to drive pro-inflammatory responses during the metabolic switch for glycolysis with a broken tricarboxylic acid (TCA) cycle (32, 33). The pentose phosphate pathway and NAD+ salvage pathway are essential for generation of mitochondrial reactive oxygen species (ROS), which induces DNA damage following M1 macrophage activation (34). In addition to massive glycolysis with an impaired TCA cycle, glucose-derived carbons are incorporated into fatty acids or sterol via lipogenesis in activated M1 macrophages (35).
Macrophages also play pro-resolving or anti-inflammatory roles that depend on signal transducer and activator of transcription-6 (STAT-6) and IL-10 release (27–29). These macrophages are involved in the phagocytosis of apoptotic cells, induction of collagen deposition, and coordination of tissue integrity to reinforce tissue repair, regeneration, and fibrosis (27, 36). There is also a method to polarize anti-inflammatory macrophages through the activation of IL-4/IL-13 or IL-10 in vitro; these alternatively activated macrophages are generally named as M2 macrophages (31). M2 macrophages preferentially utilize oxidative phosphorylation and fatty acid oxidation (FAO) to drive anti-inflammatory responses (37). In M2 macrophage activation with IL-4 treatment, mitochondrial metabolism is regulated by polyamine biosynthesis to regulate the integrity of the TCA cycle and electron transport chain (38). Collectively, M1 and M2 macrophages may undergo distinct lipid metabolic reprogramming, followed by their differential immune responses.
When infected with Mtb, both alveolar and interstitial macrophages play important roles in defending against TB and modulate immune responses (39). Mtb-infected alveolar macrophages exhibit enrichment in gene sets such as those involved in lipid uptake, oxidative phosphorylation and fatty acid oxidation, similar to M2 macrophages (39, 40). Mtb-infected alveolar macrophages express an antioxidant transcriptional signature via NRF2-dependent pathways, resulting in impaired control of Mtb growth with reduced inflammatory responses in the early stage of infection (40, 41). Mtb enhances the expression of genes in alveolar macrophages that are involved in Mtb division, growth, ribosomal protein synthesis, cell wall synthesis, fatty acid import, mycolic acid biosynthesis, the TCA cycle, and β-oxidation (40). Because the features of interstitial macrophages are determined by immunological stimuli, TB lesions have been investigated using M1 and M2 macrophages to dissect the immune responses occurring during TB progression (42, 43). In the early stage of Mtb infection, interstitial macrophages are typically differentiated to M1 macrophages. Mtb-infected interstitial macrophages express gene sets for cell adhesion, chemotaxis, ROS biosynthesis, nuclear factor-κB responses, hypoxia, and glycolysis in vivo (39, 40). In M1-like interstitial macrophages, Mtb shows a gene signature related to the response to environmental stresses and a non-replicative state (40). Mtb-infected M1 macrophages are transformed into M2 macrophages over time by the 6-kDa early secretory antigenic target (ESAT-6), which is a major virulence factor of Mtb (43). In addition to controlling the bacterial burden, modulating granuloma formation, and immune responses, Mtb-infected macrophages contribute to TB dissemination (44, 45). Mtb was shown to translocate from the phagolysosome to the cytosol, thereby eliciting host cell apoptosis in an ESAT-6–dependent manner (44). Apoptotic cells are then phagocytosed by newly infiltrating macrophages to generate the primary granuloma, and Mtb-infected macrophages egress to the distal tissues, contributing to the initiation of secondary granuloma formation (45) (Figure 1A).
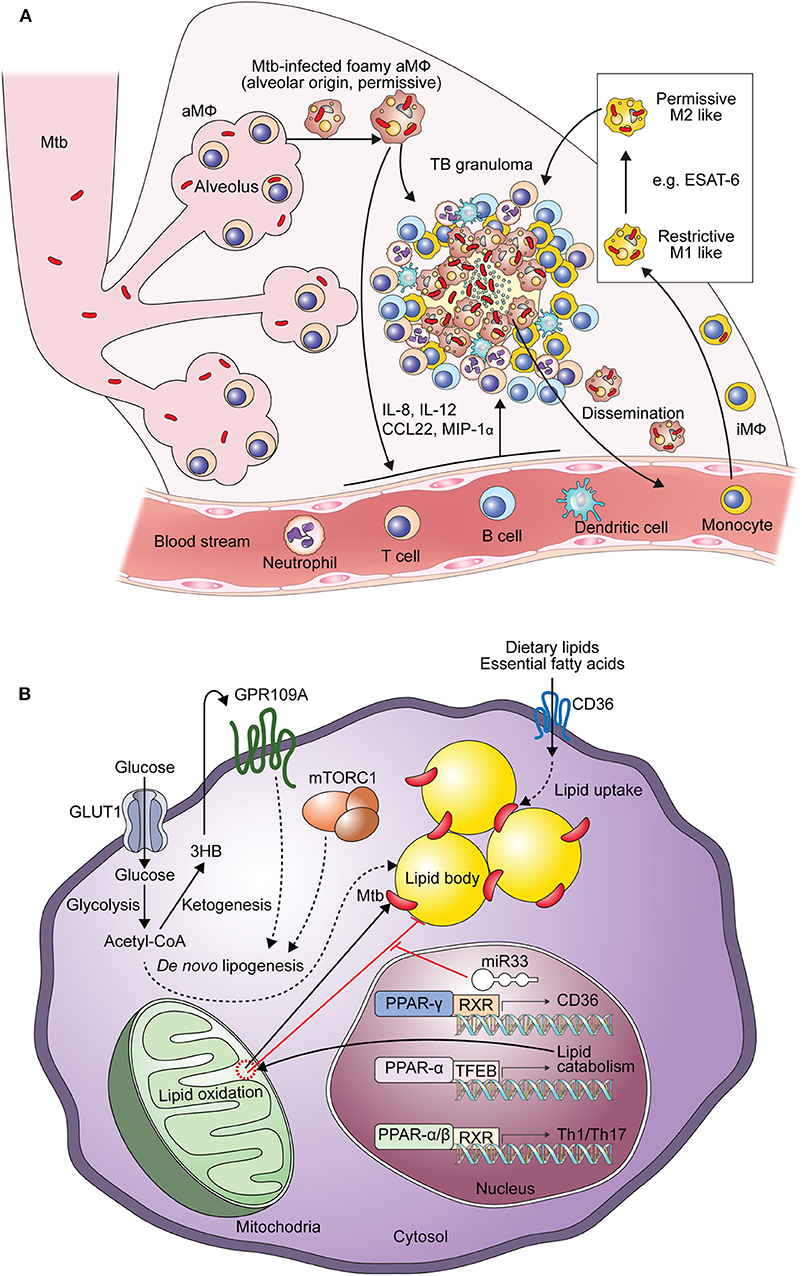
