 Emma Boberg
Emma Boberg Kristina Johansson
Kristina Johansson Carina Malmhäll
Carina Malmhäll Jenny Calvén
Jenny Calvén Julie Weidner
Julie Weidner Madeleine Rådinger
Madeleine Rådinger- Krefting Research Centre, Department of Internal Medicine and Clinical Nutrition, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
Background: Eosinophils develop from CD34+ progenitor cells in the bone marrow under the influence of interleukin (IL)-5. Several cell types produce IL-5, including type 2 innate lymphoid cells (ILC2s). The alarmin cytokine IL-33 is known to activate ILC2s in mucosal tissues, but little is known about IL-33-responsive ILC2s in the bone marrow in allergen-induced airway inflammation.
Methods: Wild type (WT) and Rag1 deficient (Rag1−/−) mice, which lack mature T and B cells, received intranasal doses of papain to induce acute allergic inflammation. In some experiments, mice were pre-treated with anti-IL-5 prior to the papain challenge. Furthermore, recombinant IL-33 was administered to WT mice, Rag1−/− mice, lymphocyte deficient mice (Rag2−/−Il2rg−/−) and to ex vivo whole bone marrow cultures. Bone marrow eosinophils and ILC2s were analyzed by flow cytometry. Eosinophil count was assessed by differential cell count and secreted IL-5 from bone marrow cells by ELISA.
Results: Intranasal administration of papain or IL-33 increased the number of mature eosinophils in the bone marrow despite the absence of adaptive immune cells in Rag1−/− mice. In parallel, an increased number of eosinophils was observed in the airways together with elevated levels of Eotaxin-2/CCL24. Bone marrow ILC2s were increased after papain or IL-33 administration, whereas ILC2s was found to be increased at baseline in Rag1−/− mice compared to WT mice. An upregulation of the IL-33 receptor (ST2) expression on bone marrow ILC2s was observed after papain challenge in both Rag1−/− and WT mice which correlated to increased number of bone marrow eosinophilia. Furthermore, an increased number of ST2+ mature eosinophils in the bone marrow was observed after papain challenge, which was further dependent on IL-5. In addition, bone marrow-derived ILC2s from both mouse strains produced large amounts of IL-5 ex vivo after IL-33 stimulation of whole bone marrow cultures. In contrast, IL-33-induced bone marrow and airway eosinophilia were abolished in the absence of ILC2s in Rag2−/−Il2rg−/− mice and no production of IL-5 was detected in IL-33-stimulated bone marrow cultures.
Conclusion: These findings establish bone marrow ILC2s and the IL-33/ST2 axis as promising targets for modulation of uncontrolled IL-5-dependent eosinophilic diseases including asthma.
Introduction
Eosinophils are terminally differentiated granulocytes that contribute to tissue damage and remodeling in asthmatic airways by releasing toxic inflammatory mediators (1, 2). Previous studies have shown that airway allergen challenge promotes bone marrow eosinophilopoiesis in individuals with asthma and in animal models of allergic airway inflammation (3–11). Importantly, high levels of eosinophils have been shown to correlate with increasing asthma severity (12). Eosinophils develop from CD34+ progenitor cells in the bone marrow under the influence of the interleukin family (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (13, 14). IL-5 plays an essential role in eosinophil biology by controlling several key features, including terminal differentiation, proliferation, and cell migration (15–17). It was previously shown that the alarmin cytokine IL-33 induces eosinophil lineage commitment in murine bone marrow (18). This was suggested to be regulated by an IL-5-dependent mechanism since blocking IL-5 prevented IL-33-induced eosinophil expansion. However, the source of IL-5 was not addressed in the study (18). Furthermore, Anderson et al. (11) showed that murine allergen-challenge with Alternaria triggered IL-33 release from the airways to induce IL-5 production by lung type 2 innate lymphoid cells (ILC2s). Elevated levels of IL-5 were shown to reach the circulation and promote eosinophilopoiesis in the bone marrow (11). Indeed, ILC2s are producers of type 2 cytokines such as IL-5 and IL-13 at sites of inflammation and have been implicated in the pathogenesis of several inflammatory diseases, including asthma (19, 20). In addition, Nussbaum et al. proposed that the predominant source of circulating IL-5 is from tissue-resident ILC2s which constitutively produce IL-5 (21). Several studies have suggested that CD4+ T cells and CD34+ progenitor cells produce IL-5 locally in the bone marrow at both homeostatic conditions and after airway allergen challenge (22–24). Recently, we showed that CD34+ progenitors and ILC2s, but not CD4+ T cells produce IL-5 locally in the bone marrow of IL-33 challenged mice (25). Interestingly, bone marrow ILC2s were the predominant source of IL-5 which coincided with the expansion of IL-5-responsive CD34+ progenitors following IL-33 challenge (25). Indeed, a positive relationship between IL-33 and eosinophilia has been demonstrated in several studies, including reports of lower baseline levels of eosinophils in peripheral blood in knock out mice that lack IL-33 or the IL-33 receptor (ST2) (18). Moreover, studies of ST2 deficient mice in allergic inflammatory settings revealed that disruption of the IL-33 signaling pathway resulted in impaired eosinophilic airway inflammation and reduced levels of type 2 cytokines upon allergen challenge (26, 27). However, the contribution of IL-33-responsive ILC2s in allergen-induced bone marrow eosinophilia remains to be determined. Thus, in the current study we sought to assess the role of ILC2s in the regulation of allergen- and IL-33-induced bone marrow eosinophilia utilizing wild type (WT) mice, Rag1−/− mice lacking mature T and B cells but retain ILCs, and mice lacking all lymphocytes (Rag2−/−Il2rg−/−).
Methods
Mice
All animal experiments were approved by the Gothenburg County Regional Ethical Committee (permit number 126/14 and 2459/19). WT C57BL/6J and C57BL/6JRj mice were purchased from Charles River (Sulzfeld, Germany) and Janvier Labs (Le Genest-Saint-Isle, France), respectively. Rag1−/− mice and WT C57BL/6 mice, used in some experiments, were obtained from in-house breeding (University of Gothenburg, Sweden). Rag2−/−Il2rg−/− mice were purchased from Taconic (Germantown, NJ, USA). Age and sex-matched mice at 7–12 weeks of age were used in all experiments. Mice were housed in pathogen-free conditions and were given food and water ad libitum.
Allergen-Induced Airway Inflammation
To induce allergic airway inflammation mice were administered 10 μg papain (Sigma-Aldrich) intranasally on days 1–3 and samples were collected 24 hours (h) after final exposure as previously described (28). In some experiments mice received an intraperitoneal single dose of 25 μg anti-mouse IL-5 or isotype control (BD Biosciences, San Jose, CA, USA) 1 h before the first exposure. In the kinetic study, mice received an intranasal single dose of 10 μg papain and samples were collected at 3, 6, 12, 24, and 48 h. Control mice received phosphate buffered saline (PBS) vehicle.
IL-33-Induced Airway Inflammation
IL-33-induced airway inflammation was performed as previously described (25, 29). In brief, WT, Rag1−/−, and Rag2−/−Il2rg−/− mice were exposed to 1 μg recombinant murine IL-33 (rmIL-33, PeproTech, Rocky Hill, NJ, USA) by intranasal administration on day 1, 3, and 5. Samples were collected 24 h after the final exposure. Control mice received PBS vehicle.
Sample Collection
Bronchoalveolar lavage (BAL), blood, and bone marrow were collected in this study. A detailed description of the sample collection is provided in Johansson et al. (29). Cells in BAL were processed for differential cell count analysis. Furthermore, cell-free BAL and serum were processed for mediator analysis by ELISA. Bone marrow cells were isolated from left and right femurs by flushing with wash buffer (2% fetal bovine serum, Sigma-Aldrich, in PBS). The samples were filtered through a 100 μm cell strainer (CellTrics®, Sysmex, Goerlitz, Germany) and red blood cells were lysed (0.1 mM EDTA in distilled water/0.15 M NH4Cl, Sigma-Aldrich/Merck Chemicals) by 10 minutes (min) incubation on ice. The cells were further processed for flow cytometric analysis, ex vivo stimulation, and differential cell count as previously described (25).
Differential Cell Count
Approximately, 10,000–50,000 cells were used for slides (425 × g, 6 min, Shandon Cytospin 3 centrifuge) and stained with Hemacolor® Rapid stain (Merck, Darmstadt, Germany) according to the manufacturer's protocol. Eosinophils were assessed by histological examination as previously described (29).
Ex vivo Stimulation of Bone Marrow Cells
Bone marrow cells from PBS exposed WT mice were seeded at a concentration of 2.5 x 106/ml in complete cell culture medium: RPMI-1640 (HyClone™; GE Healthcare Life Sciences, South Logan, UT, USA), 10% fetal bovine serum (Sigma-Aldrich), 2 mM L-glutamine (HyClone), 100 U/ml penicillin, 100 μg/ml streptomycin (HyClone), 1 mM sodium pyruvate (Sigma-Aldrich). Cells were stimulated with rmIL-33 (100 ng/ml) for 24 h or kept in complete culture medium as control. Monensin (BD GolgiStop™, BD Biosciences) was added to all samples (4 μl/6 ml) during the last 3 h of the incubation. Newly produced IL-5 by ILC2s (SSCloLin−CD45+CD127+ST2+) was measured by intracellular flow cytometry. Bone marrow cells from IL-33 and PBS exposed WT mice, PBS exposed Rag1−/− and Rag2−/−Il2rg−/− mice were cultured as described above (2.5 × 106/ml in complete cell culture medium) with or without rmIL-33 (100 ng/ml) for 44 h and cell-free culture supernatants were collected for measurement of secreted IL-5 by ELISA.
