 Ankit Gupta
Ankit Gupta Bineet Narayan
Bineet Narayan Subodh Kumar
Subodh Kumar Shailendra Kumar Verma
Shailendra Kumar Verma- Microbiology Division, Defence Research and Development Establishment, Gwalior, India
To counteract the deadly pathogens, i.e., Y. pestis, Y. enetrocolitica, and Y. pseudotuberculosis, we prepared a recombinant DNA construct lcrV-hsp70 encoding the bivalent fusion protein LcrV-HSP70. The lcrV gene of Y. pestis and hsp70 domain II DNA fragment of M. tuberculosis were amplified by PCR. The lcrV amplicon was first ligated in the pET vector using NcoI and BamHI restriction sites. Just downstream to the lcrV gene, the hsp70 domain II was ligated using BamHI and Hind III restriction sites. The in-frame and the orientation of cloned lcrV-hsp70 were checked by restriction analysis and nucleotide sequencing. The recombinant bivalent fusion protein LcrV-HSP70 was expressed in E. coli and purified by affinity chromatography. The vaccine potential of LcrV-HSP70 fusion protein was evaluated in formulation with alum. BALB/c mice were vaccinated, and the humoral and cellular immune responses were studied. The fusion protein LcrV-HSP70 induced a strong and significant humoral immune response in comparison to control animals. We also observed a significant difference in the expression levels of IFN-γ and TNF-α in LcrV–HSP70-immunized mice in comparison to control, HSP70, and LcrV groups. To test the protective efficacy of the LcrV–HSP70 fusion protein against plague and Yersiniosis, the vaccinated mice were challenged with Y. pestis, Y. enterocolitica, and Y. pseudotuberculosis separately. The bivalent fusion protein LcrV–HSP70 imparted 100% protection against the plague. In the case of Yersiniosis, on day 2 post challenge, there was a significant reduction in the number of CFU of Y. enterocolitica and Y. pseudotuberculosis in the blood (CFU/ml) and the spleen (CFU/g) of vaccinated animals in comparison to the LcrV, HSP70, and control group animals.
Introduction
The Yersinia genus contains three pathogenic species, i.e., Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica, which cause fatal infections to human beings (1). Of these, Y. enterocolitica and Y. pseudotuberculosis are responsible for Yersiniosis, a self-limiting infection. The bacilli responsible for Yersiniosis are passed on through oral or fecal routes mainly from water, soil, and food (1). The symptoms of Yersiniosis are typically mesenteric lymphadenitis, mild diarrhea, acute gastroenteritis, and reactive arthritis (1, 2).
Plague is a highly lethal and rapid disease caused by Y. pestis. This pathogen is responsible for millions of deaths throughout the world. According to the World Health Organization (WHO), the plague is a re-emerging disease, and it remains an important public health issue (3, 4). The Center for Disease Control and Prevention (CDC) has classified Y. pestis as a group-3 risk pathogen. Plague is a zoonotic infection, and infected wild rats exist as reservoirs in endemic areas throughout the world. The human population is highly susceptible to Y. pestis infection, and the manifestation of the infection is mainly dependent on the route of transmission and infection source. Consequentially, the plague develops in one of the three main clinical forms bubonic, septicaemic, and pneumonic (5). Mostly in nature, Y. pestis transmitted to humans accidentally after the bite of an infected flea. However, it can also be transmitted via inhalation of aerosolized plague bacilli (6, 7). Transmission of bacilli after bite of a flea develops into a bubonic form of the disease, which is typically characterized by the rapid dissemination of bacilli into the lymph nodes, and their replication is responsible for the development of swollen buboes, an identifying characteristic of the disease. The bubonic form can develop into septicemic or secondary pneumonic plague if the disease is not treated in time (8, 9).
In this modern world, the intentional use of aerosolized Y. pestis is a serious threat because of its high fatality rate and its rapid individual-to-individual transmission competence. For the treatment of plague, antibiotics are available. The effectiveness of these antibiotics has been confirmed in humans as well as in animal models (10, 11). However, according to some reports, multidrug-resistant strains of Y. pestis have been isolated (12, 13). By genetic engineering, antibiotic-resistant strains of virulent Y. pestis may be engineered by manipulating the plasmids harboring the antibiotic-resistant genes (13, 14). In these circumstances, the development of a new generation drugs or vaccines is of the utmost importance to control the disease.
F1 and LcrV are the major vaccine antigens that have been targeted by various scientists to develop a potential vaccine. However, there is no approved vaccine yet. In Y. pestis, LcrV is one virulence factor and an essential part of the type III secretion system (T3SS) (15). In continuation of our efforts to develop an anti-plague vaccine, here, we designed a recombinant construct lcrV–hsp70 encoding a fusion protein LcrV-HSP70 of 60 kDa. This recombinant protein was successfully expressed in E. coli and purified by immobilized metal affinity chromatography. In order to evaluate the vaccine potential of bivalent fusion protein LcrV-HSP70, Balb/C mice were immunized. The humoral and cellular immune responses were studied, and, ultimately, the protective efficacy against challenges with Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica were evaluated.
Materials and Methods
Ethics Statement
All the protocols for conducting the experiments (MB-44/57/SKV) using BALB/c mice were approved by the Institutional Animal Ethics Committee (IAEC) of Defense Research and Development Establishment (DRDE). This study was carried out in strict accordance with recommendations from the Care and Use of Laboratory Animals committee for the purpose of control and supervision of experiments on animals (CPCSEA), Govt. of India.
Bacterial Strains, Plasmids, and Reagents
Escherichia coli bacterial strains, i.e., DH5α and BL21 (DE3), were procured from Invitrogen, USA. The plasmid pET28a was purchased from Novagen, USA. The bacterial strains, i.e., Y. pestis (S1 strain), Y. pseudotuberculosis (A87 strain), and Y. enterocolitica (O:8 serotype) were collected from DRDE repository. All the challenge experiments using Y. pestis were conducted in a biosafety level-3 facility at DRDE, Gwalior.
Cloning of lcrV–hsp70 Construct in pET Vector
Y. pestis (S1 strain) was grown on a Brain Heart Infusion (BHI) agar plate at 28°C for 48 h. In order to isolate the genomic DNA, one colony from the BHI agar plate was picked up, inoculated in BHI broth (5 ml), and grown at 28°C for 48 h. The culture was pelleted at 10,000 × g for 1 min. The genomic DNA was isolated using commercially available kit (Qiagen, Germany). To clone the lcrV-hsp70 construct, the lcrV gene of Y. pestis was amplified by PCR using the forward 5′-ATACCATGGGCATGATTAGAGCCTACGAACAAAAC-3′and reverse 5′-TAGGATCCTTTACCAGACGTGTCATCTAGCA-3′ primers. The PCR amplicon was cloned in the pET28a vector using the NcoI and BamHI restriction sites (underlined nucleotide sequence). Similarly, the hsp70 domain (II) of M. tuberculosis was also amplified using forward 5′-TAGGATCCGAGAAGGAGCAGCGAATCCTG-3′ and reverse 5′-TAAAGCTTCGGGGTAACATCAAGCAGCAG-3′ primers. This amplified DNA product encoding HSP70 (domain II) was ligated to just downstream to the cloned lcrV gene using the BamHI and Hind III restriction sites (underlined nucleotide sequence) in the same plasmid. The pET plasmid carrying the in-frame of the lcrV-hsp70 DNA construct was transformed into DH5α cells. The positive transformants were selected on LB-agar plates containing kanamycin (50 μg/ml).
