 Marcela Garzón-Tituaña1†Maykel A. Arias2†José L. Sierra-Monzón1,3Elena Morte-Romea1,3Llipsy Santiago1
Marcela Garzón-Tituaña1†Maykel A. Arias2†José L. Sierra-Monzón1,3Elena Morte-Romea1,3Llipsy Santiago1 Ariel Ramirez-Labrada4
Ariel Ramirez-Labrada4 Luis Martinez-Lostao1,3,5José R. Paño-Pardo1,3
Luis Martinez-Lostao1,3,5José R. Paño-Pardo1,3 Eva M. Galvez2
Eva M. Galvez2 Julián Pardo1,5,6,7,8*
Julián Pardo1,5,6,7,8*- 1Fundación Instituto de Investigación Sanitaria Aragón (IIS Aragón), Biomedical Research Centre of Aragón (CIBA), Zaragoza, Spain
- 2Instituto de Carboquímica ICB-CSIC, Zaragoza, Spain
- 3Hospital Clínico Universitario Lozano Blesa, Zaragoza, Spain
- 4Nanotoxicology and Immunotoxicology Unit (UNATI), Fundación Instituto de Investigación Sanitaria Aragón (IIS Aragón), Zaragoza, Spain
- 5Nanoscience Institute of Aragon (INA), University of Zaragoza, Zaragoza, Spain
- 6Aragon I + D Foundation (ARAID), Zaragoza, Spain
- 7Department of Biochemistry and Molecular and Cell Biology and Department of Microbiology, Preventive Medicine and Public Health, University of Zaragoza, Zaragoza, Spain
- 8Centro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Madrid, Spain
Sepsis is a serious global health problem. In addition to a high incidence, this syndrome has a high mortality and is responsible for huge health expenditure. The pathophysiology of sepsis is very complex and it is not well-understood yet. However, it is widely accepted that the initial phase of sepsis is characterized by a hyperinflammatory response while the late phase is characterized by immunosuppression and immune anergy, increasing the risk of secondary infections. Granzymes (Gzms) are a family of serine proteases classified according to their cleavage specificity. Traditionally, it was assumed that all Gzms acted as cytotoxic proteases. However, recent evidence suggests that GzmB is the one with the greatest cytotoxic capacity, while the cytotoxicity of others such as GzmA and GzmK is not clear. Recent studies have found that GzmA, GzmB, GzmK, and GzmM act as pro-inflammatory mediators. Specially, solid evidences show that GzmA and GzmK function as extracellular proteases that regulate the inflammatory response irrespectively of its ability to induce cell death. Indeed, studies in animal models indicate that GzmA is involved in the cytokine release syndrome characteristic of sepsis. Moreover, the GZM family also could regulate other biological processes involved in sepsis pathophysiology like the coagulation cascade, platelet function, endothelial barrier permeability, and, in addition, could be involved in the immunosuppressive stage of sepsis. In this review, we provide a comprehensive overview on the contribution of these novel functions of Gzms to sepsis and the new therapeutic opportunities emerging from targeting these proteases for the treatment of this serious health problem.
Introduction
The immune system is the most important line of defense against pathogens. The immune response to infection is initiated after the recognition of pathogen derived molecules known as Pathogen Associated Molecular Patterns (PAMPs) by specific receptors known as PAMP receptors. This process triggers the production of inflammatory, angiogenic, and chemotactic factors involved in the activation of innate and adaptive immune mechanisms leading to pathogen clearance, tissue repair, and resolution of the inflammatory response. However, if not properly regulated, inflammation may act as a double-edged sword that can trigger serious complications like sepsis. This syndrome is an important health problem, defined as “a life-threatening organ dysfunction caused by a dysregulated host response to infection” (1). Recent epidemiological studies estimate an annual incidence of 48.9 million sepsis cases with 11 million sepsis-related deaths representing 20% of all global deaths (2).
The pathophysiology of sepsis is very complex and it is not well-understood yet. Simultaneous and interrelated inflammatory and anti-inflammatory responses are developed (3, 4), although it is widely accepted that the initial phase of sepsis is characterized by a hyperinflammatory response while the late phase is characterized by immunosuppression and immune anergy that increases the risk of other opportunistic infections, specially bacterial and fungal. These opposite reactions hamper the development of effective treatments to reduce the inflammatory damage, and, at the same time, do not increase the risk or even help to prevent secondary infections. Indeed, most experimental trials to reduce the inflammatory response with general or specific anti-inflammatory molecules targeting PAMPs like endotoxin, PAMPRs like TLR4, complement, cytokines, or coagulation factors have not positively impacted on patient survival (5).
The reasons for the low efficacy of anti-inflammatory therapy are not clear since reduction of tissue damage and coagulation syndrome together with an effective antimicrobial therapy should be beneficial to improve disease outcome. One reason could be that anti-inflammatory therapy might reduce host pathogen clearance due to inhibition of inflammatory pathways important for the protective immune response against the infection responsible of sepsis (5). In addition, it is not clear yet whether the anti-inflammatory therapies tested up to now are able to prevent immunosuppression and anergy during sepsis. Thus, although molecules involved in exacerbated inflammation and coagulation like inflammatory cytokines (i.e., IL6, IL1, TNFα) have shown a good prognostic value for sepsis progression and severity, targets to regulate inflammation without compromising host immune response against the sepsis-inducing pathogen or against secondary infections have not been found yet. Recently, some members of a family of serine-proteases named granzymes (Gzms), that are expressed by immune and non-immune cells (6), have been found in extracellular human fluids of different inflammatory disorders like sepsis. In addition, they have been found to regulate the inflammatory response in vitro and in vivo during different inflammatory disorders related to infections like sepsis.
The granule exocytosis pathway is a specialized form of intracellular protein delivery by which lymphocytes release perforin and Gzms. Perforin exerts its action allowing the passage of granzymes into the cytosol of target cells to carry out their effector functions, including cytotoxic and non-cytotoxic functions (7). On the other hand, Gzms can be released into the extracellular milieu where they will exert some extracellular functions including regulation of inflammation, pathogen inactivation, or extracellular matrix remodeling (6, 8). Gzms are a family of serine proteases classified according to their cleavage specificity. Five Gzms in humans (A, B, H, K, and M) and 10 in mice (A, B, C, D, E, F, G, K, M, and N) have been described. GzmA and GzmB are the most abundant and best characterized (6). Traditionally, it was assumed that all Gzms acted as cytotoxic proteases. However, recent evidence suggests that intracellulary-delivered GzmB is the one with the greatest cytotoxic capacity, while the cytotoxicity of others such as GzmA and GzmK is in controversy (7, 9–13). A recent study has shown that GzmA might mediate pyroptotic cell death in human and mouse tumor cells (14), although the relevance of this finding needs to be confirmed in different experimental models, since it has been previously reported that the cytotoxic potential of intracellulary-delivered GzmA might differ between mouse and human (15). Regarding human GzmH, a few studies have shown that it can induce cell death and inactivate viral proteins (7). However, at present there is not any study correlating GzmH with the regulation of the inflammatory response or sepsis, and, thus, GzmH will not be further discussed in this review.
Recent studies have found that GzmA, GzmB, GzmK, and GzmM act as pro-inflammatory mediators and could be involved in the pathophysiology of sepsis (7, 10–12) (Table 1 and Figure 1). Thus, a detailed study of Gzms in different sepsis models as well as in patients undergoing different types of sepsis might provide new prognostic factors and therapeutic targets to overcome some of the limitations observed for other inflammation-related targets. This hypothesis is supported by different previous experimental findings indicating that deficiency in any single Gzm does not significantly reduce pathogen clearance, and, thus, their inhibition during sepsis would reduce inflammation without compromising host anti-pathogen immunity (6).