Flow Cytometry
Bone marrow cells were resuspended in 2% mouse serum (Dako, Glostrup, Denmark) and antibodies for surface receptors were added (30 min, 4°C). Cells were washed and fixed (BD CellFix™, BD Biosciences, Erembodegem, Belgium) for 15 min in the dark at room temperature (RT). Cells were washed and analyzed on a BD FACSVerse™ Flow Cytometer running BD FACSuite™ software (BD Biosciences). Collected data were analyzed by FlowJo Software (tree Star Inc, Ashland, OR, USA). Linage negative cells were determined as CD45+CD3−CD45R/B220−CD11b−TER-119−Ly-G6/Gr1−CD11c−CD19−NK-1.1−FceR1−.
Eosinophil progenitors, mature eosinophils, and ILC2s were defined as SSCloCD45+CD34+IL5Rα+, SSChiCD45+CD34−IL5RαloCCR3+Siglec-F+, and SSCloLin−CD45+CD127+CD25+ST2+, respectively. Antibodies used are listed in Table S1. The IL-33 receptor (ST2) expression was estimated by mean fluorescence intensity (MFI) values. Relative MFI (rMFI) equals MFI of monoclonal antibody divided by MFI of corresponding fluorescence minus one (FMO) control.
Intracellular Staining
Cultured bone marrow cells were stained with surface antibodies (as described above) and fixed with 4% Paraformalaldehyde (Sigma-Aldrich) in PBS for 15 min at RT in the dark. All solutions used before fixation were supplemented with Monensin (BD GolgiStop™, 4 μl/6 ml). Cells were permeabilized with 0.1% saponin (Sigma-Aldrich) in Hank's balanced salt solution (HyClone). Anti-IL-5- or isotype control antibodies were added and cells were incubated for 40 min at RT in the dark and washed before flow cytometric analysis.
Cytokine and Chemokine Measurements
Cytokine and chemokine quantification was performed using mouse ELISA Kits (DuoSet®, R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. IL-5 was measured in cell-free bone marrow culture supernatants and serum. CCL24/Eotaxin-2 and IL-33 was measured in cell-free BAL. Absorbance or luminescence was measured on a Varioskan™ LUX multimode microplate reader (ThermoFisher Scientific Vantaa, Finland). Samples below detection limit was set to zero.
Statistical Analysis
The statistical analysis was performed with GraphPad Prism 8 Software (GraphPad Software Inc, La Jolla, CA, USA). Kruskal-Wallis test was used to determine the variance among more than two groups. If significant variance was found, the nonparametric Mann-Whitney U test was used for analysis between two independent groups. Paired Student's t-tests were applied for the analysis of in vitro studies. Statistical significance was defined as *P < 0.05, **P < 0.01, ***P < 0.001 and, ****P < 0.0001.
Results
IL-33-induced Bone Marrow Eosinophilia Develops Normally in the Absence of Adaptive Immune Cells
Investigations of the requirement of ILC2s in IL-33-mediated bone marrow eosinophilopoiesis were carried out using Rag1−/− mice which have an intact innate immune system, but lack mature T and B cells. Naïve WT and Rag1−/− mice were challenged with rmIL-33 (Figure 1A) which resulted in increased eosinophil numbers in both bone marrow (Figures 1B,C) and BAL (Figures 1B,D) compared to control mice that received PBS vehicle (25). Furthermore, IL-33 challenge caused elevated levels of the eosinophil chemokine CCL24/Eotaxin-2 in BAL in both mouse strains where higher levels of CCL24/Eotaxin-2 were observed in Rag1−/− mice compared to WT mice (Figure 1E). Mature eosinophils were significantly increased in the bone marrow of mice exposed to IL-33 (Figure 1F) whereas the relative number of eosinophil progenitors (Figure 1G) remained unchanged. Importantly, WT and Rag1−/− mice responded with a similar increase of eosinophils after IL-33 challenge, suggesting that adaptive immunity is dispensable in both IL-33-driven eosinophil production and recruitment of eosinophils from the bone marrow. Thus, in the current study by using Rag1−/− mice, we confirm our previous findings suggesting that bone marrow ILC2s support eosinophil development in WT mice in IL-33-induced inflammation (25).
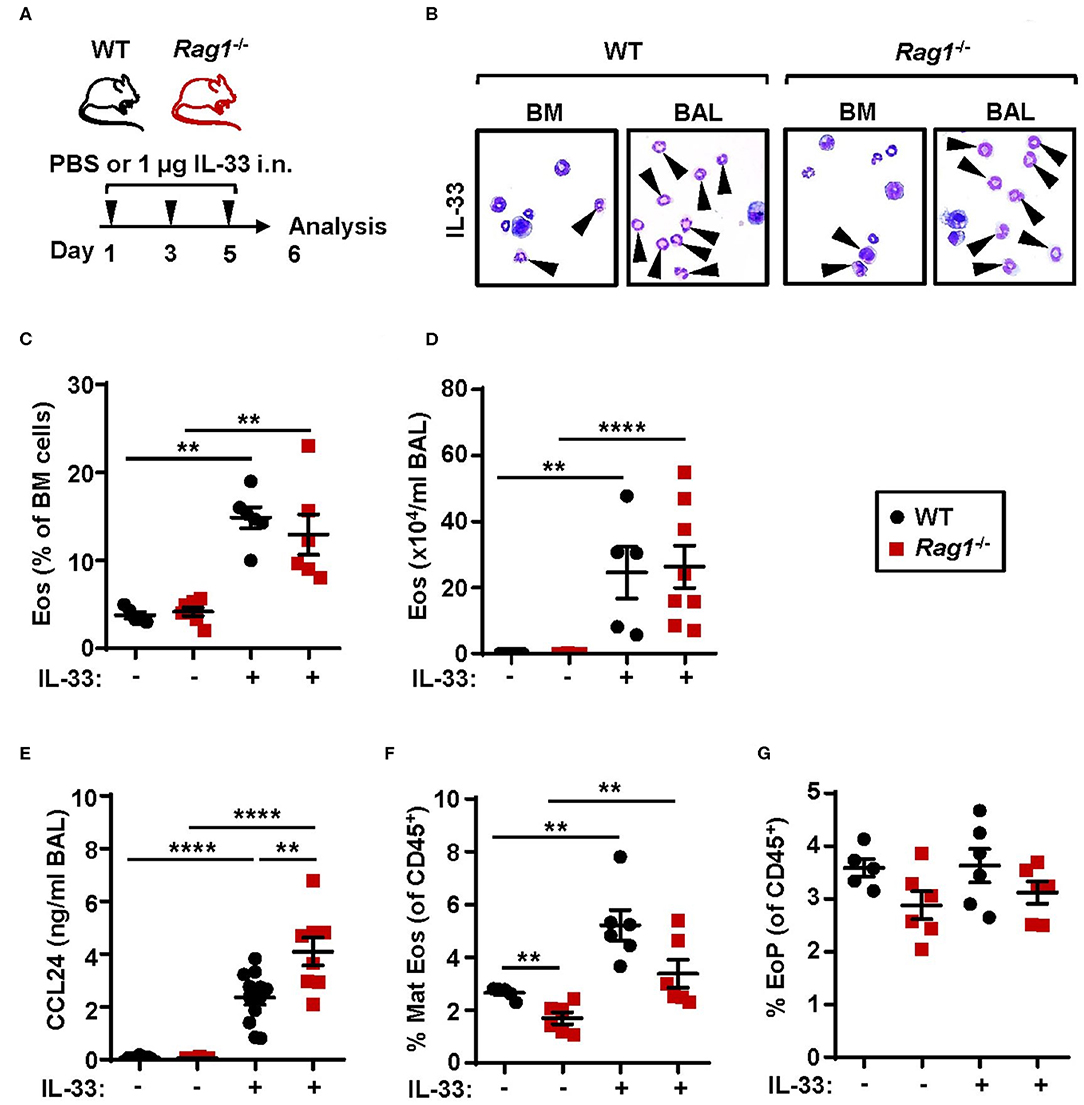
Figure 1. Adaptive immunity is dispensable for IL-33-induced bone marrow eosinophilia. (A) Wild type (WT) and Rag1−/− mice received intranasal (i.n.) challenges of rmIL-33 or PBS vehicle and were sacrificed 24 h after the final challenge. (B) Representative cytospin preparations for quantification of eosinophils (eosin positive cells, indicated by arrows) in bone marrow (BM) and bronchoalveolar lavage (BAL). (C) Number of eosinophils (Eos) in BM and (D) total number of Eos in BAL analyzed by differential cell count. (E) Concentration of CCL24/Eotaxin-2 in BAL measured by ELISA. (F) Number of mature eosinophils (Mat Eos) and (G) eosinophil progenitors (EoPs) among all CD45+ BM leukocytes. Data are representative of two to four independent experiments (n = 5–12/group) and displayed as the mean ± SEM. Mann-Whitney U test. **P < 0.01, and ****P < 0.0001. ns, not significant.