Preparations of Recombinant Proteins
Expression
One of the positive clones encoding the bivalent fusion protein LcrV-HSP70 was inoculated in 5 ml of LB broth containing 50 μg/ml of kanamycin. As reported earlier, LcrV of Yersinia pestis and HSP70 domain II of M. tuberculosis were also expressed individually (16). In brief, one full loop from stock cultures corresponding to LcrV and HSP70 (stored at −80°C) was inoculated individually into 5 ml of LB broth containing kanamycin. All three cultures were grown individually at 37°C at 200 rpm overnight. The next day, 5 ml LB broth was inoculated with 1% (v/v) individually to each overnight grown culture, and all cultures were grown at 37°C. The cultures were induced with IPTG and grown further for 4 h. One milliliter of uninduced culture was collected individually from each culture prior to adding IPTG. The induced and uninduced cultures were centrifuged at 10,000 × g for 1 min. For SDS-PAGE analysis, the cultures were lysed in 1X sample buffer (0.313 m Tris-HCL; pH 6.8; 50% glycerol; 10% SDS and 0.05% bromophenol blue; 100 mM DDT), and they were electrophoresed on SDS-PAGE.
Purification
The recombinant proteins LcrV-HSP70, LcrV, and HSP70 fused with histidine tags were purified under native conditions using Ni-NTA columns (Qiagen, Germany). One full loop of cultures corresponding to LcrV-HSP70, LcrV, and HSP70 were inoculated individually into 5 ml of LB broth containing 50 μg/ml kanamycin, grown for overnight at 37°C at 200 rpm. The next day, 500 ml of LB broth was inoculated individually with 1% overnight grown culture for each protein and grown at 37°C at 200 rpm. Cultures were induced with IPTG and grown further for 4 h. All the cultures were pelleted individually at 8,000 × g for 10 min at 4°C. Each pellet was suspended individually in 20 ml of lysis buffer (50 mM NaH2PO4, 250 mM NaCl; 10 mm imidazole; pH 8.0). The suspended pellets were sonicated, centrifuged at 20,000 × g for 30 min at 4°C, and supernatants were separated individually. Three Ni-NTA columns were prepared for each protein. Each column was equilibrated with lysis buffer (~10 ml). The supernatant of individual protein was poured on to the Ni-NTA column and allowed to pass. Each column was washed with 50 ml of wash buffer containing 50 mM NaH2PO4, pH-8.0; 250 mM NaCl; 30 mM imidazole. The proteins, LcrV-HSP70, LcrV, and HSP70, were eluted individually by applying 15 ml of elution buffer (50 mM NaH2PO4, 250 mM NaCl; 200 mM imidazole; pH 8.0). The collected fractions for each protein were analyzed by SDS-PAGE. The fractions containing each purified protein were pooled individually and subjected to dialysis by gradually lowering the salt concentration against dialysis buffer (50 mM NaH2PO4; 50 mm NaCl; pH 8.0). The concentration of each purified protein was estimated using a BCA kit (Sigma Aldrich, USA). The endotoxin contents were measured in each purified recombinant protein using Limulus Amoebocyte Lysates (LAL) QCL-1000 kit (Cambrex Biosciences, USA) as per the instructions.
Western Blot
All three prepared recombinant proteins, i.e., LcrV, HSP70, and LcrV-HSP70, were analyzed by SDS-PAGE for their purity, as shown in Figure 2Da. In order to confirm the presence of proteins of interest, an immunoblot experiment was performed. The recombinant proteins were separated by SDS-PAGE and transferred onto the nitrocellulose membrane electrophoretically. The nitrocellulose membrane was blocked overnight at 4°C in 5% skim milk. The membrane was probed with Penta-His HRP conjugate anti-histidine antibody (Cat No./ID: 34460, Qiagen) at a dilution of 1:1,000 in 5% skimmed milk for 1 h at 37°C. The membrane was washed thrice with PBS- containing 0.05% Tween 20 (PBS-T). The reaction was developed in 20 ml of PBS containing 8.8 mM H2O2 and 10 mg of 3,3′-diaminobenzidine/ml. Development was carried out for 2–3 min until bands of the desired intensity appeared, and thereafter the membrane was washed.
Animal Immunization
To test the protective efficacy and immune responses of the bivalent fusion protein LcrV-HSP70, 5-week-old female BALB/c mice were taken from DRDE animal facility. Mice were divided into four batches, and each batch was divided into four groups (10 mice/group): the control group, HSP70 group, LcrV group, and the LcrV-HSP70 group (Figure 3A). All the groups, except for the control group, were vaccinated subcutaneously with 15 μg/mouse of each vaccine antigen in formulation with alum adjuvant. The animals of batch I, II, and III were used for the evaluation of humoral immune response and protection studies against Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica challenges, respectively. The animals of batch IV were used for the study of the cellular immune response. The animals of the control group in each batch received sterile PBS only. The animals were immunized on day 0 followed by two boosters on day 14 and 21. In order to study the IgG response, blood was collected on day 0, 21, and 28 (Figure 3A).
Anti-LcrV-HSP70 IgG Response
ELISA plates (Nunc, USA) were coated individually with LcrV, HSP70, and LcrV–HSP70 proteins using 1.0 μg/well and incubated for overnight at 4°C. Plates were washed with 0.05% Tween 20 in PBS (PBS-T), blocked with 3% bovine serum albumin (BSA), and incubated for 2 h at 37°C. After washing the plates thrice with PBS-T, test sera were added in 2-fold serial dilutions in triplicate wells (100 μl/well) and incubated for 1 h at 37°C. After three washes, the wells were probed with rabbit anti-mouse IgG-HRP (Sigma, USA) at 1:15000 dilutions in PBS and incubated for 1 h at 37°C. The ELISA plates were washed, and the reaction was developed with OPD substrate. 2N H2SO4 was added to terminate the reaction, and the absorbance was read at 490 nm by a multimode reader (Biotek, USA). The IgG titers were represented as log10 titers of the highest serial dilution with a mean of OD490 value >2-fold OD490 value of control serum at the same dilution.
Cytokines
The animals of batch IV were sacrificed 1 month after the second booster to measure the cytokine levels. The spleens were removed aseptically and homogenized to prepare single-cell suspension. The splenocytes were counted, and 1 × 106 cells/well were seeded in triplicate in a 96-well-plate. The cultures were stimulated with the same vaccine antigen the animal groups were vaccinated with, i.e., HSP70, LcrV, LcrV–HSP70, or ConA (5 μg/ml each). Concanavalin A (Sigma, USA) was used as a positive control. After 48 h, the supernatants of the cultured splenocytes were harvested and stored at −80°C for further use. The expression levels of IFN-γ and TNF-α in the supernatants were measured using ELISA kit (BD Biosciences, USA).