Figure 1. Generation of Mtb-infected foamy macrophages during the formation of TB granulomas. (A) Composition of Mtb-infected foamy macrophages during TB pathogenesis. Alveolar macrophages initially infected by Mtb and translocated into the interstitial space to generate immune responses. With the extravasation of immune cells Mtb-infected alveolar macrophages differentiate into foamy macrophages. Infiltrated interstitial macrophages are also infected with Mtb and further differentiate into foamy macrophages. In the early stage of Mtb infection, macrophages show pro-inflammatory responses like M1 macrophages contributing to the restriction of Mtb survival. ESAT-6, a representative virulence factor of Mtb, polarizes these M1 macrophages into M2 macrophages to induce permissive responses in Mtb survival in the chronic stage of TB. These Mtb-infected foamy macrophages are hallmarks of TB granulomas; translocation of Mtb-infected foamy macrophages induces dissemination of Mtb. “aMΦ” and “iMΦ” indicate “alveolar macrophage” and “interstitial macrophage,” respectively. (B) Metabolic perturbation by Mtb infection to generate foamy macrophages with Mtb infection, lipid accumulation leads to the generation foamy macrophages via metabolic reprogramming. In the early stage of Mtb infection, excessive glycolysis with defective mitochondrial respiration contributes to de novo lipogenesis. Acetyl-CoA, a product of glycolysis, is metabolized to 3-hydroxybutyrate (3-HB) by ketogenesis to induce GPR109A signaling. De novo lipogenesis is also induced by signal transduction of GPR109A and mTORC1 signaling, which is induced by macrophage activation. Nuclear receptors, such as those in the PPAR and LXR family, also contribute to both metabolic reprogramming and immune responses. The expression of miR33 is induced by Mtb and miR33 inhibits lipid catabolism, supporting Mtb survival. Direct and indirect processes are indicated by arrows and dotted arrows, respectively.
From the perspective of immunometabolism, the Warburg effect in M1 macrophages generates pro-inflammatory responses, such as the secretion of IL-1β, which is known as a beneficial cytokine against Mtb (46, 47). Induced Warburg effects in the early phase of Mtb infection also generate ROS with the activation of hypoxia inducible factor 1α, which is necessary to induce Mtb specific IFN-γ dependent immunity (48, 49). IL-12, another cytokine secreted by M1 macrophages, can generate protective immune responses against Mtb (50). In contrast, M2 macrophages possess less bactericidal activities compared to M1 macrophages against Mtb infection (42). IL-10, a representative cytokine secreted from M2 macrophages, suppresses antimycobacterial immunity and promotes Mtb survival (51). It has been reported that another M2 macrophage-secreted cytokine, TGF-β, suppresses IFN-γ responses from T cells against Mtb in the lungs of mice and humans infected with TB; deletion of TGF-β signaling decreases the bacterial burden via the generation of cytotoxic T cell responses in TB granulomas (52).
Collectively, these investigations suggest the dynamic activation status of Mtb-infected macrophages (Figure 1A). Alveolar macrophages show enhanced lipid metabolism via PPAR-γ and inflammatory properties that facilitate infection and persistence of Mtb bacilli (18). In the progression of TB granulomas, blood monocyte-derived macrophages infiltrate and polarize to M1 macrophages. In the early phase of Mtb infection, excessive glycolysis with increasing lipids drives fatty acid synthesis in M1 macrophages. Further differentiation to M2 macrophages by ESAT-6 is closely linked to FAO with anti-inflammatory responses, providing favorable environments for Mtb survival (43). Therefore, these lipid metabolic reprogramming pathways may be considered targets for supporting host-directed therapy (HDT) to elicit anti-TB immune responses.