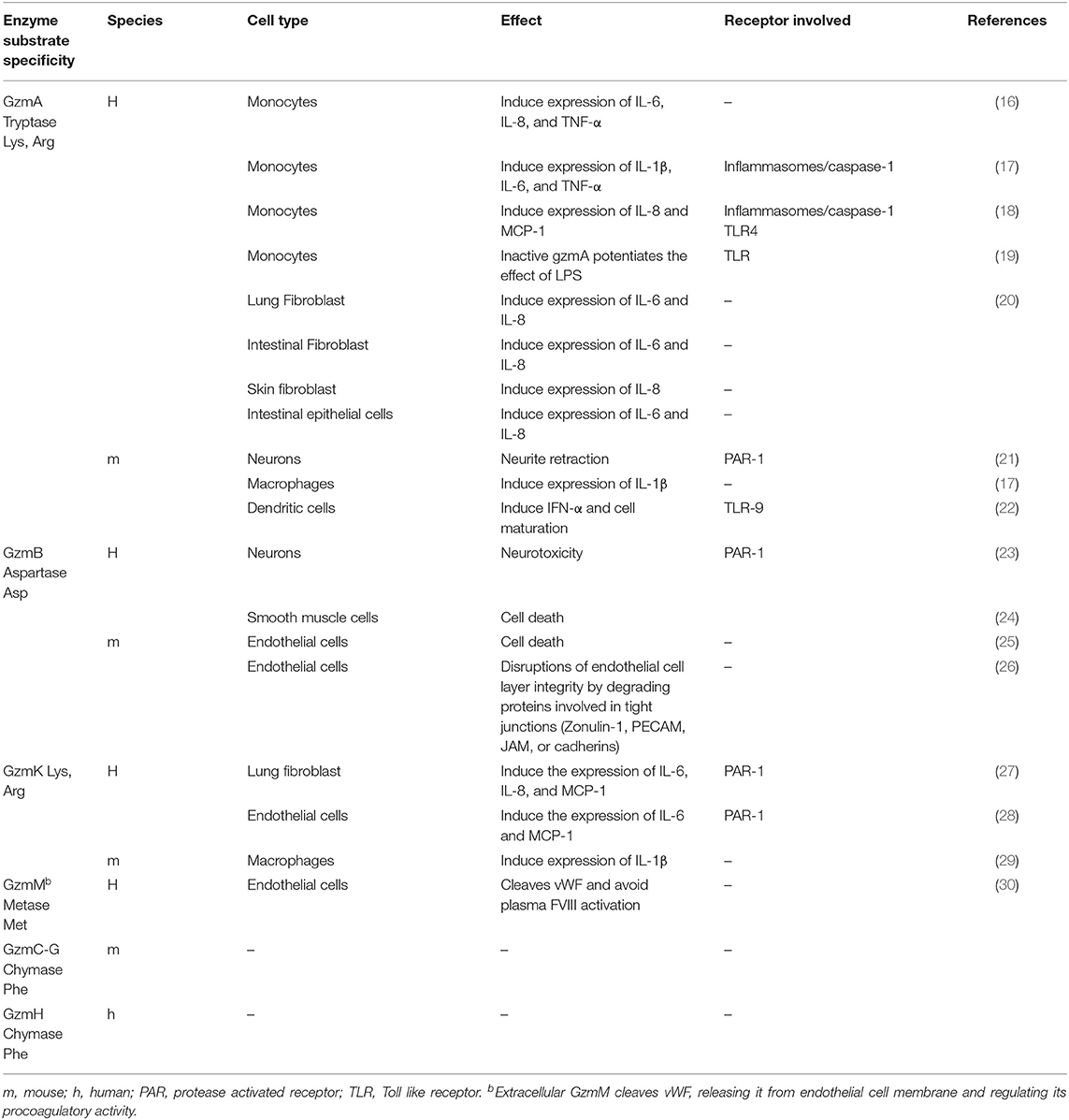
Table 1. Extracellular activity of granzymes on different cell types.
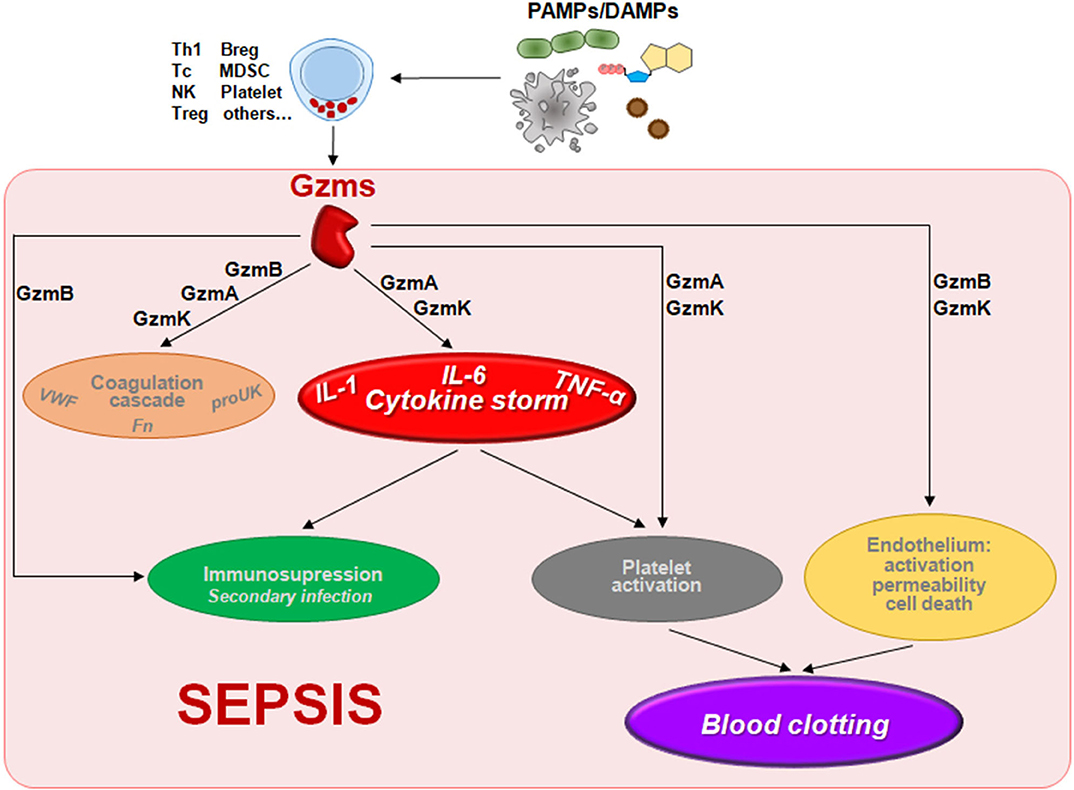
Figure 1. The potential contribution of the non-cytotoxic functions of granzymes to the pathophysiology of sepsis. Gzms can be released to the extracelular millieu by different cell types including NK cells, T cells, mast cells, platelets, or other cells stimulated by molecules presented in pathogens (fungal, bacterial, or viral PAMPs) or released by damaged cells (DAMPs) during sepsis. There different Gzms could contribute to different alterations found during sepsis like the inflammatory cytokine storm, blood clotting, cardiovascular disorders (coagulation and endotelial permeability), or immunosuppresion as described in the text. Abbreviations: Breg, regulatory B cell; MDSC, myeloid-derived suppressor cell; NK, natural killer; Tc, cytotoxic T cell; Th1, type 1 T helper cell; Treg, regulatory T cell; VWF, Von Willebrand factor; proUK, prourokinase; Fn, fibrin.
In this review, we will present the available evidence in animal models and humans that suggests that Gzms might have a relevant role in sepsis pathophysiology. In addition, we will discuss the potential advantages of using Gzms as therapeutic targets to reduce organ damage without compromising pathogen clearance and to prevent immunosuppression-associated secondary infections. Since novel functions of Gzms, non-related with its ability to induce cell death, have emerged in the last years, we will focus on the potential impact of these non-cytotoxic functions in the pathophysiology of sepsis. Some of them are originated in in vitro studies in non-sepsis models. However, since several of these non-cytotoxic functions (explained in more detail in the following sections) form part of the pathophysiology of sepsis, we think that it will be useful to discuss its potential impact on sepsis, indicating when appropriate the needed to specifically confirm their relevance in sepsis.
Granzymes are Elevated in Patients Suffering From Sepsis
Some studies have analyzed the presence of soluble Gzms in fluids from patients suffering from sepsis. These data show an increase in cellular and serum levels of some Gzms like GzmA, GzmB, and GzmK. Increased levels of GzmA and GzmB have been found in patients with severe gram negative bacterial infections as well as in healthy volunteers with an experimental endotoxemia with lipopolysaccharide (LPS) (31). GzmM and GzmK were also found in serum from volunteers with experimental endotoxemia (32) and GzmM was also elevated in serum from meningococcal sepsis (30). In contrast, GzmA levels were reduced in plasma during sepsis in burned patients (33). This result suggests that the level of GzmA in septic patients might depend on the underlying cause of sepsis. Supporting this hypothesis, Wensink et al. also showed that the release of GzmK and GzmM from human peripheral blood lymphocytes depended on the bacteria used for the stimulation (32). GzmK has also been found in the plasma of patients with sepsis as well as in bronchioalveolar fluid of patients with viral pneumonia (34, 35). However, the clinical significance of all these observations remains unknown.