Bacterial Challenge
To determine the protective efficacy, on day 60 after priming, all the animal groups of batch I were challenged with 100 LD50 (1 × 105 CFU/mouse) of Y. pestis (S1 strain) via i.p. route (16, 17). The infected animals were observed for 20 days for their survival (Figure 3A). The protection experiments were repeated thrice.
Similarly, the animals of batch II and III were challenged (1 × 108 CFU/mouse) with Y. enterocolitica (O:8 serotype) and (1 × 109 CFU/mouse) and with Y. pseudotuberculosis (A87 strain), respectively, via i.p. route (Figure 3A). To count the bacterial load in the blood and spleen, on days 1, 2, 3, 4, and 5 post-challenge, two mice from each group of batch II and III were sacrificed. Their spleens were removed aseptically and weighed. Each spleen was homogenized, and serial dilutions were made in PBS. One hundred microliter suspensions from each countable dilution were spread on BHI agar plates in duplicate. Y. pseudotuberculosis culture plates were incubated at 28°C for 48 h, and the bacterial colonies were counted. Y. enterocolitica culture plates were incubated at 37°C for 24 h, and the bacterial colonies were counted. Prior to sacrifice the remaining animals, the blood was collected. Each blood sample was serially diluted in PBS. One hundred microliter from each countable dilution was spread on BHI agar plates in duplicate. The bacterial load (CFU/ml) and (CFU/g) was determined in the blood and spleen, respectively. The protection experiments were repeated thrice, and the results were expressed as the mean log CFU ± SD per group of three experiments.
Statistical Analysis
Data were analyzed using One Way Analysis of Variance (ANOVA) to compare IgG titers, CFU count, and cytokines response. Log-rank (Mantel-Cox) test was used to generate survival curves using GraphPad Prism 5.0. Statistically significant p-values are represented as *** p < 0.0001; **p < 0.01; * p < 0.05.
Results
Cloning of lcrV–hsp70 in pET Vector
For the expression of a bivalent fusion protein LcrV-HSP70, a 981 bp gene of Y. pestis encoding LcrV and a 630 bp DNA fragment encoding HSP70 domain II of M. tuberculosis were amplified by PCR (Figures 1A,B). The amplicon of the lcrV gene was first ligated in pET28a plasmid using NcoI and BamHI restriction sites. Just downstream to lcrV gene, a 630 bp fragment of hsp70 domain II was ligated using BamHI and HindIII restriction sites, as shown in Figure 1C. The pET vector harboring the in-frame of lcrV-hsp70 DNA construct (pET-lcrV-hsp70) was transformed into competent DH5α cells. The cells were grown on LB agar plates containing kanamycin (50 μg/ml). Positive clones were selected by restriction analysis using NcoI and HindIII restriction enzymes. The digested products were analyzed by agarose gel electrophoresis. A DNA insert of 1,611 bp was observed, as shown in Figure 1D. The in-frame and the orientation of the ligated construct were further confirmed by nucleotide sequencing (Chromous Biotech, Bangalore, India).
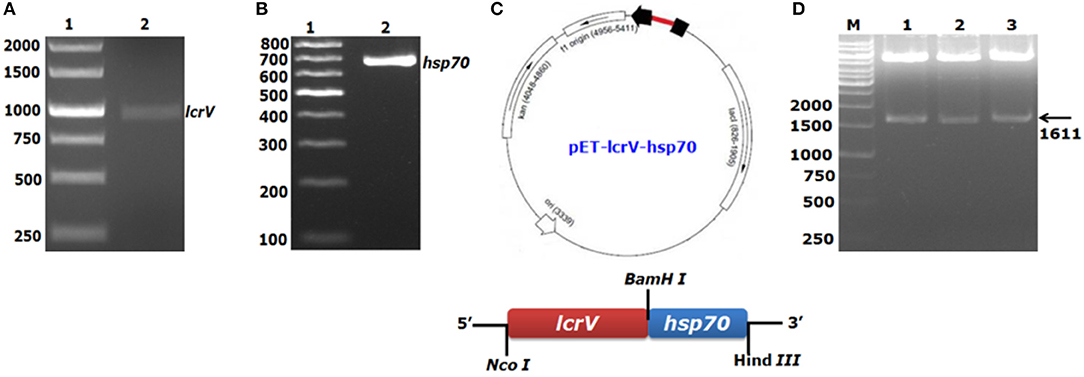
Figure 1. PCR amplification and agarose gel electrophoresis of lcrV gene and hsp70 (domain II) of M. tuberculosis and ligation of lcrV-hsp70 in pET vector: LcrV gene (A) Lane-1, 1Kb DNA ladder; Lane-2, lcrV gene product of 981 bp. (B) Lane-1, 1 Kb DNA ladder; Lane-2, hsp70 gene product of 630 bp. (C) The restriction map of cloned lcrV-hsp70 construct in pET vector. (D) Screening of positive clones of lcrV-hsp70 construct after ligation in pET28 vector, Lane-M, 1 Kb DNA ladder; Lane 1–3, released inserts of lcrV-hsp70 (1,611 bp) after restriction digestion using NcoI and Hind III restriction enzymes.
Expression and Purification of Recombinant Proteins
LcrV-HSP70
To express and purify the bivalent fusion protein LcrV-HSP70 (60 kDa), one of the positive clones of lcrV-hsp70 was transformed into E. coli expression host strain BL-21 (DE3). The colonies were selected on LB-agar plates containing kanamycin. One colony was inoculated in 5 ml LB broth, and the culture was induced with 1 mM IPTG. The IPTG-induced and uninduced E. coli cell lysates were analyzed by SDS-PAGE, as shown in Figure 2Aa. For purification of LcrV-HSP70 protein, the culture was inoculated in 500 ml LB broth. The cells were pelleted by centrifugation, and the lysis under native conditions revealed the association of LcrV-HSP70 protein in the soluble fraction. The recombinant construct of lcrV-hsp70 was engineered to carry a histidine tag at the carboxyl terminus of the LcrV-HSP70 protein. The purification was performed under native conditions by affinity chromatography using Ni-NTA resin. All the eluted fractions were analyzed by SDS-PAGE, as shown in Figure 2Ab. The eluted fractions were pooled, dialyzed, and the protein concentration was estimated. The obtained yield of the purified LcrV-HSP70 was 30 mg/L of shake flask culture. The endotoxin level was observed <5 endotoxin unit (EU)/15 μg of each purified protein.