IL-33-Responsive ILC2s Produce Large Amounts of IL-5 in Both Rag1-/- and WT Bone Marrow
The number of bone marrow ILC2s in Rag1−/− mice were significantly increased when compared to WT controls (Figures 2A,B) at baseline. Thus, IL-33 challenge resulted in a significant increase of ILC2s in WT bone marrow only, and not in bone marrow from Rag1−/− mice (Figure 2A). Moreover, ILC2s from both mouse strains responded to IL-33 challenge with increased ST2 expression (Figure 2C). Of note, 100% of bone marrow ILC2s (SSCloLin−CD45+CD127+CD25+) in PBS and IL-33 challenged mice were ST2+, while IL-33 challenge further increased the ST2 receptor expression (Figure 2C). Furthermore, these results were consistent with an approximate 2-fold upregulation of ST2 expression evaluated after IL-33 stimulation of WT and Rag1−/− bone marrow cells ex vivo (Figure 2D). In addition, stimulation with IL-33 in ex vivo cultures generated high levels of IL-5+ ILC2s in both mouse strains (Figures 2E,F), which suggests that bone marrow ILC2s contribute to IL-33-induced eosinophil development in vivo independent of adaptive immunity.
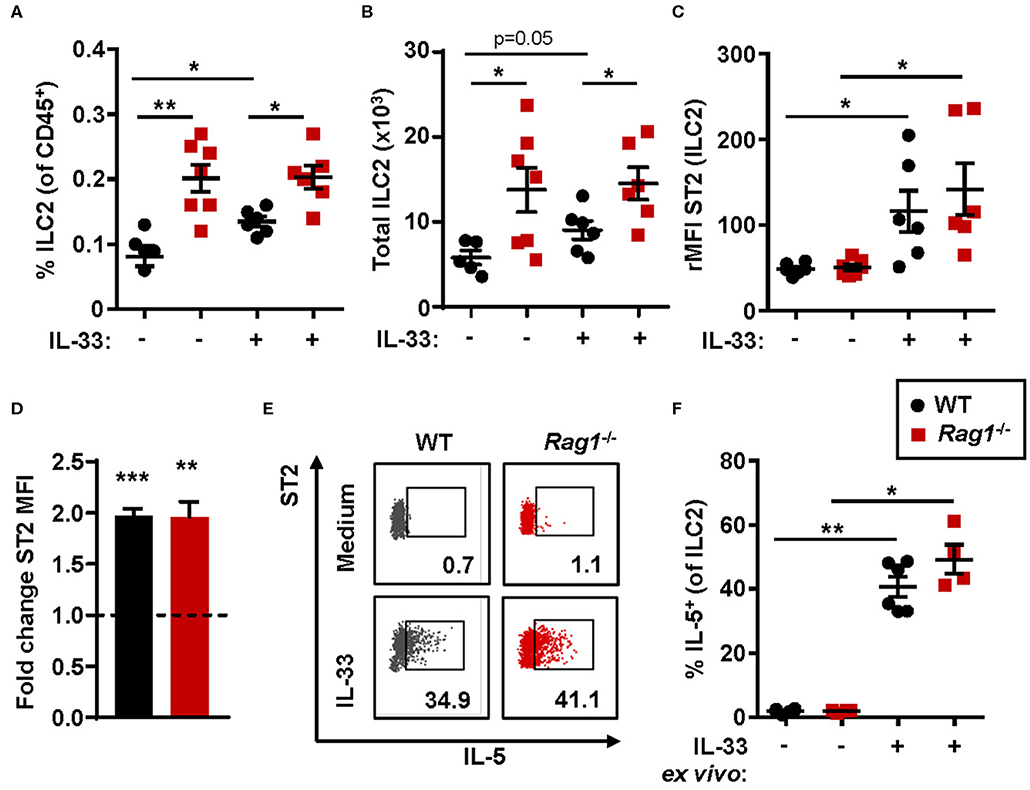
Figure 2. IL-33-responsive ILC2s produce IL-5 in both Rag1−/− and WT bone marrow. (A) Number of type 2 innate lymphoid cells (ILC2s) among all CD45+ leukocytes, and (B) total number of ILC2s in the bone marrow. (C) IL-33 receptor (ST2) expression on ILC2s showed as relative mean fluorescence intensity (rMFI) in the bone marrow of wild type (WT) and Rag1−/− mice exposed to IL-33 or PBS control. (D) Representative dot plots of IL-5+ ILC2s following ex vivo stimulation with IL-33 or unstimulated medium controls (values indicate percent of the parent population). (E) Fold change ST2 MFI (MFI of IL-33 stimulated cells divided by MFI of unstimulated control cells). (F) Number of IL-5+ cells among ILC2s. Data are representative of two to four independent experiments (n = 4–7/group) and displayed as the mean ± SEM. Mann-Whitney U test and paired t-test (D). *P < 0.05, **P < 0.01, and ***P < 0.001. ST2 = IL-33 receptor.
Intranasal Administration of Papain Induces Bone Marrow Eosinophilia in Rag1–/– Mice
To investigate eosinophil development and the downstream effects of IL-33 in allergic inflammation WT and Rag1−/− mice were subjected to a model of allergic inflammation using the protease allergen, papain (Figure 3A). Increased numbers of eosinophils in both bone marrow (Figure 3B) and BAL (Figure 3C) were detected in WT and Rag1−/− mice following papain challenge. Furthermore, both WT and Rag1−/− mice demonstrated elevated levels of CCL24/Eotaxin-2 and IL-33 in BAL following papain challenge (Figures 3D,E) allowing mature eosinophils to migrate toward the airways. A marginally decreased relative number of eosinophil progenitors was observed (Figure 3F and Figure S1A) whereas an approximate 2-fold increase of mature eosinophils was found in papain-challenged WT and Rag1−/− mice compared to saline exposed control mice (Figure 3G and Figure S1B). We have previously reported a higher amount of ST2+ mature bone marrow eosinophils in IL-33 induced inflammation (25). We next investigated the amount of ST2+ mature bone marrow eosinophils during allergic inflammation and whether adaptive immunity is required for the generation of ST2-expressing eosinophils. An increase of ST2+ mature eosinophils was detected in WT and importantly, elevated levels of ST2+ mature eosinophils were also detected in papain challenged Rag1−/− mice (Figure 3H and Figure S1C). Altogether, these results indicate that adaptive immunity is not required for induction of papain-driven eosinophilic inflammation in the bone marrow where IL-33 is an important mediator.
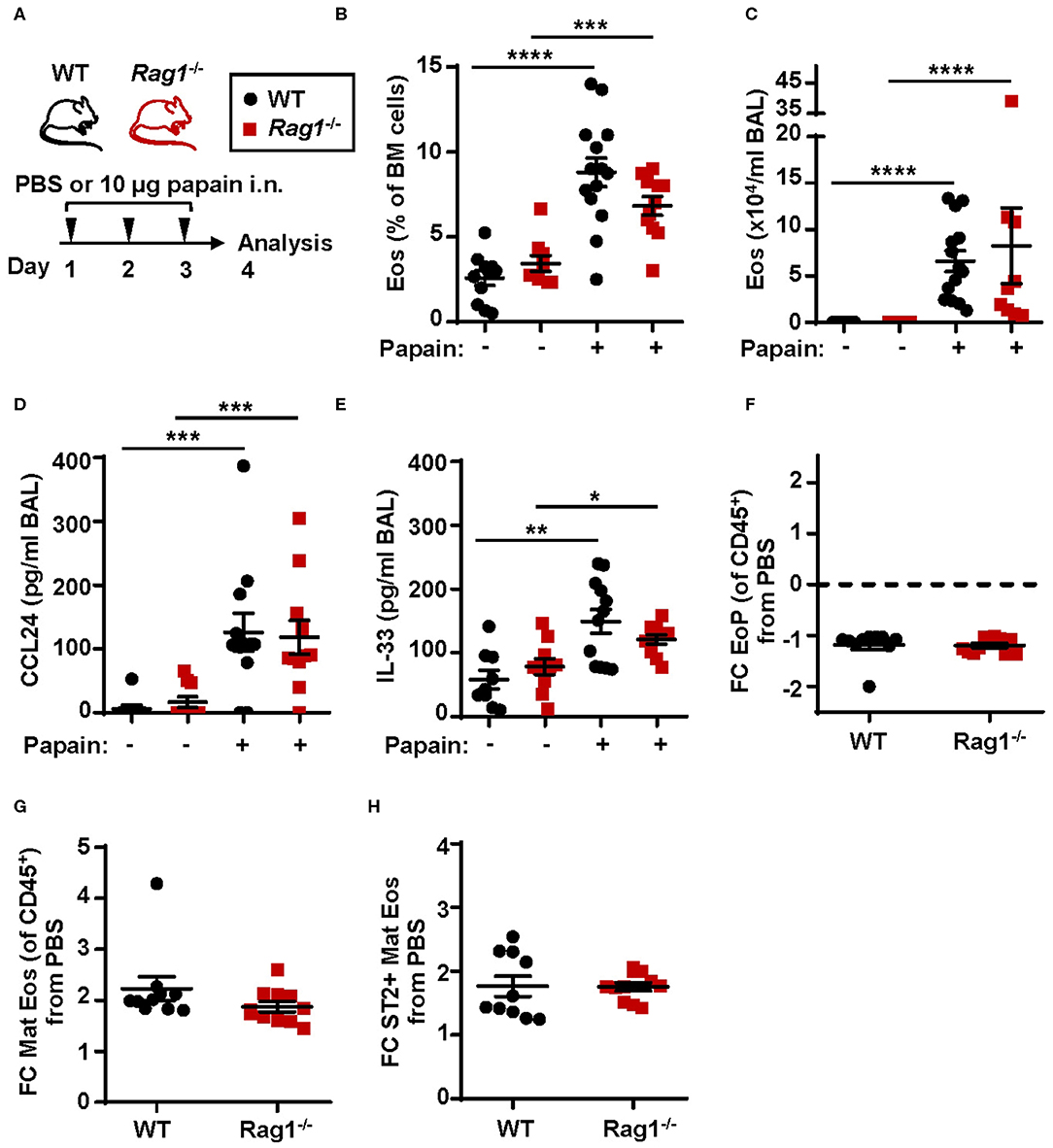
Figure 3. Papain induced IL-33-driven eosinophilic inflammation is similar in Rag1−/− and WT mice. (A) Wild type (WT) and Rag1−/− mice received intranasal (i.n.) challenges of papain or PBS vehicle and were sacrificed 24 h after the final challenge. (B) Number of eosinophils (Eos) in bone marrow (BM) and (C) total number of Eos in BAL analyzed by differential cell count. (D) Concentration of CCL24/Eotaxin-2 and (E) IL-33 in BAL measured by ELISA. (F) Fold change (FC) in numbers of eosinophil progenitors (EoPs), (G) mature eosinophils (Mat Eos) and (H) ST2+ Mat Eos among all CD45+ BM leukocytes in mice exposed to papain compared to PBS control. Data are representative of at least three independent experiments (n = 7–14/group) and displayed as the mean ± SEM. Mann-Whitney U test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ST2 = IL-33 receptor.