A key question to understand the potential significance of soluble Gzms in sepsis is the regulation of its activity at the extracellular milieu. Like other proteases intracellular and extracellular Gzms might be tightly regulated to avoid unregulated proteolysis and cell/tissue damage. The main natural inhibitors that control Gzm activity are members of the Serpin (Serineprotease inhibitor) family. Different serpins have been found to inhibit extracellular Gzms like Inter-alpha-trypsin inhibitors (IaIs) (GzmA and GzmK), antithrombin III (GzmA), or bikunin (GzmK) (36). Thus, if extracellular Gzms are expected to play any role during sepsis it should be expected that they are not counteract by those inhibitors. Confirming this hypothesis and supporting a role for extracellular Gzms in sepsis it has been described that the levels of these inhibitors in plasma are reduced during sepsis. Indeed its concentration inversely correlated with mortality (37, 38).
Regarding the clinical utility of Gzms in sepsis prognosis, it was found that increased levels of GzmA and GzmB in CD8+T cells were associated with a worse prognosis in severe sepsis patients (39). Pending of validating the clinical significance of the presence of soluble Gzms in septic patients, studies in animal models in vivo indicate a key role for some Gzms like GzmA and GzmM in inflammation and sepsis. GzmA deficiency has been shown to protect against LPS-induced endotoxicosis (17, 40) as well as against sepsis induced by different bacterial pathogens including gram negative Brucella microti (41) or gram positive Streptococcus pneumoniae (42). These results suggest that the involvement of GzmA in sepsis is not related with Gram bacterial characteristics. Similarly, GzmM deficiency protects against LPS-induced endotoxicosis (40). In contrast, GzmB deficiency did not affect LPS-induced endotoxicosis (17, 40) or bacterial-induced sepsis (41). Importantly, increased survival in GzmA or GzmM deficient mice correlated with a reduction of the cytokine storm as measured by the levels of inflammatory cytokines in serum, organ damage, and coagulation activity confirming amelioration of the main sepsis pathological consequences. The course of sepsis in GzmK deficient mice remains to be elucidated.
The activity of Gzms during infectious diseases and sepsis seems to differ between anatomical site as well as the type of pathogen involved. For instance, a study showed that there was low level of GzmA in peritoneal lavage fluid of healthy mice while this level increased overtime during sepsis induced by E. coli (43). On the other hand, in a pneumonia-induced sepsis using a gram positive bacteria (Streptococcus pneumoniae) model the opposite was found (42). Here a small remark between the differences found in the pathophysiology of sepsis induced by gram negative and gram positive bacteria should be included. The onset and severity of sepsis may be defined by the molecular pathway involved in the activation of the immune system and the cytokines that can be induced in each case, both of which depend on the PAMPs expressed by the pathogen. Traditionally, the main responsible pathogens of sepsis have been considered gram negative bacteria. Nevertheless, it has been reported that gram positive bacteria are responsible for an ever-increasing number of septic events and have become the most frequent cause of sepsis, matching or even exceeding the sepsis caused by gram negative bacteria (44). It is known that the main receptors involved in the recognition of gram positive and negative bacteria are TLRs 2 and 4, respectively. This may be helpful for future studies and developments of sepsis treatments (44) and might help to explain the differences found regarding GzmA. For instance, a higher expression of IL-1β and IL-18 may indicate that the infection is due to a gram + bacteria while IL-6 and TNF would have a higher expression in gram negative infections (44). Indeed, GzmA has been found to produce very high levels of IL6 and TNF (Santiago et al. Cell Reports, Under review).
The inflammatory effect of GzmA does not seem to regulate bacterial control as GzmA deficient mice efficiently control infections like Mycobacterium tuberculosis, Listeria monocytogenes, Brucella microti, Klebsiella pneumoniae, Streptococcus pneumoniae, or Escherichia coli (6). Based on these experimental evidences it could be hypothesized that by targeting GzmA, bacteria-associated sepsis could be ameliorated without compromising the ability of the immune system to control infection. Thus, these findings open the opportunity to treat sepsis without causing immunosuppression and provide a new opportunity to overcome some of the limitations of other inflammatory targets unsuccessfully tested in clinical trials up to now.
Which could be the Mechanisms Behind the Inflammatory Action of Gzms in Sepsis?
Several biological functions of Gzms that could contribute to the different alterations found in sepsis have been described. Mainly those related with the regulation of inflammatory responses, the coagulation cascade, and changes in vascular permeability (summarized in Figure 1). In this section we will describe the mechanisms activated by Gzms that could potentially contribute to the pathophysiology of sepsis with a special focus on the hyperinflammatory stage.
Regulation of Inflammatory Cytokine Networks and Cytokine Storm
The hyperinflammatory response in sepsis is characterized by high levels of pro- and anti-inflammatory cytokines like IL-1β, IL-1α, IL-18, IL-17, TNFα, IL-6, MIF, HMGB1, IL-10, IL-4, and IL-13 (3, 45). This stage is also characterized by coagulation disorders, interstitial edema, hypotension, reduced perfusion, tissue hypoxia, mitochondrial dysfunction, and cell death (3, 4). It has been shown that extracellular GzmA is able to induce the production of different inflammatory cytokines such as IL-1β, TNF-α IL-6, or IL-8 in both mouse and human cells like monocytes, macrophages, fibroblasts, endothelial and epithelial cells (17, 20) (Table 1) which might contribute to the cytokine storm observed during sepsis. Indeed, most cytokines are reduced in GzmA deficient mice suffering from sepsis as indicated above. One of the first described substrates for GzmA is pro-interleukin 1β (pro IL-1 β), which after removal of the N-terminal part, generates the active inflammatory form IL-1β (46). However, direct cleavage of proIL1β and activation of IL1β has not been subsequently confirmed by different authors. Alternatively it has been reported that GzmA regulate the generation of inflammatory cytokines in human monocytes by caspase-1 and TLR4 dependent pathways (17, 18). Some other studies have reported that mouse GzmA is able to activate PAR1, a member of the Protease Activated Receptor (PAR) family, in mouse neurons (21). Since PARs are involved in platelet activation and thrombosis it could be suggested that PARs are involved in GzmA-mediated inflammation and coagulation alterations. However, as discussed below, the roles of PAR1 and 2 in sepsis are not clear yet and thus, in vivo extrapolation of all these results generated in vitro should be done with caution (47).
Alternatively, GzmA has been proposed to potentiate the effects of LPS on human monocytes by a mechanism dependent on TLR4 (19). Here authors showed that enzyme activity was not required for this effect, albeit the underlying mechanism was not clarified. This result contradicts previous findings that showed that human inactive GzmA was not able to induce the production of inflammatory cytokines in human monocytes (17). In addition, it has been shown that the inflammatory activity of mouse GzmA in vivo is significantly affected when GzmA inactivated with an inhibitor is used (48). The reasons for this discrepancy are not clear yet and will require further investigation.
GzmK displays a tryptase-like activity similar, but not identical (49, 50) to GzmA, and it might also induce the production of proinflammatory cytokines (Table 1). Nanomolar concentrations of GzmK induce the maturation and secretion of IL-1β suggesting that GzmK may augment GzmA-induced proinflammatory processes by differentially cleaving the same or different specific substrates (29). GzmK has also been found to activate PAR-1 in endothelial and fibroblast cells and to induce the generation of inflammatory cytokines (27, 28). However, as indicated above, its role in sepsis remains to be elucidated. Finally, as it has been previously discussed, GzmM is involved in endotoxicosis (40) although it is not known if GzmM is able to directly modulate inflammation on target cells (51).