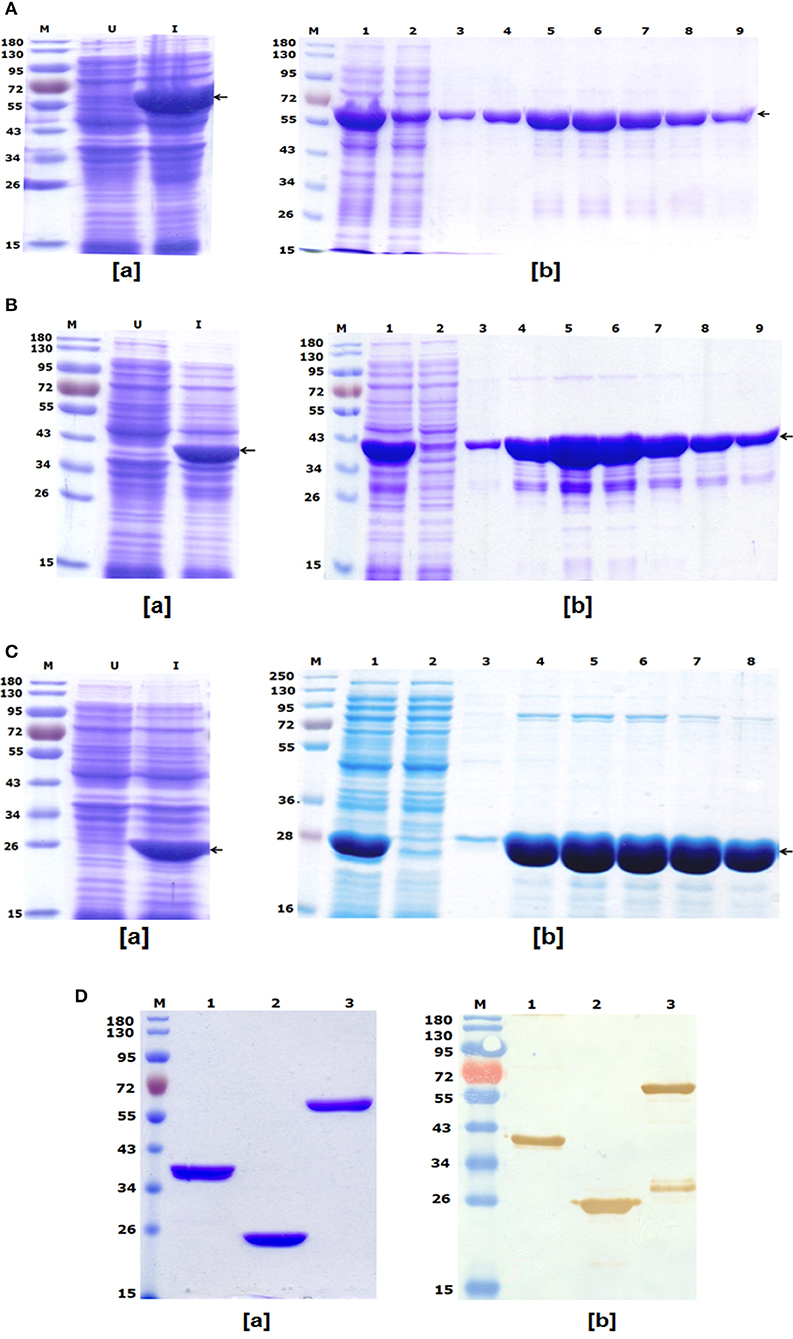
Figure 2. (A) Recombinant bivalent fusion protein LcrV-HSP70 expression profile and purification: (a) SDS–PAGE profile of LcrV-HSP70 protein expression; Lane-M, Pre-stained protein marker; Lane-U, Uninduced cell lysate; Lane-I, Induced cell lysate. The arrow at the right side of the SDS-PAGE profile indicates the position of LcrV-HSP70 protein of 60 kDa. (b) Purification of LcrV-HSP70 protein. Lane-M, Protein marker; Lane-1, Cell lysate; Lane-2, Flow through; Lane-3, Wash; Lane 4–9, eluted fractions of LcrV-HSP70 protein. The arrow at the right side of the SDS-PAGE profile indicates the position of LcrV-HSP70 protein of 60 kDa. (B) Recombinant LcrV protein expression profile and purification: (a) SDS–PAGE profile of LcrV protein expression; Lane-M, Pre-stained protein marker; Lane-U, Uninduced cell lysate; Lane-I, Induced cell lysate. The arrow at the right side of the SDS-PAGE profile indicates the position of LcrV protein of 37 kDa. (b) Purification of LcrV protein. Lane-M, Protein marker; Lane-1, Cell lysate; Lane-2, Flow through; Lane-3, Wash; Lane 4–9, eluted fractions of LcrV protein. The arrow at the right side of the SDS-PAGE profile indicates the position of LcrV protein of 37 kDa. (C) Recombinant HSP70 protein expression and purification: (a) SDS–PAGE profile of HSP70 protein expression; Lane-M, Pre-stained protein marker; Lane-U, Uninduced cell lysate; Lane-I, Induced cell lysate. The arrow at the right side of the SDS-PAGE profile indicates the position of HSP70 protein of 23 kDa. (b) Purification of HSP70 protein. Lane-M, Protein marker; Lane-1, Cell lysate; Lane-2, Flow through; Lane-3, Wash; Lane 4–8, eluted fractions of HSP70 protein. The arrow at the right side of the SDS-PAGE profile indicates the position of HSP70 protein of 23 kDa. (D) Western blot analysis of purified recombinant proteins LcrV, HSP70, and LcrV-HSP70: (a) SDS–PAGE profile of purified proteins, Lane-M, Pre-stained protein marker; Lane-1, LcrV, Lane-2, HSP70 and Lane-3, LcrV-HSP70 protein. (b) Western blot analysis of LcrV, HSP70, and LcrV-HSP70 proteins showing reaction with anti-HIS antibody: Lane-M, Pre-stained protein marker; Lane-1, LcrV, Lane-2, HSP70 and Lane-3, LcrV-HSP70 protein.
LcrV and HSP70
The recombinant LcrV and HSP70 proteins of 37 and 23 kDa, respectively, were also prepared using earlier published protocols (16). Recombinant LcrV protein was expressed and analyzed by SDS-PAGE, as shown in Figure 2Ba. The LcrV protein was purified using native conditions, and the eluted fractions of purified recombinant LcrV were analyzed by SDS-PAGE, as shown in Figure 2Bb. Similarly, recombinant HSP70 protein was expressed and analyzed by SDS-PAGE (Figure 2Ca), and it was purified using native conditions. The eluted fractions of purified recombinant HSP70 was analyzed by SDS-PAGE, as shown in Figure 2Cb. The eluted fractions of each protein were pooled, dialyzed, and analyzed by SDS-PAGE (Figure 2Da). The protein concentrations were estimated and the obtained yield of LcrV and HSP70 were 35 and 25 mg/L of shake flask culture, respectively. The endotoxin level was shown to be <4 endotoxin unit (EU)/15 μg of each purified protein.
Western Blot
In a Western blot experiment, anti-histidine antibody recognized all three proteins, i.e., LcrV, HSP70, and LcrV-HSP70, corresponding to their molecular weights, as shown in Figure 2Db. In lane-3 of Figure 2Db, we observed an extra band below the main band of LcrV-HSP70. This band is seen only on the Western blot and not on the SDS-PAGE and therefore considered to be a minor contaminant. This may have been the result of some degradation.