Protease Allergen Challenge Induces an Increased ST2 Expression on Bone Marrow ILC2s Which Correlates to Bone Marrow Eosinophilia
An increased relative number of bone marrow ILC2s was observed in WT mice airway challenged with papain compared to PBS exposed control mice whereas Rag1−/− mice had a higher number of bone marrow ILC2s already at baseline (i.e., PBS exposed control) (Figure 4A). Bone marrow ILC2s from both WT and Rag1−/− mice upregulated ST2 expression after papain challenge, which is crucial for the activation of ILC2 effector functions (Figure 4B). Moreover, high ST2 expression on ILC2s demonstrated a positive correlation with both increased bone marrow eosinophilia (Figure 4C) and an increased number of ST2+ mature eosinophils (Figure 4D). Collectively, these data indicate that IL-33-responsive bone marrow ILC2s contribute to allergen-induced bone marrow and airway eosinophilia.
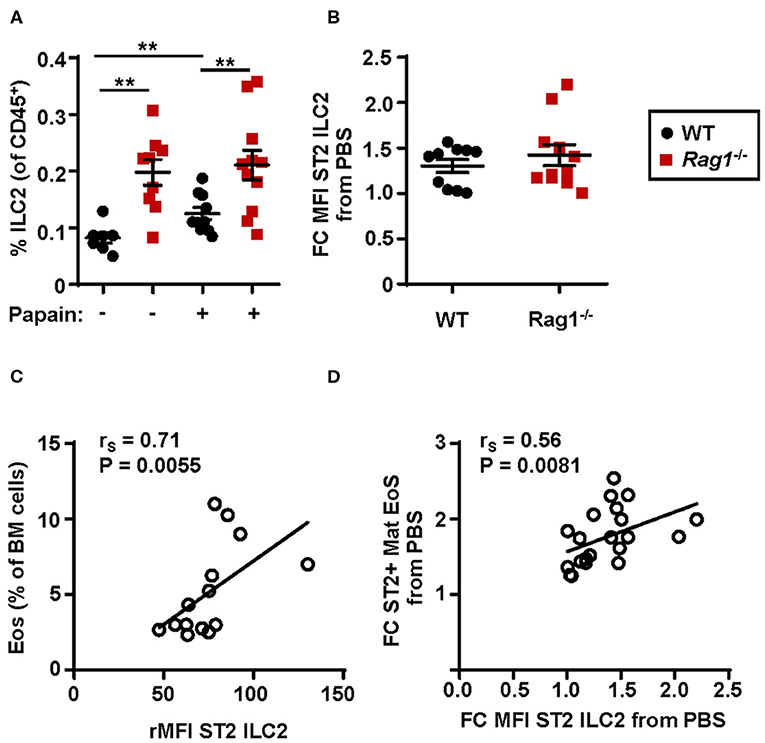
Figure 4. Protease allergen challenge makes bone marrow ILC2s susceptible to IL-33 by upregulation of the ST2 receptor. (A) Number of type 2 innate lymphoid cells (ILC2s) among all CD45+ leukocytes, and (B) fold change MFI (mean fluorescence intensity) IL-33 receptor (ST2) expression on ILC2s in the bone marrow (BM) of wild type (WT) and Rag1−/− mice exposed to papain compared to PBS control. (C) Correlation plot of relative MFI (rMFI) ST2 on ILC2s and the number of BM eosinophils (Eos) in WT and Rag1−/− mice from two representative experiments (D) Correlation plot of ST2+ mature eosinophils and ST2 expression on ILC2s in the BM of papain challenged WT and Rag1−/− mice compared to PBS controls presented as fold change (FC). Data are representative of three independent experiments unless stated otherwise (n = 7–11/group) and shown as the mean ± SEM. Correlation were performed by using Spearman's rho, with rS indicating the Spearman correlation coefficient. Mann-Whitney U test. **P < 0.01. ST2 = IL-33 receptor.
Bone Marrow ILC2s Upregulate ST2 Expression Within 24 h After Airway Papain Challenge
We next performed a time course to identify when bone marrow ILC2s respond to the inhaled protease allergen papain. WT mice were exposed to a single dose of papain intranasally (Figure 5A). Elevated levels of eosinophils in bone marrow were detected at 48 h (Figure 5B) whereas a higher relative number of BAL eosinophils was seen at 24 h (Figure 5C). In line with increased eosinophil levels in the airways there was a significant increase of ST2 expression on ILC2s in the bone marrow (Figure 5D). Additionally, high levels of BAL CCL24/Eotaxin-2 (Figure 5E) and a trend toward elevated IL-33 levels in BAL (Figure 5F) were detected at this time point. At 48 h, the ST2 expression on bone marrow ILC2s was significantly lower and CCL24/Eotaxin-2 and IL-33 levels in BAL showed a trend toward decreased levels. At 72 h the inflammation in the airways was resolved where BAL eosinophil levels were similar to control mice (Figure 5C). In line with these data, the ST2 expression on bone marrow ILC2s was significantly decreased along with lower BAL CCL24/Eotaxin-2 and IL-33 levels. These data suggest that eosinophils are no longer recruited to the airways at 72 h post airway challenge. Collectively, our results suggest that bone marrow ILC2s act rapidly in response to an inhaled allergen by upregulating its ST2 receptor expression in the bone marrow at the onset of eosinophil development.
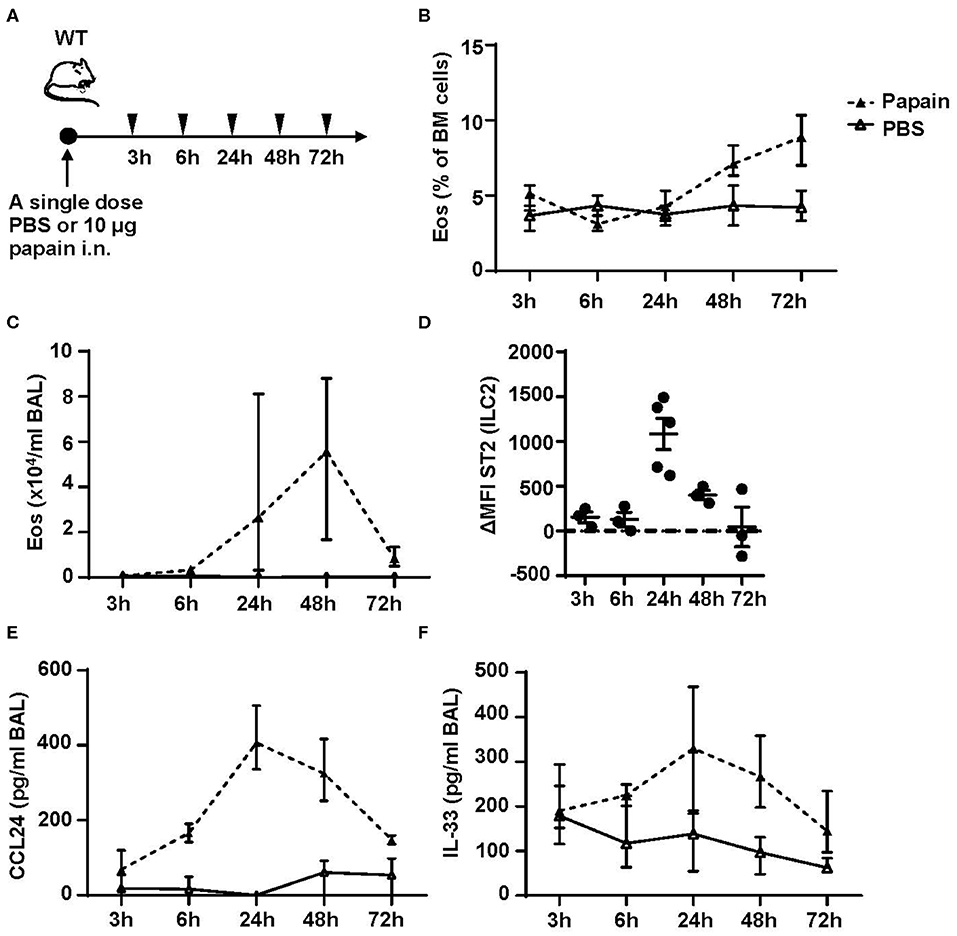
Figure 5. Kinetic profile of bone marrow ILC2s and eosinophilia post protease allergen challenge. (A) Wild type (WT) mice received one dose PBS or papain intranasally (i.n.) and were sacrificed at 3, 6, 24, 48, and 72 h. (B) Number of eosinophils (Eos) in bone marrow (BM) and (C) total number of Eos in BAL analyzed by differential cell count. (D) IL-33 receptor (ST2) expression on ILC2s shown as delta mean fluorescence intensity (ΔMFI) in the BM of wild type (WT) mice challenged with papain compared to PBS vehicle mice where PBS equals zero (dotted line). (E) Concentration of CCL24/Eotaxin-2 and (F) IL-33 in BAL measured by ELISA. Data are representative of one experiment (n = 3–5/group) and displayed as the mean ± SEM (D) or mean with min/max values represented by error bars (B,C,E,F).