The Coagulation Cascade
Systemic inflammation activates coagulation and inhibits anticoagulant and fibrinolytic mechanisms which leads to a dysregulated procoagulant state (52). The tissue factor (TF), expressed in monocytes and microparticles from living or apoptotic cells, is the responsible of the coagulation cascade activating factor VII forming the FT/VIIa complex which activates complexes IX and X. All of these play an important role in platelet activation (53, 54). TF is the main initiator of in vivo coagulation and it is not expressed in circulation under healthy conditions. Nevertheless, under inflammatory conditions, TF is expressed in endothelial and fibroblast cells and released to the circulatory system due to the action of proinflammatory cytokines, C reactive protein, and final products of advanced glycation (55–57). GzmA and GzmK could contribute to enhanced activation of the coagulation cascade in sepsis by generating cytokines involved in endothelial cell activation and coagulation like TNF-α or IL6 (58–60). GzmB is able to enhance the proinflammatory activity of IL-1α by proteolytic cleavage (61), although as indicated above, GzmB deficiency does not protect from sepsis.
Gzms can also modulate the coagulation cascade irrespectively of its ability to induce inflammatory cytokines. It has been reported that GzmB and GzmM can degrade coagulation-related substrates such as Von Willebrand factor (VWF) (30) or fibrinogen (only GzmB) (62). VWF is a homeostatic plasma protein that promotes platelet adhesion. Furthermore, VWF stabilizes coagulation factor VIII (FVIII) in plasma protecting it from proteolytic degradation and prolonging FVIII half-life. In septic patients, increased levels of VWF have been reported, so this protein could be a link between inflammation and thrombosis (63). VWF is regulated by the metalloprotease ADAMTS13, which specifically cleaves VWF in its A2 domain (64). It has been reported that GzmB and GzmM could mimic the action of ADAMTS13 by cleaving VWF regulating its adhesive function and preventing its binding to FVIII although did not affect platelet aggregation (30, 62). If these processes are relevant in vivo during sepsis, GzmB and GzmM would be expected to attenuate coagulation, preventing systemic coagulation and organ failure. This would explain why GzmB deficiency does not protect from sepsis, but would not explain the increased resistance of GzmM deficient mice to endotoxicosis and the reduced coagulation activity observed in these mice. It should be expected that VWF lost procoagulatory activity after cleavage and, thus, GzmM would protect from systemic coagulation disorders. A similar paradox has been found for GzmA. GzmA has been found to activate pro-urokinase, an enzyme that presents anticoagulant activity (65). However, the detrimental role of GzmA in sepsis has been confirmed by several independent groups, suggesting that the in vitro function of Gzms in regulating proteins involved in coagulation might not be relevant during sepsis in vivo. However, this hypothesis should be clarified by analyzing specifically the role of Gzms in regulating the coagulation system in vivo.
Regulation of Platelet Function
Platelets also play an important role in sepsis pathophysiology since the presence of thrombocytopenia is frequent in these patients. The role of platelets in sepsis is very complex as they can contribute to sepsis pathophysiology at different levels beyond regulation of haemostasis (66). Platelets can be directly activated by endotoxins, proinflammatory cytokines, or proteases such as thrombin (52, 67, 68). They contribute to the generation of inflammatory responses and, among other mediators, it has been shown that platelets can express GzmA and GzmB (18, 69, 70). Platelets can form clots on the endothelial damaged surface which produces elevated levels of VWF which is used as a marker for endothelial damage. It has been reported that the presence of proinflammatory cytokines such as IL-6, IL-8, and TNF-α (all of them induced by GzmA) are capable of induce the liberation of large VWF multimers that is a potent platelet aggregator. In septic patients, it has been observed elevated levels of VWF antigen which is related to a poor sickness prognosis (71). Here, as indicated above, since GzmB and GzmM have been found to cleave VWF and interfere with its procoagulant activity, it is tempting to speculate that these Gzms might have regulatory activities to prevent excessive VWF activity and clotting.
Curiously, GzmA was found to interact with the thrombin receptor (PAR1) in platelets and block thrombin-mediated responses (72). Although this interaction was not sufficient to induce per se platelet activation and aggregation, it might be possible that it can potentiate the effect of other ligands like endotoxin, a suggestion that has not been experimentally validated.
More recently it has been shown that during aging human platelets acquire GzmA expression which contributes to the generation of proinflammatory cytokines in monocytes (18). In light of these recent findings it will be interesting to analyse if Gzm expression is increased in older patients suffering from sepsis and its correlation with disease severity. Finally, as it will be discussed in the next section, it has been found that GzmB is expressed in platelets during sepsis using a mouse polymicrobial peritoneal sepsis model and might contribute to some of the alterations in vascular biology observed during sepsis (70).
Endothelial Barrier Permeability: Any Role for Protease Activating Receptors (PARs)?
The fluid redistribution to the extravascular space observed during sepsis is a consequence of an increase in the endothelial permeability or of the barrier function loss. Previous studies suggest that the change in the membrane permeability is a consequence of the enzymatic cleavage of intercellular junction proteins, which results in a structural damage of endothelial cells. In vivo and in vitro studies have demonstrated that TNF-α is one of the responsible cytokines of this disorder and also that thrombin could potentiate them (73). Since some Gzms like GzmA and GzmM seem to be inflammatory mediators responsible of the detrimental effects of LPS in sepsis, including TNF-α production, they also could contribute to altered endothelial permeability.
The link between coagulation and inflammation has shown that the family of PARs play an important role because various proteases activated in the coagulation cascade will induce inflammation through PAR receptors. Activation of these receptors in endothelial cells critically contributes to the regulation of endothelial permeability. This family consists of four members, PAR-1,-2,-3,-4, that are expressed in cells present in the vasculature such as endothelial cells, monocytes, macrophages, platelets, fibroblasts and smooth muscle cells. PAR-1 is activated by thrombin, FXa (Activated factor X), trypsin and APC (Activated Protein C). It has also been reported that GzmA, B, and K are able to activate PAR1 in different cell types (21, 23, 27, 28). In contrast, PAR-2,-3 or−4 have not been found to be activated by any Gzm, albeit PAR2 participation in colitis has been indirectly proposed (74). Here it should be noted that neither activation of PAR2 by GzmA nor GzmA contribution to colitis were analyzed in the later study. Thus, it is not clear yet if PAR2 is activated by GzmA and the role of PAR2 in GzmA-mediated cytokine induction. A recent study found that a PAR-2 inhibitor (I-343) inhibited foot swelling induced by in vivo administration of GzmA (48). This result suggests that PAR-2 could be directly or indirectly involved in some of the biological functions of GzmA, but its specific role in the regulation of GzmA-mediated inflammatory cytokine production will require further experimental validation.
Despite the role of PAR-1 and 2 in the regulation of vascular biology and endothelial cell activation, its contribution to the pathophysiology of sepsis is complex and not completely clarified (47). Some data suggest that multiple activation of PAR receptors by coagulation proteases may contribute to inflammation in endotoxemia and in sepsis (75). However, it was found that PAR1 and/or PAR2 deficiency neither reduce the inflammatory response nor increase survival in a mouse model of endotoxemia, indicating that PAR1 and PAR2 are dispensable for LPS-induced sepsis (76, 77). In a later study, using PAR antagonists in wild type mice, it was shown that the role of PAR1 and PAR2 in sepsis is reversal, being detrimental in the early phase of sepsis and beneficial in later stages and thus, the net balance of PAR deficiency during sepsis was indistinguishable between wild type and PAR deficient mice (78). In contrast to PARs deficient mice, GzmA deficient mice are protected from endotoxemia and sepsis and, thus, PAR1 and 2 should not play an important role in GzmA-induced inflammation during sepsis.
PARs are expressed by a variety of cells and the function in each cell type might differ, which would explain the differences between the in vitro and the in vivo observations regarding PAR-1, GzmA, and sepsis. GzmA has been shown to activate PAR1 in neurons (21), but not in platelets (72) or monocytes (16). We have found that albeit a PAR-1 inhibitor, Vorapaxar, reduced IL6 production in mouse macrophages stimulated with GzmA, the protease was able to induce the same level of IL6 in macrophages from wt or PAR1 deficient mice (Santiago et al. Cell Reports, Accepted), indicating that PAR1 is dispensable for GzmA-induced inflammation in macrophages. This apparent contradictory result might be explained by the potential unspecific effects of Vorapaxar including toxicity (79). Whatever it is, it supports the previous findings suggesting that PAR1 is not activated by GzmA in platelets or monocytes (16, 72). In addition, it should be taken into account that even PAR1 could be activated in some cell types, its relative contribution to GzmA-mediated inflammation should be validated by using adequate inhibitors or genetic deficient cell models (KO or siRNA).
Regarding PAR-3 and−4, although they can be activated by thrombin and other proteases its role during sepsis has not been confirmed yet (80) and in addition there is not any evidence relating Gzms and them.