Interaction Between Mtb and Mtb–Driven Foamy Macrophages
Characteristics of Mtb-Infected Foamy Macrophages and the Utilization of Their Lipids by Mtb
Alterations of metabolic pathways are involved not only in the inflammatory responses of macrophages but also in the transformation of Mtb-infected macrophages into foamy macrophages, which are the major contributors to TB pathogenesis (Figure 1B), as previously described (5, 6). Foamy macrophages are named based on their morphology as they contain bubble-like lipid bodies in their cytoplasm (53). Mtb-infected foamy macrophages have different features compared to non-foamy macrophages (Table 1, upper). Specifically, Mtb-infected foamy macrophages express higher levels of MHCII, CD11c, CD40, and CD205, similar to dendritic cells, but show reduced capacity for antigen processing (54). Moreover, Mtb-infected foamy macrophages induce nitric oxide with elevated secretion of TGF-β to suppress T cell responses (55–57).
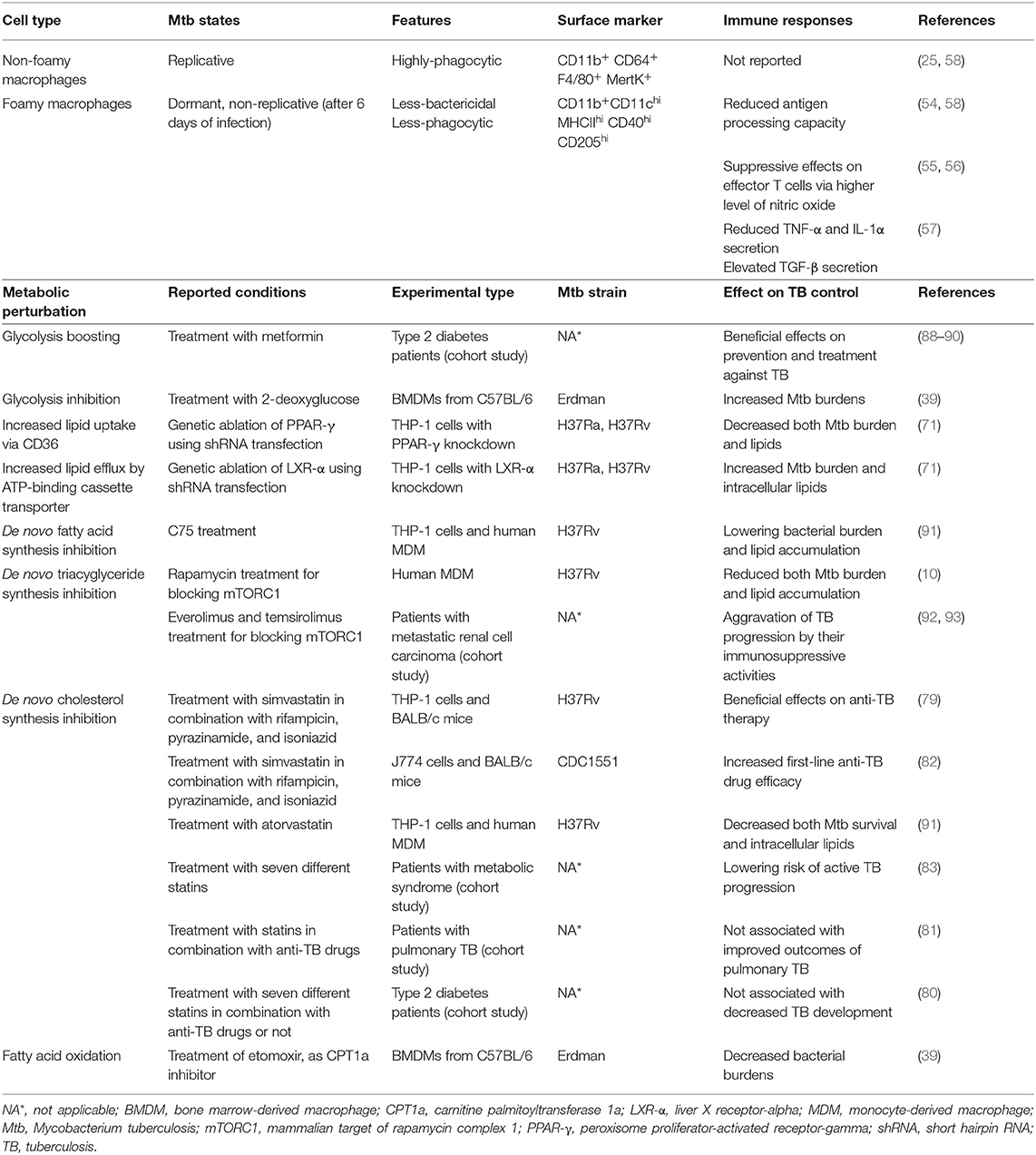
Table 1. Comparison of general characteristics between foamy and non-foamy macrophages, and metabolic interventions of Mtb-infected foamy macrophages for host-directed therapy.