Mature Eosinophils are Unable to Develop in Absence of ILC2s in Rag2–/–Il2rg–/– Bone Marrow
To determine whether IL-33-induced eosinophilia can develop in the absence of ILC2s, we challenged naïve lymphocyte deficient Rag2−/−Il2rg−/− mice with rmIL-33 (Figure 6A). No induction of eosinophils was detected by differential cell count analysis of the bone marrow (Figures 6B,C) and BAL (Figures 6B,D) samples from Rag2−/−Il2rg−/− mice. The relative number of eosinophil progenitors was lower in Rag2−/−Il2rg−/− bone marrow compared to WT (Figure 6E). The most dramatic difference was observed in the number of mature eosinophils which was severely impaired in the bone marrow of both IL-33 and PBS challenged Rag2−/−Il2rg−/− mice (Figure 6F). Together, these data suggest that innate lymphocytes are essential for the maintenance of eosinophils under homeostatic conditions as well as for the maturation of eosinophils in response to inflammatory signals.
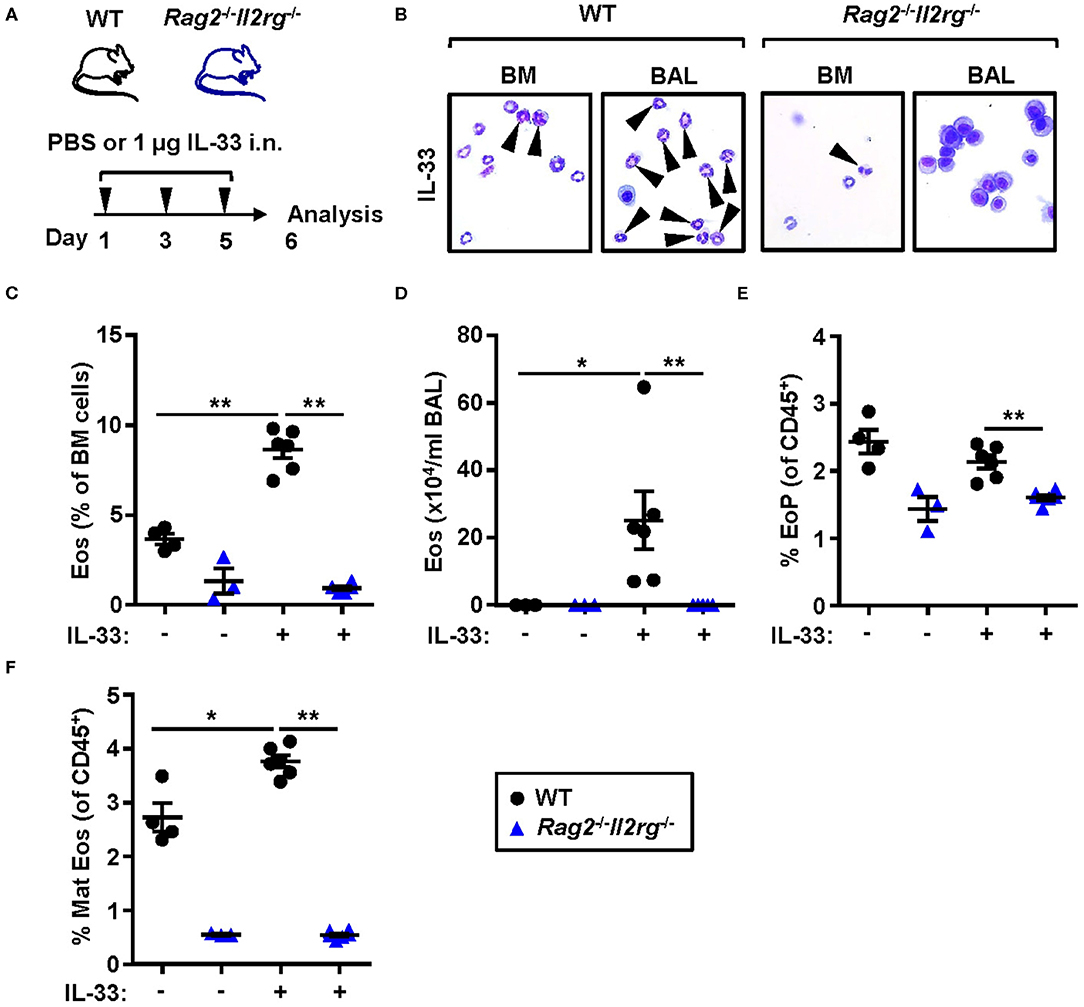
Figure 6. Lymphocyte deficient mice are unable to develop IL-33-induced bone marrow and BAL eosinophilia. (A) Wild type (WT) and Rag2−/−Il2rg−/− mice received intranasal (i.n.) challenges of rmIL-33 or PBS vehicle and were sacrificed 24 h after the final challenge. (B) Representative cytospin preparations for quantification of eosinophils (eosin positive cells, indicated by arrows) in bone marrow (BM) and bronchoalveolar lavage (BAL). (C) Number of eosinophils (Eos) in BM and (D) total number of Eos in BAL analyzed by differential cell count. (E) Number of eosinophil progenitors (EoPs) and (F) mature eosinophils (Mat Eos) among all CD45+ BM leukocytes. Data are representative of one experiment (n = 3–6/group) and displayed as the mean ± SEM. Mann-Whitney U test. *P < 0.05 and **P < 0.01.
Absence of the Adaptive Immune System Results in Higher Levels of IL-5 in IL-33-Induced Inflammation
Both systemic IL-5 and local IL-5 production have been suggested to regulate eosinophil development and maturation in the bone marrow (11, 21, 25). Next, we analyzed the concentration of IL-5 in serum from WT, Rag1−/− and Rag2−/−Il2rg−/− mice. IL-33 challenge significantly increased levels of IL-5 in serum of WT and Rag1−/− mice but not in serum of Rag2−/−Il2rg−/− mice (Figure 7A). Notably, the level of serum IL-5 was higher in IL-33 challenged Rag1−/− mice compared to WT mice (Figure 7A), which might be associated with a greater number of ILC2s in the bone marrow of Rag1−/− mice. Furthermore, bone marrow cultures from WT and Rag1−/− mice stimulated with IL-33 accumulated IL-5 in the culture media, but no IL-5 was detected in cultures from Rag2−/−Il2rg−/− mice (Figure 7B). Interestingly, we found that bone marrow cultures generated from in vivo IL-33 challenged mice produced significantly more IL-5 upon IL-33 re-stimulation ex vivo (Figure 7C). This result suggests that in IL-33 challenged mice presenting an increased expression of ST2 on bone marrow ILC2s makes the cells more susceptible to IL-33, thus, potentially leading to a higher production of IL-5, which in turn drives eosinophil development.