It seems that in light of the different results pointing to a complex regulation of PARs during sepsis, the involvement of PARs in the detrimental effects of GzmA on vascular permeability during sepsis will require further clarification and specific experimental validation.
Endothelial Barrier Permeability: Extracellular Matrix Degradation, Matrikines and More
In addition to inflammatory cytokines and direct endothelial cell activation, other factors involved in the maintenance of vascular permeability could contribute to the loss of endothelial barrier function during sepsis. Some of these biological processes have been shown to be activated by Gzms in vitro like extracellular matrix (ECM) degradation, generation of products from ECM degradation with biological activity (matrikines) or killing of endothelial and smooth muscle cells (Table 1). Although the involvement of these processes in the mechanisms activated by Gzms during sepsis has not been directly analyzed, we would like to speculate on the potential implications of some of them.
Although GzmB deficiency does not increase survival during endotoxemia (40) or bacterial sepsis (41, 81), some of the biological functions of this protease might contribute to some of the vascular alterations observed in sepsis. For example, it has been shown that extracellular GzmB is able to directly kill cells involved in the maintenance of vascular architecture like endothelial and smooth muscle cells (25).
Gzm B Induces Smooth Muscle Cell Apoptosis in the Absence of Perforin. This process is based in its ability to induce ECM degradation and cell detachment (82, 83), which might activate cell death by anoikis (26, 82). Alternatively GzmB could disrupt endothelial cell barrier integrity by degrading proteins involved in tight junctions like Zonulin-1, PECAM, JAM, or Cadherins as previously shown (26). Later on it was found a role for GzmB in VE-cadherin cleavage and endothelial permeability in vitro and in vivo (84). In addition, ECM degradation by GzmB has been shown to release vascular endothelial growth factor which could affect vascular permeability (85). GzmB has also been found to affect wound healing, which was related to the ability of this protease to degrade fibronectin (86). Last but not least, it has been reported that GzmB may act on some of components of the EC involved in fibrillogenesis such as fibrillin-1 or decorin, increasing vascular permeability (87, 88), one of the most important pathological events that occur in sepsis.
Among other cell sources, platelets (70) and mast cells (26) have been found to express GzmB, but not perforin or GzmA, and thus, these cells could be the source of extracellular GzmB leading to its detrimental functions related with ECM degradation. In addition, it was found that GzmB of CD8+T and NK cells could modulate endothelial cell permeability and immune cells transmigration as a mechanism involved in host protection against viral infections (89). NK cells have been involved in sepsis (90, 91) and thus unregulated NK cells responses leading to a high release of GzmB due to PAMP-induced activation of NK cell receptors could transform a physiological protective mechanism into a pathological insult affecting endothelial cell permeability.
We would like to reiterate that the roles of GzmB in coagulation or in vascular permeability have not been studied yet in the context of a septic response and thus, all these hypotheses will require experimental validation.
Regarding GzmA, this protease can also degrade some proteins of the ECM like fibronectin or collagen IV (92, 93) and also could be released by NK cells during sepsis (41), although the biological functions of these processes remain unexplored.
Granzymes at the Immunosuppressive Edge
With the improvement of intensive care services, the majority of patients with sepsis are able to survive the hyperinflammatory phase but enter a prolonged immunosuppression stage that has been called "immunoparalysis” in which they will be susceptible to secondary infections (94).
In the late stages of sepsis, murine studies reveal a relatively constant balance in proinflammatory and anti-inflammatory cytokines, although with a smaller magnitude when compared to the acute phase. One of the key features of immunosuppression in sepsis is the state of cellular anergy that can begin to occur even in the early stages of the disease (3, 95, 96). This immunosuppression stage is characterized by a decrease in antigen presentation, alteration in the expression of costimulatory molecules, and changes in lymphocytes populations. Here some experimental evidences acquired in different disease models suggest that some Gzms might also contribute to the different alterations leading to immunosuppression.
One of the causes of anergy is the decrease in antigen presentation and dendritic cell maturation leading to impaired protective cellular responses (97, 98). The function (expression for HLA-II and co-stimulatory molecules) and number of several antigen presenting cells has been found to be compromised in sepsis correlating with reduced survival like dendritic cells or monocytes (99–103). These alterations contribute to changes in the cytokine profiles and alterations in lymphocyte populations.
Although a direct impact of Gzms on antigen cell presentation has not been reported, GzmB might contribute to T cell immunosuppression by different means. All the hypotheses proposed below will require experimental validation in order to show if Gzms can contribute to the immunosuppressive stage in sepsis.
GzmB has been found to be expressed by human and mouse Treg cells (104, 105) and contribute to immunosuppression and cancer immune-evasion by inhibiting Tc and NK cell responses in tumor models (106) or to control viral-induced lung inflammation (107). Thus, it could be speculated that GzmB of Treg cell could contribute to the cellular immunosuppression observed in sepsis patients. T regulatory cells are known to contribute to immune homeostasis preventing reactions against self-healthy tissue and commensal microbiota. They inhibit the immune response at different levels like B, CD4 Th1, CD8 T cell, and NK cell activation or dendritic cell maturation and it has been found that sepsis patients present reduced numbers of CD4 Th1, CD8 T cell, and NK cell (108) and increased Treg cell number and activity (109–111), which contributes to secondary infection.
Alternatively, GzmB is able to cleave the zeta chain of the T cell antigen receptor (TCR) which renders un-functional T cells (112), being another potential mechanism by which GzmB could contribute to immunosuppression in septic patients.
More direct evidence on the role of Gzms in immunosuppression was recently provided by Freishtat et al., who reported that acute sepsis-induced alterations in the megakaryocyte-platelet transcriptional axis result in strongly cytotoxic platelets expressing GzmB. These platelets used GzmB to kill CD4+T cells contributing to lymphodepletion (69). Later on the same group reported that the mechanism of platelet-mediated GzmB-dependent lymphotoxicity required cell to cell contact and was perforin-independent (70).
Conclusions and Future Perspectives
Sepsis is a global serious health problem for which early specific diagnosis and optimized treatment is crucial in order to reduce organ damage and improve survival. It is well-known the importance and influence of inflammation in the development and pathogenesis of sepsis (3, 5). However, the molecular bases to understand the dysregulated inflammatory response during sepsis are complex and often not well-understood. Indeed, most experimental trials to reduce inflammation during sepsis have not been successful and a positive impact on patient survival has not been reported. Among other reasons the impact of anti-inflammatory therapy on pathogen control by host immune response and the risk of secondary infections could contribute to the low efficacy observed during the trials with these therapies. During the last years, cumulative experimental evidences indicate that some members of the Gzm family, especially GzmA, have a key role in modulating inflammation and contribute to sepsis. Indeed, elevated levels of Gzms have been found in patients suffering from sepsis albeit the clinical significance remains unclear. In addition, in vivo mouse models show that GzmA deficiency protects from bacterial sepsis and endotoxemia indicating that GzmA have the potential to be used as biomarker and/or therapeutic target in sepsis (17, 40, 41). A current limitation of these studies is that the therapeutic potential of GzmA should be validated using a GzmA inhibitor in animal models expressing the protease. In addition, all in vivo studies have been performed using purified endotoxin or a single bacterial agent, and sepsis is often induced by more than a single microbial agent like peritoneal sepsis, one of the most common causes of sepsis. Moreover, the model of endotoxemia induced by LPS, albeit useful to study septic shock, does not provide information on the impact of regulation of inflammation on microbial infection.
Most studies in this field has been focused on the ability of some Gzms like GzmA, GzmK, and GzmM to induce the production of inflammatory cytokines in different cell types in vitro including monocytes, macrophages, endothelial cells, or fibroblasts. However, the role of GzmK in vivo has not been analyzed yet and GzmM has only been validated in the LPS-induced endotoxemia in vivo model. Apart from the ability to regulate the generation of the cytokine storm associated to the septic process and the consequences of this response to coagulation disorders and organ damage, in vitro evidences suggest that Gzms might also directly regulate other alterations found in sepsis like coagulation cascades, platelets function, or vascular permeability. Further experimental studies will be required to find out the relevance of these results in animal models in vivo, although it can be anticipated that it will not be easy to separate the effects due to Gzm-induced inflammatory cytokines from those that could be directly regulated by these proteases.