Upon infection, intracellular Mtb is located in the phagosomes, and the membrane of Mtb-containing phagosomes is enclosed and interacts with lipid bodies (58). A recent study demonstrated that the phagosome–lipid body interaction is regulated by the mycobacterial cell wall components lipoarabinomannan and phosphatidylinositol mannoside mediated by the late endosome marker Rab7 (59). After the phagosomes surrounded lipid bodies, Mtb translocated to lipid bodies for the utilization of lipids, such as cholesterols, fatty acids, and triglycerides, as carbon sources for survival. Mtb takes advantage of cholesterol and fatty acids in foamy macrophages to generate energy and metabolic intermediates via the expression of isocitrate lyases for the glyoxylate cycle (60). To utilize lipids, Mtb contains abundant genes encoding lipid transporters and lipolytic enzymes. It has been reported that LucA and Mce1 are expressed in Mtb to import fatty acids (61). Rv0200/OmamB, Rv0172/Mce1D, Rv0655/MceG, and Rv0966c are also known as fatty acid transporters of Mtb in foamy macrophages (62). Rv2672 is the membrane-associated Mtb protein mycobacterial secreted hydrolase 1 and is required for Mtb persistence via utilization of host lipids under hypoxic conditions (63). In addition to Mtb survival, fatty acids are required for the synthesis of virulence-associated lipids, including polyketide lipids phiocerol-dimycoseroic acid, poly-acylated trehaloses, sulfolipids, and mycolic acids (60). Increased accumulation of triglycerides and elevated levels of triglyceride synthetase 2 have been reported in the modern Mtb Beijing strain compared to the ancient Mtb Beijing strain. Moreover, these elevated levels of triglyceride are associated with rapid disease development (64).
Furthermore, it has been suggested the use of lipids in host cells is related to dormancy of Mtb (58, 65–67). It is reported that Mtb persists in a dormant non-replicative state in foamy macrophages compared to infect non-foamy macrophages (58). Mtb utilize fatty acids from host cells to generate intracellular lipid inclusions, which are lipid bodies in the cytoplasm of mycobacteria. Bacilli with these inclusions show persistently arrested growth in response to stress (65, 66). Particularly, utilization of host triglycerides is required to obtain the dormancy-like phenotypes of Mtb in foamy macrophages (68). The Mtb mutant with reduced triglyceride synthesis is more sensitive to antibiotics compared to wild-type Mtb (65). The region of difference 1 protein in Mtb contributes to increasing the levels of intracellular triglycerides in Mtb by enhancing the expression of diacylglycerol O-acyltransferase, a key enzyme in triglyceride synthesis (67). Therefore, reducing lipid bodies in Mtb-infected foamy macrophages may control the intracellular survival of Mtb.
Mechanism of Lipid Accumulation of Foamy Macrophages by Mtb Infection and Their Implications for HDT
Recently, it has been reported that several bacterial factors of Mtb have additive modulatory effects on the lipid metabolism of host cells (Figure 1B). For example, ESAT-6 stimulates the translocation of glucose transporter GLUT-1, resulting in the active transport of glucose with the perturbation of metabolic flux (69, 70). Enhanced glucose metabolism leads to increased de novo lipid synthesis, which elevates the accumulation of lipids in foamy macrophages (69). It has been reported that activation of the G protein-coupled receptor GPR109A, an anti-lypolytic receptor, by ESAT-6 leads to the accumulation of lipid bodies in foamy macrophages, contributing to Mtb survival (70). In contrast, blockage of glycolysis with 2-deoxyglucose is detrimental to defense against Mtb infection because it disturbs M1-like activation in interstitial macrophages (39). Overall, glycolysis has contradictory roles against Mtb infection. By inducing pro-inflammatory immune responses, glycolysis with a broken TCA cycles supports anti-microbial responses in macrophages (39). However, excessive glycolysis is associated with elevated lipid accumulation by bacterial factors, including ESAT-6, to generate a niche that is suitable for Mtb (69, 70). Therefore, metabolic links between excessive glycolysis and lipogenesis are potential targets for reducing bacterial burdens with effective immune responses (Table 1).
Mtb also modulates nuclear transcription receptors of host cells involved in metabolic reprogramming and immune responses (71, 72). The Mtb-induced PPAR-γ pathway, which is a prominent signaling pathway that induces the activation of M2 macrophages, leads lipid accumulation to support the intracellular survival of Mtb. In this processes, PPAR-γ, with testicular receptor 4, increases the level of CD36 to contribute to lipid uptake (71). Another PPAR family member, PPAR-α, is a transcriptional inducer of FAO that prevents lipid accumulation in Mtb-infected foamy macrophages and promotes autophagy with transcription factor EB to reduce intracellular Mtb growth (73). Liver X-receptor (LXR)-α and LXR-β are also critical regulators of oxysterol metabolism that modulate immune responses in Mtb infection (74, 75). In mice with Mtb infection, LXR-α and LXR-β signaling is required to generate protective T cell responses (74). It has been reported that LXR signaling plays a role in regulating antimicrobial peptide expression to restrict Mtb growth via IL-36 (75).