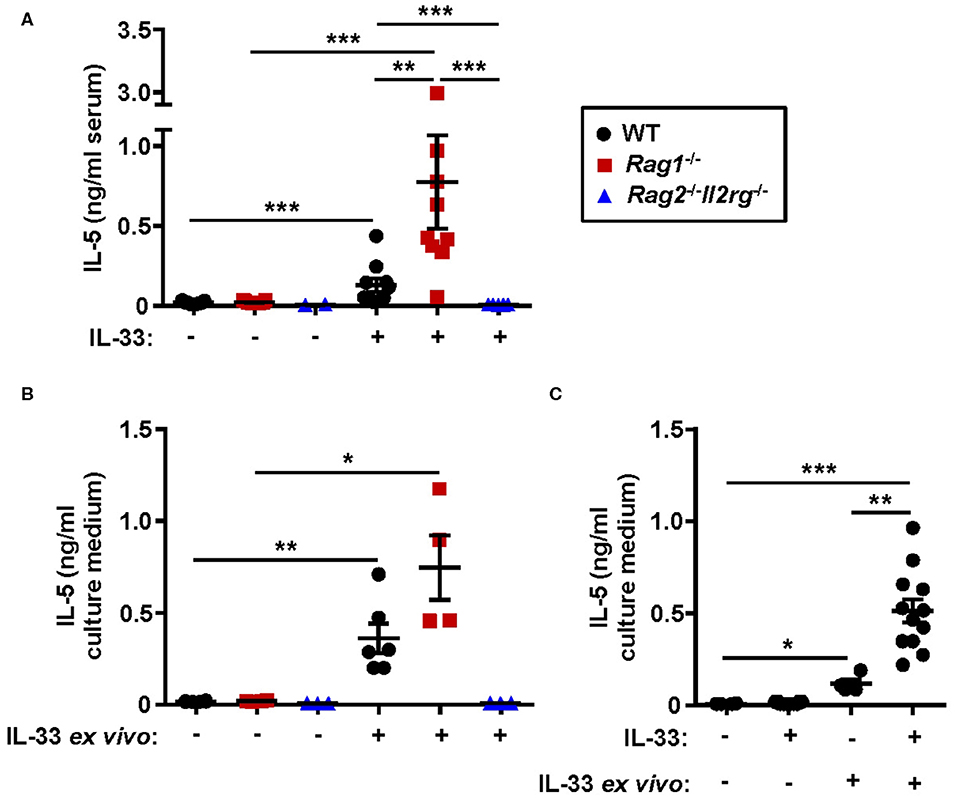
Figure 7. Absence of IL-5 in IL-33 challenged lymphocyte deficient mice. (A) Concentration of IL-5 in serum of wild type (WT), Rag1−/− and Rag2−/−Il2rg−/− mice exposed to IL-33 or PBS. (B) IL-5 in culture medium of bone marrow (BM) cells stimulated with rmIL-33 (100 ng/ml) for 44 h or unstimulated medium controls. (C) IL-5 in the culture medium of BM cells from IL-33 challenged or PBS controls (in vivo) followed by restimulation with rmIL-33 (100 ng/ml) ex vivo for 44 h or unstimulated medium controls. Data are representative of four independent experiments (n = 2–12/group) and shown as the mean ± SEM. Mann-Whitney U test and paired t-test (B). *P < 0.05, **P < 0.01, and ***P < 0.001.
IL-5 is Required for Allergen-Induced Eosinophilic Inflammation Despite the Absence of an Adaptive Immune System
To assess whether IL-5 is required in the development and maturation of bone marrow eosinophilia in Rag1−/− and WT mice, mice were pre-treated with anti-IL-5 antibodies 1 h before the dose regimen of intranasal papain exposures (Figure 8A). Anti-IL-5 treated papain challenged Rag1−/− and WT mice were unable to induce eosinophilia in the bone marrow (Figures 8B,D) and BAL (Figure 8C). Moreover, a decreased number of ST2+ mature eosinophils was seen in both mouse strains treated with anti-IL-5 (Figure 8E). Interestingly, a significant decrease of the ST2 expression on mature eosinophils was observed in both Rag1−/− and WT mice (Figure 8F). These findings suggest link between IL-5 levels and ST2 expression on eosinophils in the bone marrow. The relative number of eosinophil progenitors (data not shown) and ILC2s were not affected by anti-IL-5 treatment (Figure S2). Collectively, these data show that papain induced eosinophilic inflammation is dependent on IL-5 and that IL-5 levels might regulate ST2 expression on bone marrow eosinophils which in turn drives the maturation of eosinophil within the bone marrow. In addition, the absence of IL-5-producing T cells in Rag1−/− mice make bone marrow ILC2s a potential local source of IL-5.
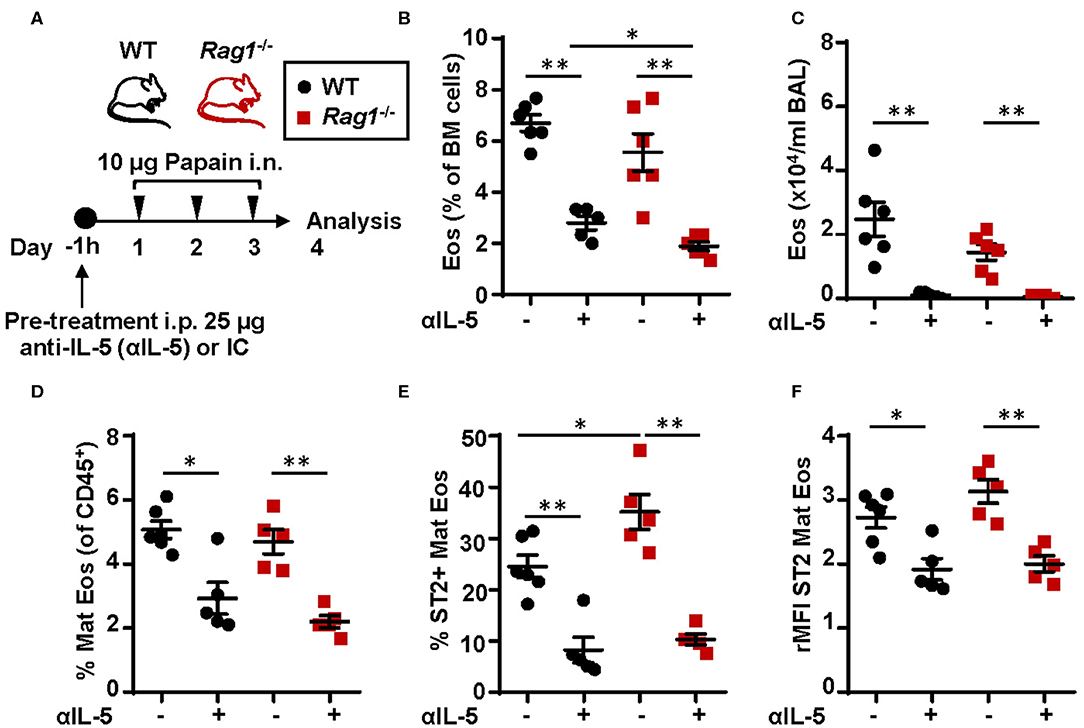
Figure 8. Neutralization of IL-5 disrupts ST2 expression on bone marrow eosinophils and abolish protease allergen-induced eosinophilic inflammation (A) Wild type (WT) and Rag1−/− mice were pre-treated with anti-IL-5 (αIL-5) or isotype control (IC), received intranasal (i.n.) challenges of papain and sacrificed 24 h after the final challenge. (B) Number of eosinophils (Eos) in bone marrow (BM) and (C) total number of Eos in BAL analyzed by differential cell count. (D) Number of mature eosinophils (Mat Eos) among all CD45+ BM leukocytes. (E) Number of ST2+ Mat Eos in BM. (F) ST2 expression on BM Mat Eos shown as relative mean fluorescence intensity (rMFI). Data are representative of three independent experiments (n = 4–6/group) and displayed as the mean ± SEM. Mann-Whitney U test. *P < 0.05, **P < 0.01. IC is isotype control. ST2 = IL-33 receptor.
Discussion
A growing body of evidence links IL-33 to initiating events that occur during allergen exposure. Important findings include the identification of IL-33 and ST2 as major susceptibility loci in several genome-wide association studies of allergic diseases (30–35). In addition, a loss-of-function mutation in the Il33 gene has been reported to be associated with lower number of blood eosinophils and a reduced risk of developing asthma (36). Moreover, vaccination against IL-33 reduced airway eosinophilia and airway hyperresponsiveness in a mouse model of HDM-induced asthma, propose IL-33 as a promising target for asthma intervention (37). A major finding in our study is that ST2+ ILC2s in the bone marrow is activated upon one intranasal dose of the protease allergen papain within 24 h. This was followed by an increased number of eosinophils in the bone marrow and airways. Many studies of the ILC2 potent alarmin IL-33 in allergic diseases have focused on barrier tissues such as the lung, but our findings suggest that IL-33 has additional roles in the bone marrow. Analysis of ILC2s in naïve mice show that bone marrow-derived ILC2s exhibit a 6-fold higher ST2 expression compared to lung-derived ILC2s (29). It is possible that the expression level of ST2 is related to differential stages of ILC2 development in the bone marrow (i.e., ILC2 precursors) (38) vs. the lung, but more importantly, it indicates that IL-33 signaling plays a role in both compartments. Constantly exposed to the inhaled environment, ILC2s in the airway mucosa are indeed located in a prime position to react to IL-33. The bone marrow on the other hand is privileged in this respect and the high ST2 expression may suggest that bone marrow ILC2s are more sensitive to IL-33, thus even low levels of IL-33 might be enough to activate the cells at a distance.
Several immune cells express ST2 including eosinophils and can thereby produce proinflammatory mediators upon activation (39). We show that both the number of mature eosinophils in the bone marrow and the ST2 expression on these cells increases upon protease allergen challenge (Figures 3G,H). These results are in line with a recent study demonstrating an increased number of ST2-expressing sputum and blood eosinophils post 24 h allergen challenge in allergic asthmatics (40). Furthermore, our study show that the ST2 expression was dependent on IL-5 demonstrated by the use of anti-IL-5 treatment (Figures 8E,F) suggesting a link between IL-5 and ST2 expression on eosinophils. Prospective studies have to be carried out in order to determine whether IL-5 directly or indirectly regulate the ST2 expression on eosinophils. We further demonstrate that ST2+ mature eosinophils positively correlate with an increased ST2 receptor expression on ILC2s, suggesting both cell types being IL-33-responsive simultaneously in allergen-induced eosinophilic inflammation. Previous studies have investigated IL-33-responsive blood eosinophils in atopic subjects in terms of effects on adhesion, degranulation and chemotaxis (39, 41). Eosinophils were more adherent and potent in degranulation upon IL-33 stimulation whereas no effects was observed on chemotaxis (39, 41). Moreover, the IL-33/ST2/IL-5 axis was recently shown to influence eosinophilopoiesis where ST2 deficient mice challenged with the fungal Alternaria exhibited lower levels of IL-5 in serum and a decreased relative number of eosinophils in the bone marrow (11). In addition, it has been shown that the increased number of IL-5+ blood eosinophils from allergic asthmatics observed after IL-33 stimulation was attenuated by treatment with either anti-ST2 monoclonal antibody, soluble ST2 or the combination of the two (40). An emerging interest in targeting IL-33 and its receptor ST2 with pharmacological treatments in individuals with respiratory diseases including asthma can be seen with examples such as SAR440340 (ClinicalTrials.gov identifier, NCT03546907) from Regeneron Pharmaceuticals and Sanofi, MSTT1041A (ClinicalTrials.gov Identifier: NCT02918019) from Hoffmann-La Roche, GSK3772847 (ClinicalTrials.gov Identifier: NCT03207243) from GlaxoSmithKline and etokimab (ClinicalTrials.gov Identifier: NCT03469934) from AnaptysBio. Moreover, the anti-IL-33 or anti-ST2 strategy may hold a potential clinical future outside the field of respiratory diseases with allergy or atopic dermatitis as examples.