Humoral Immune Response
In order to test the humoral immune response evoked by LcrV, HSP70, and LcrV-HSP70, IgG endpoint titers were determined by ELISA in the sera taken 7 days after first and second boosters, respectively. The IgG titers were represented as log10 titers after first and second booster sera. There was a significant difference in the IgG titer in the sera collected after first and second boosters for each antigen, as shown in the Figure 3B. However, there was no significant difference in the IgG titers in the vaccinated sera of LcrV and LcrV-HSP70.
Figure 3. (A) Representation of animal groups and schedule of vaccination activities: Animal groups (10 mice/group) for vaccination studies; Vaccine formulations of LcrV, HSP70 and LcrV–HSP70 vaccine antigens with alum. Schematic representation of vaccination schedule, blood collection for humoral and cell-mediated studies, and challenge experiments. (B) The ELISA plates were coated by the same antigens, which were used as immunizing antigens. IgG titers were determined by ELISA and represented as log10 titers. Serum samples were collected after first and second boosters from vaccinated animal groups, i.e., LcrV, HSP70, and LcrV–HSP70. Analysis was done by one-way ANOVA, ***p < 0.0001; **p < 0.01; *p < 0.05. (C) Cytokine profile of IFN-γ and (D) TNF-α; the induced levels of cytokines in vaccinated mice were measured in pg/ml, as represented in graphs. Splenocytes were prepared from all the vaccinated and control groups. The cells were induced with the same vaccine antigens as the immunizing antigens, i.e., LcrV, HSP70, and LcrV-HSP70 (5 μg/ml each) and grown for 48 h. All the statistical comparisons were done by one-way ANOVA, ***p < 0.0001; **p < 0.01; *p < 0.05. (E) Recombinant bivalent fusion protein LcrV–HSP70 imparts protection against challenge with Y. pestis. The immunized and control group mice were challenged against 100 LD50 of Y. pestis. All the animals were observed for 20 days after the challenge for their survival. Log-rank (Mantel-Cox) test was used to compare the survival against plague infection amongst different vaccinated groups (***p < 0.0001).
Cellular Immune Response
The expression levels of IFN-γ and TNF-α in the collected supernatants of splenocytes were measured. We observed a significant difference in the expression levels IFN-γ (Figure 3C) and TNF-α (Figure 3D) in LcrV, HSP70, and LcrV-HSP70 vaccinated mice in comparison to the control group. ConA was used to induce the spleen cells of all the groups, and it responded non-specifically. There was a significant difference in the expression of IFN-γ (***P < 0.0001) and TNF-α (*P < 0.05) in the LcrV-HSP70 vaccinated group in comparison to LcrV and HSP70 alone (Figures 3C,D).
Protection Studies
All the animals of batch I were challenged against 100 LD50 of Y. pestis. The survival of the infected animals was observed for 20 days after the challenge. There was 100% survival in the LcrV-HSP70 vaccinated group, whereas a 70% survival rate was observed in the LcrV vaccinated group. No protection was observed in the control and HSP70 groups. A Log-rank (Mantel-Cox) test was used to compare the survival amongst different vaccinated groups (***P < 0.0001) (Figure 3E).
The animals infected with Y. enterocolitica and Y. pseudotuberculosis of Batch II and III, respectively, were sacrificed to determine the bacterial loads. The level of infection was evaluated by determining CFU count in the blood (CFU/ml) and spleens (CFU/g). The control animal groups of each batch had the highest bacterial counts. The CFUs were counted on day 1, 2, 3, 4, and 5 of post-challenge (Figure S1). On day 2 after challenge, there was a significant reduction in CFUs of Y. enterocolitica (Figure 4) and Y. pseudotuberculosis (Figure 5) in LcrV-HSP70 vaccinated mice in comparison to LcrV and HSP70 vaccinated animals. In Y. enterocolitica challenged mice, the protection unit of LcrV-HSP70 was 1.1905, whereas the protection unit of LcrV and HSP70 were 0.9875 and 0.0215, respectively, in the spleen. In the blood, the protection units of LcrV-HSP70, LcrV, and HSP70 were observed to be 0.9465, 0.468, and 0 respectively (Table 1). In Y. pseudotuberculosis challenged mice, the protection unit of LcrV-HSP70 was 0.984, whereas the protection unit of LcrV and HSP70 were 0.886 and 0, respectively, in the spleen. In the blood, the protection unit of LcrV-HSP70, LcrV, and HSP70 were observed to be 0.9815, 0.49, and 0.027, respectively, (Table 2). We did not find any CFU on day 4 in the spleen and on day 3 in the blood. Units of protection were obtained by subtracting the mean log CFU of the vaccinated group from the mean log CFU of the control group.
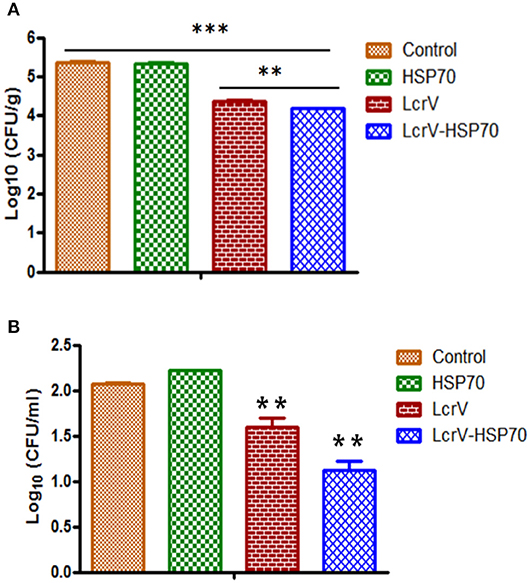
Figure 4. (A) CFU count in the spleens of Y. enterocolitica-challenged mice. Culture plates were incubated at 37°C for 24 h, and the bacterial colonies were counted. The number of bacteria in the spleens is represented as the mean log CFU ± SD per group. Protection was found statistically significant in LcrV-HSP70 vaccinated group when compared to LcrV (**p < 0.01), HSP70 and control group (***p < 0.0001). (B) CFU count in the blood of Y. enterocolitica challenged mice: Y. enterocolitica culture plates were incubated at 37°C for 24 h and the bacterial colonies were counted. The number of bacteria in the blood is represented as the mean log CFU ± SD per group.