In addition to modulating nuclear transcription factor signaling pathways, infection by Mtb induces tumor necrosis factor receptor signaling, followed by the activation of its downstream mTOR complex 1 (mTORC1) and caspase pathways in human primary macrophages (10). Activation of mTORC1 results in the accumulation of triglycerides in Mtb-infected macrophages. Mtb also induces miR-33 expression in foamy macrophages to inhibit lipid catabolism and autophagy, enabling intracellular survival and persistence (76).
Additionally, accumulating evidence has suggested that the disruption of lipid homeostasis, including hypercholesterolemia, leads to increased TB susceptibility. It is reported that apolipoprotein E-deficient mice have an increased susceptibility to hypercholesterolemia, showing defective priming of IFN-γ responses (77). Oxidized low-density lipoproteins, which are modified under hyperlipidemic conditions, also contribute to enhancing TB susceptibility via lysosomal dysfunction, impairing the control of Mtb survival (78). Statins, the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors for reducing cholesterol synthesis, have been attempted to treat Mtb infection (79–82). Clinically, treatment with statins has been reported to elicit improved outcomes for patients with TB (82, 83), but showed no additional effects in some cases (80, 81). By considering the characteristics of each patient, it is necessary for novel HDT approaches to modulate the immune response and/or cholesterol metabolism to be effective against TB. Collectively, these observations suggest that lipid metabolism and/or homeostatic pathways are promising targets for HDT against TB (Table 1, lower).
Future Direction and Perspectives
To discover new therapeutic strategies against TB, many researches have attempted to alleviate Mtb infection through HDT. Foamy macrophages provide a niche for bacilli survival, maintenance, and persistence using their enriched cytosolic lipids. Even worse, necrosis of Mtb-infected foamy macrophages results in the generation of the necrotic core of TB granulomas, causing extracellular growth of Mtb. As Mtb actively utilizes lipids to generate energy and virulent factors from host cells, the regulation of host lipid metabolism pathways is a potential therapeutic strategy for alleviating TB by preventing vicious cycles between Mtb and macrophages.
In summary, alveolar and interstitial macrophages exert opposite roles against Mtb infection; therefore, understanding these macrophages is important for establishing strategies against TB pathogenesis. In TB pathogenesis, alveolar macrophages are susceptible to Mtb with elevated fatty acid uptake and FAO. In contrast, interstitial macrophages show resistance features like M1 macrophages in the early stage of Mtb infection. Pro-inflammatory M1 macrophages exhibit massively increased glycolysis and fatty acid synthesis with bactericidal activities. M2 macrophages show elevated lipid catabolism with suppressive activities in their antibacterial responses against Mtb. Thus, the Mtb-induced transformation processes from M1 to M2 macrophages are potential immunological targets of TB. Some recent revolutionary investigations have been conducted using in vitro granuloma and organoid culture systems to gain further insights into TB (42, 84–86). Using in vitro granuloma culture systems, M1 macrophages were shown to transform into M2 macrophages following Mtb infection (42). Although these techniques have only provided limited information of Mtb-infected macrophages to date, they have potential for further detailed investigations of Mtb-infected foamy macrophages to suggest more promising targets of HDT. Moreover, the metabolic pathways of Mtb-infected foamy macrophages perturbed by Mtb infection are important targets of HDT against TB. A recent study indicated that Mtb-infected foamy macrophages could uptake more fluoroquinolones compared to non-foamy macrophages in vivo (87), suggesting that it is important to consider the functions of foamy macrophages for anti-TB drug treatment. With consideration of the physiology of macrophages, it is expected that HDT strategy targeting immunological functions and/or metabolic perturbation of Mtb-infected foamy macrophages can be developed.
Author Contributions
DS, HK, and SS wrote and revised the manuscript. SS conceptualized and edited the manuscript. All authors read and gave final approval of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2019R1A2C2003204), Republic of Korea.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank MID (Medical Illustration & Design), a part of the Medical Research Support Services of Yonsei University College of Medicine, for all artistic support related to this work.