Interestingly, we observed an increased relative number of bone marrow ILC2s and an upregulation of ST2 expression on these cells after intranasal challenges with the allergen protease papain. These findings were also observed in IL-33 challenged mice which are in line with previous reports (42–44). In contrast, Stier et al. recently reported a decreased number of progenitor ILC2s in the bone marrow upon acute exposure to the fungal allergen Alternaria (45). This process was indeed dependent on IL-33 signaling as both ST2 and IL-33 deficient mice demonstrated lower numbers of ILC2s in peripheral tissues while progenitor ILC2s accumulated in the bone marrow (45). Furthermore, we have unpublished data revealing an increased ST2 expression on bone marrow ILC2s in HDM-sensitized mice, thus strengthen a role for bone marrow ILC2s at an early stage of eosinophilic inflammation caused by a variety of allergens. In line with our results, Brickshawana et al. showed that IL-33 stimulation increased ST2 expression and IL-5 production in an early study of bone marrow ILC2s (42). However, the functional implication of IL-33-responsive ILC2s in the development of eosinophilia was not addressed in their study.
Importantly, we identified IL-5+ bone marrow ILC2s in IL-33 induced inflammation and we demonstrate by neutralizing IL-5 in vivo, that IL-5 is essential in eosinophilic inflammation in the bone marrow in response to the protease allergen papain. We further demonstrate that bone marrow ILC2s are able to respond within 24 h by increased ST2 expression to an intranasal single dose of papain. At 48 h, the ST2 expression on ILC2s was starting to decrease and simultaneously eosinophils were increased in the bone marrow. In addition, a similar increased relative number of mature bone marrow eosinophils was found after papain challenge in Rag1−/− mice lacking adaptive immune cells, but with an intact innate immune system including functional ILC2s. Therefore, bone marrow ILC2s, in addition to airway ILC2s, are a possible early source of IL-5 in allergic diseases including asthma (46). Likewise, restimulation with IL-33 ex vivo revealed that bone marrow cultures from IL-33 exposed mice produced larger amounts of IL-5 compared to bone marrow cultures from PBS control mice (Figure 7C). Indeed, it has previously been discovered that allergen-experienced airway ILC2s responded more vigorously to a second challenge compared to naïve ILC2s (47) and that ILC2s previously exposed to papain or rmIL-33 displayed higher responsiveness even to unrelated allergens (47). Innate immune memory is an emerging field with important implications in ILC2 biology that might extend to ILC2 functions in the bone marrow, with potential consequences such as increased eosinophil hematopoiesis in allergic subjects (4, 7, 9, 48).
In our continued analysis of the requirement of ILC2s in IL-33-driven eosinophilic inflammation, we demonstrate for the first time that Rag1−/− mice display normal eosinophil development in the bone marrow in both direct IL-33- and in protease allergen-induced eosinophilic inflammation. This result eliminates a contribution from the adaptive immune system during acute airway inflammation and adds a functional effect of ILC2s in the bone marrow. Interestingly, we observed a significant increased relative number of ILC2s in the bone marrow of Rag1−/− mice compared to WT mice. In addition, IL-33 challenged Rag1−/− mice also demonstrated higher levels of serum IL-5 and CCL24/Eotaxin-2 in BAL compared to WT. These excessive responses might reflect a lack of inhibitory signals that are mediated by adaptive immune cells and should be further assessed in prospective studies. For instance, ST2+ T regulatory cells have been described to be superior to ST2− T regulatory cells in suppressing type 2 inflammation (49). Further, Rag1−/− mice also exhibited a wide range of bone marrow ILC2s at both baseline and upon airway challenge. These data might indicate heterogeneity in the ILC2 population.
A striking result in our study was that Rag2−/−Il2rg−/− mice displayed a reduced number of bone marrow eosinophils at baseline and were unable to develop bone marrow eosinophilia in response to IL-33 challenge. These findings clearly show that innate lymphocytes are essential for the development of eosinophils and we found it interesting that the difference was more apparent in the number of mature eosinophils compared to eosinophil progenitors, which might reflect the critical role of IL-5 for terminal differentiation and maturation of eosinophils. Indeed, no IL-5 production was detected in bone marrow cultures from Rag2−/−Il2rg−/− mice and the level of serum IL-5 was below detection limit. Moreover, we show that Rag2−/−Il2rg−/− mice are unable to develop airway eosinophilia, which is consistent with a previous study where airway eosinophil numbers were restored upon adoptive transfer of ILC2s to Rag2−/−Il2rg−/− mice (28).
We conclude that IL-33 plays important roles for the initiation of bone marrow eosinophilia where IL-33-responsive bone marrow ILC2s contribute to allergen-induced IL-5-dependent eosinophilic inflammation. Identification of mechanisms that regulate eosinophilic inflammation is critical for developing new therapies of uncontrolled eosinophilic asthma.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Gothenburg County Regional Ethical Committee Kammarrätten i Göteborg Box 1531 401 50 Gothenburg Sweden.
Author Contributions
EB, KJ, and MR designed experiments and wrote the manuscript. EB, KJ, JW, JC, MR, and CM performed the experiments and approved the final manuscript. EB and KJ analyzed data. JW, JC, and CM provided comments to the manuscript.
Funding
This work was supported by grants from the Swedish Research Council (2017-02302), the Swedish Heart and Lung Foundation (20170392) and the VBG Group Herman Krefting Foundation for Asthma and Allergy research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Karin Schön and Nils Lycke, Department of Microbiology and Immunology at University of Gothenburg for providing us with C57BL/6 and Rag1−/− mice used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01058/full#supplementary-material
Figure S1. (A) number of eosinophil progenitors (EoPs) and (B) mature eosinophils (Mat Eos) among all CD45+ BM leukocytes. (C) Number of ST2+ Mat Eos in BM. Data are representative of three independent experiments (n = 7–11/group) and displayed as the mean ± SEM. Mann-Whitney U test. *P < 0.05, ***P < 0.001, and ****P < 0.0001. ST2 = IL-33 receptor.
Figure S2. Number of type 2 innate lymphoid cells (ILC2s) among all CD45+ leukocytes in wild type (WT) and Rag1−/− mice pre-treated with anti-IL-5 (αIL-5) or isotype control (IC) 1 h prior to intranasal (i.n.) challenges of papain (Figure 8A).
Table S1. Antibodies used in flow cytometry.
Abbreviations
BAL, Bronchoalveolar lavage; ILC2, type 2 innate lymphoid cell; IL, Interleukin; WT, Wild type.