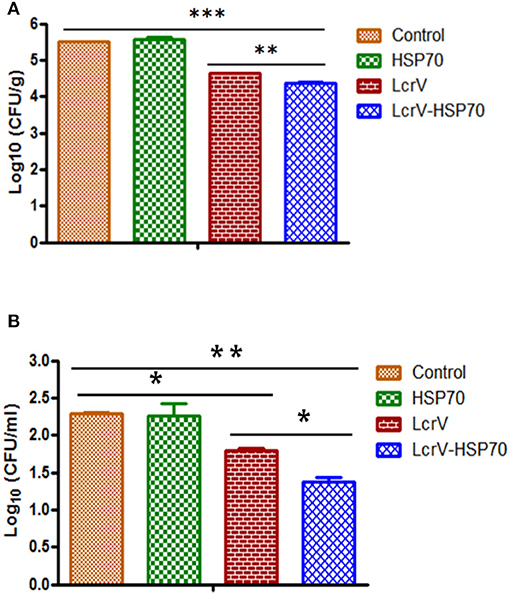
Figure 5. (A) CFU count in the spleens of Y. pseudotuberculosis-challenged mice. Culture plates were incubated at 28°C for 48 h, and the bacterial colonies were counted. The number of bacteria in the spleens is represented as the mean log CFU ± SD per group. Protection was found statistically significant in LcrV-HSP70 vaccinated group when compared to LcrV (**p < 0.01), HSP70 and control group (***p < 0.0001). (B) CFU count in the blood of Y. pseudotuberculosis challenged mice: Y. pseudotuberculosis culture plates were incubated at 28°C for 48 h, and the bacterial colonies were counted. The number of bacteria in the blood is represented as the mean log CFU ± SD per group (*p = 0.05).
Table 1. Protection on day 2 post-challenge in LcrV-HSP70 vaccinated mice against Y. enterocolitica.
Table 2. Protection on day 2 post-challenge in LcrV-HSP70 vaccinated mice against Y. pseudotuberculosis.
Discussion
Despite continuous and dedicated efforts by the scientific community, an ideal plague vaccine is yet to come. Many plague vaccine formulations based on the live attenuated and killed whole-cell of Y. pestis have been developed. Despite having ethical issues and safety concerns, i.e., high fever, headache, and inflammation at the site of injection, these formulations are in use in some countries (18). The main limitations of these vaccine formulations are partial or incomplete protective efficacy, poor memory, and repeated boosters (9, 19, 20). In the recent past, many reports have been published on F1/LcrV based vaccine formulations. The F1/LcrV vaccine antigens adjuvanted with alum induce robust humoral immune responses and impart 100% protection in a mouse model (16, 21–24) with no side effects in humans (25). F1/LcrV antigen-based vaccine formulations protect cynomolgus macaques against aerosolized Y. pestis but failed to protect African Green monkeys (26, 27). The immune mechanisms responsible for this poor and inconsistent protection in the African Green monkey model yet to be analyzed. To improve the efficacy of these vaccine formulations by inducing stronger cellular immunity might be the best solution. A number of strategies are in development to advance the efficacy of F1/LcrV antigens, e.g., use of novel and competent adjuvants (28–30), vaccine antigen delivery systems (31, 32), and genetically modified antigens (33). To counteract the pathogen effectively, addition of one or more vaccine antigens might be considered which can significantly augment the immune response.
In the recent years, the efficacy of F1/LcrV based vaccines have been improved by adding molecular adjuvants, i.e., F1/V adjuvanted with flagellin (Flagellin/F1/V) for phase I safety and immunogenicity trial in healthy adult volunteer (34); the recombinant flagellin of Salmonella typhi also improved the protective potential of YopE (35); the SA-4-1BBL adjuvant, a strong inducer of the Th1 response, improved the vaccine potential of F1-LcrV (30); and the HSP70 domain II of M. tuberculosis augments the humoral and cellular immune response of F1/LcrV vaccine (16, 24). HSP70 proteins have been characterized to immunomodulate the immune response of vaccine antigens (36–42). The domain II of heat shock protein HSP70(II), when formulated with vaccine antigen/s, evoked the T-cell responses. A bivalent fusion protein ovalbumin-HSP70(II) induces CD8 cytotoxic T lymphocytes specific to ovalbumin (36). HSP70(II) of M. tuberculosis modulated the immune responses of vaccine antigen p24 of HIV-1 (36). The amino acids (359–610) of the carboxy-terminus of HSP70 evoke the expression of IFN-γ, IL-2, and TNF-α and a high titer of IgG2a and IgG3 antibodies (43).
Earlier, we have also characterized the potential role of HSP70(II) of M. tuberculosis to augment the vaccine efficacy of F1/LcrV in a mouse model (16, 24). Our findings showed that HSP70(II) improved the cellular immune response (IL-2, IFN-γ, and TNF-α) of F1+LcrV+HSP70(II)-vaccinated mice. It also enhanced the IFN-γ secreting CD4+ and CD8+ T cells. Earlier, we developed a recombinant trivalent fusion protein F1-LcrV-HSP70(II) and evaluated as a vaccine candidate against plague in a mouse model (24). We experienced difficulties in the expression and purification of this trivalent fusion protein in E. coli. The expression was not found to be optimal, making inclusion bodies during expression. Hence the protein can only be purified under denaturing conditions. Moreover, this trivalent fusion protein is highly degradable at room temperature. In this study, we prepared a recombinant bivalent fusion protein LcrV-HSP70, and we evaluated its vaccine potential in BALB/c mice. The expression of the bivalent fusion protein is up to the mark in E. coli, and this protein does not make inclusion during the expression. The bivalent protein can be easily purified under native conditions to the optimum yield. All the three antigens, i.e., LcrV-HSP70, LcrV, and HSP70, were adjuvanted individually with alum, and the animals were vaccinated. There was a significantly higher anti-LcrV IgG titer in the sera of LcrV-HSP70 vaccinated mice in comparison to HSP70 and control group. No significant difference was observed in the anti-LcrV IgG titer between LcrV alone and fusion protein LcrV-HSP70 vaccinated sera. However, there was a significant difference in the IgG titers in the sera after first and second booster. We also observed a significant difference in the expression of IFN-γ and TNF-α in LcrV-HSP70 vaccinated mice in comparison to groups vaccinated with LcrV and HSP70 alone.
Yersinia species use Type III secretion system (T3SS) to translocate effector proteins into the host target cells to breach host immune barriers (44). LcrV is an essential virulent factor that makes the tip of T3SS. Before contact with the host cells, LcrV protein is expressed on the cell membrane of Y. pestis, enterocolitica, and pseudotuberculosis (45). LcrV essentially helps in the regulation and translocation of other virulence factors into the host cell (46). It also helps Yersiniae to defeat the host immune response via IL-10 mediated immunomodulatory role by suppressing the expression of proinflammatory cytokines (47, 48). As the LcrV is a common virulence factor of Y. pestis, enterocolitica, and pseudotuberculosis, we took advantage of this to test the protective efficacy of LcrV-HSP70 against these human pathogens. However, our primary focus is to develop the subunit vaccine against plague.