References
1. The Lancet. End the tuberculosis emergency: a promise is not enough. Lancet. (2019) 394:1482. doi: 10.1016/S0140-6736(19)32459-6
2. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–46.e4. doi: 10.1016/j.chom.2018.08.001
3. Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. (2019) 43:341–61. doi: 10.1093/femsre/fuz006
4. Meena LS, Rajni. Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. (2010) 277:2416–27. doi: 10.1111/j.1742-4658.2010.07666.x
5. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:411. doi: 10.3389/fimmu.2012.00411
6. Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. (2014) 26:601–9. doi: 10.1016/j.smim.2014.09.009
7. Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol. (2017) 17:691–702. doi: 10.1038/nri.2017.69
8. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunolgy. (2009) 10:943–8. doi: 10.1038/ni.1781
9. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. (2012) 12:352–66. doi: 10.1038/nri3211
10. Guerrini V, Prideaux B, Blanc L, Bruiners N, Arrigucci R, Singh S, et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. (2018) 14:e1007223. doi: 10.1371/journal.ppat.1007223
11. McClean CM, Tobin DM. Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog Dis. (2016) 74:ftw068. doi: 10.1093/femspd/ftw068
12. Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. (2014) 262:56–73. doi: 10.1111/imr.12224
13. Ginhoux F, Guilliams M. Tissue-Resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
14. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
15. Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development. (2016) 143:1318–27. doi: 10.1242/dev.129122
16. Malur A, McCoy AJ, Arce S, Barna BP, Kavuru MS, Malur AG, et al. Deletion of PPAR gamma in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J Immunol. (2009) 182:5816–22. doi: 10.4049/jimmunol.0803504
17. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
18. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
19. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
20. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. (2014) 159:1327–40. doi: 10.1016/j.cell.2014.11.023
21. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
22. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, et al. mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science. (2017) 357:1014–21. doi: 10.1126/science.aaj2155
23. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol. (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
24. Fathi M, Johansson A, Lundborg M, Orre L, Skold CM, Camner P. Functional and morphological differences between human alveolar and interstitial macrophages. Exp Mol Pathol. (2001) 70:77–82. doi: 10.1006/exmp.2000.2344
25. Yu YR, O'Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, et al. A Protocol for the comprehensive flow cytometric analysis of immune cells in normal and inflamed murine non-lymphoid tissues. PLoS ONE. (2016) 11:e0150606. doi: 10.1371/journal.pone.0150606
26. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
27. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. (2014) 5:491. doi: 10.3389/fimmu.2014.00491
28. Ley K. M1 Means Kill; M2 Means Heal. J Immunol. (2017) 199:2191–3. doi: 10.4049/jimmunol.1701135
29. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
30. Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol. (2008) 181:5829–35. doi: 10.4049/jimmunol.181.9.5829
31. Murray PJ. Macrophage polarization. Ann Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
32. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
33. Wculek SK, Khouili SC, Priego E, Heras-Murillo I, Sancho D. Metabolic control of dendritic cell functions: digesting information. Front Immunol. (2019) 10:775. doi: 10.3389/fimmu.2019.00775
34. Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat Immunol. (2019) 20:420–32. doi: 10.1038/s41590-019-0336-y
35. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. (2012) 92:829–39. doi: 10.1189/jlb.1111537
36. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
37. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. (2019) 19:291–304. doi: 10.1038/s41577-019-0124-9
38. Puleston DJ, Buck MD, Klein Geltink RI, Kyle RL, Caputa G, O'Sullivan D, et al. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. (2019) 30:352–63.e8. doi: 10.1016/j.cmet.2019.05.003
39. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
40. Pisu D, Huang L, Grenier JK, Russell DG. Dual RNA-Seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. (2020) 30:335–50.e4. doi: 10.1016/j.celrep.2019.12.033
41. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci Immunol. (2019) 4:eaaw6693. doi: 10.1126/sciimmunol.aaw6693
42. Huang Z, Luo Q, Guo Y, Chen J, Xiong G, Peng Y, et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS ONE. (2015) 10:e0129744. doi: 10.1371/journal.pone.0129744
43. Refai A, Gritli S, Barbouche MR, Essafi M. Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Front Cell Infect Microbiol. (2018) 8:327. doi: 10.3389/fcimb.2018.00327
44. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. (2007) 129:1287–98. doi: 10.1016/j.cell.2007.05.059
45. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. (2009) 136:37–49. doi: 10.1016/j.cell.2008.11.014
46. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
47. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. doi: 10.1038/nature13489
48. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. (2015) 5:18176. doi: 10.1038/srep18176
49. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an essential mediator of IFN-γ-dependent immunity to Mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. doi: 10.4049/jimmunol.1600266
50. Cooper AM, Solache A, Khader SA. Interleukin-12 and tuberculosis: an old story revisited. Curr Opin Immunol. (2007) 19:441–7. doi: 10.1016/j.coi.2007.07.004
51. Abdalla AE, Lambert N, Duan X, Xie J. Interleukin-10 family and tuberculosis: an old story renewed. Int J Biol Sci. (2016) 12:710–7. doi: 10.7150/ijbs.13881
52. Warsinske HC, Pienaar E, Linderman JJ, Mattila JT, Kirschner DE. Deletion of TGF-β1 increases bacterial clearance by cytotoxic T cells in a tuberculosis granuloma model. Front Immunol. (2017) 8:1843. doi: 10.3389/fimmu.2017.01843
53. Mori M, Itabe H, Higashi Y, Fujimoto Y, Shiomi M, Yoshizumi M, et al. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J Lipid Res. (2001) 42:1771–81.