References
1. Pegorier S, Wagner LA, Gleich GJ, Pretolani M. Eosinophil-derived cationic proteins activate the synthesis of remodeling factors by airway epithelial cells. J Immunol. (2006) 177:4861–9. doi: 10.4049/jimmunol.177.7.4861
2. Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem. (2014) 289:17406–15. doi: 10.1074/jbc.R113.546218
3. Woolley MJ, Denburg JA, Ellis R, Dahlback M, O'Byrne PM. Allergen-induced changes in bone marrow progenitors and airway responsiveness in dogs and the effect of inhaled budesonide on these parameters. Am J Respir Cell Mol Biol. (1994) 11:600–6. doi: 10.1165/ajrcmb.11.5.7946389
4. Sehmi R, Howie K, Sutherland DR, Schragge W, O'Byrne PM, Denburg JA. Increased levels of CD34+ hemopoietic progenitor cells in atopic subjects. Am J Respir Cell Mol Biol. (1996) 15:645–55. doi: 10.1165/ajrcmb.15.5.8918371
5. Gaspar Elsas MI, Joseph D, Elsas PX, Vargaftig BB. Rapid increase in bone-marrow eosinophil production and responses to eosinopoietic interleukins triggered by intranasal allergen challenge. Am J Respir Cell Mol Biol. (1997) 17:404–13. doi: 10.1165/ajrcmb.17.4.2691
6. Ohkawara Y, Lei XF, Stampfli MR, Marshall JS, Xing Z, Jordana M. Cytokine and eosinophil responses in the lung, peripheral blood, and bone marrow compartments in a murine model of allergen-induced airways inflammation. Am J Respir Cell Mol Biol. (1997) 16:510–20. doi: 10.1165/ajrcmb.16.5.9160833
7. Sehmi R, Wood LJ, Watson R, Foley R, Hamid Q, O'Byrne PM, et al. Allergen-induced increases in IL-5 receptor alpha-subunit expression on bone marrow-derived CD34+ cells from asthmatic subjects. A novel marker of progenitor cell commitment towards eosinophilic differentiation. J Clin Invest. (1997) 100:2466–75. doi: 10.1172/JCI119789
8. Wood LJ, Inman MD, Denburg JA, O'Byrne PM. Allergen challenge increases cell traffic between bone marrow and lung. Am J Respir Cell Mol Biol. (1998) 18:759–67. doi: 10.1165/ajrcmb.18.6.3006
9. Wood LJ, Inman MD, Watson RM, Foley R, Denburg JA, O'Byrne PM. Changes in bone marrow inflammatory cell progenitors after inhaled allergen in asthmatic subjects. Am J Respir Crit Care Med. (1998) 157:99–105. doi: 10.1164/ajrccm.157.1.9704125
10. Tomaki M, Zhao LL, Lundahl J, Sjostrand M, Jordana M, Linden A, et al. Eosinophilopoiesis in a murine model of allergic airway eosinophilia: involvement of bone marrow IL-5 and IL-5 receptor alpha. J Immunol. (2000) 165:4040–50. doi: 10.4049/jimmunol.165.7.4040
11. Anderson EL, Kobayashi T, Iijima K, Bartemes KR, Chen CC, Kita H. IL-33 mediates reactive eosinophilopoiesis in response to airborne allergen exposure. Allergy. (2016) 71:977–88. doi: 10.1111/all.12861
12. Lemiere C, Ernst P, Olivenstein R, Yamauchi Y, Govindaraju K, Ludwig MS, et al. Airway inflammation assessed by invasive and noninvasive means in severe asthma: eosinophilic and noneosinophilic phenotypes. J Allergy Clin Immunol. (2006) 118:1033–9. doi: 10.1016/j.jaci.2006.08.003
13. Ema H, Suda T, Nagayoshi K, Miura Y, Civin CI, Nakauchi H. Target cells for granulocyte colony-stimulating factor, interleukin-3, and interleukin-5 in differentiation pathways of neutrophils and eosinophils. Blood. (1990) 76:1956–61. doi: 10.1182/blood.V76.10.1956.1956
14. Weller PF. The immunobiology of eosinophils. New Engl J Med. (1991) 324:1110–8. doi: 10.1056/NEJM199104183241607
15. Sanderson CJ. Interleukin-5, eosinophils, and disease. Blood. (1992) 79:3101–9. doi: 10.1182/blood.V79.12.3101.bloodjournal79123101
16. Humbles AA, Conroy DM, Marleau S, Rankin SM, Palframan RT, Proudfoot AE, et al. Kinetics of eotaxin generation and its relationship to eosinophil accumulation in allergic airways disease: analysis in a guinea pig model in vivo. J Exp Med. (1997) 186:601–12. doi: 10.1084/jem.186.4.601
17. Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. (2006) 24:147–74. doi: 10.1146/annurev.immunol.24.021605.090720
18. Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol. (2016) 197:3445–53. doi: 10.4049/jimmunol.1600611
19. Kortekaas Krohn I, Shikhagaie MM, Golebski K, Bernink JH, Breynaert C, Creyns B, et al. Emerging roles of innate lymphoid cells in inflammatory diseases: clinical implications. Allergy. (2018) 73:837–50. doi: 10.1111/all.13340
20. Kato A. Group 2 innate lymphoid cells in airway diseases. Chest. (2019) 156:141–9. doi: 10.1016/j.chest.2019.04.101
21. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. (2013) 502:245–8. doi: 10.1038/nature12526
22. Minshall EM, Schleimer R, Cameron L, Minnicozzi M, Egan RW, Gutierrez-Ramos JC, et al. Interleukin-5 expression in the bone marrow of sensitized Balb/c mice after allergen challenge. Am J Respir Crit Care Med. (1998) 158:951–7. doi: 10.1164/ajrccm.158.3.9709114
23. Wood LJ, Sehmi R, Dorman S, Hamid Q, Tulic MK, Watson RM, et al. Allergen-induced increases in bone marrow T lymphocytes and interleukin-5 expression in subjects with asthma. Am J Respir Crit Care Med. (2002) 166:883–9. doi: 10.1164/rccm.2108015
24. Johansson AK, Sergejeva S, Sjostrand M, Lee JJ, Lotvall J. Allergen-induced traffic of bone marrow eosinophils, neutrophils and lymphocytes to airways. Eur J Immunol. (2004) 34:3135–45. doi: 10.1002/eji.200425043
25. Johansson K, Malmhall C, Ramos-Ramirez P, Radinger M. Bone marrow type 2 innate lymphoid cells: a local source of interleukin-5 in interleukin-33-driven eosinophilia. Immunology. (2018) 153:268–78. doi: 10.1111/imm.12842
26. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. (2008) 181:4780–90. doi: 10.4049/jimmunol.181.7.4780
27. Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA. (2010) 107:18581–6. doi: 10.1073/pnas.1003059107
28. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
29. Johansson K, Malmhall C, Ramos-Ramirez P, Radinger M. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J Allergy Clin Immunol. (2017) 139:1007–16.e1009. doi: 10.1016/j.jaci.2016.06.035
30. Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. (2009) 41:342–7. doi: 10.1038/ng.323
31. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
32. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. (2011) 43:887–92. doi: 10.1038/ng.888
33. Ramasamy A, Kuokkanen M, Vedantam S, Gajdos ZK, Couto Alves A, Lyon HN, et al. Genome-wide association studies of asthma in population-based cohorts confirm known and suggested loci and identify an additional association near HLA. PLoS ONE. (2012) 7:e44008. doi: 10.1371/journal.pone.0044008
34. Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. (2014) 46:51–5. doi: 10.1038/ng.2830
35. Johansson A, Rask-Andersen M, Karlsson T, Ek WE. Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum Mol Genet. (2019) 28:4022–41. doi: 10.1093/hmg/ddz175
36. Smith D, Helgason H, Sulem P, Bjornsdottir US, Lim AC, Sveinbjornsson G, et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLoS Genet. (2017) 13:e1006659. doi: 10.1371/journal.pgen.1006659
37. Lei Y, Boinapally V, Zoltowska A, Adner M, Hellman L, Nilsson G. Vaccination against IL-33 inhibits airway hyperresponsiveness and inflammation in a house dust mite model of asthma. PLoS ONE. (2015) 10:e0133774. doi: 10.1371/journal.pone.0133774
38. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
39. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. (2008) 121:1484–90. doi: 10.1016/j.jaci.2008.04.005
40. Mitchell PD, Salter BM, Oliveria JP, El-Gammal A, Tworek D, Smith SG, et al. IL-33 and its receptor ST2 after inhaled allergen challenge in allergic asthmatics. Int Arch Allergy Immunol. (2018) 176:133–42. doi: 10.1159/000488015
41. Angulo EL, McKernan EM, Fichtinger PS, Mathur SK. Comparison of IL-33 and IL-5 family mediated activation of human eosinophils. PLoS ONE. (2019) 14:e0217807. doi: 10.1371/journal.pone.0217807
42. Brickshawana A, Shapiro VS, Kita H, Pease LR. Lineage(-)Sca1+c-Kit(-)CD25+ cells are IL-33-responsive type 2 innate cells in the mouse bone marrow. J Immunol. (2011) 187:5795–804. doi: 10.4049/jimmunol.1102242
43. Lei AH, Xiao Q, Liu GY, Shi K, Yang Q, Li X, et al. ICAM-1 controls development and function of ILC2. J Exp Med. (2018) 215:2157–74. doi: 10.1084/jem.20172359
44. Li Q, Li D, Zhang X, Wan Q, Zhang W, Zheng M, et al. E3 ligase VHL promotes group 2 innate lymphoid cell maturation and function via glycolysis inhibition and induction of interleukin-33 receptor. Immunity. (2018) 48:258–70.e255. doi: 10.1016/j.immuni.2017.12.013
45. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med. (2018) 215:263–81. doi: 10.1084/jem.20170449
46. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. (2016) 137:75–86.e78. doi: 10.1016/j.jaci.2015.05.037
47. Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. (2016) 45:198–208. doi: 10.1016/j.immuni.2016.06.017
48. Salter BM, Sehmi R. Hematopoietic processes in eosinophilic asthma. Chest. (2017) 152:410–6. doi: 10.1016/j.chest.2017.01.021
Keywords: allergy, papain, bone marrow, eosinophilia, interleukin-33, interleukin-5, ILC2, asthma
Citation: Boberg E, Johansson K, Malmhäll C, Calvén J, Weidner J and Rådinger M (2020) Interplay Between the IL-33/ST2 Axis and Bone Marrow ILC2s in Protease Allergen-Induced IL-5-Dependent Eosinophilia. Front. Immunol. 11:1058. doi: 10.3389/fimmu.2020.01058
Received: 20 December 2019; Accepted: 01 May 2020;
Published: 02 June 2020.
Edited by:
Jean Sylvia Marshall, Dalhousie University, CanadaReviewed by:
Jorg Hermann Fritz, McGill University, CanadaManali Mukherjee, McMaster University, Canada
Copyright © 2020 Boberg, Johansson, Malmhäll, Calvén, Weidner and Rådinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madeleine Rådinger, bWFkZWxlaW5lLnJhZGluZ2VyQC5ndS5zZQ==
†These authors have contributed equally to this work