Apart from the disorders related with the inflammatory response leading to disseminated coagulation and organ damage, as discussed above, Gzms might also contribute to the immunosuppressive stage commonly observed in septic patients, responsible for a high morbidity/mortality due to secondary infections. Indirect evidences from other experimental models suggest that some Gzms might contribute to immunosuppression and, thus, it might be worth to dedicate some efforts to find out the role of Gzms in promoting immunosuppression and/or anergy during sepsis.
Despite the limitations mentioned above and all the hypotheses and speculations pending of experimental validation, some Gzms like GzmA or GzmK might present an important advantage in comparison with other inflammatory molecules that have been proposed and tested as therapeutic targets. GzmA or GzmK deficiency does not predispose to infection and these animals are able to efficiently clear most experimental infections (6, 113). Thus, targeting Gzms might reduce sepsis pathology without compromising the host immune response against the offending pathogen/s and, thus, enhancing the chances of efficient antimicrobial treatment to re-establish immune homeostasis and reduce immune-associated damage. Before these therapies can be developed and tested in potential clinical trials, it will be required to better understand the biology of granzymes during sepsis and the mechanisms involved.
Author Contributions
MG-T, MA, and JP-P designed and wrote the first draft. LM-L prepared Figure 1. All authors wrote and revised the manuscript.
Funding
This work was supported in part by FEDER/Gobierno de Aragón (group B29), Ministerio de Economia y Competitividad [SAF2014-54763-C2-1 and SAF2017-83120-C2-1-R (JP-P), SAF2014-54763-C2-2-R (EG)] and Instituto de Salud Carlos III (PI16-00526, LM-L; PI18/00527, JP-P). Predoctoral grants/contracts from Fundacion Santander/Universidad de Zaragoza (LS and MA), Ministerio de Ciencia, Innovación y Universidades (MG-T). MA has a Juan de la Cierva Contract (Ministerio de Ciencia, Innovación y Universidades) and JS-M a Rio Hortega Contract (Instituto de Salud Carlos III). JP was supported by Fundación Aragon I+D (ARAID).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock. (sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
3. Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, et al. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev. (2013) 93:1247–88. doi: 10.1152/physrev.00037.2012
4. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. (2016) 353:i1585. doi: 10.1136/bmj.i1585
5. Nedeva C, Menassa J, Puthalakath H. Sepsis: inflammation is a necessary evil. Front Cell Dev Biol. (2019) 7:108. doi: 10.3389/fcell.2019.00108
6. Arias M, Martinez-Lostao L, Santiago L, Ferrandez A, Granville DJ, Pardo J. The untold story of granzymes in oncoimmunology: novel opportunities with old acquaintances. Trends Cancer. (2017) 3:407–22. doi: 10.1016/j.trecan.2017.04.001
7. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. (2015) 15:388–400. doi: 10.1038/nri3839
8. Turner CT, Lim D, Granville DJ. Granzyme B in skin inflammation and disease. Matrix Biol. (2019) 75–6, 126–140. doi: 10.1016/j.matbio.2017.12.005
9. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
10. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. (2010) 17:616–23. doi: 10.1038/cdd.2009.206
11. Joeckel LT, Bird PI. Are all granzymes cytotoxic in vivo? Biol Chem. (2014) 395:181–202. doi: 10.1515/hsz-2013-0238
12. Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
13. Wensink AC, Hack CE, Bovenschen N. Granzymes regulate proinflammatory cytokine responses. J Immunol. (2015) 194:491–7. doi: 10.4049/jimmunol.1401214
14. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
15. Kaiserman D, Bird CH, Sun J, Matthews A, Ung K, Whisstock JC, et al. The major human and mouse granzymes are structurally and functionally divergent. J Cell Biol. (2006) 175:619–30. doi: 10.1083/jcb.200606073
16. Sower LE, Froelich CJ, Allegretto N, Rose PM, Hanna WD, Klimpel GR. Extracellular activities of human granzyme A. Monocyte activation by granzyme A versus alpha-thrombin. J Immunol. (1996) 156:2585–90.
17. Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. (2008) 29:720–33. doi: 10.1016/j.immuni.2008.08.014
18. Campbell RA, Franks Z, Bhatnagar A, Rowley JW, Manne BK, Supiano MA, et al. Granzyme A in Human Platelets Regulates the Synthesis of Proinflammatory Cytokines by Monocytes in Aging. J Immunol. (2018) 200:295–304. doi: 10.4049/jimmunol.1700885
19. Wensink AC, Kok HM, Meeldijk J, Fermie J, Froelich CJ, Hack CE, et al. Granzymes A and K differentially potentiate LPS-induced cytokine response. Cell Death Discov. (2016) 2:16084. doi: 10.1038/cddiscovery.2016.84
20. Sower LE, Klimpel GR, Hanna W, Froelich CJ. Extracellular activities of human granzymes. I. granzyme A induces IL6 and IL8 production in fibroblast and epithelial cell lines. Cell Immunol. (1996) 171:159–63. doi: 10.1006/cimm.1996.0187
21. Suidan HS, Bouvier J, Schaerer E, Stone SR, Monard D, Tschopp J. Granzyme A released upon stimulation of cytotoxic T lymphocytes activates the thrombin receptor on neuronal cells and astrocytes. Proc Natl Acad Sci USA. (1994) 91:8112–6.
22. Shimizu K, Yamasaki S, Sakurai M, Yumoto N, Ikeda M, Mishima-Tsumagari C, et al. Granzyme A stimulates pDCs to promote adaptive immunity via induction of type I IFN. Front Immunol. (2019) 10:1450. doi: 10.3389/fimmu.2019.01450
23. Lee PR, Johnson TP, Gnanapavan S, Giovannoni G, Wang T, Steiner JP, et al. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1beta. J Neuroinflammation. (2017) 14:131. doi: 10.1186/s12974-017-0901-y
24. Choy JC, Hung VH, Hunter AL, Cheung PK, Motyka B, Goping IS, et al. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin: involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol. (2004) 24:2245–50. doi: 10.1161/01.ATV.0000147162.51930.b7
25. Choy JC, Cruz RP, Kerjner A, Geisbrecht J, Sawchuk T, Fraser SA, et al. Granzyme B induces endothelial cell apoptosis and contributes to the development of transplant vascular disease. Am J Transplant. (2005) 5:494–9. doi: 10.1111/j.1600-6143.2004.00710.x
26. Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. (2007) 14:1768–79. doi: 10.1038/sj.cdd.4402183
27. Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, Granville DJ. Granzyme K activates protease-activated receptor-1. PLoS ONE. (2011) 6:e21484. doi: 10.1371/journal.pone.0021484
28. Sharma M, Merkulova Y, Raithatha S, Parkinson LG, Shen Y, Cooper D, et al. Extracellular granzyme K mediates endothelial activation through the cleavage of protease-activated receptor-1. FEBS J. (2016) 283:1734–47. doi: 10.1111/febs.13699
29. Joeckel LT, Wallich R, Martin P, Sanchez-Martinez D, Weber FC, Martin SF, et al. Mouse granzyme K has pro-inflammatory potential. Cell Death Differ. (2011) 18:1112–9. doi: 10.1038/cdd.2011.5
30. Hollestelle MJ, Lai KW, van Deuren M, Lenting PJ, de Groot PG, Sprong T, et al. Cleavage of von Willebrand factor by granzyme M destroys its factor VIII binding capacity. PLoS ONE. (2011) 6:e24216. doi: 10.1371/journal.pone.0024216
31. Lauw FN, Simpson AJ, Hack CE, Prins JM, Wolbink AM, van Deventer SJ, et al. Soluble granzymes are released during human endotoxemia and in patients with severe infection due to gram-negative bacteria. J Infect Dis. (2000) 182:206–13. doi: 10.1086/315642
32. Wensink AC, Wiewel MA, Jongeneel LH, Boes M, van der Poll T, Hack CE, et al. Granzyme M and K release in human experimental endotoxemia. Immunobiology. (2016) 221:773–7. doi: 10.1016/j.imbio.2016.02.006