Clinical symptoms of yersiniosis by Y. enterocolitica first appear after an incubation period of about 5 days (range 1–11 days) and include diarrhea, fever, vomiting, tenesmus, and abdominal pain. The incubation period of Y. pseudotuberculosis is 5–10 days; however, durations of 2–20 days have been reported in occasional outbreaks, with the average time being 4 days after exposure to the bacterium when symptoms are present (https://www.cdc.gov/yersinia/healthcare.html). Therefore, in our studies, we determined the presence of both of the pathogens in the spleen and in the blood to know whether our vaccine formulation is effective or not. We challenged all the vaccinated and control mice on day 60, and we determined the CFUs in the blood and spleen on day 61–65. On day 2 post-challenge, we observed a significant reduction in the number of CFUs in the spleen and blood of Y. enterocolitica- and Y. pseudotuberculosis-challenged mice that were immunized with LcrV-HSP70 in comparison to LcrV and HSP70 alone. Since the oral route is the natural route of infection for Y. enterocolitica or Y. pseudotuberculosis, it would be relevant in the future to determine the protective efficacy of the above vaccine candidate by a mucosal route. In the case of plague, the bivalent fusion protein LcrV-HSP70 imparts 100% protection against plague, whereas LcrV alone provided 70% protection only. There was no protection in control and HSP70-vaccinated mice. Taken together, the recombinant bivalent fusion protein LcrV-HSP70 has the scope for further evaluation of its mucosal immune response in animal models, thus increasing its potential to become a vaccine against plague and Yersiniosis.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Ethics Committee (IAEC) of Defence Research and Development Establishment.
Author Contributions
SV initiated this project and was responsible for the overall design of the study, conducting challenge experiments, interpretation of data and writing of this manuscript. AG and BN conducted the laboratory experiments. SK contributed in the interpretation of results and reviewing of this manuscript before submission.
Funding
The necessary funding and facilities to complete these studies were provided by Defense Research and Development Establishment (DRDE), Ministry of Defense, Govt. of India.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to the Director of the Defense Research and Development Establishment (DRDE), Ministry of Defense, Govt. of India, for providing the necessary facilities to complete these studies. The article has been assigned an accession No. DRDE/MB/37/2019.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00988/full#supplementary-material
References
1. Brubaker RR. Factors promoting acute and chronic diseases caused by yersiniae. Clin Microbiol Rev. (1991) 4:309–24. doi: 10.1128/CMR.4.3.309
2. Bottone EJ. Yersinia enterocolitica: the charisma continues. Clin Microbiol Rev. (1997) 10:257–76. doi: 10.1128/CMR.10.2.257
4. Sun W, Singh AK. Plague vaccine: recent progress and prospects. NPJ Vaccines. (2019) 4:11. doi: 10.1038/s41541-019-0105-9
6. Dennis DT, Chow CC. Plague. Pediatr Infect Dis J. (2004) 23:69–71. doi: 10.1097/01.inf.0000106918.18570.dd
7. Pechous RD, Sivaraman V, Stasulli NM, Goldman WE. Pneumonic plague: the darker side of Yersinia pestis. Trends Microbiol. (2016) 24:190–7. doi: 10.1016/j.tim.2015.11.008
8. Prentice MB, Rahalison L. Plague. Lancet. (2007) 369:1196–207. doi: 10.1016/S0140-6736(07)60566-2
9. Verma SK, Tuteja U. Plague vaccine development: current research and future trends. Front Immunol. (2016) 7:602. doi: 10.3389/fimmu.2016.00602
10. Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, et al. Plague as a biological weapon: medical and public health management. working group on civilian biodefense. JAMA. (2000) 283:2281–90. doi: 10.1001/jama.283.17.2281
11. Levy Y, Flashner Y, Tidhar A, Zauberman A, Aftalion M, Lazar S, et al. T cells play an essential role in anti-F1 mediated rapid protection against bubonic plague. Vaccine. (2011) 29:6866–73. doi: 10.1016/j.vaccine.2011.07.059
12. Galimand M, Guiyoule A, Gerbaud G, Rasoamanana B, Chanteau S, Carniel E, et al. Multi drug resistance in Yersinia pestis mediated by a transferable plasmid. N Engl J Med. (1997) 337:677–80. doi: 10.1056/NEJM199709043371004
13. Guiyoule A, Gerbaud G, Buchrieser C, Galimand M, Rahalison L, Chanteau S, et al. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg Infect Dis. (2001) 7:43–8. doi: 10.3201/eid0701.010106
14. Chain PSG, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci USA. (2004) 101:13826–31. doi: 10.1073/pnas.0404012101
15. Skrzypek E, Straley SC. Differential effects of deletions in lcrV on secretion of V-antigen, regulation of the low-Ca++ response and virulence of Yersinia pestis. J Bacteriol. (1995) 177:2530–42. doi: 10.1128/JB.177.9.2530-2542.1995
16. Batra L, Verma SK, Nagar DP, Saxena N, Pathak PP, Pant SC, et al. HSP70 domain II of Mycobacterium tuberculosis modulates immune response and protective potential of F1 and LcrV antigens of Yersinia pestis in a mouse model. PLoS Neg Trop Dis. (2014) 8:e3322. doi: 10.1371/journal.pntd.0003322
17. Verma SK, Batra L, Athmaram TN, Pathak P, Katram N, Agarwal GS, et al. Characterization of immune responses to Yersinia pestis (Indian isolate) infection in mouse model. J Clin Cell Immunol. (2013) 4:151. doi: 10.4172/2155-9899.1000151
18. Hart MK, Saviolakis GA, Welkos SL, House RV. Advanced development of the rF1V and rBV A/B vaccines: progress and challenges. Adv Prev Med. (2012) 2012:731604. doi: 10.1155/2012/731604
19. Wang X, Zhang X, Zhou D, Yang R. Live-attenuated Yersinia pestis vaccines. Expert Rev Vaccines. (2013) 12:677–86. doi: 10.1586/erv.13.42
20. Chu K, Hu J, Meng F, Li J, Luo L, Xu J, et al. Immunogenicity and safety of subunit plague vaccine: a randomized phase. 2 a clinical trial. Hum Vaccine Immunother. (2016) 12:2334–40. doi: 10.1080/21645515.2016.1175261
21. Williamson ED, Eley SM, Stagg AJ, Green M, Russell P, Titball RW. A sub-unit vaccine elicits IgG in serum, spleen cell cultures and bronchial washings and protects immunized animals against pneumonic plague. Vaccine. (1997) 15:1079–84. doi: 10.1016/S0264-410X(96)00303-9
22. Lin JS, Park S, Adamovicz JJ, Hill J, Bliska JB, Cote CK, et al. TNF-α and IFN-γ contribute to F1/LcrV-targeted immune defense in mouse models of fully virulent pneumonic plague. Vaccine. (2010) 29:357–62. doi: 10.1016/j.vaccine.2010.08.099