54. Ordway D, Henao-Tamayo M, Orme IM, Gonzalez-Juarrero M. Foamy macrophages within lung granulomas of mice infected with Mycobacterium tuberculosis express molecules characteristic of dendritic cells and antiapoptotic markers of the TNF receptor-associated factor family. J Immunol. (2005) 175:3873–81. doi: 10.4049/jimmunol.175.6.3873
55. Ríos-Barrera VA, Campos-Peña V, Aguilar-León D, Lascurain LR, Meraz-Ríos MA, Moreno J, et al. Macrophage and T lymphocyte apoptosis during experimental pulmonary tuberculosis: their relationship to mycobacterial virulence. Eur J Immunol. (2006) 36:345–53. doi: 10.1002/eji.200535202
56. Nabeshima S, Nomoto M, Matsuzaki G, Kishihara K, Taniguchi H, Yoshida S, et al. T-Cell hyporesponsiveness induced by activated macrophages through nitric oxide production in mice infected with Mycobacterium tuberculosis. Infect Immun. (1999) 67:3221–6. doi: 10.1128/IAI.67.7.3221-3226.1999
57. Hernandez-Pando R, Orozco H, Arriaga K, Sampieri A, Larriva-Sahd J, Madrid-Marina V. Analysis of the local kinetics and localization of interleukin-1 alpha, tumour necrosis factor-alpha and transforming growth factor-beta, during the course of experimental pulmonary tuberculosis. Immunology. (1997) 90:607–17. doi: 10.1046/j.1365-2567.1997.00193.x
58. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. (2008) 4:e1000204. doi: 10.1371/journal.ppat.1000204
59. Roque NR, Lage SL, Navarro R, Fazolini N, Maya-Monteiro CM, Rietdorf J, et al. Rab7 controls lipid droplet-phagosome association during mycobacterial infection. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158703. doi: 10.1016/j.bbalip.2020.158703
60. Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. (2006) 60:1109–22. doi: 10.1111/j.1365-2958.2006.05155.x
61. Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, et al. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. ELife. (2017) 6:e26969. doi: 10.7554/eLife.26969.019
62. Nazarova EV, Montague CR, Huang L, La T, Russell D, VanderVen BC. The genetic requirements of fatty acid import by Mycobacterium tuberculosis within macrophages. ELife. (2019) 8:e43621. doi: 10.7554/eLife.43621.015
63. Singh KH, Jha B, Dwivedy A, Choudhary E, N AG, Ashraf A, et al. Characterization of a secretory hydrolase from Mycobacterium tuberculosis sheds critical insight into host lipid utilization by M. tuberculosis. J Biol Chem. (2017) 292:11326–35. doi: 10.1074/jbc.M117.794297
64. Tong J, Liu Q, Wu J, Jiang Y, Takiff HE, Gao Q. Mycobacterium tuberculosis strains of the modern Beijing sublineage excessively accumulate triacylglycerols in vitro. Tuberculosis. (2019) 120:101892. doi: 10.1016/j.tube.2019.101892
65. Baek SH, Li AH, Sassetti CM. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. (2011) 9:e1001065. doi: 10.1371/journal.pbio.1001065
66. Rodriguez JG, Hernandez AC, Helguera-Repetto C, Aguilar Ayala D, Guadarrama-Medina R, Anzola JM, et al. Global adaptation to a lipid environment triggers the dormancy-related phenotype of Mycobacterium tuberculosis. mBio. (2014) 5:e01125–14. doi: 10.1128/mBio.01125-14
67. Jaisinghani N, Dawa S, Singh K, Nandy A, Menon D, Bhandari PD, et al. Necrosis driven triglyceride synthesis primes macrophages for inflammation during Mycobacterium tuberculosis infection. Front Immunol. (2018) 9:1490. doi: 10.3389/fimmu.2018.01490
68. Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. (2011) 7:e1002093. doi: 10.1371/journal.ppat.1002093
69. Singh V, Kaur C, Chaudhary VK, Rao KV, Chatterjee S. M. tuberculosis secretory protein ESAT-6 induces metabolic flux perturbations to drive foamy macrophage differentiation. Sci Rep. (2015) 5:12906. doi: 10.1038/srep12906
70. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. (2012) 12:669–81. doi: 10.1016/j.chom.2012.09.012
71. Mahajan S, Dkhar HK, Chandra V, Dave S, Nanduri R, Janmeja AK, et al. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARgamma and TR4 for survival. J Immunol. (2012) 188:5593–603. doi: 10.4049/jimmunol.1103038
72. Arnett E, Weaver AM, Woodyard KC, Montoya MJ, Li M, Hoang KV, et al. PPARγ is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLoS Pathog. (2018) 14:e1007100. doi: 10.1371/journal.ppat.1007100
73. Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, et al. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J Immunol. (2017) 198:3283–95. doi: 10.4049/jimmunol.1601920
74. Korf H, Vander Beken S, Romano M, Steffensen KR, Stijlemans B, Gustafsson JA, et al. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest. (2009) 119:1626–37. doi: 10.1172/JCI35288