33. Accardo-Palumbo A, D'Amelio L, Pileri D, D'Arpa N, Mogavero R, Amato G, et al. Reduction of plasma granzyme A correlates with severity of sepsis in burn patients. Burns. (2010) 36:811–8. doi: 10.1016/j.burns.2009.11.009
34. Rucevic M, Fast LD, Jay GD, Trespalcios FM, Sucov A, Siryaporn E, et al. Altered levels and molecular forms of granzyme k in plasma from septic patients. Shock. (2007) 27:488–93. doi: 10.1097/01.shk.0000246905.24895.e5
35. Bratke K, Klug A, Julius P, Kuepper M, Lommatzsch M, Sparmann G, et al. Granzyme K: a novel mediator in acute airway inflammation. Thorax. (2008) 63:1006–11. doi: 10.1136/thx.2007.091215
36. Kaiserman D, Bird PI. Control of granzymes by serpins. Cell Death Differ. (2010) 17:586–95. doi: 10.1038/cdd.2009.169
37. Lim YP, Bendelja K, Opal SM, Siryaporn E, Hixson DC, Palardy JE. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J Infect Dis. (2003) 188:919–26. doi: 10.1086/377642
38. Opal SM, Lim YP, Siryaporn E, Moldawer LL, Pribble JP, Palardy JE, et al. Longitudinal studies of inter-alpha inhibitor proteins in severely septic patients: a potential clinical marker and mediator of severe sepsis. Crit Care Med. (2007) 35:387–92. doi: 10.1097/01.CCM.0000253810.08230.83
39. Napoli AM, Fast LD, Gardiner F, Nevola M, Machan JT. Increased granzyme levels in cytotoxic T lymphocytes are associated with disease severity in emergency department patients with severe sepsis. Shock. (2012) 37:257–62. doi: 10.1097/SHK.0b013e31823fca44
40. Anthony DA, Andrews DM, Chow M, Watt SV, House C, Akira S, et al. A role for granzyme M in TLR4-driven inflammation and endotoxicosis. J Immunol. (2010) 185:1794–803. doi: 10.4049/jimmunol.1000430
41. Arias MA, Jimenez de Bagues MP, Aguilo N, Menao S, Hervas-Stubbs S, de Martino A, et al. Elucidating sources and roles of granzymes A and B during bacterial infection and sepsis. Cell Rep. (2014) 8:420–9. doi: 10.1016/j.celrep.2014.06.012
42. van den Boogaard FE, van Gisbergen KP, Vernooy JH, Medema JP, Roelofs JJ, van Zoelen MA, et al. Granzyme A impairs host defense during Streptococcus pneumoniae pneumonia. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L507–16. doi: 10.1152/ajplung.00116.2016
43. Garcia-Laorden MI, Stroo I, Terpstra S, Florquin S, Medema JP, van TVC, et al. Expression and function of granzymes A and B in Escherichia coli peritonitis and sepsis. Mediators Inflamm. (2017) 2017:4137563. doi: 10.1155/2017/4137563
44. Feezor RJ, Oberholzer C, Baker HV, Novick D, Rubinstein M, Moldawer LL, et al. Molecular characterization of the acute inflammatory response to infections with gram-negative versus gram-positive bacteria. Infect Immun. (2003) 71:5803–13. doi: 10.1128/iai.71.10.5803-5813.2003
45. Matsumoto H, Ogura H, Shimizu K, Ikeda M, Hirose T, Matsuura H, et al. The clinical importance of a cytokine network in the acute phase of sepsis. Sci Rep. (2018) 8:13995. doi: 10.1038/s41598-018-32275-8
46. Irmler M, Hertig S, MacDonald HR, Sadoul R, Becherer JD, Proudfoot A, et al. Granzyme A is an interleukin 1 beta-converting enzyme. J Exp Med. (1995) 181:1917–22. doi: 10.1084/jem.181.5.1917
47. Asehnoune K, Moine P. Protease-activated receptor-1: key player in the sepsis coagulation-inflammation crosstalk. Crit Care. (2013) 17:119. doi: 10.1186/cc12502
48. Schanoski AS, Le TT, Kaiserman D, Rowe C, Prow NA, Barboza DD, et al. Granzyme A in chikungunya and other arboviral infections. Front Immunol. (2019) 10:3083. doi: 10.3389/fimmu.2019.03083
49. Bovenschen N, Quadir R, van den Berg AL, Brenkman AB, Vandenberghe I, Devreese B, et al. Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A. J Biol Chem. (2009) 284:3504–12. doi: 10.1074/jbc.M806716200
50. Plasman K, Demol H, Bird PI, Gevaert K, Van Damme P. Substrate specificities of the granzyme tryptases A and K. J Proteome Res. (2014) 13:6067–77. doi: 10.1021/pr500968d
51. de Poot SA, Bovenschen N. Granzyme M: behind enemy lines. Cell Death Differ. (2014) 21:359–68. doi: 10.1038/cdd.2013.189
52. Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. (2008) 83:536–45. doi: 10.1189/jlb.0607373
53. Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci. (2006) 11:1349–59. doi: 10.2741/1888
54. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. (2007) 27:1687–93. doi: 10.1161/ATVBAHA.107.141911
55. Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. (1983) 49:5–7.
56. Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA Jr. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci USA. (1986) 83:4533–7.
57. Camerer E, Kolsto AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res. (1996) 81:1–41.
58. Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. (2002) 4(Suppl. 3):S109–16. doi: 10.1186/ar576
59. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. (2004) 104:100–6. doi: 10.1182/blood-2004-01-0107
60. Hou T, Tieu BC, Ray S, Recinos Iii A, Cui R, Tilton RG, et al. Roles of IL-6-gp130 signaling in vascular inflammation. Curr Cardiol Rev. (2008) 4:179–92. doi: 10.2174/157340308785160570
61. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. (2011) 44:265–78. doi: 10.1016/j.molcel.2011.07.037
62. Buzza MS, Dyson JM, Choi H, Gardiner EE, Andrews RK, Kaiserman D, et al. Antihemostatic activity of human granzyme B mediated by cleavage of von willebrand factor. J Biol Chem. (2008) 283:22498–504. doi: 10.1074/jbc.M709080200
63. Kremer Hovinga JA, Zeerleder S, Kessler P, Romani de Wit T, van Mourik JA, Hack CE, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. (2007) 5:2284–90. doi: 10.1111/j.1538-7836.2007.02743.x
64. Gao W, Anderson PJ, Sadler JE. Extensive contacts between ADAMTS13 exosites and von Willebrand factor domain A2 contribute to substrate specificity. Blood. (2008) 112:1713–9. doi: 10.1182/blood-2008-04-148759
65. Brunner G, Simon MM, Kramer MD. Activation of pro-urokinase by the human T cell-associated serine proteinase HuTSP-1. FEBS Lett. (1990) 260:141–4. doi: 10.1016/0014-5793(90)80087-y
66. Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, et al. Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critically [corrected] ill patients? Ann Intensive Care. (2017) 7:115. doi: 10.1186/s13613-017-0337-7
67. Zielinski T, Wachowicz B, Saluk-Juszczak J, Kaca W. Polysaccharide part of Proteus mirabilis lipopolysaccharide may be responsible for the stimulation of platelet adhesion to collagen. Platelets. (2002) 13:419–24. doi: 10.1080/0953710021000024385
68. Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. (2002) 30:S294–301. doi: 10.1097/00003246-200205001-00020
69. Freishtat RJ, Natale J, Benton AS, Cohen J, Sharron M, Wiles AA, et al. Sepsis alters the megakaryocyte-platelet transcriptional axis resulting in granzyme B-mediated lymphotoxicity. Am J Respir Crit Care Med. (2009) 179:467–73. doi: 10.1164/rccm.200807-1085OC
70. Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, et al. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS ONE. (2012) 7:e41549. doi: 10.1371/journal.pone.0041549
71. Martin K, Borgel D, Lerolle N, Feys HB, Trinquart L, Vanhoorelbeke K, et al. Decreased ADAMTS-13. (A disintegrin-like and metalloprotease with thrombospondin type 1 repeats) is associated with a poor prognosis in sepsis-induced organ failure. Crit Care Med. (2007) 35:2375–82. doi: 10.1097/01.ccm.0000284508.05247.b3
72. Suidan HS, Clemetson KJ, Brown-Luedi M, Niclou SP, Clemetson JM, Tschopp J, et al. The serine protease granzyme A does not induce platelet aggregation but inhibits responses triggered by thrombin. Biochem J. (1996) 315:939–45.
73. Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. (2003) 60:49–57. doi: 10.1016/s0008-6363(03)00397-3
74. Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci USA. (2005) 102:8363–8. doi: 10.1073/pnas.0409535102
75. Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. (2003) 16:43–7. doi: 10.1023/B:THRO.0000014592.27892.11
76. Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, et al. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. (2004) 103:1342–7. doi: 10.1182/blood-2003-09-3051
77. Camerer E, Cornelissen I, Kataoka H, Duong DN, Zheng YW, Coughlin SR. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood. (2006) 107:3912–21. doi: 10.1182/blood-2005-08-3130
78. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. Role reversal for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. (2007) 8:1303–12. doi: 10.1038/ni1525
79. Flaumenhaft R, De Ceunynck K. Targeting PAR1: now what? Trends Pharmacol Sci. (2017) 38:701–16. doi: 10.1016/j.tips.2017.05.001
80. Heuberger DM, Schuepbach RA. Protease-activated receptors. (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J. (2019) 17:4. doi: 10.1186/s12959-019-0194-8
81. Garcia-Laorden MI, Stroo I, Blok DC, Florquin S, Medema JP, de Vos AF, et al. Granzymes A and B regulate the local inflammatory response during klebsiella pneumoniae pneumonia. J Innate Immun. (2016) 8:258–68. doi: 10.1159/000443401
82. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. (2005) 280:23549–58. doi: 10.1074/jbc.M412001200
83. Hendel A, Granville DJ. Granzyme B cleavage of fibronectin disrupts endothelial cell adhesion, migration and capillary tube formation. Matrix Biol. (2013) 32:14–22. doi: 10.1016/j.matbio.2012.11.013
84. Shen Z, Liu Y, Dewidar B, Hu J, Park O, Feng T, et al. Delta-like ligand 4 modulates liver damage by down-regulating chemokine expression. Am J Pathol. (2016) 186:1874–89. doi: 10.1016/j.ajpath.2016.03.010
85. Hendel A, Hsu I, Granville DJ. Granzyme B releases vascular endothelial growth factor from extracellular matrix and induces vascular permeability. Lab Invest. (2014) 94:716–25. doi: 10.1038/labinvest.2014.62
86. Hiebert PR, Wu D, Granville DJ. Granzyme B degrades extracellular matrix and contributes to delayed wound closure in apolipoprotein E knockout mice. Cell Death Differ. (2013) 20:1404–14. doi: 10.1038/cdd.2013.96
87. Chamberlain CM, Ang LS, Boivin WA, Cooper DM, Williams SJ, Zhao H, et al. Perforin-independent extracellular granzyme B activity contributes to abdominal aortic aneurysm. Am J Pathol. (2010) 176:1038–49. doi: 10.2353/ajpath.2010.090700
88. Ang LS, Boivin WA, Williams SJ, Zhao H, Abraham T, Carmine-Simmen K, et al. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. (2011) 2:e209. doi: 10.1038/cddis.2011.88
89. Prakash MD, Munoz MA, Jain R, Tong PL, Koskinen A, Regner M, et al. Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity. (2014) 41:960–72. doi: 10.1016/j.immuni.2014.11.012
90. Sherwood ER, Enoh VT, Murphey ED, Lin CY. Mice depleted of CD8+ T and NK cells are resistant to injury caused by cecal ligation and puncture. Lab Invest. (2004) 84:1655–65. doi: 10.1038/labinvest.3700184
91. Etogo AO, Nunez J, Lin CY, Toliver-Kinsky TE, Sherwood ER. NK but not CD1-restricted NKT cells facilitate systemic inflammation during polymicrobial intra-abdominal sepsis. J Immunol. (2008) 180:6334–45. doi: 10.4049/jimmunol.180.9.6334
92. Simon MM, Prester M, Nerz G, Kramer MD, Fruth U. Release of biologically active fragments from human plasma-fibronectin by murine T cell-specific proteinase 1. (TSP-1). Biol Chem Hoppe Seyler. (1988) 369:107–12.
93. Simon MM, Kramer MD, Prester M, Gay S. Mouse T-cell associated serine proteinase 1 degrades collagen type IV: a structural basis for the migration of lymphocytes through vascular basement membranes. Immunology. (1991) 73:117–9.
94. Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. (2009) 15:496–7. doi: 10.1038/nm0509-496
95. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/S1473-3099(13)70001-X
96. Ono S, Tsujimoto H, Hiraki S, Aosasa S. Mechanisms of sepsis-induced immunosuppression and immunological modification therapies for sepsis. Ann Gastroenterol Surg. (2018) 2:351–8. doi: 10.1002/ags3.12194
97. Hynninen M, Pettila V, Takkunen O, Orko R, Jansson SE, Kuusela P, et al. Predictive value of monocyte histocompatibility leukocyte antigen-DR expression and plasma interleukin-4 and−10 levels in critically ill patients with sepsis. Shock. (2003) 20:1–4. doi: 10.1097/01.shk.0000068322.08268.b4
98. Monneret G, Lepape A, Voirin N, Bohe J, Venet F, Debard AL, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. (2006) 32:1175–83. doi: 10.1007/s00134-006-0204-8
99. Gold JA, Parsey M, Hoshino Y, Hoshino S, Nolan A, Yee H, et al. CD40 contributes to lethality in acute sepsis: in vivo role for CD40 in innate immunity. Infect Immun. (2003) 71:3521–8. doi: 10.1128/iai.71.6.3521-3528.2003
100. Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol. (2006) 79:473–81. doi: 10.1189/jlb.0705413
101. Guisset O, Dilhuydy MS, Thiebaut R, Lefevre J, Camou F, Sarrat A, et al. Decrease in circulating dendritic cells predicts fatal outcome in septic shock. Intensive Care Med. (2007) 33:148–52. doi: 10.1007/s00134-006-0436-7
102. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome. (CARS) in critically ill patients. Clin Chest Med. (2008) 29, 617–25. doi: 10.1016/j.ccm.2008.06.010
103. Roger PM, Hyvernat H, Breittmayer JP, Dunais B, Dellamonica J, Bernardin G, et al. Enhanced T-cell apoptosis in human septic shock is associated with alteration of the costimulatory pathway. Eur J Clin Microbiol Infect Dis. (2009) 28:575–84. doi: 10.1007/s10096-008-0673-5
104. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. (2004) 104:2840–8. doi: 10.1182/blood-2004-03-0859
105. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. (2005) 174:1783–6. doi: 10.4049/jimmunol.174.4.1783
106. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
107. Loebbermann J, Thornton H, Durant L, Sparwasser T, Webster KE, Sprent J, et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. (2012) 5:161–72. doi: 10.1038/mi.2011.62
108. Harjai M, Bogra J, Kohli M, Pant AB. Is suppression of apoptosis a new therapeutic target in sepsis? Anaesth Intensive Care. (2013) 41:175–83. doi: 10.1177/0310057X1304100207
109. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med. (2004) 32:2329–31. doi: 10.1097/01.ccm.0000145999.42971.4b
110. Taylor AL, Llewelyn MJ. Superantigen-induced proliferation of human CD4+CD25- T cells is followed by a switch to a functional regulatory phenotype. J Immunol. (2010) 185:6591–8. doi: 10.4049/jimmunol.1002416
111. Cao C, Ma T, Chai YF, Shou ST. The role of regulatory T cells in immune dysfunction during sepsis. World J Emerg Med. (2015) 6:5–9. doi: 10.5847/wjem.j.1920-8642.2015.01.001
112. Wieckowski E, Wang GQ, Gastman BR, Goldstein LA, Rabinowich H. Granzyme B-mediated degradation of T-cell receptor zeta chain. Cancer Res. (2002) 62:4884–9.
Keywords: sepsis, inflammatory cytokine, granzymes, coagulopathy, endothelial (dys)function, immunosuppression
Citation: Garzón-Tituaña M, Arias MA, Sierra-Monzón JL, Morte-Romea E, Santiago L, Ramirez-Labrada A, Martinez-Lostao L, Paño-Pardo JR, Galvez EM and Pardo J (2020) The Multifaceted Function of Granzymes in Sepsis: Some Facts and a Lot to Discover. Front. Immunol. 11:1054. doi: 10.3389/fimmu.2020.01054
Received: 11 March 2020; Accepted: 30 April 2020;
Published: 17 June 2020.
Edited by:
Francesca Velotti, University of Tuscia, ItalyReviewed by:
David J. Granville, University of British Columbia, CanadaVanessa Pinho, Federal University of Minas Gerais, Brazil
Copyright © 2020 Garzón-Tituaña, Arias, Sierra-Monzón, Morte-Romea, Santiago, Ramirez-Labrada, Martinez-Lostao, Paño-Pardo, Galvez and Pardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián Pardo, cGFyZG9qaW1AdW5pemFyLmVz
†These authors have contributed equally to this work