23. Levy Y, Vagima Y, Tidhar A, Aftalion M, Gur D, Nili U, et al. Targeting of the Yersinia pestis F1 capsular antigen by innate like B1b cells mediates a rapid protective response against bubonic plague. NPJ Vaccines. (2018) 3:52. doi: 10.1038/s41541-018-0087-z
24. Verma SK, Batra L, Tuteja U. A recombinant trivalent fusion protein F1-LcrV-HSP70(II) augments humoral and cellular immune responses and imparts full protection against Yersinia pestis. Front Microbiol. (2016) 7:1053. doi: 10.3389/fmicb.2016.01053
25. Price JL, Manetz TS, Shearer JD, House RV. Preclinical safety assessment of a recombinant plague vaccine (rF1V). Int J Toxicol. (2013) 32:327–335. doi: 10.1177/1091581813497405
26. Smiley ST. Current challenges in the development of vaccines for pneumonic plague. Expert Rev Vaccines. (2008) 7:209–21. doi: 10.1586/14760584.7.2.209
27. Williamson ED, Packer PJ, Waters EL, Simpson AJ, Dyer D, Hartings J, et al. Recombinant (F1+V) vaccine protects cynomolgus macaques against pneumonic plague. Vaccine. (2011) 29:4771–7. doi: 10.1016/j.vaccine.2011.04.084
28. Amemiya K, Meyers JL, Rogers TE, Fast RL, Bassett AD, Worsham PL, et al. CpG oligodeoxynucleotides augment the murine immune response to the Yersinia pestis F1-V vaccine in bubonic and pneumonic models of plague. Vaccine. (2009) 27:2220–9. doi: 10.1016/j.vaccine.2009.02.016
29. Honko AN, Sriranganathan N, Lees CJ, Mizel SB. Flagellin is an effective adjuvant for immunization against lethal respiratory challenge with Yersinia pestis. Infect Immun. (2006) 74:1113–20. doi: 10.1128/IAI.74.2.1113-1120.2006
30. Bowen W, Batra L, Pulsifer AR, Yolcu ES, Lawrenz MB, Shirwan H. Robust Th1 cellular and humoral responses generated by the Yersinia pestis rF1-V subunit vaccine formulated to contain an agonist of the CD137 pathway do not translate into increased protection against pneumonic plague. Vaccine. (2019) 37:5708–16. doi: 10.1016/j.vaccine.2019.07.103
31. Chiuchiolo MJ, Boyer JL, Krause A, Senina S, Hackett NR, Crystal RG. Protective immunity against respiratory tract challenge with Yersinia pestis in mice immunized with an adenovirus-based vaccine vector expressing V antigen. J Infect Dis. (2006) 194:1249–57. doi: 10.1086/507644
32. Huang SS, Li IH, Hong PD, Yeh MK. Development of Yersinia pestis F1 antigen-loaded microspheres vaccine against plague. Int J Nanomed. (2014) 9:813–22. doi: 10.2147/IJN.S56260
33. DeBord KL, Anderson DM, Marketon MM, Overheim KA, DePaolo RW, Ciletti NA, et al. Immunogenicity protective immunity against bubonic plague pneumonic plague by immunization of mice with the recombinant V10 antigen, a variant of LcrV. Infect Immun. (2006) 74:4910–4. doi: 10.1128/IAI.01860-05
34. Frey SE, Lottenbach K, Graham I, Anderson E, Bajwa K, May RC, et al. A phase I safety and immunogenicity dose escalation trial of plague vaccine, Flagellin/F1/V, in healthy adult volunteers (DMID 08-0066). Vaccine. (2017) 35:6759–65. doi: 10.1016/j.vaccine.2017.09.070
35. Verma SK, Batra L, Tuteja U. Escherichia coli expressed flagellin C (FliC) of Salmonella Typhi improved the protective efficacy of YopE against plague infection. Vaccine. (2019) 37:19–24. doi: 10.1016/j.vaccine.2018.11.057
36. Suzue K, Young RA. Adjuvant-free HSP70(II) fusion protein system elicits humoral and cellular responses to HIV-1 p24. J Immunol. (1996) 156:873–9.
37. Suzue K, Zhou X, Eisen HN, Young RA. Heat shock protein fusion proteins as vehicles for antigen delivery into the major histocompatibility complex class I presentation pathway. Proc Natl Acad Sci USA. (1997) 94:13146–51. doi: 10.1073/pnas.94.24.13146
38. Huang Q, Richmond JF, Suzue K, Eisen HN, Young RA. In vivo cytotoxic T lymphocyte elicitation by mycobacterial heat shock protein 70 fusion proteins maps to a discrete domain and is CD4+ T cell independent. J Exp Med. (2000) 191:403–8. doi: 10.1084/jem.191.2.403
39. Srivastava PK, Amato RJ. Heat shock proteins: the ‘Swiss army knife' vaccines against cancers and infectious agents. Vaccine. (2001) 19:2590–7. doi: 10.1016/S0264-410X(00)00492-8
40. Pockley AG. Heat shock proteins as regulators of the immune response. Lancet. (2003) 362:469–76. doi: 10.1016/S0140-6736(03)14075-5
41. Robert J. Evolution of heat shock protein and immunity. Dev Comp Immunol. (2003) 27:449–64. doi: 10.1016/S0145-305X(02)00160-X
42. Hauser H, Chen SY. Augmentation of DNA vaccine potency through secretory heat shock protein-mediated antigen targeting. Methods. (2003) 31:225–31. doi: 10.1016/S1046-2023(03)00136-1
43. Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, et al. Stimulation of Th1-polarizing cytokines, C-C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein70. J Immunol. (2002) 169:2422–9. doi: 10.4049/jimmunol.169.5.2422
44. Cornelis GR. Yersinia type III secretion send in the effectors. J Cell Biol. (2002) 158:401–8. doi: 10.1083/jcb.200205077
45. Pettersson J, Holmström A, Hill J, Leary S, Frithz-Lindsten E, von Euler-Matell A, et al. The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol Microbiol. (1999) 32:961–76. doi: 10.1046/j.1365-2958.1999.01408.x
46. Mueller CA, Broz P, Müller SA, Ringler P, Erne-Brand F, Sorg I, et al. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science. (2005) 310:674–6. doi: 10.1126/science.1118476
47. Sing A, Reithmeier-Rost D, Granfors K, Hill J, Roggenkamp A, Heesemann J. A hypervariable N-terminal region of Yersinia LcrV determines Toll-like receptor 2-mediated IL-10 induction and mouse virulence. Proc Natl Acad Sci USA. (2005) 102:16049–54. doi: 10.1073/pnas.0504728102
Keywords: cellular immune response, humoral immune response, LcrV-HSP70, plague, yersiniosis, Yersinia enterocolitica, Yersinia pestis, Yersinia pseudotuberculosis
Citation: Gupta A, Narayan B, Kumar S and Verma SK (2020) Vaccine Potential of a Recombinant Bivalent Fusion Protein LcrV-HSP70 Against Plague and Yersiniosis. Front. Immunol. 11:988. doi: 10.3389/fimmu.2020.00988
Received: 12 November 2019; Accepted: 27 April 2020;
Published: 12 June 2020.
Edited by:
Fabiano Oliveira, National Institutes of Health (NIH), United StatesReviewed by:
Rodrigo Javier Gonzalez, Harvard Medical School, United StatesAxel T. Lehrer, University of Hawaii at Manoa, United States
Copyright © 2020 Gupta, Narayan, Kumar and Verma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shailendra Kumar Verma, dmVybWFza0BkcmRlLmRyZG8uaW4=; c2t2MDAwNUBnbWFpbC5jb20=