75. Ahsan F, Maertzdorf J, Guhlich-Bornhof U, Kaufmann SHE, Moura-Alves P. IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis. Sci Rep. (2018) 8:1520. doi: 10.1038/s41598-018-19476-x
76. Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. (2016) 17:677–86. doi: 10.1038/ni.3434
77. Martens GW, Arikan MC, Lee J, Ren F, Vallerskog T, Kornfeld H. Hypercholesterolemia impairs immunity to tuberculosis. Infect Immun. (2008) 76:3464–72. doi: 10.1128/IAI.00037-08
78. Vrieling F, Wilson L, Rensen PCN, Walzl G, Ottenhoff THM, Joosten SA. Oxidized low-density lipoprotein (oxLDL) supports Mycobacterium tuberculosis survival in macrophages by inducing lysosomal dysfunction. PLoS Pathog. (2019) 15:e1007724. doi: 10.1371/journal.ppat.1007724
79. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother. (2016) 71:1570–7. doi: 10.1093/jac/dkw014
80. Kang YA, Choi NK, Seong JM, Heo EY, Koo BK, Hwang SS, et al. The effects of statin use on the development of tuberculosis among patients with diabetes mellitus. Int J Tuberc Lung Dis. (2014) 18:717–24. doi: 10.5588/ijtld.13.0854
81. Chen YT, Kuo SC, Chao PW, Chang YY. Use of lipid-lowering agents is not associated with improved outcomes for tuberculosis patients on standard-course therapy: a population-based cohort study. PLoS ONE. (2019) 14:e0210479. doi: 10.1371/journal.pone.0210479
82. Skerry C, Pinn ML, Bruiners N, Pine R, Gennaro ML, Karakousis PC. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother. (2014) 69:2453–7. doi: 10.1093/jac/dku166
83. Lai CC, Lee MT, Lee SH, Hsu WT, Chang SS, Chen SC, et al. Statin treatment is associated with a decreased risk of active tuberculosis: an analysis of a nationally representative cohort. Thorax. (2016) 71:646–51. doi: 10.1136/thoraxjnl-2015-207052
84. Kapoor N, Pawar S, Sirakova TD, Deb C, Warren WL, Kolattukudy PE. Human granuloma in vitro model, for TB dormancy and resuscitation. PLoS ONE. (2013) 8:e53657. doi: 10.1371/journal.pone.0053657
85. Fonseca KL, Rodrigues PNS, Olsson IAS, Saraiva M. Experimental study of tuberculosis: from animal models to complex cell systems and organoids. PLoS Pathog. (2017) 13:e1006421. doi: 10.1371/journal.ppat.1006421
86. Elkington P, Lerm M, Kapoor N, Mahon R, Pienaar E, Huh D, et al. In vitro granuloma models of tuberculosis: potential and challenges. J Infect Dis. (2019) 219:1858–66. doi: 10.1093/infdis/jiz020
87. Blanc L, Daudelin IB, Podell BK, Chen PY, Zimmerman M, Martinot AJ, et al. High-resolution mapping of fluoroquinolones in TB rabbit lesions reveals specific distribution in immune cell types. ELife. (2018) 7:e41115. doi: 10.7554/eLife.41115.025
88. Tseng CH. Metformin decreases risk of tuberculosis infection in type 2 diabetes patients. J Clin Med. (2018) 7:264. doi: 10.3390/jcm7090264
89. Lin SY, Tu HP, Lu PL, Chen TC, Wang WH, Chong IW, et al. Metformin is associated with a lower risk of active tuberculosis in patients with type 2 diabetes. Respirology. (2018) 23:1063–73. doi: 10.1111/resp.13338
90. Zhang M, He JQ. Impacts of metformin on tuberculosis incidence and clinical outcomes in patients with diabetes: a systematic review and meta-analysis. Eur J Clin Pharmacol. (2019) 76:149–59. doi: 10.1007/s00228-019-02786-y
91. Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, et al. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog. (2014) 10:e1004265. doi: 10.1371/journal.ppat.1004265
92. Coriat R, Mir O, Ropert S, Loulergue P, Billemont B, Goldwasser F. Reactivation of tuberculosis during temsirolimus therapy. Invest New Drugs. (2011) 29:1494–6. doi: 10.1007/s10637-010-9487-2
Keywords: Mycobacterium tuberculosis, foamy macrophage, tuberculosis, immune responses, lipid metabolism, lung inflammation, host-directed therapy
Citation: Shim D, Kim H and Shin SJ (2020) Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 11:910. doi: 10.3389/fimmu.2020.00910
Received: 07 December 2019; Accepted: 20 April 2020;
Published: 12 May 2020.
Edited by:
Charles S. Dela Cruz, Yale University School of Medicine, United StatesReviewed by:
Carmen Judith Serrano, Mexican Social Security Institute (IMSS), MexicoAude Remot, Institut National de la Recherche Agronomique (INRA), France
Zhou Xing, McMaster University, Canada
Luiz Gustavo Gardinassi, Universidade Federal de Goiás (IPTSP—UFG), Brazil
Luciana Balboa, Academia Nacional de Medicina, Argentina
Copyright © 2020 Shim, Kim and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sung Jae Shin, c2pzaGluQHl1aHMuYWM=